

Summary
Circulating natural killer (NK) cells help protect the host from lympho-hematogenous acute viral diseases by rapidly entering the draining lymph node (dLN) to curb virus dissemination. Here we identified a highly choreographed mechanism underlying this process. Using footpad infection with ectromelia virus, a pathogenic DNA virus of mice, we show that TLR9/MyD88 sensing induces NKG2D ligands in virus-infected, skin-derived migratory dendritic cells (mDCs) to induce production of IFN-γ by classical NK cells and other types of Group 1 innate lymphoid cells (ILC) already in the dLN, via NKG2D. Uninfected inflammatory monocytes, also recruited to the dLN by mDCs in a TLR9/MyD88 dependent manner, respond to this IFN-γ by secreting CXCL9 for optimal CXCR3-dependent recruitment of circulating NK cells. This work unveils a TLR9/MyD88-dependent mechanism whereby in the dLN, three cells types -mDCs, Group 1 ILC (mostly NK cells), and inflammatory monocytes-coordinately recruit protective circulating NK cells to the dLN.
Introduction
Many viruses relevant to human and animal health breach epithelial surfaces and then disseminate lympho-hematogenously through the regional draining lymph node (dLN) to produce systemic diseases (Flint and American Society for Microbiology., 2009). Ectromelia virus (ECTV), an Orthopoxvirus similar to the virus of human smallpox and its vaccine species vaccinia virus, is a pathogen of the laboratory mouse. Following footpad infection, ECTV disseminates lympho-hematogenously causing fatal mousepox to susceptible strains of mice but not to mousepox-resistant young C57BL/6 (B6) mice. Virology textbooks frequently use ECTV as the paradigm of viruses that disseminate lympho-hematogenously (Flint and American Society for Microbiology., 2009).
Lymph nodes (LNs) are organs where lymphocytes are primed before they egress to combat pathogens at the primary sites of infection (Abbas et al., 2007). Yet, LNs are also sites where immune cells restrict the spread of pathogens. For example, we have previously shown that after footpad infection, memory CD8+ T cells curb the spread of ECTV from the popliteal draining LN (dLN) to the liver and spleen (Xu et al., 2007). Furthermore, others have shown that subcapsular macrophages in the dLN limit the lympho-neuro (Iannacone et al., 2010) and lympho-hematogenous spread (Junt et al., 2007) of vesicular stomatitis virus (VSV). Moreover, we have also found that 2-3 days after footpad infection of young, mousepox-resistant B6 mice with ECTV, terminally differentiated Natural killer (NK) cells recruited from the blood, accumulate in the dLN to restrict the systemic spread of the virus. When these circulating NK cells did not accumulate in the dLN, such as in NK cell depleted (Fang et al., 2008) or aged B6 mice (Fang et al., 2010), ECTV disseminated from the dLN to the liver and spleen more rapidly, and the mice succumbed to mousepox. Hence, the early accumulation of NK cells in the dLN restricts ECTV lympho-hematogenous spread and protects mice from lethal mousepox. Yet, the specific mechanisms of NK cell recruitment to the dLN during viral infection remain mostly unknown. In addition to controlling ECTV, NK cells also play an essential role in the early control of other viruses in mice and humans such as herpesviruses, human immunodeficiency virus, influenza virus (Lodoen and Lanier, 2006). Thus, understanding the mechanisms of NK cell recruitment to dLNs has important implications for our general understanding of virus control.
Innate Lymphoid cells (ILC) derive from the common innate lymphoid cell progenitor (CILP) (Klose et al., 2014). NK cells together with ILC type 1 (ILC-1) belong to the Group 1 ILC which produce IFN-γ after stimulation. In mice, Group 1 ILC express the T-box transcription factor T-bet, the activation molecule NKp46 and, in B6 mice, the activating receptor NK1.1 (CD161). The distinction between NK cells and ILC-1 is not simple. In many cases, but not always (Robinette et al., 2015), NK cells but not ILC-1 express the transcription factor Eomesodermin (Eomes) and the integrin CD49b while ILC-1 but not NK cells express CD49a and CD127 (the IL-7 receptor alpha chain). Functionally, ILC-1 are thought to be tissue resident while NK cells circulate between the blood and secondary lymphoid organs, migrating to tissues during inflammation. In mesenteric LNs, the CD3-NK1.1+ NKp46+ cells includes circulating Eomes+ NK cells as well as resident Eomes− ILC-1 (Gasteiger et al., 2015). In skin-draining LNs such as the popliteal LN, 0.2-0.5% of the cells are CD3-NK1.1+ NKp46+ at the uninflamed steady-state. These cells can be broadly classified as Group 1 ILC. While it has been suggested that most of them are NK cells (Kim et al., 2016), unequivocal distinction between NK cells and ILC-1 in peripheral LNs is compromised by their incomplete characterization.
Toll like receptor 9 (TLR9) recognizes double-stranded DNA (Hemmi et al., 2000) and signals through the adapter MyD88 to activate the transcription factors nuclear factor kappa B (Nf-κB) and interferon regulatory factor 7 (IRF7) (Hemmi et al., 2000). Mice deficient in TLR9 (Tlr9−/−) or MyD88 (Myd88−/−) are susceptible to mousepox (Rubio et al., 2013; Samuelsson et al., 2008). CD11c+ cells (presumably skin-derived mDC) require TLR9 and MyD88 to express CCR2-binding chemokines, which are necessary for the recruitment of inflammatory monocytes (iMo) to the dLN. These recruited iMo are major targets of ECTV infection and, when infected, produce Type I interferon (IFN-I) in the dLN (Xu et al., 2015). Given that both, NK cells and iMo are recruited to the dLN soon after infection and are critical for anti-viral defense, we investigated possible shared mechanisms for their recruitment and whether there is cross-talk between iMo, NK cells, and mDCs.
Results
Recruitment of NK cells into the dLN requires non-autonomous TLR9 and MyD88
At 3 days post infection (dpi) with ECTV expressing firefly luciferase (ECTV-Luc) (Xu et al., 2012), Myd88−/− and Tlr9−/− mice had higher luminescent signal than WT B6 mice in the liver, indicating that the virus spreads faster in the mutant strains (Figure 1A). Given that NK cells that accumulate in the dLN control the spread of ECTV to the liver and spleen at this time point (Fang et al., 2008), we hypothesized that the TLR9-MyD88 could be involved in the accumulation of NK cells in the dLN. Accordingly, at 2.5 dpi with ECTV, there were significantly fewer NK1.1+ cells in the dLN of Tlr9−/− and Myd88−/− than in the dLN of wild type (WT) B6 mice. Indeed, the absolute numbers of NK1.1+ cells in the dLN of ECTV-infected Myd88−/− and Tlr9−/− mice did not differ significantly from those found in the popliteal LN of uninfected mice (uLN) (Figure 1B). However, to accumulate in the dLN following ECTV infection, NK cells did not need intrinsic MyD88 (and likely TLR9), because in co-adoptive transfer experiments of CFSE-labeled splenocytes from WT (B6-CD45.1) and Myd88−/− (CD45.2) mice into WT mice (WT+Myd88−/−→WT) (Figure 1C), the ratio of CFSE-labeled WT/Myd88−/− NK1.1+ cells in dLN and non-draining LN (ndLN) were similar (Figure 1D). Also, significantly more WT and Myd88−/− NK cells accumulated in the dLN than in the ndLN of infected WT+Myd88−/−→WT mice, while the few WT and Myd88−/− NK1.1+ cells in the dLN and ndLN of WT+Myd88−/−→Myd88−/− mice did not differ significantly in number (Figure 1E). Additionally, significantly more WT and Myd88−/− NK1.1+ cells were activated in the dLN than in the ndLN of WT+Myd88−/−→WT mice while few NK1.1+ cells of either type were activated in the dLN of WT+Myd88−/−→Myd88−/− mice, as determined by intracellular staining for IFN-γ (Figure 1F) or granzyme B (GzmB) (Figure 1G). Thus, while MyD88 and TLR9 were required for the recruitment of NK cells to the dLN, autonomous expression of TLR9 and MyD88 in NK cells was not required for their accumulation or activation. This indicates that cells other than NK cells must express MyD88 and TLR9 to promote the recruitment of NK cells to the dLN.
Figure 1. Recruitment of NK cells into the dLN requires non-autonomous TLR9 and MyD88:

A) The indicated mice were infected with ECTV-Luc. At 2 dpi the mice were injected with luciferin, and imaged for luminescence and x-rayed. The original images (left) and a dot plot depicting the radiance values for individual mice (n=3) with mean ± SEM (right) are shown. B) Absolute numbers of NK cells in the dLN at 2 dpi and uLN of the indicated mice. Individual mice (n=4-10) with mean ± SEM are shown. C-D) Flow cytometry analysis of dLN and ndLN at 2 dpi of B6 mice that had been co-transferred with equal numbers of CFSE labeled splenocytes from B6-CD45.1 (Myd88+/+) and Myd88−/− mice. Representative flow cytometry plots showing the gating strategy (C) and a dot plot displaying the ratio of Myd88+/+/Myd88−/− NK cells for individual mice (n=5) with mean ±SEM (D). E) Total number of donor NK cells of the indicated genotype at 2 dpi in the dLN and ndLN of individual B6 or Myd88−/− mice (n=5) that had been transferred with equal numbers of CFSE labeled splenocytes from B6 or Myd88−/− mice. Mean ±SEM are also shown. F) As in A but for the number of donor NK cells that express IFN-γ. G) As in E but for the number of NK cells that express GzmB. H) Number of NK cells in the uLN or the dLN at 2 dpi of individual Myd88fl/fl mice (n=4-9) expressing cre recombinase from the indicated promoters. Mean ±SEM are also shown. All experiments were performed a minimum of two times and repeats yielded similar results.
To determine which cells need MyD88 to promote the accumulation of NK cells in the dLN, we used mice homozygous for a floxed Myd88 allele (Myd88fl/fl) expressing cre recombinase from various promoters. NK cells accumulated normally in the dLN of Myd88fl/fl mice expressing cre from the albumin promoter (Alb-cre+) causing MyD88 deficiency in hepatocytes, or the lysozyme 2 promoter (Lyz2-cre+), causing MyD88 deficiency in macrophages, monocytes, and granulocytes. On the other hand, the accumulation of NK cell was impaired in Myd88fl/fl mice expressing cre from the vav guanine nucleotide exchange factor 1 (Vav1) promoter (Vav1-cre+) which lack MyD88 in all cells of hematopoietic origin, or in mice expressing cre from the promoter the CD11c gene (Itgax-cre+), which lack MyD88 in dendritic cells (DC) (Figure 1H). Similar results were obtained in mice with a floxed TLR9 gene and the various cre-driven promoters (data not shown). We concluded that the recruitment of NK cells to the dLN requires MyD88 and TLR9 in CD11c+ cells of hematopoietic origin, suggesting that TLR9 and MyD88 must be expressed by some type of dendritic cell (DC). Importantly, we have previously shown that Vav1-Cre Myd88fl/fl or -Tlr9fl/fl and Itgax-Cre Myd88fl/fl or -tlr9fl/fl mice, which succumb to mousepox due to unrestrained viral replication, also fail to recruit iMo to the dLN (Xu et al., 2015). This suggests that the recruitment of iMo and NK cells to the dLN could be mechanistically linked.
Optimal recruitment of NK cells to the dLN requires autonomous expression of the chemokine receptor CXCR3
Recruitment of immune cells from the blood to LNs requires the interaction of chemokine receptors on the migrating cell with homeostatic and pro-inflammatory chemokines on endothelial cells (Rot and von Andrian, 2004; Schulz et al., 2016). The chemokine receptor CXCR3 is known to be involved in the migration of NK cells to the dLN of mice inoculated in the footpad with lipopolysaccharide-matured DCs, with several adjuvants (Martin-Fontecha et al., 2004; Walzer and Vivier, 2011) or with cowpox virus (Pak-Wittel et al., 2013). Accordingly, at 2.5 dpi with ECTV, CXCR3 deficient (Cxcr3−/−) mice recruited significantly fewer NK cells to the dLN than WT B6 mice (Figure 2A). Moreover, at 2.5 dpi, WT (B6-CD45.1) mice adoptively transferred with a 1:1 mixture of Cxcr3−/− (CD45.2) and WT (B6-CD45.1) splenocytes labeled with CFSE (WT+Cxcr3−/−→WT, Figure 2B) had significantly higher frequencies (Figure 2C) and absolute numbers (Figure 2D) of WT than Cxcr3−/− NK cells in the dLN but not in the ndLN. Thus, in vivo, CXCR3 in NK cells is important for their optimal recruitment into the dLN of ECTV infected mice, but not for their constitutive entry into the uninflamed LN. However, it should be noted that Figure 2A and 2D both show significantly more Cxcr3−/− NK cells in the dLN than in the ndLN or uLN, findings that suggest that an alternative, CXCR3-independent mechanism of NK cell recruitment to the dLN. This alternative mechanism could at least partially explain why Cxcr3−/− mice do not succumb to mousepox and control virus loads similar to B6 mice (Figure S1).
Figure 2. Optimal migration of NK cells to the dLN requires autonomous expression of the chemokine receptor CXCR3.

A) Total number of NK cells in the indicated LNs of individual B6 and Cxcr3−/− mice (n=5) with mean ± SEM. B-D) Donor NK cells in dLN and ndLN at 2 dpi or uLN from B6 mice that had been co-transferred with equal numbers of CFSE labeled splenocytes from B6-CD45.1 (Cxcr3+/+) and Cxcr3−/− mice. Representative flow cytometry plots showing the gating strategy (B), frequencies of Cxcr3+/+ and Cxcr3−/− NK cells in the indicated LNs of of individual mice (n=3-5) with mean ± SEM (C), and total number of the indicated donor NK cells ± SEM in the indicated LNs of the same mice (D). All experiments were performed a minimum of two times and repeats yielded similar results.
Efficient production of the CXCR3 ligand CXCL9 in the dLN requires TLR9 and MyD88
Consistent with the finding that CXCR3 is important for the efficient accumulation of NK cells in the dLN, both purified splenic total NK cells (Figure 3A) and mature (CD27− CD11b+) NK cells (Figure 3B) migrated towards the CXCR3 ligands CXCL9 and CXCL10 in in vitro migration assays. Furthermore, RT-qPCR (quantitative polymerase chain reaction analysis of reverse transcribed RNA) analysis at 2.5 dpi showed that the dLN of WT mice had a significant increase in mRNA for CXCL9 (Figure 3C) and CXCL10 (Figure 3D) while the small increase in the dLN of Tlr9−/− or Myd88−/− mice did not achieve statistical significance. Moreover, CXCL9 protein was detected by Western Blot (WB) in the dLN of B6 mice but not in the uLN or dLN of Tlr9−/− and Myd88−/− mice (Figure 3E). The CXCL10 protein was not detected with various commercial Abs and at several time points (data not shown), suggesting that CXCL10 mRNA either was not translated into protein or that the tested Abs were not sensitive enough for the amount of CXCL10 produced during ECTV infection. Nevertheless, the results indicate that TLR9 and MyD88 in some cell types, are necessary for the detectable production in the dLN of CXCL9, a CXCR3 ligand capable of recruiting NK cells.
Figure 3. Efficient production of the CXCR3 ligand CXCL9 in the dLN requires TLR9 and MyD88:

A-B) Purified NK cells were obtained by FACS sorting GFP+ splenocytes from NKp46-GFP mice and placed in the upper chambers of trans-well plates containing the indicated chemokines in the lower chamber. Data shows the mean ± SEM of total number (A) or CD27−− CD11b+ (mature, B) NK cells that migrated to the lower chambers. Statistics are compared to media and correspond to three wells/condition. C-D) Transcripts for CXCL9 (C) and CXCL10 (D) in the indicated LNs at 2 dpi. Data is a combination of two independent experiments (n=2/experiment). E) CXCL9 protein expression was determined by Western Blot in pools of 10 uLN or 5 dLN at 2 dpi of the indicated mice. All experiments were performed a minimum of two times and repeats yielded similar results.
CXCL9 induced by ECTV infection localizes to high endothelial venules and the stromal cell network of the dLN
We infected mice with fully virulent ECTV expressing the red-fluorescent protein mCherry (ECTV-mCherry) and used confocal microscopy to visualize infected cells and CXCL9 at various dpi (Figure 4A). In control uLN, neither virus nor CXCL9 were seen. At 1 dpi, CXCL9 was present in discrete areas of the subcapsular sinus and paracortex of the dLN, indicating that viral recognition had occurred. However, the fluorescent signal of viral infection was not yet visible, perhaps because only a few cells were infected at this time, or because mCherry expression was still too low. At 2 dpi, CXCL9 distributed throughout the dLN with particularly strong fluorescence in structures suggestive of fibroblastoid reticular cells (FRC) that form the LN stroma and in high endothelial venules (HEV) of capillary vessels, where circulating leukocytes enter LNs. On the other hand, the localization of virus-infected cells was restricted to distinct subcapsular and paracortical areas. At 3 dpi, CXCL9 expression appeared restricted to HEVs while the virus was present throughout the dLN. Co-staining with Abs to CXCL9 and the HEV marker PNAd at 2 dpi with WT ECTV confirmed localization of CXCL9 to HEV (Figure 4B) while co-staining with Abs to CXCL9 and the FRC marker ERTR-7 confirmed that CXCL9 is also present in FRC (Figure 4C). Hence, soon after infection, CXCL9 is produced rapidly and distributed throughout the dLN. Of note, NK cell accumulation in the dLN temporarily coincided with CXCL9 localizing to HEV and the stromal cell network. These structures could respectively be used by NK cells to enter the dLN (Chen et al., 2005; Martin-Fontecha et al., 2004) and for guided intranodal migration (Sung et al., 2012).
Figure 4. CXCL9 induced by ECTV infection localizes to high endothelial venules and the stromal cell network of the dLN:

A) Confocal microscopy (original magnification 10×) of uLN and dLN at the indicated dpi from mice infected with ECTV-mCherry (red) and stained with CXCL9 Ab (green). LN edges are marked with a dashed line. Arrows at 1 dpi show an area of CXCL9 staining and at 2 dpi an area with ECTV-mCherry infected cells. B) Confocal microscopy (60x original magnification) of a dLN at 2 dpi stained with PNAd Ab, an HEV marker (green) and CXCL9 Ab (red). C) As in B but stained with ER-TR7 Ab, an FRC/stroma marker (red), and CXCL9 Ab (green). Scale bars represent 100 μM. Experiment was performed twice with n=3 in each experiment and the results were comparable.
Uninfected iMo are the major producers of CXCL9 in the dLN
Secreted chemokines can bind to glycosaminoglycans at the surface of cells (Schulz et al., 2016). Therefore, while CXCL9 is localized to endothelial and fibroblastoid reticular cells, it is not necessarily produced by these cells. Thus, we sought to identify the CXCL9-producing cells. When we sorted by flow cytometry at 2.5 dpi from pooled dLNs (Figure 5A), we found that iMo (CD3−, B220−, NK1.1− MHC II+ CD11c+ CD11b+) and to a lesser extent classical DC (CD3−, B220−, NK1.1− MHC II+ CD11c+ CD11b−) but not total lymphocytes (stained with a cocktail of cascade blue-labeled Abs to CD3e, B220 and NK1.1) transcribed CXCL9 (Figure 5B) and also CXCL10 (data not shown). Similarly, we found that iMo (CD11c+, CD11b+) and to a lesser degree DC (CD11c+ CD11b−) specifically stained intracellularly with CXCL9 mAb in the dLN at 2.5 dpi but not in the uLN (Figure 5C and Figure S2A) (note that we classified the cells in the CD11c+, CD11b+ gate in the dLN plots as iMo, based on our previous extensive analysis of this population (Xu et al., 2015) using markers for DC and monocytic lineages as described by publications from the Immunolgical Genomic consortium (Gautier et al., 2012; Miller et al., 2012) and by morphology of sorted cells at the microscope). Given the post-infection excess of iMo over DC in the dLN, we conclude that iMo are the major producers of CXCL9 in the dLN and to a lesser extent, DC. Additional flow cytometry and RT-qPCR experiments showed that stromal cells are not producers of CXCL9 during ECTV infection (Figure S2 B and C).
Figure 5. Uninfected iMO produce CXCL9 in the dLN.

A-B) Gating strategy (A) and expression of Cxcl9 mRNA in uLN and dLN cells before sorting or the indicated sorted cells from dLNs (from 40 pooled dLNs, B). Data shows three technical replicates with mean ± SEM. C) Gating strategy and expression of intracellular CXCL9 protein in the dLN at 2 dpi and in control uLN. D) Gating strategy and expression of intracellular CXCL9 protein in dLN of B6 mice at 2 dpi with ECTV-GFP. Note that Figure 5D shows the data as contour plot for visualization, but the uLN as well as the dLN from Vav1+-cre Myd88fl/fl and Itgax-cre Myd88fl/fl mice contained 10-20 times fewer iMo than the dLN from Alb-cre+ myd88fl/fl and Lyz2-cre+ myd88fl/fl mice. E) Gating strategy and expression of intracellular CXCL9 protein by either infected (GFP+) or uninfected (GFP−) iMo or DCs at 2 dpi from 3 pooled LNs. All experiments were performed a minimum of two times and yielded similar results.
Next, we investigated whether iMo require intrinsic MyD88/TLR9 to produce CXCL9. We found that iMo produced CXCL9 not only in Alb-cre+ myd88fl/fl and Lyz2-cre+ myd88fl/fl mice, but also in the few iMo present in the dLN of Vav1+-cre Myd88fl/fl, Itgax-cre Myd88fl/fl (Figure 5D), Tlr9−/−, and Myd88−/− mice (data not shown). Consistently, sorted iMo from Itgax-Cre+TLR9fl/fl or TLR9−/− iMo expressed significantly higher CXCL9 transcripts than unsorted dLN cells, albeit at significantly lower levels than B6 iMo (Figure S3). Note that in this case, where we used pooled LNs, there were significant differences in the levels of Cxcl9 expression between ndLN and dLN (presort) from Tlr9−/− and Itgax-Cre+Tlr9fl/fl mice. This suggest that the absence of a significant increase in CXCL9 in the dLN of Tlr9−/− and Myd88−/− mice by RT-qPCR in Figures 3C was likely due to insufficient statistical power for an experiment where there was intra-group variability, in which the n (=4) was small, and where inter-group differences were minor likely due to the very small number of iMo present in the dLN of these mice, which is 10-20 times lower than in WT mice (Xu et al., 2015). Together, these experiments show that iMo do not need autonomous TLR9 or MyD88 to produce CXCL9, and that the low amount of CXCL9 in the dLN of mice with global or CD11c+ cell-restricted MyD88/TLR9 deficiency is due to their inability to efficiently recruit iMo into the dLN.
We have previously shown that ECTV infection targets iMo (Xu et al., 2015). When we used ECTV-GFP, we found that uninfected (GFP−), rather than infected (GFP+) iMo predominantly produced CXCL9 (Figure 5E). Concordant results were observed by RT-qPCR and we observed a similar result for DC (data not shown). The findings contrast with our earlier observations that infected (GFP+) but not uninfected (GFP−) iMo are major producers of IFN-I (Xu et al., 2015). Hence, uninfected iMo in the dLN produce CXCL9 upon receiving extracellular signals that require MyD88-TLR9, and they downregulate CXCL9 production once they become infected.
iMo require IFN-γ but not IFN-I to produce CXCL9
IFN-I and IFN-γ are both required for resistance to mousepox (Karupiah et al., 1993; Panchanathan et al., 2005; Xu et al., 2008). During infection with Lymphocytic Choriomeningitis virus (LCMV), IFN-I induces the expression of CXCL10 while IFN-γ induces the expression of CXCL9 (Sung et al., 2012). Thus, we compared the expression of CXCL9 and CXCL10 in the dLN and ndLN of mice deficient in the IFN-I receptor (Ifnar1−/−) or in IFN-γ (Ifng−/−) at 2.5 dpi with ECTV. WT and Ifnar1−/− mice had significantly more Cxcl9 and Cxcl10 RNA in the dLN compared to the ndLN (Figure 6A). Of note, Cxcl9 was significantly more upregulated in Ifnar1−/− mice, possibly due to the increased viral replication in these mice. On the other hand, Ifng−/− mice did not significantly upregulate Cxcl9 expression in their dLN, and they upregulated Cxcl10 but to significantly lower levels than WT mice (Figure 6B). Consistently, CXCL9 was produced by a large proportion of iMo in the dLN of Ifnar1−/− mice but not in the dLN of Ifng−/− mice (Figure 6C). In WT+Ifngr−/− →WT mixed bone marrow chimeras, a significantly lower frequency of Ifngr−/− than WT iMo produced CXCL9 (Figure 6D), indicating that iMo need direct IFN-γ signals to efficiently produce CXCL9. Of note, while significantly fewer NK cells accumulated in the dLN of Ifng−/− than WT mice (Figure 6E), NK cells did not need direct IFN-γ signals to accumulate in the dLN because in WT+Ifngr−/−→WT mixed bone marrow chimeras, WT and Ifngr−/− NK cells accumulated at similar ratios in the dLN and ndLN (Figure 6F). Hence, to efficiently produce CXCL9, iMo need direct IFN-γ signaling. On the other hand, to efficiently accumulate in the dLN, NK cells do not require direct IFN-γ signaling, but they do require CXCL9 and likely other cytokines that iMo produce in response to IFN-γ stimulation.
Figure 6. iMo require IFN-γ but not IFN-I to produce CXCL9:
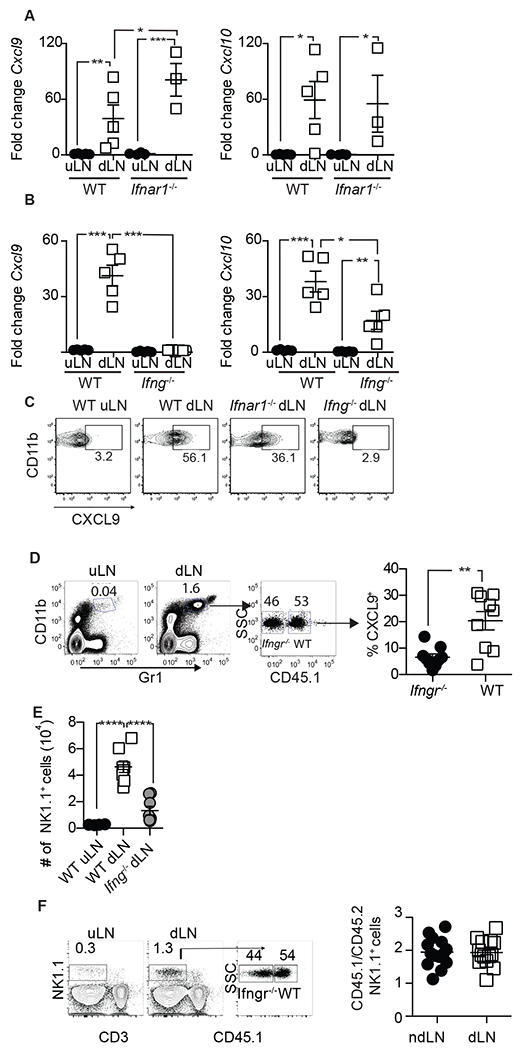
A) RT-qPCR for CXCL9 (left) and CXCL10 (right) in the dLN of individual (n=3-5) WT or Ifnar1−/− mice. Mean ± SEM are also shown B) As in a but for Ifng−/− mice. C) Expression of CXCL9 in iMo from the indicated LNs from a pool of three LNs. D) Gating strategy and frequency of iMo in the indicated LNs of individual WT+Ifngr−/− →WT chimeric mice (n=9 in two experiments combined) with mean ± SEM. E) Absolute number with with mean ± SEM of NK cells from the indicated LNs of WT B6 and Ifng−/− mice (n=6 in two combined experiments). F) Gating strategy and ratio of Ifng−/− and WT NK cells in the indicated LNs of In WT+Ifngr−/− →WT chimeric mice (n=14 from 3 combined experiments). All experiments were performed a minimum of two times and repeats yielded similar results.
Early IFN-γ is produced by steady-state dLN NK1.1+ cells in response to NKG2D stimulation
Early expression of IFN-γ is directly necessary for the the induction of CXCL9 in uninfected iMo and indirectly required for the accumulation of NK cells in the dLN. Thus, we next aimed to identify the cell type responsible for the early expression of IFN-γ in the dLN. We sorted various cell populations at 16-24 hours post infection (hpi), and found that only NK1.1+ cells transcribed Ifng (Figure 7A). Depletion of NK1.1+ cells with mAb PK136 inhibited this early transcription of Ifng (Figure 7B). Furthermore, a significant increased frequency NK1.1+ cells produced IFN-γ protein in the dLN than in the uLN as determined by flow cytometry (Figure 7C). These cells represented the NK1.1+ cells typically present in the LN at steady-state and not actively recruited NK cells because, consistent with our previously published data (Fang et al., 2008), there were similar frequencies of NK1.1+ cells in the dLN and ndLN (Figure 7D).
Figure 7. Early IFN-γ is produced by steady-state NK cells and other G1-ILCs in response to NKG2D stimulation:

A) IFN-γ transcripts (mean ± SEM of three technical replicates) in the indicated sorted cells from pools of 3 uLNs or dLN at 16 hpi (P relative to unsorted ndLN). B) Mean ± SEM of Ifng transcripts at 16 hpi in the dLN of mice (n=5) that had been treated with isotype control or depleted of NK cells with PK136 mAb. C) Frequency of IFN-γ+ NK1.1+ cells in the ndLN or the dLN of individual mice (with Mean ± SEM) at 16 hpi (accumulated data from two experiments with n=5 each). D) Frequency of NK1.1+ cells in the dLN and ndLN at 24 hpi. Individual mice with mean ± SEM for two independent experiments with n=3 each is shown. E) Gating strategy to identify classical NK cells vs. other types of G1-ILC in the dLN. Plots are concatenated for five mice/group. Black contour plots represent staining with the indicated mAb and red contour plots with the corresponding isotype control. F) Mean ± SEM (n=5) for IFN-γ transcripts (fold increase over ndLN) in the dLN at 16 hpi of B6 mice treated with rat IgG1 (isotype control) or the NKG2D blocking mAb CX5. G) Frequency of IFN-γ+ NK1.1+ cells in the dLN at 24 hpi in individual mice with Mean ± SEM (n=5) treated as indicated. H) Frequency of NK1.1+ cells at 2.5 dpi in the uLN or the dLN of individual mice (with mean ± SEM, n=4-8) treated as indicated. Comparison of uLN vs anti-NKG2D was not significant. I) Expression of the indicated transcript in the indicated cell populations sorted at 24 hpi from pools of five dLNs (data expressed as relative to unsorted ndLN) All comparisons between mDCs vs. all other cell types P<0.01 to P<0.0001. All other comparisons were not significant. Data correspond to 3 technical replicates. J) Gating strategy for the sorting of infected and uninfected skin-derived mDCs from dLN at 2.5 dpi with ECTV-GFP, and expression of the indicated transcripts in the indicated cells from pooled LNs from 5 B6 and Tlr9−/− mice. Data is for three technical replicates. Indicated P values vs. GFP+ K) Gating strategy to determine MULT1 in uninfected (dsRed−) and infected (dsRed+) mDC in the dLN at 2 dpi with ECTV-dsRed and dot plot showing the MFI with mean for MULT1 at the surface of dsRed− and dsRed+ mDC in individual mice (accumulated data from two experiments with n=4 each, with mean ± SEM).
Next, we determined whether the cells producing IFN-γ were NK cells and/or other Group 1 ILC. We found that the vast majority of NK1.1+ cells that produced IFN-γ also expressed CD49b+, a typical NK cell marker. Of these, ~62% could definitively be classified as NK cells because they were Eomes+. The reminder 38% Eomes− were difficult to classify because the majority did not express the ILC1 marker CD127 and a minority did express it but at low levels (Figure 7E). From this, we conclude that the majority of the cells producing IFN-γ in the dLN at 24 hpi are NK cells while the reminder are atypical Group 1 ILCs, either Eomes− NK cells, or CD49b+ CD49alow/- ILC1.
We have previously shown that the activating receptor NKG2D is important to control the early dissemination of ECTV and resistance to mousepox (Fang et al., 2008). Thus, we next investigated the possible involvement of NKG2D in early IFN-γ production. In vivo, NKG2D blockade with mAb CX5 resulted in decreased IFN-γ transcripts (Figure 7F) and decreased frequency of IFN-γ+ NK1.1+ cells (Figure 7G) in the dLN at 24 hpi; and significantly reduced recruitment of NK cells to the dLN at 2.5 dpi (Figure 7H). Thus, NKG2D is required for optimal production of early IFN-γ and for optimal NK cell recruitment.
Activation through NKG2D requires interaction with NKG2D ligands such as Rae-1δ (encoded by Raet1d), Rae-1ε (Raet1e) and MULT-1 (Ulbp1) in B6 mice. Because it is known that viral infection can increase the expression of NKG2D ligands in antigen presenting cells (Raulet et al., 2013), we determined whether virus infection of skin-derived mDCs (CD11c+ MHC-IIhi) or other LN cell populations such as B cell (CD19+), myeloid CD11b+ cells, myeloid CD11b− Gr1+ cells and CD8α+ DCs upregulate NKG2D ligands at early stages of infection. Unfortunately, at 24 hpi, the low frequency of ECTV infected cells (as determined by green or red fluorescense when using ECTV-GFP or ECTV-dsRed, respectively) did not allow us to directly analyze individual cells for infection and surface NKG2D ligands by flow cytometry. However, by RT-qPCR, mDC sorted from the dLN at 24 hpi had high and significantly more transcripts of the viral gene EVM003 than any other cell population. Moreover, compared to the other cells types, mDCs also had significantly higher expression of Raet1d and Raet1e and Ulbp1 (Figure 7I). These data suggest that mDCs carrying virus from the footpad to the dLN upregulate NKG2D ligands to stimulate IFN-γ production in NK cells and other Group 1 ILC already present in the dLN at the initial times post infection. To determine whether direct infection was necessary for NKG2D ligand upregulation, we performed experiments at 2.5 dpi, when infection with ECTV-GFP or ECTV-dsRed allows for the identication of infected cells. We found that in B6 mice infected with ECTV-GFP, infected (GFP+) but not uninfected (GFP−) mDC upregulated Raet1d and Raet1e. This upregulation was TLR9 dependent because infected mDCs did not upregulate Raet1e or Raet1d in Tlr9−/− mice (Figure 7J). In addition, in B6 mice infected with ECTV-dsRed, dsRed+ mDC had higher expression of MULT1 at the cell surface than dsRed− mDC from the same dLN (Figure 7K). At this time point, other types of infected cells, such as iMo and lymph node resident CD4+ and CD8α+ DCs, also upregulated NKG2D ligands, albeit at significantly lower levels than mDCs (Figure S4). This suggests that iMo and other DC types could also play a role in the NKG2D-dependent activation of NK1.1+ cells, but at later times post infection.
Discussion
We previously used the natural mouse pathogen ECTV to show that NK cells recruited to the dLN use direct anti-viral effector functions, such as IFN-γ production and cytolytic killing of infected cells, to control virus dissemination. In this way, NK cells play a fundamental role in resistance to lethal mousepox (Fang et al., 2008; Fang et al., 2011; Fang et al., 2010). However, the specific mechanisms involved in the recruitment of NK cells to the dLN has remained obscure. Here, we have deconstructed a choreographed mechanism whereby mDC, Group 1 ILC (mostly NK cells), and iMo communicate with each other at very early stages of the infection to rapidly recruit NK cells to the dLN of ECTV-infected mice.
We show that soon after ECTV enters through the footpad, infected skin-derived mDC in the dLN, which likely emigrate from the skin, use TLR9 sensing to upregulate NKG2D ligands. In the dLN, NKG2D signaling induces classical NK cells and other Group 1 ILCs to produce IFN-γ. Uninfected iMo respond to this IFN-γ by producing the CXCR3 ligand CXCL9 and possibly other chemokines, which distribute throughout the stromal cell network and HEVs. NK cells then use CXCR3 for optimal migration into the infected dLN, presumably by recognizing CXCL9 on HEVs.
Our work here and previously published complementary work (Xu et al., 2015) demonstrates that infected mDCs use intrinsic TLR9 and MyD88 to perform at least two functions required for the optimal recruitment of NK cells: 1) They upregulate NKG2D ligands to induce IFN-γ production in NK cells and other Group 1 ILCs (this paper); and 2) they produce CCL2 and CCL7 to recruit iMo to the dLN (Xu et al., 2015). In addition, mDCs perform some of the same functions of iMo but to a lower extent, including CXCL9 production when uninfected, and IFN-I production when infected (data not shown). Of interest, we found that NK cells do not need intrinsic TLR9 or MyD88 expression to migrate into the dLN. This finding differs from previous work showing impaired function of Tlr2−/− and Myd88−/− NK cells in mice infected intraperitoneally with the related vaccinia virus (Martinez et al., 2010). It should be noted, however, that TLR2 is not required for the control of ECTV (O’Gorman et al., 2010; Sutherland et al., 2011). The reasons for these differences remain undetermined, but the disparate results highlight the fact that the mechanisms of control can vary, even for highly related viruses.
Our work shows that steady-state NK cells and other Group 1 ILCs in the dLN play an important indirect role in anti-viral protection. It would be of interest to further elucidate and distinguish the mechanisms and functions of steady-state NK cells and other Group 1 ILC present in the dLN during the early anti-viral immune response as they may have an underappreciated foundational role in dictating the effectiveness and breadth of the anti-viral response. For example, in addition, to stimulation through NKG2D and the resulting production of IFN-γ, other cytokines produced by mDC could affect ILC1 and NK cells differently, and they could produce other cytokines that could have direct and indirect anti-viral effects. In this regard, we have found in the dLN at 24 hpi a large increase of TNFα and modest but significant increase in other proinflammatory cytokines such as IL-1α and IL-12 (Figure S5). However, the contribution of these cytokines to the early Group 1 ILC response remains to be elucidated.
We previously showed that mDCs use TLR9/MyD88 to recruit iMo to the dLN, and that these iMo are major producers of T1-IFN but only after they become infected (Xu et al., 2015). We now show that iMo produce CXCL9 in response to IFN-γ before but not after becoming infected. Thus, depending on their infection status, iMo play at least two essential roles at protecting form viral dissemination: 1) Uninfected iMo receiving direct IFN-γ signals from Group 1 ILC help recruit NK cells by upregulating CXCL9 and possibly other chemokines (this paper); and 2) infected iMo detecting virus in the cytosol through STING-IRF7 upregulate transcription of anti-viral T1-IFN (Xu et al., 2015). These finding are interesting because they indicate that iMo differentially activate anti-viral gene networks according to their infection status. Future work should identify the mechanisms that trigger this transcriptional switch, determine which are the anti-viral genes that are induced or inhibited by co-lateral and self-infection, and seek to understand the cross-talk between infected and uninfected cells in vivo.
We found increase transcription but did not detect protein for CXCL10, the other CXCR3 ligand in B6 mice. The reason we failed to detect CXCL10 is not clear. One possibility is that the Abs to CXCL10 are not as sensitive as those to CXCL9. However, it is also possible that CXCL10 is not efficiently produced, and that CXCL9 is the only CXCR3 ligand that invokes NK cells to the dLN during ECTV infection. In support of this latter possibility, Ifng−/− mice did not produce CXCL9 and failed to efficiently recruit NK cells to the dLN even though they transcribed some CXCL10. Furthermore, others have shown that after administration of complete Freund’s adjuvant, CXCL9 but not CXCL10 localizes to HEV (Janatpour et al., 2001).
Previous research showed that CXCR3 is necessary for the migration of NK cells to the dLN following immunization with LPS-matured DCs or administration of various adjuvants, but not with CpG DNA, a form of double stranded DNA that is a well-known ligand for TLR9 (Martin-Fontecha et al., 2004). That CpG DNA does not recruit NK cells while TLR9 and MyD88 are essential to recruit NK cells during ECTV infection suggests important differences of NK cell recruitment for inert adjuvants and natural viral infections. Our work demonstrates that during ECTV infection, CXCR3 is important for NK cell recruitment but it is not absolutely essential, suggesting that NK cells can use alternative pathways to migrate into dLNs. Additional work will be necessary to identify these alternative pathways.
Our finding that indirect viral induction of CXCR3 ligands in the dLN is important to curb systemic viral dissemination aligns with two previous reports. Pak-Wittel et al. showed a role for CXCR3 in NK cell recruitment for the control of cowpox virus (Pak-Wittel et al., 2013), while Sung et al. showed that CXCL9 and CXCL10 were major players in promoting virus control in the context central memory CD8+ T cell protection from LCMV (Sung et al., 2012). However, most of the specific details of our findings differ strikingly from these two reports, and the depth of the analysis also differed. Pak-Wittel et al. showed that during cowpox virus infection, NK cells migrated to the dLN in a process that was fully dependent on CXCR3. Based on clodronate-depletion experiments, this group also concluded that it is subcapsular macrophages that produce CXCL9 and CXCL10 in response to IFN-γ. However, the source of IFN-γ was not identified and a possible role for mDCs and TLR9/MyD88 was not investigated. In Sung’s report, CXCL9 and CXCL10 were restricted to the outer areas of the dLN and favored the intranodal migration of central memory CD8+ T cells towards them. In this model, CXCL9 and CXCL10 were produced by subcapsular macrophages in response to IFN-γ or T1-IFN, respectively. It is not clear however, whether cowpox and LCMV penetrate deeply into the dLN soon after infection or whether they remain confined to the subcapsular sinus and medulla (Sung et al., 2012). On the other hand, our data shows that ECTV, a natural pathogen of the mouse, initially localizes not only to subcapsular but also to paracortical areas of the dLN, to then spread swiftly throughout the whole dLN and then systemically. Our data suggests that even before this massive intranodal spread of ECTV occurs, uninfected iMo in the dLN produce CXCL9 in an IFN-γ-dependent manner. These iMo are recruited to the dLN in a process that requires MyD88 and TLR9 in mDC, which presumably carry the infectious virus into the dLN. Within the dLN, CXCL9 rapidly distributes to HEV, probably through the conduits formed by the FRC. CXCL9, likely presented at the lumen of HEV, could now be recognized by CXCR3 on NK, resulting in their entry into the dLN. In this model T1-IFN appear to protect the integrity of the dLN but not to induce the expression of CXCL10. Our report also suggests that, in addition to CXCR3, NK cells can use additional, still unidentified mechanisms to enter the dLN.
Experimental Procedures
Mice:
All experiments were approved by the FCCC or TJU Institutional Animal Care and Use Committee (IACUC). Mice of both sexes at 5-12 weeks of age were used. Additional mouse information can be found in the Online Supplemental methods section.
Viruses:
ECTV-GFP (Xu et al., 2008), ECTV-Luc (Xu et al., 2012) and ECTV-dsRED (Roscoe et al., 2012) have been described. ECTV-mCherry was generated using the same procedures and found to be fully virulent in LD50 experiments. Stocks of the recombinant ECTV or WT ECTV Moscow (ATCC VR-1374) were propagated and titered as previously described (Fang et al., 2006; Fang et al., 2008). Infections were performed in the footpad with 3,000 pfu ECTV unless otherwise indicated.
Bioluminescent imaging:
ECTV-Luc loads in infected mice were measured in vivo as described (Xu et al., 2012) but using an IVIS LXR system (Caliper Life Sciences; Hopkinton, MA, USA). To avoid the overexposure signal effect from the infected foot, the lower part of the mice was covered with black cardboard. X-ray and radiance images (represented as pseudocolor) were taken and overlayed. The photons/second emitted from the liver region of the mice were quantified using a fixed region of interest (ROI) of 2×1.5 cm.
Bone marrow chimeras:
2×105 Ifngr−/− and 2×105 B6.CD45.1 bone marrow cells were injected into irradiate B6 mice (doses of 600 and 500 rads 4 hs apart) intravenously to generate WT+Ifngr−/− →WT mice. 45 days after reconstitution, the mice were infected with ECTV.
Adoptive transfers:
CFSE-labeled splenocytes from B6-CD45.1+ and mutant mice (107 each) were injected intravenously into B6 or mutant (CD45.2+) recipient mice. The next day, mice were infected with ECTV in the footpad. At 2 dpi, dLN and ndLN were harvested, the cells counted, stained and analyzed by flow cytometry (see below) as indicated in the Figures.
NK cell migration assay:
NK cells were enriched from the spleens of 10 NKp46-GFP mice by negative selection using the NK cell isolation kit II (Miltenyi Biotech) as directed by the manufacturer. These cells were further purified with a FACS Vantage sorter (BD) according to Forward scatter and GFP expression without any staining. Cells were 99% pure NK cells as determined by flow cytometry in aliquots stained with NK1.1 and CD3 Abs. Mouse CK CCL5, CCL12, CXCL9 and CXCL10 (Peprotech) were reconstituted in sterile PBS containing 0.1% BSA, diluted as indicated, and loaded in the lower chambers of 24-well trans-well plates (Corning). Purified NK cells (2.5×107 in 10 ml) were place in the upper chambers of the trans-wells and incubated at 37ºC for 2h. The cells in the lower chambers were then harvested, counted, stained and analyzed by flow cytometry.
Western blot:
LNs were homogenized in PBS containing Aprotinin, 10 μg/mL Leupeptin, and 10 μg/mL Pepstatin using a Tissuelyser instrument (Qiagen). After homogenization, Triton X-100 was added to a final concentration of 1%. Samples were frozen at −80 °C, thawed and centrifuged at 10,000 x g for 5 minutes to remove cellular debris. Supernatants were collected, protein quantified, and 10 μg protein in SDS sample buffer was run in a precast 10% Tri-Tricine minigel (Biorad). Proteins were transferred to a PVDF membrane, incubated with goat anti-mouse CXCL9 or b-actin antibody (R&D) followed by HRP labeled anti-goat IgG. Bands were detected by chemilumminiscence.
Confocal microscopy:
uLN and dLN were harvested at different dpi, fixed in paraformaldehyde, dehydrated, embedded in OCT-freezing media (Tissue-Tek), cut into 8µ frozen sections, stained with anti-CXCL9 (R&D), anti-PNAD (MECA-79, BD) or anti-ER-TR7 (Santa Cruz). Alexa flour 488 donkey anti-rat and Dyelight 549 Bovine anti-goat (Jackson) were used as secondary antibodies. Images were collected with a Nikon C1 laser Scanning microscope (LSCM) and all the micrographs assembled into a single file using Photoshop software. Contrast and brightness was adjusted in the assembled files to improve visualization in the printed document.
Flow Cytometry:
Flow cytometry was done as described previously (Fang et al., 2008; Fang et al., 2011; Fang et al., 2010; Xu et al., 2008). Briefly, LNs were obtained from mice and made into single-cell suspensions. After blocking nonspecific binding to FC receptor by antibodies 2.4G2 (anti-Fc-γ II/III receptor; American Type Culture Collection), the cells were stained with. Cells were analyzed/and or sorted by flow cytometry at the Fox Chase and TJU Flow Cytometry Facilities using LSR II, Fortessa and FACSARIA instruments (all from Becton Dickinson).
Quantitative Reverse transcript (qRT-PC):
for CXCL9, CXCL10 and GAPDH was performed as before using the Universal Library (Roche) library probes as well as 2X FastStart Universal Probe Master with ROX (Roche) (Xu et al., 2012) or, alternatively, using iTaq Universal SYBR Green with the primers indicated in Supplemental experimental Methods. Samples were analyzed using a Biorad CFX96 system.
Statistical analysis:
Data were analyzed using Prism 5 software (GraphPad Software Inc.). We analyzed the survival curves with a Log-rank (Mantel-Cox) Test and the qPCR experiments with unpaired two-tailed or ANOVA as necessary. In all figures * = P<0.05; ** = P<0.01; *** = P<0.001, **** = P<0.0001. All experiments were repeated a minimum of two times with similar results.
Supplementary Material
Acknowledgments
We thank Fox Chase Cancer Center and Thomas Jefferson University Laboratory Animal and Flow Cytometry Facilities for their services and Jennifer Wilson for editing the manuscript. This work was supported by NIAID grants R01AI110457, R01AI065544 and AG048602 to LJS. Eric Wong was partially supported by F32AI129352.
Footnotes
Declaration of Interests
The authors declare no competing interests
References
- Abbas AK, Lichtman AH, and Pillai S (2007). Cellular and molecular immunology, 6th edn (Philadelphia: Saunders Elsevier; ). [Google Scholar]
- Chen S, Kawashima H, Lowe JB, Lanier LL, and Fukuda M (2005). Suppression of tumor formation in lymph nodes by L-selectin-mediated natural killer cell recruitment. The Journal of experimental medicine 202, 1679–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang M, Cheng H, Dai Z, Bu Z, and Sigal LJ (2006). Immunization with a single extracellular enveloped virus protein produced in bacteria provides partial protection from a lethal orthopoxvirus infection in a natural host. Virology 345, 231–243. [DOI] [PubMed] [Google Scholar]
- Fang M, Lanier LL, and Sigal LJ (2008). A role for NKG2D in NK cell-mediated resistance to poxvirus disease. PLoS pathogens 4, e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang M, Orr Mark T, Spee P., Egebjerg T., Lanier Lewis L., and Sigal Luis J. (2011). CD94 Is Essential for NK Cell-Mediated Resistance to a Lethal Viral Disease. Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang M, Roscoe F, and Sigal LJ (2010). Age-dependent susceptibility to a viral disease due to decreased natural killer cell numbers and trafficking. The Journal of experimental medicine 207, 2369–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint SJ, and American Society for Microbiology. (2009). Principles of virology, 3rd edn (Washington, DC: ASM Press; ). [Google Scholar]
- Gasteiger G, Fan X, Dikiy S, Lee SY, and Rudensky AY (2015). Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 350, 981–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, et al. (2012). Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nature immunology 13, 1118–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, and Akira S (2000). A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745. [DOI] [PubMed] [Google Scholar]
- Iannacone M, Moseman EA, Tonti E, Bosurgi L, Junt T, Henrickson SE, Whelan SP, Guidotti LG, and von Andrian UH (2010). Subcapsular sinus macrophages prevent CNS invasion on peripheral infection with a neurotropic virus. Nature 465, 1079–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janatpour MJ, Hudak S, Sathe M, Sedgwick JD, and McEvoy LM (2001). Tumor necrosis factor-dependent segmental control of MIG expression by high endothelial venules in inflamed lymph nodes regulates monocyte recruitment. The Journal of experimental medicine 194, 1375–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junt T, Moseman EA, Iannacone M, Massberg S, Lang PA, Boes M, Fink K, Henrickson SE, Shayakhmetov DM, Di Paolo NC, et al. (2007). Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature 450, 110–114. [DOI] [PubMed] [Google Scholar]
- Karupiah G, Fredrickson TN, Holmes KL, Khairallah LH, and Buller RM (1993). Importance of interferons in recovery from mousepox. J Virol 67, 4214–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Hashimoto-Hill S, and Kim M (2016). Migration and Tissue Tropism of Innate Lymphoid Cells. Trends in immunology 37, 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose CSN, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, Fabiunke C, Pfeifer D, Sexl V, Fonseca-Pereira D, et al. (2014). Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 157, 340–356. [DOI] [PubMed] [Google Scholar]
- Lodoen MB, and Lanier LL (2006). Natural killer cells as an initial defense against pathogens. Current opinion in immunology 18, 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Fontecha A, Thomsen LL, Brett S, Gerard C, Lipp M, Lanzavecchia A, and Sallusto F (2004). Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nature immunology 5, 1260–1265. [DOI] [PubMed] [Google Scholar]
- Martinez J, Huang X, and Yang Y (2010). Direct TLR2 signaling is critical for NK cell activation and function in response to vaccinia viral infection. PLoS pathogens 6, e1000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, Pandey G, Leboeuf M, Elpek KG, Helft J, et al. (2012). Deciphering the transcriptional network of the dendritic cell lineage. Nature immunology 13, 888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Gorman WE, Sampath P, Simonds EF, Sikorski R, O’Malley M, Krutzik PO, Chen H, Panchanathan V, Chaudhri G, Karupiah G, et al. (2010). Alternate mechanisms of initial pattern recognition drive differential immune responses to related poxviruses. Cell host & microbe 8, 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak-Wittel MA, Yang L, Sojka DK, Rivenbark JG, and Yokoyama WM (2013). Interferon-gamma mediates chemokine-dependent recruitment of natural killer cells during viral infection. Proceedings of the National Academy of Sciences of the United States of America 110, E50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchanathan V, Chaudhri G, and Karupiah G (2005). Interferon function is not required for recovery from a secondary poxvirus infection. Proceedings of the National Academy of Sciences of the United States of America 102, 12921–12926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raulet DH, Gasser S, Gowen BG, Deng W, and Jung H (2013). Regulation of ligands for the NKG2D activating receptor. Annual review of immunology 31, 413–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, Gilfillan S, Colonna M, and Immunological Genome C (2015). Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nature immunology 16, 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscoe F, Xu RH, and Sigal LJ (2012). Characterization of ectromelia virus deficient in EVM036, the homolog of vaccinia virus F13L, and its application for rapid generation of recombinant viruses. J Virol 86, 13501–13507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rot A, and von Andrian UH (2004). Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annual review of immunology 22, 891–928. [DOI] [PubMed] [Google Scholar]
- Rubio D, Xu RH, Remakus S, Krouse TE, Truckenmiller ME, Thapa RJ, Balachandran S, Alcami A, Norbury CC, and Sigal LJ (2013). Crosstalk between the type 1 interferon and nuclear factor kappa B pathways confers resistance to a lethal virus infection. Cell host & microbe 13, 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelsson C, Hausmann J, Lauterbach H, Schmidt M, Akira S, Wagner H, Chaplin P, Suter M, O’Keeffe M, and Hochrein H (2008). Survival of lethal poxvirus infection in mice depends on TLR9, and therapeutic vaccination provides protection. The Journal of clinical investigation 118, 1776–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz O, Hammerschmidt SI, Moschovakis GL, and Forster R (2016). Chemokines and Chemokine Receptors in Lymphoid Tissue Dynamics. Annual review of immunology 34, 203–242. [DOI] [PubMed] [Google Scholar]
- Sung JH, Zhang H, Moseman EA, Alvarez D, Iannacone M, Henrickson SE, de la Torre JC, Groom JR, Luster AD, and von Andrian UH (2012). Chemokine guidance of central memory T cells is critical for antiviral recall responses in lymph nodes. Cell 150, 1249–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland DB, Ranasinghe C, Regner M, Phipps S, Matthaei KI, Day SL, and Ramshaw IA (2011). Evaluating vaccinia virus cytokine co-expression in TLR GKO mice. Immunology and cell biology 89, 706–715. [DOI] [PubMed] [Google Scholar]
- Walzer T, and Vivier E (2011). G-protein-coupled receptors in control of natural killer cell migration. Trends in immunology 32, 486–492. [DOI] [PubMed] [Google Scholar]
- Xu RH, Cohen M, Tang Y, Lazear E, Whitbeck JC, Eisenberg RJ, Cohen GH, and Sigal LJ (2008). The orthopoxvirus type I IFN binding protein is essential for virulence and an effective target for vaccination. The Journal of experimental medicine 205, 981–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu RH, Fang M, Klein-Szanto A, and Sigal LJ (2007). Memory CD8+ T cells are gatekeepers of the lymph node draining the site of viral infection. Proceedings of the National Academy of Sciences of the United States of America 104, 10992–10997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu RH, Rubio D, Roscoe F, Krouse TE, Truckenmiller ME, Norbury CC, Hudson PN, Damon IK, Alcami A, and Sigal LJ (2012). Antibody inhibition of a viral type 1 interferon decoy receptor cures a viral disease by restoring interferon signaling in the liver. PLoS pathogens 8, e1002475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu RH, Wong EB, Rubio D, Roscoe F, Ma X, Nair S, Remakus S, Schwendener R, John S, Shlomchik M, and Sigal LJ (2015). Sequential Activation of Two Pathogen-Sensing Pathways Required for Type I Interferon Expression and Resistance to an Acute DNA Virus Infection. Immunity 43, 1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.