

Abstract
Heart valves are dynamic, highly organized structures required for unidirectional blood flow through the heart. Over an average lifetime, the valve leaflets or cusps open and close over a billion times, however in over 5 million Americans, leaflet function fails due to biomechanical insufficiency in response to wear-and-tear or pathological stimulus. Calcific aortic valve disease (CAVD) is the most common valve pathology and leads to stiffening of the cusp and narrowing of the aortic orifice leading to stenosis and insufficiency. At the cellular level, CAVD is characterized by valve endothelial cell dysfunction and osteoblast-like differentiation of valve interstitial cells. These processes are associated with dysregulation of several molecular pathways important for valve development including Notch, Sox9, Tgfβ, Bmp, Wnt, as well as additional epigenetic regulators. In this review, we discuss the multifactorial mechanisms that contribute to CAVD pathogenesis and the potential of targeting these for the development of novel, alternative therapeutics beyond surgical intervention.
Keywords: aortic valve calcification, extracellular matrix, valve interstitial cell, valve endothelial cell, hemodynamics, epigenetics, signaling, development
Introduction
Two sets of cardiac valves open and close over 100,000 times a day to maintain unidirectional blood flow through the heart. The atrioventricular (AV) valves (mitral, tricuspid) regulate flow from the atria into the ventricular chambers, while the semilunar valves (aortic, pulmonary) guide flow out of the ventricles into the pulmonary and systemic circulation. This necessitate function of the valves is largely facilitated by a highly organized connective tissue, composed of stratified layers of extracellular matrix (ECM), and valve interstitial and endothelial cell populations that molecularly communicate (Figure 1A). Establishing and maintaining heart valve connective tissue is essential for structure-function relationships, and aberrations in embryonic development or adult homeostasis underlie dysfunction and disease that affect more than 5 million Americans each year (1, 2).
Figure 1.

Schematic representation of a healthy and calcified aortic valve cusp. Cross sectional representation of a heart highlighting the aortic valve (box). (A) A healthy aortic valve cusp structure consists of three layers of extracellular matrix (ECM); the ventricularis (elastin fibers, black); the spongiosa (proteoglycans, blue); and the fibrosa (collagens, yellow). In addition to the matrix, the valve cusp is encapsulated by a single layer of valve endothelial cells (VECs), while valve interstitial cells (VICs) populate the core of the cusp. (B) A calcified valve cusp is thickened with abrogations in ECM organization including fragmented elastin fibers and increased collagen content. In addition, calcific nodules form on the fibrosa surface of the cusp (off-white).
In contrast to healthy valve structures, diseased valves are characterized by loss of ECM organization and injury to, or dysfunction of valve cell populations (1). Calcific aortic valve disease (CAVD) is the most common form of valvular pathology and the third most common cardiovascular disease following hypertension and ischemic heart disease (3–5). The disease is a significant economic and healthcare burden contributing to over 15,000 deaths in the US annually (6). The pathogenesis of affected aortic valves is characterized by ectopic development of calcium nodules on the aortic surface of the valve cusp, annulus, or both (Figure 1B). The resulting sclerotic aortic valve exhibits compromised function by virtue of limited movement which progresses to calcific aortic stenosis over time (7). Over 2.5 million individuals are affected by aortic stenosis which can lead to significantly impaired cardiac function and if left untreated, death. CAVD and aortic stenosis is most prevalent in the aging population but is also observed during earlier stages of life in individuals born with bicuspid aortic valve (BAV) disease.
The underlying etiology of CAVD is largely unknown. While BAV is proven hereditary, there is limited information regarding genetic contributions to CAVD [reviewed in (8)]. Risk factors are known and similar to most other cardiovascular diseases (hypertension, tobacco-use, hypercholesterolemia, etc.), however the mechanisms that underlie calcific nodule formation on the aortic cusp surface following exposure to risk factors are largely unknown. The hemodynamic environment of the aortic valve is known to play a role in valve pathogenesis, and studies using animal and in vitro models have identified aberrations in critical signaling pathways required for valve formation in CAVD [reviewed in (8)]. However, the field has yet to delineate cause and effect of these multifactorial contributors. The current limitations in understanding the etiology of CAVD has hindered the development of alternative therapeutics beyond surgery, to prevent or regress CAVD. Therefore, further basic science research is needed to decipher the cellular and molecular processes underlying the pathology of CAVD and translate these discoveries into mechanistic-based pharmacological therapies to reestablish valve structure-function relationships.
Healthy heart valve structure-function relationships
The mature valve structures are composed of leaflets (AV) or cusps (semilunar) with supporting structures. In the AV position, the mitral valve consists of two leaflets, while the tricuspid possesses three, and both display external supporting chordae tendineae that attach the underside of the valve leaflet to the papillary muscles within the ventricle (9). The three cusps of the semilunar valves (aortic, pulmonic) lack external support, but a unique supporting structure within the aortic roots in the form of a fibrous annulus has been described (9). The “Lub-Dub” noise of the heart beat is attributed to sequential closing of the AV and semilunar valve leaflets/cusps, respectively, during the cardiac cycle and this is driven by the valve hemodynamics. In systole, the aortic valve cusps open and experience oscillatory flow patterns on the aortic surface and laminar shear on the ventricular side with overall low stress, while the mitral valve leaflets are closed to prevent back flow into the left atrium and therefore pressure is high on the ventricular side. In contrast during diastole, the closed aortic cusps create high pressure and tensile stretch on the aortic and ventricular surfaces, respectively, while open mitral leaflets experience laminar shear flow and reduced pressure (10). This coordinated movement of the valve leaflets/cusps and their supporting structures in response to the hemodynamic environment is attributed to a highly specialized connective tissue that provides all the necessary biomechanical properties during diastole and systole. The extracellular component of the valve connective tissue is largely composed of three stratified layers of matrix arranged according to blood flow (see Figure 1A) (1, 11, 12). The cross-sectional structure of healthy valve leaflets contains the fibrosa layer located on the ventricular side of the AV valve leaflets and atrial side of the semilunar valves, away from blood flow. This layer is predominantly composed of bundles of collagen fibers aligned along the circumferential direction of the free edge of the leaflets (13–16). This arrangement provides tensile strength and flexibility to the valve leaflet/cusp during opening, while transmitting forces to promote coaptation of the leaflets in the closed position (17–19). Adjacent to the fibrosa is the spongiosa layer, with a lower abundance of collagens, high prevalence of proteoglycans, and water retention. This composition provides a more compressible matrix, allowing the valve to geometrically “flex” and absorb high force (16, 20). Finally, the layer adjacent to blood flow is termed the atrialis (AV) or ventricularis (semilunar) and largely consists of radially orientated elastin fibers that allow for high deformations to facilitate tissue movement as the valve leaflet opens and recoils during closure (21–23). In the mitral position, histological studies of human tissue report an additional fourth layer of elastin on the opposing side to the atrialis, which presumably allows for further flexibility (11). The AV chordae tendinae are composed of a cylindrical collagen core within an elastin sheath and exhibit high viscoelastic properties, while the “built-in” supporting structures of the semilunar valves contain similar extracellular matrix (ECM) components only arranged within the underside of the cusp structure (1, 24, 25). The overall protein contents of the valve matrix is adaptive and has been shown to remodel in response to normal wear-and-tear and aging and this is thought be beneficial in maintaining structure-function relationships throughout life (26).
In addition to the extracellular component of the valve, the mature leaflet/cusp contains several differential cell populations. The valve interstitial cells (VICs) are the most abundant cell type and have been described as heterogeneous and fibroblast-like in nature (27). In the healthy adult, VICs express markers such as Vimentin (28) and are considered quiescent as proliferation rates are comparatively low (~1%), functioning to mediate turnover of the valve ECM in response to general wear-and-tear (27, 29, 30). Previous studies have identified sub-populations of VICs based on molecular profiles (28), and this may be attributed to differential embryonic origins of this cell population (described below), or potential mechanical influences from the ECM or hemodynamic environment as described (31). Although VICs from the four heart valves all exhibit a fibroblast-like phenotype (32), those isolated from left-side valves (aortic and mitral) exhibit a greater stiffness compared to those isolated from right-side valves (33, 34). Furthermore, it was shown that aortic VICs exhibit greater capability of contracting the ECM relative to those isolated from pulmonary valves (34), highlighting the influence of the hemodynamic environment on VIC behavior.
In addition to VICs, the valve leaflet/cusp is encapsulated by a single layer of valve endothelial cells (VECs) that form a tight barrier between the circulating blood, and the underlying cellular and extracellular contents of the valve. VECs are dynamic, and molecular and phenotypic profiles are side-, and age-dependent (30, 35–38). Furthermore, there is increasing evidence to suggest that VEC function has important implications in regulating VIC behavior to maintain valve homeostasis and prevent disease (39–41). A relatively minor contributor to the valve cell population includes extra-cardiac hematopoietic or bone marrow-derived cells (42, 43). Under homeostatic conditions, these cells make up ~1–18% of the total valve cell population, depending on age and valve position, and maintain CD45 expression with co-expression of VIC markers including Vimentin (44). The function of these extra-cardiac cells in healthy valves remains elusive but numbers almost double in mouse models of valve disease (45, 46). Together, it's the integrated network of ECM components and valve cell populations that provide the necessary architecture for efficient valve function throughout life.
Heart valve development
Formation of highly organized mature heart valves begins during embryogenesis when the primitive heart tube undergoes rightward looping and the cardiac jelly expands at the outflow tract (OFT) and the AV canal forming local swellings of endocardial cushions. At this time, a subset of endocardial cells overlying the cushions undergo endothelial-to-mesenchymal transformation (EMT) giving rise to a population of highly proliferative and migratory, valve precursor cells [reviewed in (47)]. EMT is a tightly regulated process important not only for valvulogensis, but embryonic development and differentiation in general. Early experiments employing the chick embryo as a model system demonstrated that explants of AV cushion consisting of endocardium and myocardium exhibited EMT when grown on collagen gels (48). Furthermore, there seems to be a need for specialized spatial signaling to induce EMT, as it was shown that only the endocardium of AV and OFT cushions, but not ventricular endocardium, undergoes EMT (49, 50). While the mechanisms of cushion development are conserved between AV and OFT valves, EMT in OFT cushions lags behind that occurring in the AV cushions. While endothelial-derived cells contribute the majority of mesenchyme cells within the cushions (51, 52), there is additional contribution from the cardiac neural crest and secondary heart field cells in the OFT position (53–55), and epicardial-derived cells are present in the parietal leaflets of the AV valves (56).
Many signaling pathways have been implicated in EMT and these cross-talk with each other to form complex molecular networks between differential cell types. The process is largely initiated by Transforming Growth Factor-β (Tgfβ) (47, 57–59), and Bone Morphogenetic Protein (BMP) (57, 60–68) signaling pathways emanating from the adjacent myocardium, as well as Notch predominant in valve endothelial cells (69–77). Wnt signaling in the endothelial-lineage is also critical for early stages of EMT (58, 78). Similar pathways (Tgfβ, Bmp, Wnt) also play a role in remodeling and sculpting the endocardial cushions as they morph into valve primordia during later stages of development (79–83). Aside from growth factors, the transcription factor Sox9 is required for proliferation of newly transformed mesenchyme cells and remodeling of the matrix, but not initiation of EMT (84, 85). In addition to molecular signaling, the hemodynamic environment is important for endocardial cushion formation and it is well-established that the endothelium, via mechanotransduction, senses and responds to hemodynamics (86) via the rearrangement of the cytoskeleton and alignment of these cells in a direction parallel to flow (87). In vitro studies employing a 3D tubular culture have identified the role of shear stress in the expression and deposition of fibrous ECM proteins in both AV and OFT cushions (88, 89). Furthermore, several in vivo studies reinforce the importance of maintaining normal hemodynamic stimuli during valve development, as demonstrated by the molecular/cellular responses that go awry when intracardiac hemodynamics are perturbed leading to a myriad of congenital heart defects (90–106). Defects in EMT or failure to establish the valve precursor cell pool during embryogenesis as a result of molecular abrogations or hemodynamic disturbances is lethal in mice and likely detrimental to human development. However, those affected by more subtle defects in post-EMT growth and maturation survive, but such disturbances could underlie valve malformations present at birth or acquired disease manifested later in life.
Calcific aortic valve disease
Calcific aortic valve disease (CAVD) is the most predominant form of valve pathology affecting more than 5.2 million people in the US, particularly those over the age of 65 (107). In 2013, 50,222 deaths occurred due to valvular heart diseases in the USA, out of which 67.5% were due to aortic valve disorders (6). CAVD is an active cellular-driven pathological process beginning with alterations in the aortic valve cusps (sclerosis) and culminating in stenosis (108, 109). Risk factors for CAVD have been well-established and are shared with many other cardiovascular diseases (110). Histologically, sclerotic calcified valves are thickened with alterations in the composition and distribution of ECM components including fragmented elastin fibers and increased collagen fiber content (111). In addition, deposits of calcium and hydroxyapatite develop on the aortic surface of the cusp leading to bone-like, rigid nodules that limit cusp movement (Figure 1B) (112). The cellular mechanisms underlying the formation of calcific nodules within the valve tissue are not clear and likely diverse. Based on expression studies, it is considered that residing VICs are activated similar to myofibroblasts, and undergo transdifferentiation toward an osteoblast-like cell via a process similar to endochondral ossification. This is concluded from studies in calcified valves excised from humans that show ectopic expression of osteogenic transcription factors including Runx2, as well as mineralized matrix proteins (Matrix Gla Protein, Osteopontin, Bone Sialoprotein) commonly observed in bone (113–115). Histological analysis indicates that in CAVD, osteogenic-like changes (or increased Runx2) occurs in subset of VICs and not the entire population. Some attribute the pathological cell specificity to embryonic origin (116), others suggest that exogenous extra-cardiac cells play a role (117, 118), and there is no doubt that the hemodynamic environment influences formation of calcific nodules (119–121). While the field has been given a taster to these ideas, more in-depth studies are needed. In parallel with VIC pro-osteogenic fate changes in CAVD, studies have shown a significant contribution of extracellular vesicles (EVs) to the formation of calcific nodules at localized sites within the valve structure, similar to that observed in medial arterial calcifications and atherosclerotic intimal plaques (122), which suggest that the process of VIC differentiation and calcific nodule formation is multi-faceted.
CAVD is largely considered an acquired disease that develops later in life, however studies in mice might suggest that pathogenesis might stem from perturbations in embryonic development. Homozygous loss of Notch1, associated ligands or signaling mediators during gastrulation leads to embryonic lethality due to endocardial cushion defects as a result of defective EMT (69–77). Heterozygous mice are viable but when fed Western diet, develop CAVD (123), and cross breeding with the Nos3−/− line increases this incidence with the additional development of BAV (40). In addition, Notch1 is the most predominant disease-causing mutation in humans affected by calcification of the aortic valves (124). Similarly, loss of Sox9 in endothelially-derived cells of the embryo causes early lethality by embryonic day (E)12.5 associated with hypoplasia of the endocardial cushions due to defects in proliferation of newly transformed mesenchyme cells following otherwise normal EMT (85). Despite this, heterozygotes have a normal lifespan, but develop CAVD phenotypes by 3 months of age, even without the addition of Western diet (85, 125). In vivo mutations, or targeted loss of function in vitro are sufficient to cause EMT defects [reviewed in (8)], and endothelial-specific deletion of Tgfβ1 promotes osteogenic differentiation of adult VICs and calcification (41). These studies suggest that Notch1, Sox9, and Tgfβ1 are required for valvulogenesis and viable mice with targeted reduced function develop valve disease later in life, potentially due to subtle embryonic defects that are manifested over time. BMP and Wnt are two signaling pathways previously shown to be activated in calcified valves from human patients, mouse models and cultured VICs (113, 126–137). However, mutations in Wnt or BMP signaling mediators have not been shown to be causative of CAVD, although tissue-specific deletion of the BMP receptor Alk2 underlies BAV in mice (138). The question beckons if “re-activation” of these developmental programs play a role in CAVD pathogenesis or if these are a read out of the pathological state.
While several molecular pathways have been implicated in CAVD, the cellular mechanisms that underlie osteoblast-like changes in resident VICs have not been extensively explored. During early stages of CAVD, endothelial dysfunction has been reported (30, 139). In the vasculature, this pathological process has been well characterized, largely in the setting of atherosclerosis and plaque formation, however, in the valves, endothelial dysfunction is less well described. In a recent study, it was demonstrated that aging, a known risk factor of CAVD, is associated with reduced nitric oxide bioavailability (or increased reactive oxygen species) and decreased proliferation of VECs, in addition to enhanced permeability of the endothelial cell lining. Morphologically, VECs change in size and shape, and express distinct molecular profiles with aging (30). In the last 5 years, there have been an increasing number of studies to show that VECs are protective against calcific nodule formation by porcine aortic valve interstitial cells (pAVICs) in vitro and perturbations in VEC-VIC communication promote the onset of calcification (40, 41, 140). Protective paracrine factors emanating from VECs include nitric oxide (40) and Tgfβ1 (41) and their targets in VICs (and potentially VECs) have been identified as, but are not limited to Notch1 (40) and Sox9 (41), respectively (Figure 2A). There are likely many other pathways that exist between these two cell types that regulate cellular function. A previous study by Hjortnaes et al. demonstrated in vitro that VECs prevent activation of VICs through currently unknown mechanisms, while VICs prevent EMT of VECs suggesting two-way communications (Figure 2B) (140).
Figure 2.
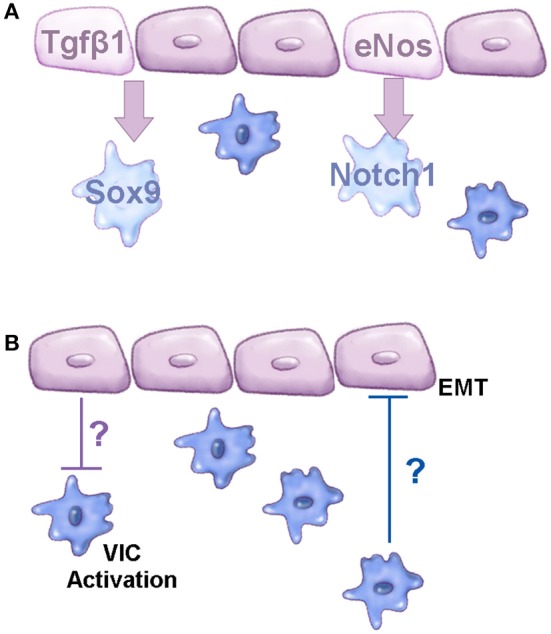
Molecular communications between VEC and VIC populations. (A) Schematic to show known signaling pathways active between VEC and VIC populations that prevent CAVD. (B) Diagram to demonstrate known cellular functions regulated by VEC-VIC communications; the mechanisms underlying these functions are currently unknown, based on studies by Hjortnaes et al. (140). Purple cells represent valve endothelial cells, blue cells are valve interstitial cells.
In addition to the molecular role of VECs communicating with underlying VICs, the endothelium is in direct contact with the circulation and it is known that VECs lining the ventricular and aortic cusp surface sense the complex, fluidic environment (35, 36, 39, 141, 142). There is also strong data to suggest that altered spatio-temporal flow patterns correlate with aortic valve disease. More specifically, it has been suggested that the local hemodynamic environment dictates the localization of calcified nodules, being present exclusively on the aortic aspect (fibrosa) of the aortic valve cusp (35, 143, 144). Furthermore, patients with bicuspid aortic valve (BAV) exhibit different hemodynamic profiles relative to that seen with tricuspid aortic valve (145, 146) and have an increased propensity to develop CAVD as early as 20–30 years of age (2, 147, 148). In addition to alterations in the hemodynamics surrounding the adult valve, studies have also demonstrated that altered intracardiac hemodynamics during early avian development perturbs key molecular pathways involved in valve development and disease, including Tgfβ signaling (100, 101, 106). While correlations between the valve hemodynamics and disease have been made, little is known about how mechanical stress on the endothelium translates into differentiation of VICs toward an osteoblast-like lineage. Previous studies have shown that the complex fluid mechanics surrounding the aortic valve is initially sensed by VECs lining the ventricular and aortic cusp surface (35, 36, 39, 141, 142). Compared to static flow conditions, unidirectional shear stress (20 dynes/cm) (or increased flow) promotes expression of inflammatory response genes and pro-calcification signaling, both of which are known contributors of CAVD pathogenesis (36, 39, 142). Interestingly, these pathogenic responses are greater in cells isolated from the aortic surface of the valve cusp, the side most susceptible to calcification (35, 36, 39, 142). With this in mind, it is appreciated how “endothelial cell dysfunction” has detrimental effects on relaying molecular cues to VICs and promoting pathogenic changes.
Epigenetic regulation of aortic stenosis and calcific aortic valve disease
Dysregulation of critical signaling pathways are detrimental to valve cell function and structure-function relationships and there is emerging work to show that the epigenetic landscape can further influence gene regulation via multiple mechanisms including DNA methylation and non-coding RNAs (149, 150). Micro RNAs (MiRs) and long non-coding RNAs (LncRNAs) lack protein-coding function. MiRs are 18–26 bases long and predominantly inhibit expression of target genes either by directly preventing protein translation through target 3′-UTR binding or inducing mRNA degradation (151). In contrast, LncRNAs are more than 200 nucleotides long and can localize to different sub-cellular depots (152). While there are limited studies on the epigenetic regulation of cardiogenesis and valvulogenesis (153–155), gene methylation status, and LncRNAs and MiRs profiles have shown to be altered in CAVD and aortic stenosis and these are summarized in Tables 1, 2, respectively (181–183).
Table 1.
The Role of DNA methylation in CAVD/Aortic Stenosis.
| Observation in CAVD/AS | Effect | Species | References |
|---|---|---|---|
| Increased levels of DNMT3B in human stenotic aortic valves | Increase in global DNA methylation. More than 6,000 differentially methylated sites were identified between normal and stenotic valves. Expression of the osteogenic marker Sp7 was increased four-fold in stenotic vs. normal tissue | Human | (156) |
| Genetic inactivation of DNMT3B | Protects against activation of osteogenic pathways and slows the progression of aortic stenosis | Mouse | (157) |
| Altered CpG methylation in newborns with congenital aortic stenosis | Differentially methylated CpG sites | Human | (158) |
| Altered methylation of CpG sites | Contribution to regulation of left ventricular hypertrophy due to aortic stenosis -induced pressure overload | Human | (159) |
| Hypomethylation of LncRNA H19 | Promotes VIC osteogenic-like changes by NOTCH1 silencing | Human | (160) |
| 5-methylcytosine (5meC) in intron 1 in a mammalian interspersed repeat element (MIR) was increased by 2.2-fold in CAVD compared to control aortic valves | Phospholipid phosphatase (PLPP3) gene and enzymatic activity were downregulated in mineralized aortic valves | Human | (161) |
| Decreased promoter methylation of the gene encoding the proinflammatory enzyme 5-lipoxygenase (5-LO) | Increased 5-LO mRNA levels. (Aortic stenosis is associated with increased leukotriene production, in part, due to induction of 5-LO in VICs) | Human | (162) |
Table 2.
Role of non-coding RNAs in CAVD/Aortic Stenosis.
| Non-coding RNA | Mechanism in CAVD/AS | Target | Species | References |
|---|---|---|---|---|
| LncRNA MALAT1 | Upregulated. Positive regulator of osteodifferentiation by sponging miR-204 | miR-204 | Human | (163) |
| miR-214 | Upregulation, increased fibrosa thickness and calcification were observed when porcine fibrosa was exposed to oscillatory shear. | TGFβ1 | Porcine | (164) |
| LncRNA H19 | Upregulated | NOTCH1 | Human | (160) |
| miR-30c | Upregulated | ITGB1, CXCL12, FLT1, CAMTA1, COL9A3 | Human | (165) |
| miR-486 | Upregulated in TGFβ1 and BMP2-stimulated VICs and VICs from calcified aortic valves | miR-204, Smurf2 | Human | (137) |
| miR-181b | Upregulated | TIMP3, SIRT1, GATA6 | Human | (166) |
| miR-125b | Upregulated | CCL4 | Human | (167) |
| miR-21-5p, miR-221-3p | Upregulated | TGFβ, MAPK, Wnt signaling pathways | Human | (168) |
| hsa-miR-193a-3p, hsa-miR-29b-1-5p, hsa-miR-505-5p, hsa-miR-194-5p, hsa-miR-99b-3p, and hsa-miR-200b-3p | Upregulated | Unknown | Human | (169) |
| LncRNA TUG1 | Highly expressed. Sponges miR-204-5p | miR-204-5p | Human | (170) |
| miR-92a | Overexpressed in calcified bicuspid aortic valves | Unknown | Human | (171) |
| miR-204 | Downregulated | Smad4, Runx2 | Human | (137, 163, 172) |
| miR-141 | Downregulated | BMP2 | Porcine | (173) |
| miR-106a, miR-148a, miR-204, miR-211, miR-31 and miR-424 | Downregulated | Runx2, BMPR2, BMP2, BMP3, BMP8B, CBFB | Human | (137, 163, 165) |
| miR-195 | Downregulated | Runx2, BMP2, Smad7, Smad1, Smad3, Smad5, Jag2 | Human | (174, 175) |
| miR-30b | Downregulated | Smad1, Smad3, Runx2, caspase-3, Jag2, Smad7, Notch1 | Human | (174, 176) |
| miR-26a | Downregulated | BMP2, ALP, Smad1, Jag2, Smad7, Runx2, Smad5 | Human | (174) |
| miR-122-5p | Downregulated | Lipid metabolism, TGFβ | Human | (168, 177) |
| miR-625-5p | Downregulated | Unknown | Human | (168) |
| miR-30e-5p | Downregulated | PI3K-Akt, MAPK signaling pathway | Human | (168) |
| hsa-miR-3663-3p, hsa-miR-513a-5p, hsa-miR-146b-5p, hsa-miR-1972, hsa-miR-718, hsa-miR-3138, hsa-miR-21-5p, hsa-miR-630, hsa-miR-575, hsa-miR-301a-3p, hsa-miR-636, hsa-miR-34a-3p, hsa-miR-21-3p, and hsa-miR-516a-5p | Downregulated | Unknown | Human | (169) |
| miR-10b | Downregulated | Inhibition of miR-10b in HL-1 cardiomyocytes caused over expression of Apaf-1 | Human | (178) |
| miR-1, miR-133, miR-378 | Downregulated | Unknown | Human | (179) |
| miR-616 | SNP in PON1 affects miRNA-mRNA interaction. Patients with the CT or TT genotype at loci rs3735590 were associated with a lower risk of CAVD than the patients harboring the CC genotype | PON1 | Human | (180) |
Calcific aortic valve disease therapeutics; past, present, and future
Decalcification of aortic cusps with moderate aortic stenosis has been unsuccessful (184), and valve replacement surgery, either with mechanical or biological prostheses, is currently the treatment option of choice for CAVD patients. The ideal prosthetic valve would have hemodynamics similar to the native valve, grow and remodel with the individual, and not require additional therapy such as anticoagulation. However, to date we have yet to develop such an ideal prosthetic and there is a high rate of failure due to progressive deterioration including calcification and non-calcific damage that limits their effectiveness (185). This is likely related to the incompatibility of prosthetic valves with the native environment, leading to increased infiltration of inflammatory cells and disturbances in the hemodynamic environment (186, 187). Furthermore, the implanted valve will be similarly exposed to the underpinning risk factors of CAVD that were present before surgery. As a result, aortic stenosis patients are at risk of secondary left ventricular hypertrophy, and mortality rates of 2–3% have been reported (188–192). Transcatheter aortic valve replacement (TAVR) was pioneered in the early 2000s and is considered a less-invasive option for aortic stenosis patients (193, 194). Therefore, this procedure is common in patients deemed too “high risk” for conventional valve replacement surgery, which is advantageous for those suffering from senile-related CAVD. However, TAVR is also costly and not risk-free (195). In order to advance the field, tissue engineering approaches need to address the structural degeneration and thromboembolism risks of current prosthetics, and consider the need for growth and remodeling of implanted valves in the pediatric population. In parallel, there is a need to discover alternative therapeutics that go beyond surgery to halt or reverse the pathogenesis of CAVD.
Attempts have been made to pharmacologically treat CAVD. Statins, 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors, have been successfully used at reducing cardiovascular events (196, 197) by virtue of several pleiotropic beneficial effects including decreasing low-density lipoprotein (LDL) cholesterol, improving endothelial cell function, reducing inflammation, and decreasing thrombus formation (198). In vitro studies have shown pharmacological statins to be successful in curbing calcific nodule formation VICs (199), however, treatment with Simvastatin led to increased VIC osteoblast markers (200). The use of statins have been shown to successfully prevent CAVD in preclinical studies (201–204), however, these drugs do not seem to reduce CAVD or aortic stenosis in humans (205). Moreover, metabolic syndrome patients on statin treatment exhibited an increased rate of CAVD progression relative to placebo-treated control subjects (206). A major challenge in the use of statins for CAVD treatment is that the valve-specific molecular targets remain unclear, and hyperlipidemia/inflammation are not always causal of CAVD (207, 208). Furthermore, CAVD is often clinically detected as aortic stenosis, an advanced stage of the disease at which statins may not be useful in regressing or modulating pathology (208). Therefore, the utilization of statins in the treatment of CAVD remains unsubstantiated.
It has been shown that stenotic aortic valves have increased expression of the renin angiotensin (ag) system including ag-converting enzyme (ACE) and the type I ag II receptor (209). Animal studies have shown that ag-II receptor blocker (ARB) treatment inhibited sclerotic changes in the aortic valve, inhibited transdifferentiating of quiescent VICs to myofibroblasts and/or osteo-VICs while maintaining endothelial integrity (210). While ARBs are used to treat Marfan Syndrome (211), reports of therapeutic effects of ARBs for CAVD are mixed. It has been shown that ARBs could not prevent the progression of CAVD in elderly, high-risk hypertensive patients (212), however, other patients on ARBs had reduced aortic valve tissue remodeling (213) and, furthermore, treatment with ACE inhibitors and ARBs led to improved survival and a reduced risk of cardiovascular events in aortic stenosis patients (214), and might also abrogate CAVD pathogenesis in a hypertensive setting (215).
As discussed, the lncRNA and miRNA signatures are altered in the setting of CAVD, thus miRNA-based therapeutics maybe beneficial in restoring their expression and normalizing levels of downstream target genes that may play a role in CAVD pathogenesis. Strategies to increase miR binding specificity to target mRNAs include the utilization of locked nucleic acids or 2′-O-methylation of the antisense oligonucleotides, while circulation time and cellular uptake of miRNAs can be enhanced by cholesterol conjugation (216). miR-204 expression is significantly dysregulated in CAVD and aortic stenosis, as are the miR-34 family members that are known to suppress BMP2; making these attractive miR-based therapeutic targets (137, 163, 165, 183, 217). In addition, LncRNA H19 promotes osteogenic-like changes in VICs by silencing NOTCH1. Given its success as a therapeutic strategy for pancreatic cancer (218), LncRNA H19 may also serve as potential CAVD therapy (160).
While a handful of genes have been identified largely in mice, as playing a causative role in CAVD, manipulating gene function therapeutically to attenuate calcific nodule progression in humans is challenging, largely due to the off-target effects in non-valvular structures. In susceptible mouse models, pharmacologically targeting enzymes important in signaling pathways increased in calcified valves has been successful in promoting the regression or reducing the formation of nodules, including Carbonic Anhydrase (219) and Cyclooxygenase 2 (116), respectively. As discussed here, the epigenetic regulation of CAVD is evolving and miRNA- or LncRNA-based therapeutics to target key drivers of pathogenesis could potentially be beneficial in restoring the expression of affected genes. In addition to genes, risk factors for CAVD are also environmental, or influenced by life style habits. Interestingly, while CAVD is observed in old, hypercholesterolemic mice (220), studies to increase exercise and reduce dietary cholesterol intake did not influence aortic valve disease (221), although lowering plasma cholesterol genetically did alleviate calcific nodule formation, but not stenosis (222). Thus, once end-stage calcification and stenosis occur, eliminating the cause/risk-factor for CAVD may not abrogate the disease, supporting the notion that CAVD is irreversible and early intervention is needed.
In summary, current therapies for the treatment of CAVD are limiting and long-term ineffective. Moving forward there is a critical need to increase our understanding of the etiology of CAVD, and the molecular and cellular process that contribute to onset and progression of calcific nodule formation. From this we will be poised to advance the discovery of mechanistic-based therapies that treat the underlying cause, rather than just the symptoms.
Author contributions
VM and JL wrote the text. JL generated figures and edited the final submission.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Mandy Root-Thompson (MedDraw Studio) for illustrations.
Footnotes
Funding. This work is supported by R01HL142685, R01HL132801 (JL) and The Research Institute at Nationwide Children's Hospital IDEA award (VM).
References
- 1.Hinton RBJr, Lincoln J, Deutsch GH, Osinska H, Manning PB, Benson DW, et al. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res. (2006) 98:1431–8. 10.1161/01.RES.0000224114.65109.4e [DOI] [PubMed] [Google Scholar]
- 2.Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez-Sarano M. Burden of valvular heart diseases: a population-based study. Lancet (2006) 368:1005–11. 10.1016/S0140-6736(06)69208-8 [DOI] [PubMed] [Google Scholar]
- 3.Ferreira-Gonzalez I, Pinar-Sopena J, Ribera A, Marsal JR, Cascant P, Gonzalez-Alujas T, et al. Prevalence of calcific aortic valve disease in the elderly and associated risk factors: a population-based study in a Mediterranean area. Eur J Prev Cardiol. (2013) 20:1022–30. 10.1177/2047487312451238 [DOI] [PubMed] [Google Scholar]
- 4.Thanassoulis G, Williams K, Altobelli KK, Pencina MJ, Cannon CP, Sniderman AD. Individualized statin benefit for determining statin eligibility in the primary prevention of cardiovascular disease. Circulation (2016) 133:1574–81. 10.1161/CIRCULATIONAHA.115.018383 [DOI] [PubMed] [Google Scholar]
- 5.Lamprea-Montealegre JA, Otto CM. Health behaviors and calcific aortic valve disease. J Am Heart Assoc. (2018) 7:e008385. 10.1161/JAHA.117.008385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation (2016) 133:e38–60. 10.1161/CIR.0000000000000350 [DOI] [PubMed] [Google Scholar]
- 7.Otto CM, Prendergast B. Aortic-valve stenosis–from patients at risk to severe valve obstruction. N Engl J Med. (2014) 371:744–56. 10.1056/NEJMra1313875 [DOI] [PubMed] [Google Scholar]
- 8.Dutta P, Lincoln J. Calcific aortic valve disease: a developmental biology perspective. Curr Cardiol Rep. (2018) 20:21. 10.1007/s11886-018-0968-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson RH. Clinical anatomy of the aortic root. Heart (2000) 84:670–3. 10.1136/heart.84.6.670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balachandran K, Sucosky P, Yoganathan AP. Hemodynamics and mechanobiology of aortic valve inflammation and calcification. Int J Inflam. (2011) 2011:263870. 10.4061/2011/263870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gross L, Kugel MA. Topographic anatomy and histology of the valves in the human heart. Am J Pathol. (1931) 7:445–474.7. [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia-Martinez V, Sanchez-Quintana D, Hurle JM. Histochemical and ultrastructural changes in the extracellular matrix of the developing chick semilunar heart valves. Acta Anat. (1991) 142:87–96. 10.1159/000147166 [DOI] [PubMed] [Google Scholar]
- 13.Kunzelman KS, Cochran RP, Murphree SS, Ring WS, Verrier ED, Eberhart RC. Differential collagen distribution in the mitral valve and its influence on biomechanical behaviour. J Heart Valve Dis. (1993) 2:236–44. [PubMed] [Google Scholar]
- 14.Icardo JM, Colvee E. Atrioventricular valves of the mouse: III. Collagenous skeleton and myotendinous junction. Anat Rec. (1995) 243:367–75. 10.1002/ar.1092430311 [DOI] [PubMed] [Google Scholar]
- 15.Rabkin-Aikawa E, Mayer Jr JE, Schoen FJ. Heart valve regeneration. Adv Biochem Eng Biotechnol. (2005) 94:141–79. 10.1007/b100003 [DOI] [PubMed] [Google Scholar]
- 16.Lincoln J, Lange AW, Yutzey KE. Hearts and bones: shared regulatory mechanisms in heart valve, cartilage, tendon, and bone development. Dev Biol. (2006) 294:292–302. 10.1016/j.ydbio.2006.03.027 [DOI] [PubMed] [Google Scholar]
- 17.Aldous IG, Veres SP, Jahangir A, Lee JM. Differences in collagen cross-linking between the four valves of the bovine heart: a possible role in adaptation to mechanical fatigue. Am J Physiol Heart Circul Physiol. (2009) 296:H1898–906. 10.1152/ajpheart.01173.2008 [DOI] [PubMed] [Google Scholar]
- 18.Balachandran K, Alford PW, Wylie-Sears J, Goss JA, Grosberg A, Bischoff J, et al. Cyclic strain induces dual-mode endothelial-mesenchymal transformation of the cardiac valve. Proc Natl Acad Sci USA. (2011) 108:19943–8. 10.1073/pnas.1106954108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grande-Allen KJ, Liao J. The heterogeneous biomechanics and mechanobiology of the mitral valve: implications for tissue engineering. Curr Cardiol Rep. (2011) 13:113–20. 10.1007/s11886-010-0161-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sacks MS, David Merryman W, Schmidt DE. On the biomechanics of heart valve function. J Biomech. (2009) 42:1804–24. 10.1016/j.jbiomech.2009.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scott M, Vesely I. Aortic valve cusp microstructure: the role of elastin. Ann Thorac Surg. (1995) 60(2 Suppl.):S391–4. 10.1016/0003-4975(95)00263-K [DOI] [PubMed] [Google Scholar]
- 22.Schoen FJ. Aortic valve structure-function correlations: role of elastic fibers no longer a stretch of the imagination. J Heart Valve Dis. (1997) 6:1–6. [PubMed] [Google Scholar]
- 23.Hinton RB, Yutzey KE. Heart valve structure and function in development and disease. Annu Rev Physiol. (2011) 73:29–46. 10.1146/annurev-physiol-012110-142145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim KO, Boughner DR. Mechanical properties of human mitral valve chordae tendineae: variation with size and strain rate. Can J Physiol Pharmacol. (1975) 53:330–9. 10.1139/y75-048 [DOI] [PubMed] [Google Scholar]
- 25.Lim KO, Boughner DR, Smith CA. Dynamic elasticity of human mitral valve chorade tendinease. Can J Physiol Pharmacol. (1977) 55:413–8. 10.1139/y77-058 [DOI] [PubMed] [Google Scholar]
- 26.Balaoing LR, Post AD, Liu H, Minn KT, Grande-Allen KJ. Age-related changes in aortic valve hemostatic protein regulation. Arterioscler Thromb Vasc Biol. (2014) 34:72–80. 10.1161/ATVBAHA.113.301936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol. (2007) 171:1407–18. 10.2353/ajpath.2007.070251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horne TE, VandeKopple M, Sauls K, Koenig SN, Anstine LJ, Garg V, et al. Dynamic Heterogeneity of the heart valve interstitial cell population in mitral valve health and disease. J Cardiovasc Dev Dis. (2015) 2:214–32. 10.3390/jcdd2030214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aikawa E, Whittaker P, Farber M, Mendelson K, Padera RF, Aikawa M, et al. Human semilunar cardiac valve remodeling by activated cells from fetus to adult: implications for postnatal adaptation, pathology, and tissue engineering. Circulation (2006) 113:1344–52. 10.1161/CIRCULATIONAHA.105.591768 [DOI] [PubMed] [Google Scholar]
- 30.Anstine LJ, Bobba C, Ghadiali S, Lincoln J. Growth and maturation of heart valves leads to changes in endothelial cell distribution, impaired function, decreased metabolism and reduced cell proliferation. J Mol Cell Cardiol. (2016) 100:72–82. 10.1016/j.yjmcc.2016.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ayoub S, Ferrari G, Gorman RC, Gorman JH, Schoen FJ, Sacks MS. Heart valve biomechanics and underlying mechanobiology. Compr Physiol. (2016) 6:1743–80. 10.1002/cphy.c150048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Venardos N, Nadlonek NA, Zhan Q, Weyant MJ, Reece TB, Meng X, et al. Aortic valve calcification is mediated by a differential response of aortic valve interstitial cells to inflammation. J Surg Res. (2014) 190:1–8. 10.1016/j.jss.2014.03.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Merryman WD, Youn I, Lukoff HD, Krueger PM, Guilak F, Hopkins RA, et al. Correlation between heart valve interstitial cell stiffness and transvalvular pressure: implications for collagen biosynthesis. Am J Physiol Heart Circ Physiol. (2006) 290:H224–31. 10.1152/ajpheart.00521.2005 [DOI] [PubMed] [Google Scholar]
- 34.Merryman WD, Liao J, Parekh A, Candiello JE, Lin H, Sacks MS. Differences in tissue-remodeling potential of aortic and pulmonary heart valve interstitial cells. Tissue Eng. (2007) 13:2281–9. 10.1089/ten.2006.0324 [DOI] [PubMed] [Google Scholar]
- 35.Simmons CA, Grant GR, Manduchi E, Davies PF. Spatial heterogeneity of endothelial phenotypes correlates with side-specific vulnerability to calcification in normal porcine aortic valves. Circ Res. (2005) 96:792–9. 10.1161/01.RES.0000161998.92009.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Butcher JT, Simmons CA, Warnock JN. Mechanobiology of the aortic heart valve. J Heart Valve Dis. (2008) 17:62–73. [PubMed] [Google Scholar]
- 37.El-Hamamsy I, Balachandran K, Yacoub MH, Stevens LM, Sarathchandra P, Taylor PM, et al. Endothelium-dependent regulation of the mechanical properties of aortic valve cusps. J Am Coll Cardiol. (2009) 53:1448–55. 10.1016/j.jacc.2008.11.056 [DOI] [PubMed] [Google Scholar]
- 38.Holliday CJ, Ankeny RF, Jo H, Nerem RM. Discovery of shear- and side-specific mRNAs and miRNAs in human aortic valvular endothelial cells. Am J Physiol Heart Circ Physiol. (2011) 301:H856–67. 10.1152/ajpheart.00117.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Butcher JT, Nerem RM. Valvular endothelial cells regulate the phenotype of interstitial cells in co-culture: effects of steady shear stress. Tissue Eng. (2006) 12:905–15. 10.1089/ten.2006.12.905 [DOI] [PubMed] [Google Scholar]
- 40.Bosse K, Hans CP, Zhao N, Koenig SN, Huang N, Guggilam A, et al. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J Mol Cell Cardiol. (2013) 60:27–35. 10.1016/j.yjmcc.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huk DJ, Austin BF, Horne TE, Hinton RB, Ray WC, Heistad DD, et al. Valve endothelial cell-derived Tgfβ1 signaling promotes nuclear localization of Sox9 in interstitial cells associated with attenuated calcification. Arterioscler Thromb Vasc Biol. (2015) 36:328–38. 10.1161/ATVBAHA.115.306091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Visconti RP, Ebihara Y, LaRue AC, Fleming PA, McQuinn TC, Masuya M, et al. An in vivo analysis of hematopoietic stem cell potential: hematopoietic origin of cardiac valve interstitial cells. Circ Res. (2006) 98:690–6. 10.1161/01.RES.0000207384.81818.d4 [DOI] [PubMed] [Google Scholar]
- 43.Hajdu Z, Romeo SJ, Fleming PA, Markwald RR, Visconti RP, Drake CJ. Recruitment of bone marrow-derived valve interstitial cells is a normal homeostatic process. J Mol Cell Cardiol. (2011) 51:955–65. 10.1016/j.yjmcc.2011.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anstine LJ, Horne TE, Horwitz EM, Lincoln J. Contribution of extra-cardiac cells in murine heart valves is age-dependent. J Am Heart Assoc. (2017) 6:e007097. 10.1161/JAHA.117.007097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sauls K, Toomer K, Williams K, Johnson AJ, Markwald RR, Hajdu Z, et al. Increased infiltration of extra-cardiac cells in myxomatous valve disease. J Cardiovasc Dev Dis. (2015) 2:200–13. 10.3390/jcdd2030200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hulin A, Anstine LJ, Kim AJ, Potter SJ, DeFalco T, Lincoln J, et al. Macrophage transitions in heart valve development and myxomatous valve disease. Arterioscler Thromb Vasc Biol. (2018) 38:636–44. 10.1161/ATVBAHA.117.310667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. Int Rev Cytol. (2005) 243:287–335. 10.1016/S0074-7696(05)43005-3 [DOI] [PubMed] [Google Scholar]
- 48.Bernanke DH, Markwald RR. Migratory behavior of cardiac cushion tissue cells in a collagen-lattice culture system. Dev Biol. (1982) 91:235–45. 10.1016/0012-1606(82)90030-6 [DOI] [PubMed] [Google Scholar]
- 49.Runyan RB, Markwald RR. Invasion of mesenchyme into three-dimensional collagen gels: a regional and temporal analysis of interaction in embryonic heart tissue. Dev Biol. (1983) 95:108–14. 10.1016/0012-1606(83)90010-6 [DOI] [PubMed] [Google Scholar]
- 50.Mjaatvedt CH, Lepera RC, Markwald RR. Myocardial specificity for initiating endothelial-mesenchymal cell transition in embryonic chick heart correlates with a particulate distribution of fibronectin. Dev Biol. (1987) 119:59–67. 10.1016/0012-1606(87)90206-5 [DOI] [PubMed] [Google Scholar]
- 51.de Lange FJ, Moorman AF, Anderson RH, Männer J, Soufan AT, de Gier-de Vries C, et al. Lineage and morphogenetic analysis of the cardiac valves. Circ Res. (2004) 95:645–54. 10.1161/01.RES.0000141429.13560.cb [DOI] [PubMed] [Google Scholar]
- 52.Lincoln J, Alfieri CM, Yutzey KE. BMP and FGF regulatory pathways control cell lineage diversification of heart valve precursor cells. Dev Biol. (2006) 292:292–302. [DOI] [PubMed] [Google Scholar]
- 53.Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development (2000) 127:1607–16. [DOI] [PubMed] [Google Scholar]
- 54.Jiang X, Choudhary B, Merki E, Chien KR, Maxson RE, Sucov HM. Normal fate and altered function of the cardiac neural crest cell lineage in retinoic acid receptor mutant embryos. Mech Dev. (2002) 117:115–22. 10.1016/S0925-4773(02)00206-X [DOI] [PubMed] [Google Scholar]
- 55.Verzi MP, McCulley DJ, De Val S, Dodou E, Black BL. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev Biol. (2005) 287:134–45. 10.1016/j.ydbio.2005.08.041 [DOI] [PubMed] [Google Scholar]
- 56.Wessels A, van den Hoff MJ, Adamo RF, Phelps AL, Lockhart MM, Sauls K, et al. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev Biol. (2012) 366:111–24. 10.1016/j.ydbio.2012.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakajima Y, Yamagishi T, Hokari S, Nakamura H. Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: roles of transforming growth factor (TGF)-beta and bone morphogenetic protein (BMP). (2000) Anat Rec. 258:119–27. [DOI] [PubMed] [Google Scholar]
- 58.Liebner S, Cattelino A, Gallini R, Rudini N, Iurlaro M, Piccolo S, et al. Beta-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J Cell Biol. (2004) 166:359–67. 10.1083/jcb.200403050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mercado-Pimentel ME, Runyan RB. Multiple transforming growth factor-beta isoforms and receptors function during epithelial-mesenchymal cell transformation in the embryonic heart. Cells Tissues Organs (2007) 185:146–56. 10.1159/000101315 [DOI] [PubMed] [Google Scholar]
- 60.Matzuk MM, Lu N, Vogel H, Sellheyer K, Roop DR, Bradley A. Multiple defects and perinatal death in mice deficient in follistatin. Nature (1995) 374:360–3. 10.1038/374360a0 [DOI] [PubMed] [Google Scholar]
- 61.Brunet LJ, McMahon JA, McMahon AP, Harland RM. Noggin, cartilage morphogenesis, and joint formation in the mammalian skeleton. Science (1998) 280:1455–7. 10.1126/science.280.5368.1455 [DOI] [PubMed] [Google Scholar]
- 62.Galvin KM, Donovan MJ, Lynch CA, Meyer RI, Paul RJ, Lorenz JN, et al. A role for smad6 in development and homeostasis of the cardiovascular system. Nat Genet. (2000) 24:171–4. 10.1038/72835 [DOI] [PubMed] [Google Scholar]
- 63.Bachiller D, Klingensmith J, Shneyder N, Tran U, Anderson R, Rossant J, et al. The role of chordin/Bmp signals in mammalian pharyngeal development and DiGeorge syndrome. Development (2003) 130:3567–78. 10.1242/dev.00581 [DOI] [PubMed] [Google Scholar]
- 64.Delot EC. Control of endocardial cushion and cardiac valve maturation by BMP signaling pathways. Mol Genet Metab. (2003) 80:27–35. 10.1016/j.ymgme.2003.07.004 [DOI] [PubMed] [Google Scholar]
- 65.Somi S, Buffing AA, Moorman AF, Van Den Hoff MJ. Dynamic patterns of expression of BMP isoforms 2, 4, 5, 6, and 7 during chicken heart development. Anat Rec Part A (2004) 279:636–51. 10.1002/ar.a.20031 [DOI] [PubMed] [Google Scholar]
- 66.Ma L, Lu MF, Schwartz RJ, Martin JF. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development (2005) 132:5601–11. 10.1242/dev.02156 [DOI] [PubMed] [Google Scholar]
- 67.Ma L, Martin JF. Generation of a Bmp2 conditional null allele. Genesis (2005) 42:203–6. 10.1002/gene.20132 [DOI] [PubMed] [Google Scholar]
- 68.Rivera-Feliciano J, Tabin CJ. Bmp2 instructs cardiac progenitors to form the heart-valve-inducing field. Dev Biol. (2006) 295:580–8. 10.1016/j.ydbio.2006.03.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Timmerman LA, Grego-Bessa J, Raya A, Bertran E, Perez-Pomares JM, Diez J, et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. (2004) 18:99–115. 10.1101/gad.276304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rutenberg JB, Fischer A, Jia H, Gessler M, Zhong TP, Mercola M. Developmental patterning of the cardiac atrioventricular canal by Notch and Hairy-related transcription factors. Development (2006) 133:4381–90. 10.1242/dev.02607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kokubo H, Tomita-Miyagawa S, Hamada Y, Saga Y. Hesr1 and Hesr2 regulate atrioventricular boundary formation in the developing heart through the repression of Tbx2. Development (2007) 134:747–55. 10.1242/dev.02777 [DOI] [PubMed] [Google Scholar]
- 72.Venkatesh DA, Park KS, Harrington A, Miceli-Libby L, Yoon JK, Liaw L. Cardiovascular and hematopoietic defects associated with Notch1 activation in embryonic Tie2-expressing populations. Circ Res. (2008) 103:423–31. 10.1161/CIRCRESAHA.108.177808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Luna-Zurita L, Prados B, Grego-Bessa J, Luxan G, del Monte G, Benguria A, et al. Integration of a Notch-dependent mesenchymal gene program and Bmp2-driven cell invasiveness regulates murine cardiac valve formation. J Clin Invest. (2010) 120:3493–507. 10.1172/JCI42666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Macgrogan D, Luna-Zurita L, de la Pompa JL. Notch signaling in cardiac valve development and disease. Birth Defects Res A Clin Mol Teratol. (2011) 91:449–459. 10.1002/bdra.20815 [DOI] [PubMed] [Google Scholar]
- 75.Chang AC, Garside VC, Fournier M, Smrz J, Vrljicak P, Umlandt P, et al. A Notch-dependent transcriptional hierarchy promotes mesenchymal transdifferentiation in the cardiac cushion. Dev Dyn. (2014) 243:894–905. 10.1002/dvdy.24127 [DOI] [PubMed] [Google Scholar]
- 76.Luxan G, D'Amato G, MacGrogan D, de la Pompa JL. Endocardial notch signaling in cardiac development and disease. Circ Res. (2016) 118:1–18. 10.1161/CIRCRESAHA.115.305350 [DOI] [PubMed] [Google Scholar]
- 77.MacGrogan D, D'Amato G, Travisano S, Martinez-Poveda B, Luxan G, de la Pompa G, et al. Sequential ligand-dependent notch signaling activation regulates valve primordium formation and morphogenesis. Circ Res. (2016) 118:1480–97. 10.1161/CIRCRESAHA.115.308077 [DOI] [PubMed] [Google Scholar]
- 78.Cai X, Zhang W, Hu J, Zhang L, Sultana N, Wu B, et al. Tbx20 acts upstream of Wnt signaling to regulate endocardial cushion formation and valve remodeling during mouse cardiogenesis. Development (2013) 140:3176–87. 10.1242/dev.092502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development (1997) 124:2659–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bartram U, Molin DG, Wisse LJ, Mohamad A, Sanford LP, Doetschman T, et al. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in TGF-beta(2)-knockout mice. Circulation (2001) 103:2745–52. 10.1161/01.CIR.103.22.2745 [DOI] [PubMed] [Google Scholar]
- 81.Hurlstone AF, Haramis AP, Wienholds E, Begthel H, Korving J, Van Eeden F, et al. The Wnt/beta-catenin pathway regulates cardiac valve formation. Nature (2003) 425:633–7. 10.1038/nature02028 [DOI] [PubMed] [Google Scholar]
- 82.Sridurongrit S, Larsson J, Schwartz R, Ruiz-Lozano P, Kaartinen V. Signaling via the Tgf-beta type I receptor Alk5 in heart development. Dev Biol. (2008) 322:208–18. 10.1016/j.ydbio.2008.07.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Todorovic V, Finnegan E, Freyer L, Zilberberg L, Ota M, Rifkin DB. Long form of latent TGF-beta binding protein 1 (Ltbp1L) regulates cardiac valve development. Dev Dynamics (2011) 240:176–87. 10.1002/dvdy.22521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Akiyama H, Chaboissier MC, Behringer RR, Rowitch DH, Schedl A, Epstein JA, et al. Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa. Proc Natl Acad Sci USA. (2004) 101:6502–7. 10.1073/pnas.0401711101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lincoln J, Kist R, Scherer G, Yutzey KE. Sox9 is required for precursor cell expansion and extracellular matrix organization during mouse heart valve development. Dev Biol. (2007) 305:120–32. 10.1016/j.ydbio.2007.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev. (1995) 75:519–60. 10.1152/physrev.1995.75.3.519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Culver JC, Dickinson ME. The effects of hemodynamic force on embryonic development. Microcirculation (2010) 17:164–78. 10.1111/j.1549-8719.2010.00025.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tan H, Biechler S, Junor L, Yost MJ, Dean D, Li J, et al. Fluid flow forces and rhoA regulate fibrous development of the atrioventricular valves. Dev Biol. (2013) 374:345–56. 10.1016/j.ydbio.2012.11.023 [DOI] [PubMed] [Google Scholar]
- 89.Biechler SV, Junor L, Evans AN, Eberth JF, Price RL, Potts JD, et al. The impact of flow-induced forces on the morphogenesis of the outflow tract. Front Physiol. (2014) 5:225. 10.3389/fphys.2014.00225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hu N, Clark EB. Hemodynamics of the stage 12 to stage 29 chick embryo. Circ Res. (1989) 65:1665–70. 10.1161/01.RES.65.6.1665 [DOI] [PubMed] [Google Scholar]
- 91.Hogers B, DeRuiter MC, Gittenberger-de Groot AC, Poelmann RE. Unilateral vitelline vein ligation alters intracardiac blood flow patterns and morphogenesis in the chick embryo. Circ Res. (1997) 80:473–81. 10.1161/01.RES.80.4.473 [DOI] [PubMed] [Google Scholar]
- 92.Broekhuizen ML, Hogers B, DeRuiter MC, Poelmann RE, Gittenberger-de Groot AC, Wladimiroff JW. Altered hemodynamics in chick embryos after extraembryonic venous obstruction. Ultrasound Obstet Gynecol. (1999) 13:437–45. 10.1046/j.1469-0705.1999.13060437.x [DOI] [PubMed] [Google Scholar]
- 93.Hogers B, DeRuiter MC, Gittenberger-de Groot AC, Poelmann RE. Extraembryonic venous obstructions lead to cardiovascular malformations and can be embryolethal. Cardiovasc Res. (1999) 41:87–99. 10.1016/S0008-6363(98)00218-1 [DOI] [PubMed] [Google Scholar]
- 94.Hove JR, Köster RW, Forouhar AS, Acevedo-Bolton G, Fraser SE, Gharib M. Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature (2003) 421:172–7. 10.1038/nature01282 [DOI] [PubMed] [Google Scholar]
- 95.Reckova M, Rosengarten C, de Almeida A, Stanley CP, Wessels A, Gourdie RG, et al. Hemodynamics is a key epigenetic factor in development of the cardiac conduction system. Circ Res. (2003) 93:77–85. 10.1161/01.RES.0000079488.91342.B7 [DOI] [PubMed] [Google Scholar]
- 96.Stekelenburg-de Vos S, Ursem NT, Hop WC, Wladimiroff JW, Gittenberger-de Groot AC, Poelmann RE. Acutely altered hemodynamics following venous obstruction in the early chick embryo. J Exp Biol. (2003) 206(Pt 6):1051–7. 10.1242/jeb.00216 [DOI] [PubMed] [Google Scholar]
- 97.Lucitti JL, Tobita K, Keller BB. Arterial hemodynamics and mechanical properties after circulatory intervention in the chick embryo. J Exp Biol. (2005) 208(Pt 10):1877–85. 10.1242/jeb.01574 [DOI] [PubMed] [Google Scholar]
- 98.deAlmeida A, McQuinn T, Sedmera D. Increased ventricular preload is compensated by myocyte proliferation in normal and hypoplastic fetal chick left ventricle. Circ Res. (2007) 100:1363–70. 10.1161/01.RES.0000266606.88463.cb [DOI] [PubMed] [Google Scholar]
- 99.Midgett M, Goenezen S, Rugonyi S. Blood flow dynamics reflect degree of outflow tract banding in Hamburger-Hamilton stage 18 chicken embryos. J R Soc Interface (2014) 11:20140643. 10.1098/rsif.2014.0643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Menon V, Eberth JF, Goodwin RL, Potts JD. Altered hemodynamics in the embryonic heart affects outflow valve development. J Cardiovasc Dev Dis. (2015) 2:108–24. 10.3390/jcdd2020108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Menon V, Junor L, Balhaj M, Eberth JF, Potts J. A novel ex ovo banding technique to alter intracardiac hemodynamics in an embryonic chicken system. J Vis Exp. (2016) 13:53955 10.3791/53955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ford SM, McPheeters MT, Wang YT, Ma P, Gu S, Strainic J, et al. Increased regurgitant flow causes endocardial cushion defects in an avian embryonic model of congenital heart disease. Congenit Heart Dis. (2017) 12:322–31. 10.1111/chd.12443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Midgett M, López CS, David L, Maloyan A, Rugonyi S. Increased hemodynamic load in early embryonic stages alters endocardial to mesenchymal transition. Front Physiol. (2017) 8:56. 10.3389/fphys.2017.00056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pang KL, Parnall M, Loughna S. Effect of altered haemodynamics on the developing mitral valve in chick embryonic heart. J Mol Cell Cardiol. (2017) 108:114–26. 10.1016/j.yjmcc.2017.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rennie MY, Stovall S, Carson JP, Danilchik M, Thornburg KL, Rugonyi S. Hemodynamics modify collagen deposition in the early embryonic chicken heart outflow tract. J Cardiovasc Dev Dis. (2017) 4:24. 10.3390/jcdd4040024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Menon V, Eberth JF, Junor L, Potts AJ, Belhaj M, Dipette DJ, et al. Removing vessel constriction on the embryonic heart results in changes in valve gene expression, morphology, and hemodynamics. Dev Dyn. (2018) 247:531–41. 10.1002/dvdy.24588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics−2012 update: a report from the American Heart Association. Circulation (2012) 125:e2–e220. 10.1161/CIR.0b013e31823ac046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Miller JD, Weiss RM, Heistad DD. Calcific aortic valve stenosis: methods, models, and mechanisms. Circ Res. (2011) 108:1392–412. 10.1161/CIRCRESAHA.110.234138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, et al. Calcific aortic valve disease: not simply a degenerative process: a review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation (2011) 124:1783–91. 10.1161/CIRCULATIONAHA.110.006767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.O'Brien KD. Epidemiology and genetics of calcific aortic valve disease. J Investig Med. (2007) 55:284–91. 10.2310/6650.2007.00010 [DOI] [PubMed] [Google Scholar]
- 111.Hutson HN, Marohl T, Anderson M, Eliceiri K, Campagnola P, Masters KS. Calcific aortic valve disease is associated with layer-specific alterations in collagen architecture. PLoS ONE (2016) 11:e0163858. 10.1371/journal.pone.0163858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cottignoli V, Cavarretta E, Salvador L, Valfre C, Maras A. Morphological and chemical study of pathological deposits in human aortic and mitral valve stenosis: a biomineralogical contribution. Patholog Res Int. (2015) 2015:342984. 10.1155/2015/342984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mohler ER III, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation (2001) 103:1522–8. 10.1161/01.CIR.103.11.1522 [DOI] [PubMed] [Google Scholar]
- 114.Rajamannan NM, Subramaniam M, Rickard D, Stock SR, Donovan J, Springett M, et al. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation (2003) 107:2181–4. 10.1161/01.CIR.0000070591.21548.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bosse Y, Miqdad A, Fournier D, Pepin A, Pibarot P, Mathieu P. Refining molecular pathways leading to calcific aortic valve stenosis by studying gene expression profile of normal and calcified stenotic human aortic valves. Circ Cardiovasc Genet. (2009) 2:489–98. 10.1161/CIRCGENETICS.108.820795 [DOI] [PubMed] [Google Scholar]
- 116.Wirrig EE, Gomez MV, Hinton RB, Yutzey KE. COX2 inhibition reduces aortic valve calcification in vivo. Arterioscler Thromb Vasc Biol. (2015) 35:938–47. 10.1161/ATVBAHA.114.305159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Leskela HV, Satta J, Oiva J, Eriksen H, Juha R, Korkiamaki P, et al. Calcification and cellularity in human aortic heart valve tissue determine the differentiation of bone-marrow-derived cells. J Mol Cell Cardiol. (2006) 41:642–9. 10.1016/j.yjmcc.2006.07.014 [DOI] [PubMed] [Google Scholar]
- 118.Nomura A, Seya K, Yu Z, Daitoku K, Motomura S, Murakami M, et al. CD34-negative mesenchymal stem-like cells may act as the cellular origin of human aortic valve calcification. Biochem Biophys Res Commun. (2013) 440:780–5. 10.1016/j.bbrc.2013.10.003 [DOI] [PubMed] [Google Scholar]
- 119.Sun L, Chandra S, Sucosky P. Ex vivo evidence for the contribution of hemodynamic shear stress abnormalities to the early pathogenesis of calcific bicuspid aortic valve disease. PLoS ONE (2012) 7:e48843. 10.1371/journal.pone.0048843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Marom G, Peleg M, Halevi R, Rosenfeld M, Raanani E, Hamdan A, et al. Fluid-structure interaction model of aortic valve with porcine-specific collagen fiber alignment in the cusps. J Biomech Eng. (2013) 135:101001–6. 10.1115/1.4024824 [DOI] [PubMed] [Google Scholar]
- 121.Seaman C, McNally A, Biddle S, Jankowski L, Sucosky P. Generation of simulated calcific lesions in valve leaflets for flow studies. J Heart Valve Dis. (2015) 24:115–25. [PubMed] [Google Scholar]
- 122.Krohn JB, Hutcheson JD, Martinez-Martinez E, Aikawa E. Extracellular vesicles in cardiovascular calcification: expanding current paradigms. J Physiol. (2016) 594:2895–903. 10.1113/JP271338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nigam V, Srivastava D. Notch1 represses osteogenic pathways in aortic valve cells. J Mol Cell Cardiol. (2009) 47:828–34. 10.1016/j.yjmcc.2009.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, et al. Mutations in NOTCH1 cause aortic valve disease. Nature (2005) 437:270–4. 10.1038/nature03940 [DOI] [PubMed] [Google Scholar]
- 125.Peacock JD, Levay AK, Gillaspie DB, Tao G, Lincoln J. Reduced sox9 function promotes heart valve calcification phenotypes in vivo. Circ Res. (2010) 106:712–9. 10.1161/CIRCRESAHA.109.213702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kaden JJ, Bickelhaupt S, Grobholz R, Vahl CF, Hagl S, Brueckmann M, et al. Expression of bone sialoprotein and bone morphogenetic protein-2 in calcific aortic stenosis. J Heart Valve Dis. (2004) 13:560–6. [PubMed] [Google Scholar]
- 127.Caira FC, Stock SR, Gleason TG, McGee EC, Huang J, Bonow RO, et al. Human degenerative valve disease is associated with up-regulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation. J Am Coll Cardiol. (2006) 47:1707–12. 10.1016/j.jacc.2006.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yang X, Fullerton DA, Su X, Ao L, Cleveland Jr JC, Meng X. Pro-osteogenic phenotype of human aortic valve interstitial cells is associated with higher levels of Toll-like receptors 2 and 4 and enhanced expression of bone morphogenetic protein 2. J Am Coll Cardiol. (2009) 53:491–500. 10.1016/j.jacc.2008.09.052 [DOI] [PubMed] [Google Scholar]
- 129.Miller JD, Weiss RM, Serrano KM, Castaneda LE, Brooks RM, Zimmerman K, et al. Evidence for active regulation of pro-osteogenic signaling in advanced aortic valve disease. Arterioscler Thromb Vasc Biol. (2010) 30:2482–6. 10.1161/ATVBAHA.110.211029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ankeny RF, Thourani VH, Weiss D, Vega JD, Taylor WR, Nerem RM, et al. Preferential activation of SMAD1/5/8 on the fibrosa endothelium in calcified human aortic valves–association with low BMP antagonists and SMAD6. PLoS ONE (2011) 6:e20969. 10.1371/journal.pone.0020969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Seya K, Yu Z, Kanemaru K, Daitoku K, Akemoto Y, Shibuya H, et al. Contribution of bone morphogenetic protein-2 to aortic valve calcification in aged rat. J Pharmacol Sci. (2011) 115:8–14. 10.1254/jphs.10198FP [DOI] [PubMed] [Google Scholar]
- 132.Wirrig EE, Hinton RB, Yutzey KE. Differential expression of cartilage and bone-related proteins in pediatric and adult diseased aortic valves. J Mol Cell Cardiol. (2011) 50:561–9. 10.1016/j.yjmcc.2010.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Askevold ET, Gullestad L, Aakhus S, Ranheim T, Tonnessen T, Solberg OG, et al. Secreted Wnt modulators in symptomatic aortic stenosis. J Am Heart Assoc. (2012) 1:e002261. 10.1161/JAHA.112.002261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gu GJ, Chen T, Zhou HM, Sun KX, Li J. Role of Wnt/beta-catenin signaling pathway in the mechanism of calcification of aortic valve. J Huazhong Univ Sci Technolog Med Sci. (2014) 34:33–6. 10.1007/s11596-014-1228-x [DOI] [PubMed] [Google Scholar]
- 135.Gao X, Zhang L, Gu G, Wu PH, Jin S, Hu W, et al. The effect of oxLDL on aortic valve calcification via the Wnt/ beta-catenin signaling pathway: an important molecular mechanism. J Heart Valve Dis. (2015) 24:190–6. [PubMed] [Google Scholar]
- 136.Albanese I, Yu B, Al-Kindi H, Barratt B, Ott L, Al-Refai M, et al. Role of noncanonical Wnt signaling pathway in human aortic valve calcification. Arterioscler Thromb Vasc Biol. (2017) 37:543–52. 10.1161/ATVBAHA.116.308394 [DOI] [PubMed] [Google Scholar]
- 137.Song R, Fullerton DA, Ao L, Zhao KS, Meng X. An epigenetic regulatory loop controls pro-osteogenic activation by TGF-β1 or bone morphogenetic protein 2 in human aortic valve interstitial cells. J Biol Chem. (2017) 292:8657–66. 10.1074/jbc.M117.783308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Thomas PS, Sridurongrit S, Ruiz-Lozano P, Kaartinen V. Deficient signaling via Alk2 (Acvr1) leads to bicuspid aortic valve development. PLoS ONE (2012) 7:e35539. 10.1371/journal.pone.0035539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Farrar EJ, Huntley GD, Butcher J. Endothelial-derived oxidative stress drives myofibroblastic activation and calcification of the aortic valve. PLoS ONE (2015) 10:e123257 10.1371/journal.pone.0123257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hjortnaes J, Shapero K, Goettsch C, Hutcheson JD, Keegan J, Kluin J, et al. Valvular interstitial cells suppress calcification of valvular endothelial cells. Atherosclerosis (2015) 242:251–60. 10.1016/j.atherosclerosis.2015.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Butcher JT, Nerem RM. Porcine aortic valve interstitial cells in three-dimensional culture: comparison of phenotype with aortic smooth muscle cells. J Heart Valve Dis. (2004) 13:478–85. discussion: 485–76. [PubMed] [Google Scholar]
- 142.Sucosky P, Balachandran K, Elhammali A, Jo H, Yoganathan AP. Altered shear stress stimulates upregulation of endothelial VCAM-1 and ICAM-1 in a BMP-4- and TGF-beta1-dependent pathway. Arterioscler Thromb Vasc Biol. (2009) 29:254–60. 10.1161/ATVBAHA.108.176347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yap CH, Saikrishnan N, Tamilselvan G, Yoganathan AP. Experimental measurement of dynamic fluid shear stress on the aortic surface of the aortic valve leaflet. Biomech Model Mechanobiol. (2012) 11:171–82. 10.1007/s10237-011-0301-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gould ST, Srigunapalan S, Simmons CA, Anseth KS. Hemodynamic and cellular response feedback in calcific aortic valve disease. Circ Res. (2013) 113:186–97. 10.1161/CIRCRESAHA.112.300154 [DOI] [PubMed] [Google Scholar]
- 145.Meierhofer C, Schneider EP, Lyko C, Hutter A, Martinoff S, Markl M, et al. Wall shear stress and flow patterns in the ascending aorta in patients with bicuspid aortic valves differ significantly from tricuspid aortic valves: a prospective study. Eur Heart J Cardiovasc Imaging (2013) 14:797–804. 10.1093/ehjci/jes273 [DOI] [PubMed] [Google Scholar]
- 146.Saikrishnan N, Mirabella L, Yoganathan AP. Bicuspid aortic valves are associated with increased wall and turbulence shear stress levels compared to trileaflet aortic valves. Biomech Model Mechanobiol. (2015) 14:577–88. 10.1007/s10237-014-0623-3 [DOI] [PubMed] [Google Scholar]
- 147.Stewart BF, Siscovick D, Lind BK, Gardin JM, Gottdiener JS, Smith VE, et al. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study J Am Coll Cardiol. (1997) 29:630–4. 10.1016/S0735-1097(96)00563-3 [DOI] [PubMed] [Google Scholar]
- 148.Mathieu P, Bossé Y, Huggins GS, Della Corte A, Pibarot P, Michelena HI, et al. The pathology and pathobiology of bicuspid aortic valve: State of the art and novel research perspectives. J Pathol Clin Res. (2015) 1:195–206. 10.1002/cjp2.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kim JK, Samaranayake M, Pradhan S. Epigenetic mechanisms in mammals. Cell Mol Life Sci. (2009) 66:596–612. 10.1007/s00018-008-8432-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. (2011) 12:861–74. 10.1038/nrg3074 [DOI] [PubMed] [Google Scholar]
- 151.Thomson DW, Bracken CP, Goodall GJ. Experimental strategies for microRNA target identification. Nucleic Acids Res. (2011) 39:6845–53. 10.1093/nar/gkr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cabili MN, Dunagin MC, McClanahan PD, Biaesch A, Padovan-Merhar O, Regev A, et al. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol. (2015) 16:20. 10.1186/s13059-015-0586-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chamberlain AA, Lin M, Lister RL, Maslov AA, Wang Y, Suzuki M. DNA methylation is developmentally regulated for genes essential for cardiogenesis. J Am Heart Assoc. (2014) 3:e000976. 10.1161/JAHA.114.000976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nührenberg T, Gilsbach R, Preissl S, Schnick T, Hein L. Epigenetics in cardiac development, function, and disease. Cell Tissue Res. (2014) 356:585–600. 10.1007/s00441-014-1887-8 [DOI] [PubMed] [Google Scholar]
- 155.Lewandowski SL, Janardhan HP, Trivedi CM. Histone deacetylase 3 coordinates deacetylase-independent epigenetic silencing of Transforming Growth Factor-β1 (TGF-β1) to Orchestrate second heart field development. J Biol Chem. (2015) 290:27067–89. 10.1074/jbc.M115.684753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nwachukwu N, Hagler M, Kafa R, Roos C, Miller J. Evidence for altered DNA methylation as a major regulator of gene expression in calcific aortic valve disease. FASEB J. (2014) 28. [Google Scholar]
- 157.Sritharen Y, Roos CM, Nwachukwu N, Kafa R, Hagler MA, Verzosa GC, et al. Genetic inactivation of DNMT3b protects against activation of osteogenic signaling and slows progression of aortic valve stenosis in mice. FASEB J. (2016) 30. [Google Scholar]
- 158.Radhakrishna U, Albayrak S, Alpay-Savasan Z, Zeb A, Turkoglu O, Sobolewski P, et al. Genome-wide DNA Methylation analysis and epigenetic variations associated with congenital Aortic Valve Stenosis (AVS). PLoS ONE (2016) 11:e154010. 10.1371/journal.pone.0154010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gilsbach R, Preißl S, Rühle F, Weiß S, Mühle A, Doenst T, et al. Genome-wide DNA methylation in chronic heart disease. Clin Res Cardiol. (2012) 101(Suppl. 1). [Google Scholar]
- 160.Hadji F, Boulanger MC, Guay SP, Gaudreault N, Amellah S, Mkannez G, et al. Altered DNA methylation of long noncoding RNA H19 in calcific aortic valve disease promotes mineralization by silencing NOTCH1. Circulation (2016) 134:1848–62. 10.1161/CIRCULATIONAHA.116.023116 [DOI] [PubMed] [Google Scholar]
- 161.Mkannez G, Gagné-Ouellet V, Nsaibia MJ, Boulanger MC, Rosa M, Argaud D, et al. DNA methylation of a PLPP3 MIR transposon-based enhancer promotes an osteogenic program in calcific aortic valve disease. Cardiovasc Res. (2018) 114:1525–35. 10.1093/cvr/cvy111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Nagy E, Bäck M. Epigenetic regulation of 5-lipoxygenase in the phenotypic plasticity of valvular interstitial cells associated with aortic valve stenosis. FEBS Lett. (2012) 586:1325–9. 10.1016/j.febslet.2012.03.039 [DOI] [PubMed] [Google Scholar]
- 163.Xiao X, Zhou T, Guo S, Guo C, Zhang Q, Dong N, et al. LncRNA MALAT1 sponges miR-204 to promote osteoblast differentiation of human aortic valve interstitial cells through up-regulating Smad4. Int J Cardiol. (2017) 243:404–12. 10.1016/j.ijcard.2017.05.037 [DOI] [PubMed] [Google Scholar]
- 164.Rathan S, Ankeny CJ, Arjunon S, Ferdous Z, Kumar S, Fernandez Esmerats J, et al. Identification of side- and shear-dependent microRNAs regulating porcine aortic valve pathogenesis. Sci Rep. (2016) 6:25397. 10.1038/srep25397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Takahashi K, Satoh M, Takahashi Y, Osaki T, Nasu T, Tamada M, et al. Dysregulation of ossification-related miRNAs in circulating osteogenic progenitor cells obtained from patients with aortic stenosis. Clin Sci. (2016) 130:1115–24. 10.1042/CS20160094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Heath JM, Fernandez Esmerats J, Khambouneheuang L, Kumar S, Simmons R, Jo H. Mechanosensitive microRNA-181b regulates aortic valve endothelial matrix degradation by targeting TIMP3. Cardiovasc Eng Technol. (2018) 9:141–50. 10.1007/s13239-017-0296-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ohukainen P, Syväranta S, Näpänkangas J, Rajamäki K, Taskinen P, Peltonen T, et al. MicroRNA-125b and chemokine CCL4 expression are associated with calcific aortic valve disease. Ann Med. (2015) 47:423–9. 10.3109/07853890.2015.1059955 [DOI] [PubMed] [Google Scholar]
- 168.Coffey S, Williams MJ, Phillips LV, Galvin IF, Bunton RW, Jones GT. Integrated microRNA and messenger RNA analysis in aortic stenosis. Sci Rep. (2016) 6:3604. 10.1038/srep36904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Shi J, Liu H, Wang H, Kong X. MicroRNA expression signature in degenerative aortic stenosis. Biomed Res Int. (2016) 2016:4682172. 10.1155/2016/4682172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Yu C, Li L, Xie F, Guo S, Liu F, Dong N, et al. LncRNA TUG1 sponges miR-204-5p to promote osteoblast differentiation through upregulating Runx2 in aortic valve calcification. Cardiovasc Res. (2018) 114:168–79. 10.1093/cvr/cvx180 [DOI] [PubMed] [Google Scholar]
- 171.Nader J, Metzinger-Le Meuth V, Maitrias P, Humbert JR, Brigant B, Tribouilloy C, et al. miR-92a: a novel potential biomarker of rapid aortic valve calcification. J Heart Valve Dis. (2017) 26:327–33. [PubMed] [Google Scholar]
- 172.Wang Y, Chen S, Deng C, Li F, Wang Y, Hu X, et al. MicroRNA-204 targets Runx2 to attenuate BMP-2-induced osteoblast differentiation of human aortic valve interstitial cells. J Cardiovasc Pharmacol. (2015) 66:63–71. 10.1097/FJC.0000000000000244 [DOI] [PubMed] [Google Scholar]
- 173.Yanagawa B, Lovren F, Pan Y, Garg V, Quan A, Tang G, et al. miRNA-141 is a novel regulator of BMP-2-mediated calcification in aortic stenosis. J Thorac Cardiovasc Surg. (2012) 144:256–62. 10.1016/j.jtcvs.2011.10.097 [DOI] [PubMed] [Google Scholar]
- 174.Nigam V, Sievers HH, Jensen BC, Sier HA, Simpson PC, Srivastava D, et al. Altered microRNAs in bicuspid aortic valve: a comparison between stenotic and insufficient valves. J Heart Valve Dis. (2010) 19:459–65. [PMC free article] [PubMed] [Google Scholar]
- 175.Du J, Zheng R, Xiao F, Zhang S, He K, Zhang J, et al. Downregulated MicroRNA-195 in the bicuspid aortic valve promotes calcification of valve interstitial cells via targeting SMAD7. Cell Physiol Biochem. (2017) 44:884–96. 10.1159/000485356 [DOI] [PubMed] [Google Scholar]
- 176.Zhang M, Liu X, Zhang X, Song Z, Han L, He Y, et al. MicroRNA-30b is a multifunctional regulator of aortic valve interstitial cells. J Thorac Cardiovasc Surg. (2014) 147:1073–80. 10.1016/j.jtcvs.2013.05.011 [DOI] [PubMed] [Google Scholar]
- 177.Beaumont J, López B, Hermida N, Schroen B, San José G, Heymans S, et al. microRNA-122 down-regulation may play a role in severe myocardial fibrosis in human aortic stenosis through TGF-β1 up-regulation. Clin Sci. (2014). 126:497–506. 10.1042/CS20130538 [DOI] [PubMed] [Google Scholar]
- 178.Gallego I, Beaumont J, López B, Ravassa S, Gómez-Doblas JJ, Moreno MU, et al. Potential role of microRNA-10b down-regulation in cardiomyocyte apoptosis in aortic stenosis patients. Clin Sci. (2016) 130:2139–49. 10.1042/CS20160462 [DOI] [PubMed] [Google Scholar]
- 179.Chen Z, Li C, Xu Y, Li Y, Yang H, Rao L. Circulating level of miR-378 predicts left ventricular hypertrophy in patients with aortic stenosis. PLoS ONE (2014). 9:e105702. 10.1371/journal.pone.0105702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wang Z, Chen S, Zhu M, Zhang W, Zhang H, Li H, et al. Functional SNP in the 3'UTR of PON1 is associated with the risk of calcific aortic valve stenosis via MiR-616. Cell Physiol Biochem. (2018) 45:1390–8. 10.1159/000487565 [DOI] [PubMed] [Google Scholar]
- 181.Gošev I, Zeljko M, Durić Ž, Nikolić I, Gošev M, Ivčević S, et al. Epigenome alterations in aortic valve stenosis and its related left ventricular hypertrophy. Clin Epigenet. (2017). 9:106. 10.1186/s13148-017-0406-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wang H, Shi J, Li B, Zhou Q, Kong X, Bei Y. MicroRNA expression signature in human calcific aortic valve disease. Biomed Res Int. (2017) 2017:4820275. 10.1155/2017/4820275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ni WJ, Wu YZ, Ma DH, Leng XM. Noncoding RNAs in calcific aortic valve disease: a review of recent studies. J Cardiovasc Pharmacol. (2018) 71:317–23. 10.1097/FJC.0000000000000569 [DOI] [PubMed] [Google Scholar]
- 184.McBride LR, Naunheim KS, Fiore AC, Harris HH, Willman VL, Kaiser GC, et al. Aortic valve decalcification. J Thorac Cardiovasc Surg. (1990) 100:36–42; discussion: 42–33. [PubMed] [Google Scholar]
- 185.Schoen FJ, Levy RJ. Calcification of tissue heart valve substitutes: progress toward understanding and prevention. Ann Thorac Surg. (2005) 79:1072–80. 10.1016/j.athoracsur.2004.06.033 [DOI] [PubMed] [Google Scholar]
- 186.Jegatheeswaran A, Butany J. Pathology of infectious and inflammatory diseases in prosthetic heart valves. Cardiovasc Pathol. (2006) 15:252–5. 10.1016/j.carpath.2006.05.002 [DOI] [PubMed] [Google Scholar]
- 187.Barbarash L Kudryavtsev I, Rutkovskaya N, Golovkin A. T cell response in patients with implanted biological and mechanical prosthetic heart valves. Mediators Inflamm. (2016) 2016:1937564. 10.1155/2016/1937564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Föppl M, Hoffmann A, Amann FW, Roth J, Stulz P, Hasse J, et al. Sudden cardiac death after aortic valve surgery: incidence and concomitant factors. Clin Cardiol. (1989) 12:202–7. [DOI] [PubMed] [Google Scholar]
- 189.Bloomfield P, Wheatley DJ, Prescott RJ, Miller HC. Twelve-year comparison of a Bjork-Shiley mechanical heart valve with porcine bioprostheses. N Engl J Med. (1991) 324:573–9. 10.1056/NEJM199102283240901 [DOI] [PubMed] [Google Scholar]
- 190.Barreto-Filho JA, Wang Y, Dodson JA, Desai MM, Sugeng L, Geirsson A, et al. Trends in aortic valve replacement for elderly patients in the United States, 1999-2011. JAMA (2013) 310:2078–85. 10.1001/jama.2013.282437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Mohr FW, Holzhey D, Möllmann H, Beckmann A, Veit C, Figulla HR, et al. The German Aortic Valve Registry: 1-year results from 13,680 patients with aortic valve disease. Eur J Cardiothorac Surg. (2014) 46:808–16. 10.1093/ejcts/ezu290 [DOI] [PubMed] [Google Scholar]
- 192.Thourani VH, Suri RM, Gunter RL, Sheng S, O'Brien SM, Ailawadi G, et al. Contemporary real-world outcomes of surgical aortic valve replacement in 141,905 low-risk, intermediate-risk, and high-risk patients. Ann Thorac Surg. (2015) 99:55–61. 10.1016/j.athoracsur.2014.06.050 [DOI] [PubMed] [Google Scholar]
- 193.Cribier A, Eltchaninoff H, Bash A, Borenstein N, Tron C, Bauer F, et al. Percutaneous transcatheter implantation of an aortic valve prosthesis for calcific aortic stenosis: first human case description. Circulation (2002) 106:3006–8. 10.1161/01.CIR.0000047200.36165.B8 [DOI] [PubMed] [Google Scholar]
- 194.Zahn R, Gerckens U, Grube E, Linke A, Sievert H, Eggebrecht H, et al. Transcatheter aortic valve implantation: first results from a multi-centre real-world registry. Eur Heart J. (2011) 32:198–204. 10.1093/eurheartj/ehq339 [DOI] [PubMed] [Google Scholar]
- 195.Van Brabandt H, Neyt M, Hulstaert F. Transcatheter aortic valve implantation (TAVI): risky and costly. BMJ (2012) 345:e4710 10.1136/bmj.e4710 [DOI] [PubMed] [Google Scholar]
- 196.Gould AL, Rossouw JE, Santanello NC, Heyse JF, Furberg CD. Cholesterol reduction yields clinical benefit: impact of statin trials. Circulation (1998) 97:946–52. 10.1161/01.CIR.97.10.946 [DOI] [PubMed] [Google Scholar]
- 197.Ray KK, Seshasai SR, Erqou S, Sever P, Jukema JW, Ford I, et al. Statins and all-cause mortality in high-risk primary prevention: a meta-analysis of 11 randomized controlled trials involving 65,229 participants. Arch Intern Med. (2010) 170:1024–31. 10.1001/archinternmed.2010.182 [DOI] [PubMed] [Google Scholar]
- 198.Lim SY. Role of statins in coronary artery disease. Chonnam Med J. (2013) 49:1–6. 10.4068/cmj.2013.49.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Benton JA, Kern HB, Leinwand LA, Mariner PD, Anseth KS. Statins block calcific nodule formation of valvular interstitial cells by inhibiting alpha-smooth muscle actin expression. Arterioscler Thromb Vasc Biol. (2009) 29:1950–7. 10.1161/ATVBAHA.109.195271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Monzack EL, Masters KS. A time course investigation of the statin paradox among valvular interstitial cell phenotypes. Am J Physiol Heart Circ Physiol. (2012) 303:H903–9. 10.1152/ajpheart.00263.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Aronow WS, Ahn KIC, Goldman ME. Association of coronary risk factors and use of statins with progression of mild valvular aortic stenosis in older persons. Am J Cardiol. (2001) 88:693–5. 10.1016/S0002-9149(01)01821-5 [DOI] [PubMed] [Google Scholar]
- 202.Novaro GM, Tiong IY, Pearce GL, Lauer MS, Sprecher DL, Griffin BP. Effect of hydroxymethylglutaryl coenzyme a reductase inhibitors on the progression of calcific aortic stenosis. Circulation (2001) 104:2205–9. 10.1161/hc4301.098249 [DOI] [PubMed] [Google Scholar]
- 203.Pohle K, Mäffert R, Ropers D, Moshage W, Stilianakis N, Daniel WG, et al. Progression of aortic valve calcification: association with coronary atherosclerosis and cardiovascular risk factors. Circulation (2001) 104:1927–32. [DOI] [PubMed] [Google Scholar]
- 204.Moura LM, Ramos SF, Zamorano JL, Barros IM, Azevedo LF, Rocha-Gonçalves F, et al. Rosuvastatin affecting aortic valve endothelium to slow the progression of aortic stenosis. J Am Coll Cardiol. (2007) 49:554–61. 10.1016/j.jacc.2006.07.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Teo KK, Corsi DJ, Tam JW, Dumesnil JG, Chan KL. Lipid lowering on progression of mild to moderate aortic stenosis: meta-analysis of the randomized placebo-controlled clinical trials on 2344 patients. Can J Cardiol. (2011) 27:800–8. 10.1016/j.cjca.2011.03.012 [DOI] [PubMed] [Google Scholar]
- 206.Capoulade R, Clavel MA, Dumesnil JG, Chan KL, Teo KK, Tam JW, et al. Impact of metabolic syndrome on progression of aortic stenosis: influence of age and statin therapy. J Am Coll Cardiol. (2012) 60:216–23. 10.1016/j.jacc.2012.03.052 [DOI] [PubMed] [Google Scholar]
- 207.Ortlepp JR, Schmitz F, Bozoglu T, Hanrath P, Hoffmann R. Cardiovascular risk factors in patients with aortic stenosis predict prevalence of coronary artery disease but not of aortic stenosis: an angiographic pair matched case-control study. Heart (2003) 89:1019–22. 10.1136/heart.89.9.1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hutcheson JD, Aikawa E, Merryman WD. Potential drug targets for calcific aortic valve disease. Nat Rev Cardiol. (2014) 11:218–31. 10.1038/nrcardio.2014.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Helske S, Lindstedt KA, Laine M, Mäyränpää M, Werkkala K, Lommi J, et al. Induction of local angiotensin II-producing systems in stenotic aortic valves. J Am Coll Cardiol. (2004) 44:1859–66. 10.1016/j.jacc.2004.07.054 [DOI] [PubMed] [Google Scholar]
- 210.Arishiro K, Hoshiga M, Negoro N, Jin D, Takai S, Miyazaki M, et al. Angiotensin receptor-1 blocker inhibits atherosclerotic changes and endothelial disruption of the aortic valve in hypercholesterolemic rabbits. J Am Coll Cardiol. (2007) 49:1482–9. 10.1016/j.jacc.2006.11.043 [DOI] [PubMed] [Google Scholar]
- 211.Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz, et al. Angiotensin II blockade and aortic-root dilation in Marfan's syndrome. N Engl J Med. (2008) 358:2787–95. 10.1056/NEJMoa0706585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Olsen MH, Wachtell K, Bella JN, Liu JE, Boman K, Gerdts E, et al. Effect of losartan versus atenolol on aortic valve sclerosis (a LIFE substudy). Am J Cardiol. (2004) 94:1076–80. 10.1016/j.amjcard.2004.06.074 [DOI] [PubMed] [Google Scholar]
- 213.Côté N, Couture C, Pibarot P, Després JP, Mathieu P. Angiotensin receptor blockers are associated with a lower remodelling score of stenotic aortic valves. Eur J Clin Invest. (2011) 41:1172–9. 10.1111/j.1365-2362.2011.02522.x [DOI] [PubMed] [Google Scholar]
- 214.Nadir MA, Wei L, Elder DH, Libianto R, Lim TK, Pauriah M, et al. Impact of renin-angiotensin system blockade therapy on outcome in aortic stenosis. J Am Coll Cardiol. (2011) 58:570–6. 10.1016/j.jacc.2011.01.063 [DOI] [PubMed] [Google Scholar]
- 215.Capoulade R, Clavel MA, Mathieu P, Côté N, Dumesnil JG, Arsenault M, et al. Impact of hypertension and renin-angiotensin system inhibitors in aortic stenosis. Eur J Clin Invest. (2013) 43:1262–72. 10.1111/eci.12169 [DOI] [PubMed] [Google Scholar]
- 216.Nouraee N, Mowla SJ. miRNA therapeutics in cardiovascular diseases: promises and problems. Front Genet. (2015) 6:232. 10.3389/fgene.2015.00232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Fotinos A, Nagarajan N, Martins AS, Fritz DT, Garsetti D, Lee AT, et al. Bone morphogenetic protein-focused strategies to induce cytotoxicity in lung cancer cells. Anticancer Res. (2014) 34:2095–104. [PMC free article] [PubMed] [Google Scholar]
- 218.Hanna N, Ohana P, Konikoff FM, Leichtmann G, Hubert A, Appelbaum L, et al. Phase 1/2a, dose-escalation, safety, pharmacokinetic and preliminary efficacy study of intratumoral administration of BC-819 in patients with unresectable pancreatic cancer. Cancer Gene Ther. (2012) 19:374–81. 10.1038/cgt.2012.10 [DOI] [PubMed] [Google Scholar]
- 219.Bouchareb R, Cote N, Marie Chloe B, Le Quang K, El Husseini D, Asselin J, et al. Carbonic anhydrase XII in valve interstitial cells promotes the regression of calcific aortic valve stenosis. J Mol Cell Cardiol. (2015) 82:104–15. 10.1016/j.yjmcc.2015.03.002 [DOI] [PubMed] [Google Scholar]
- 220.Weiss RM, Ohashi M, Miller JD, Young SG, Heistad DD. Calcific aortic valve stenosis in old hypercholesterolemic mice. Circulation (2006) 114:2065–9. 10.1161/CIRCULATIONAHA.106.634139 [DOI] [PubMed] [Google Scholar]
- 221.Schlotter F, Matsumoto Y, Mangner N, Schuler G, Linke A, Adams V. Regular exercise or changing diet does not influence aortic valve disease progression in LDLR deficient mice. PLoS ONE (2012) 7:e37298 10.1371/journal.pone.0037298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Miller JD, Weiss RM, Serrano KM, Brooks RM II, Berry CJ, Zimmerman K, et al. Lowering plasma cholesterol levels halts progression of aortic valve disease in mice. Circulation (2009) 119:2693–701. 10.1161/CIRCULATIONAHA.108.834614 [DOI] [PMC free article] [PubMed] [Google Scholar]