

Abstract
An 82-year-old woman developed neck weakness and dysarthria with antibodies against acetylcholine receptor (AChR) and low-density lipoprotein receptor-related protein 4 (LRP4). Myasthenia gravis (MG) was diagnosed by edrophonium and repetitive nerve stimulation tests. Her symptoms resolved completely by immunotherapy. One year later, she presented with muscle weakness and bulbar palsy accompanied by atrophy and fasciculation. Her tendon reflexes were brisk, and Babinski’s sign was positive. She was diagnosed with probable amyotrophic lateral sclerosis (ALS). Immunotherapy did not improve her symptoms, and she ultimately died of respiratory failure. MG and ALS may share a pathophysiology, including anti-LRP4 antibodies at the neuromuscular junction.
Keywords: myasthenia gravis, amyotrophic lateral sclerosis, anti-acetylcholine receptor antibody, anti-low-density lipoprotein receptor-related protein 4 antibody, neuromuscular junction
Introduction
Myasthenia gravis (MG) is an autoimmune disorder in which the neuromuscular junction (NMJ) is damaged or impaired functionally by autoantibodies targeting the acetylcholine receptor (AChR) or muscle-specific kinase (MuSK) (1). Recent studies have reported that antibody against low-density lipoprotein receptor-related protein 4 (LRP4) is detected in some patients with MG and may play a role in the pathogenesis of MG (2-4). LRP4 is a co-receptor for agrin and forms an intracellular signaling complex with MuSK to maintain the structure and function of the NMJ (5). It has been reported that LRP4 immunization induced experimental autoimmune MG with the anti-LRP4 antibody in mice (6). Therefore, the anti-LRP4 antibody is thought to have pathogenicity for MG, similar to the anti-AChR and anti-MuSK antibodies.
Anti-LRP4 antibodies have also been reported in some patients with amyotrophic lateral sclerosis (ALS), which is characterized by the selective degeneration of the upper and lower motor neurons, resulting in diffuse muscle weakness, atrophy, and fasciculation (7, 8). However, the importance of anti-LRP4 antibody in the pathophysiology of ALS has not been elucidated.
Although MG and ALS differ in their etiological and pathological features, the association between MG and ALS reported in some patients may imply a possible common pathophysiological background (9).
We herein report a rare case in which a patient had both MG and ALS with antibodies against both LRP4 and AChR in whom only the symptom of MG was successfully resolved after immunotherapy.
Case Report
An 82-year-old woman was referred to our hospital for neck weakness and dysarthria. The clinical examination revealed severe weakness in her neck muscles and moderate weakness in both orbicularis oculi muscles. She also presented mild dysarthria without dysphagia but did not present atrophy or fasciculation of the tongue. There was no muscle weakness, atrophy, or fasciculation in any of her extremities. Her tendon reflex was normal, and Babinski’s sign was negative. Sensory abnormalities were not detected. Her serum was positive for anti-AChR antibodies (20 nmol/L; normal values are <0.3 nmol/L). The MuSK antibody was not detected. An edrophonium test improved the weakness in the orbicularis oculi and neck muscles. Repetitive nerve stimulation (RNS) at 3 Hz showed a 26.3% decrease in nasal muscle amplitude (Fig. 1) and an 11.5% decrease in right trapezius muscle amplitude. Needle electromyography (EMG) showed fibrillation potentials and positive sharp waves only in the neck extensor muscles; these were not observed in the limbs or paraspinal muscles. Computed tomography of the chest was normal. Spirometry did not reveal any abnormalities. Based on these findings, she was diagnosed with seropositive late-onset generalized MG.
Figure 1.
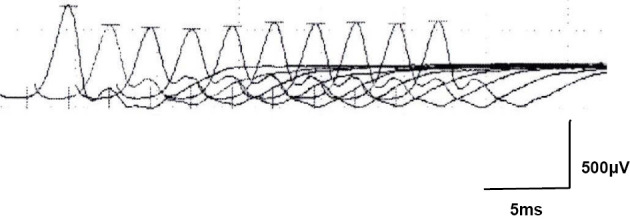
Repetitive nerve stimulation at 3 Hz showed a 26.3% decrease in the nasal muscle amplitude. The decrement response was calculated using the following formula: decrement (%)=(amplitude of 1st response-amplitude of 4th response) ×100/amplitude of 1st response.
After eight sessions of plasmapheresis, the patient showed remarkable improvement leading to remission without any clinical manifestation. Prednisolone and tacrolimus were started as maintenance therapy. One year after remission, she began to exhibit progressive weakness in her neck and limbs and dysarthria while on immunosuppressive therapy. She was then readmitted to our hospital. Her tongue showed atrophy and fasciculation. Muscle weakness was detected in the neck and all extremities. Her tendon reflexes were brisk, and Babinski’s sign was positive. Although RNS showed a decrement response in the trapezius muscle (26.4%) but not in the nasal muscle (4.4%), EMG showed fibrillation potentials, positive sharp waves, and fasciculation potentials with chronic denervation in the right trapezius, biceps brachii, first dorsal interosseous, rectus femoris, and thoracic paraspinal muscles. These clinical and EMG findings supported a diagnosis of possible ALS according to the Awaji criteria (10). We detected anti-LRP4 antibodies in serum samples obtained during the first admission (antibody index, 1.04) and second admission (antibody index, 1.4). The anti-AChR antibodies had declined (0.7 nmol/L). Anti-MuSK antibodies were not detected again. Her symptoms did not respond to plasmapheresis or immunosuppressive therapy for MG.
She was transferred to a nursing home, and her condition worsened progressively (Fig. 2). She ultimately died of respiratory failure years after the onset of the initial symptoms.
Figure 2.

The clinical course and medications. Note that the muscle weakness trace shows weakness in the extremities (upper and lower).
Discussion
We herein report a case in which the patient was diagnosed with both MG and ALS and exhibited anti-AChR and anti-LRP4 antibodies. While MG is an autoimmune disease targeting the NMJ, ALS is a neurodegenerative disease causing selective motor neuron loss; however, the two disease entities have been reported to coexist in rare cases, as in our patient (8, 9). A recent Italian study reported that 0.75% of patients with ALS were affected by MG, which was higher than expected (11). Since both MG and ALS have very low incidences in the general population, comorbidity of these diseases may not occur by mere chance. In addition, a previous study showed that patients with ALS have a relatively high incidence of preceding autoimmune diseases, including MG. These results may suggest that MG and ALS have a shared pathophysiology (12).
There is evidence that the immunological response is involved in the pathophysiology of ALS and modulates its severity and progression (13). Post-mortem studies have elucidated the activation of the innate immune response with microglial activation and the expression of a variety of inflammatory mediators in the motor cortex and spinal cord (13). ALS has also been linked to the upregulation of inflammatory cytokines in damaged tissue and the peripheral blood (13-15). These results imply that the immune response is activated not only at damaged motor neurons but also systemically and promotes a humoral immune response against a variety of antigens derived from damaged tissue in ALS. In ALS, both central and peripheral motor neurons are damaged as pathognomonic findings. Previous studies on human pathology and animal models have shown that the NMJ is damaged and accompanied by an activated immune response from the early stage of the disease (15). Thus, it is reasonable to assume that autoantibodies against components of the NMJ are induced during the early stage of the disease.
The prevalence of the anti-LRP4 antibody in ALS without MG is 9.8% to 23.1% higher than that with MG, in which the prevalence of the anti-LRP4 antibody is 1% to 3% (7, 8, 16). In contrast, the prevalence of the anti-AChR antibody in MG is around 80% (1). This may suggest a specific role of the anti-LRP4 antibody in the pathogenesis of ALS. However, a previous report showed that immunosuppressive treatment is effective only for myasthenic symptoms but not ALS symptoms in patients with the anti-LRP4 antibody, similar to our patient (17).
A deranged or activated immune response in the pathophysiology of ALS may have triggered antibody production against auto-antigens derived from the NMJ including AChR and LRP4 as secondary phenomena in our patient. However, there is a possibility that the anti-LRP4 antibody affects the manifestation of ALS, since previous reports have shown differences in the clinical features between ALS with and without the anti-LRP4 antibody. Furthermore, the anti-LRP4 antibody titer is associated with the severity of ALS (7). However, the importance of the anti-LRP4 antibody in the pathophysiology of ALS is unknown.
We reasonably speculated that the pathophysiology of ALS insidiously progressed and caused neurodegenerative changes accompanied by an immune response in the motor neuron system involving the NMJ long before the onset of MG in our patient. This may have induced autoantibodies against AChR and LRP4, leading to the development of MG. Thereafter, ALS may have become obvious at the point when the neurodegenerative mechanism exceeded the clinical threshold. Since we did not perform a post-mortem study or analyze the role of the anti-LRP4 antibody, it was impossible to elucidate how the anti-LRP4 antibody was induced and subsequently affected MG and ALS in this case.
In conclusion, the anti-LRP4 antibody seems to be associated with both ALS and MG, although the role and importance of the antibody in the pathophysiologies of these two diseases are different. The anti-LRP4 antibody may be important not only as an important diagnostic marker for MG but also as a marker of pathological changes in the NMJ in both ALS and MG. Further studies are needed to clarify the utility of the anti-LRP4 antibody titers in determining the treatment plans and evaluating the status of the two diseases.
The authors state that they have no Conflict of Interest (COI).
References
- 1. Gilhus NE, Verschuuren JJ. Myasthenia gravis: subgroup classification and therapeutic strategies. Lancet Neurol 14: 1023-1036, 2015. [DOI] [PubMed] [Google Scholar]
- 2. Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol 69: 418-422, 2011. [DOI] [PubMed] [Google Scholar]
- 3. Zisimopoulou P, Evangelakou P, Tzartos J, et al. A comprehensive analysis of the epidemiology and clinical characteristics of anti-LRP4 in myasthenia gravis. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. J Autoimmun 52: 139-145, 2014. [DOI] [PubMed] [Google Scholar]
- 4. Cordts I, Bodart N, Hartmann K, et al. Screening for lipoprotein receptor-related protein 4-, agrin-, and titin-antibodies and exploring the autoimmune spectrum in myasthenia gravis. J Neurol 264: 1193-1203, 2017. [DOI] [PubMed] [Google Scholar]
- 5. Ohno K, Ohkawara B, Ito M. Agrin-LRP4-MuSK signaling as a therapeutic target for myasthenia gravis and other neuromuscular disorders. Expert Opin Ther Targets 21: 949-958, 2017. [DOI] [PubMed] [Google Scholar]
- 6. Mori S, Motohashi N, Takashima R, Kishi M, Nishimune H, Shigemoto K. Immunization of mice with LRP4 induces myasthenia similar to MuSK-associated myasthenia gravis. Exp Neurol 297: 158-167, 2017. [DOI] [PubMed] [Google Scholar]
- 7. Tzartos JS, Zisimopoulou P, Rentzos M, et al. LRP4 antibodies in serum and CSF from amyotrophic lateral sclerosis patients. Ann Clin Transl Neurol 1: 80-87, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rivner MH, Liu S, Quarles B, et al. Agrin and low-density lipoprotein-related receptor protein 4 antibodies in amyotrophic lateral sclerosis patients. Muscle Nerve 55: 430-432, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Del Mar Amador M, Vandenberghe N, Berhoune N, et al. Unusual association of amyotrophic lateral sclerosis and myasthenia gravis: a dysregulation of the adaptive immune system? Neuromuscul Disord 26: 342-346, 2016. [DOI] [PubMed] [Google Scholar]
- 10. de Carvalho M, Dengler R, Eisen A, et al. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol 119: 497-503, 2008. [DOI] [PubMed] [Google Scholar]
- 11. de Pasqua S, Cavallieri F, D'Angelo R, et al. Amyotrophic lateral sclerosis and myasthenia gravis: association or chance occurrence? Neurol Sci 38: 441-444, 2017. [DOI] [PubMed] [Google Scholar]
- 12. Turner MR, Goldacre R, Ramagopalan S, Talbot K, Goldacre MJ. Autoimmune disease preceding amyotrophic lateral sclerosis: an epidemiologic study. Neurology 81: 1222-1225, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Puentes F, Malaspina A, van Noort JM, Amor S. Non-neuronal cells in ALS: role of glial, immune cells and blood-CNS barriers. Brain Pathol 26: 248-257, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu CH, Allen K, Oei F, et al. Systemic inflammatory response and neuromuscular involvement in amyotrophic lateral sclerosis. Neurol Neuroimmunol Neuroinflamm 3: e244, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Malaspina A, Puentes F, Amor S. Disease origin and progression in amyotrophic lateral sclerosis: an immunology perspective. Int Immunol 27: 117-129, 2015. [DOI] [PubMed] [Google Scholar]
- 16. Gilhus NE. Myasthenia gravis. N Engl J Med 376: e25, 2017. [DOI] [PubMed] [Google Scholar]
- 17. Takahashi H, Noto YI, Makita N, et al. Myasthenic symptoms in anti-low-density lipoprotein receptor-related protein 4 antibody-seropositive amyotrophic lateral sclerosis: two case reports. BMC Neurol 16: 229, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]