

Summary.
Objective:
When establishing endovascular infections, Staphylococcus aureus (S. aureus) overcomes shear forces of flowing blood by binding to von Willebrand factor (VWF). Staphylococcal VWF-binding protein (vWbp) interacts with VWF, but it is unknown how this secreted protein binds to the bacterial cell wall. We hypothesized that vWbp interacts with a staphylococcal surface protein, mediating the adhesion of S. aureus to VWF and vascular endothelium under shear stress.
Methods:
We studied the binding of S. aureus to vWbp, VWF and endothelial cells in a micro-parallel flow chamber using various mutants deficient in Sortase A (SrtA) and SrtA-dependent surface proteins, and Lactococcus lactis expressing single staphylococcal surface proteins. In vivo adhesion of bacteria was evaluated in the murine mesenteric circulation using real-time intravital vascular microscopy.
Results:
vWbp bridges the bacterial cell wall and VWF, allowing shear-resistant binding of S. aureus to inflamed or damaged endothelium. Absence of SrtA and Clumping factor A (ClfA) reduced adhesion of S. aureus to vWbp, VWF and activated endothelial cells. ADAMTS-13 and an anti-VWF A1 domain antibody, when combined, reduced S. aureus adhesion to activated endothelial cells by 90%. Selective overexpression of ClfA in the membrane of Lactococcus lactis enabled these bacteria to bind to VWF and activated endothelial cells but only in the presence of vWbp. Absence of ClfA abolished bacterial adhesion to the activated murine vessel wall.
Conclusions:
vWbp interacts with VWF and with the SrtA-dependent staphylococcal surface protein ClfA. The complex formed by VWF, secreted vWbp and bacterial ClfA anchors S. aureus to vascular endothelium under shear stress.
Keywords: endothelium, infection, shear stress, Staphylococcus aureus, von Willebrand factor
Introduction
Staphylococcus aureus (S. aureus) is the leading cause of life-threatening endovascular infections [1]. One of the most feared complications of invasive S. aureus disease is infective endocarditis. Compared with other pathogens, infective endocarditis caused by S. aureus has a higher mortality and is more frequently associated with severe complications [2,3].
Once S. aureus infects the heart valves, almost one in three patients will die, despite aggressive surgery and antibiotics [4]. The dramatic morbidity and mortality of S. aureus endocarditis have remained unchanged over the past decades. This stresses the need for new therapeutic strategies to prevent and treat infective endocarditis.
To cause endocarditis, bacteria first need to adhere to the endothelium of the heart valve. However, binding to endothelial cells in flowing blood requires mechanisms to withstand shear stress. A better understanding of the initial binding of S. aureus to the valvular endocardium will allow the development of new strategies to prevent and treat endocarditis.
We and others showed that S. aureus adheres to the vessel wall under flow by binding to von Willebrand factor (VWF) [5,6]. VWF binds to sites of vascular damage and is exposed on the endothelial surface upon activation or injury [7]. VWF multimers are cleaved by ADAMTS-13 (a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13) [8]. The binding of S. aureus to VWF is mediated by the von Willebrand factor-binding protein (vWbp); however, because vWbp is thought to be a secreted protein not anchoring to the cell wall, it remains unclear how vWbp mediates bacterial attachment.
S. aureus expresses a number of bacterial cell wall-anchored surface proteins that mediate bacterial adherence to host cells and to extracellular matrix components. Several of these S. aureus surface proteins, or MSCRAMMs (microbial surface components recognizing adhesive matrix molecules), have been proposed to contribute to the pathogenesis of endovascular infections [9–11]. Many of those MSCRAMMs, which recognize fibronectin, fibrinogen, collagen or VWF, have a conserved C-terminal cell wall sorting signal with a Leu-Pro-X-Thr-Gly (LPXTG) motif [12]. Together, more than 20 members of this family of cell wall-anchored surface proteins have been identified in the S. aureus genome [13–15]. This sorting signal triggers the covalent anchoring of these proteins to the bacterial cell wall by Sortase A (SrtA), a transpeptidase [12]. Strains with a mutation in the srtA gene lack these cell wall-anchored proteins [14].
We hypothesized that vWbp bridges VWF with a cell wall-anchored surface protein of S. aureus. In this study we identify the bacterial binding partners for vWbp and unravel the mechanism of S. aureus binding to the vascular wall under shear stress, a crucial step in the early phases of the infectious process.
Materials and methods
Bacterial strains
S. aureus strains were stored in Brain Heart Infusion with 10% glycerol at 80 °C. Bacteria were grown overnight in tryptic soy broth at 37 °C. The reference strain used in this study is S. aureus Newman [16]. Isogenic single mutants of S. aureus Newman [17–19] are listed in Table 1. Lactococcus lactis (L. lactis) strains [11,20] were grown overnight at 37 °C in M17 medium (Fluka, Sigma-Aldrich, Darmstadt, Germany) supplemented with 0.5% glucose and 5 µg mL−1 erythromycin and stored in M17 medium supplemented with 10% glycerol at 80 °C. The strains used in this study are listed in Table 1. Escherichia coli (E. coli) strains DH5α and BL21 (DE3) were cultured on Luria agar or broth at 37 °C. Ampicillin (100 µg mL−1) and erythromycin (10 µg mL−1) were used for plasmid selection.
Table 1.
List of the bacteria used in this study, including abbreviations used in the text and the strain’s origin and properties
| Abbreviation | Original strain | Properties | References |
|---|---|---|---|
| WT | S. aureus Newman | S. aureus reference strain | [17] |
| Vwb | S. aureus Newman | Deletion of vwb gene | [18,19] |
| coa/vwb | S. aureus Newman | Deletion of coa and vwb genes | [18,19] |
| srtA | S. aureus Newman | Deletion of srtA gene | [18,19] |
| Spa | S. aureus Newman | Deletion of spa gene | [18,19] |
| clfA | S. aureus Newman | Deletion of clfA gene | [18,19] |
| clfB | S. aureus Newman | Deletion of clfB gene | [18,19] |
| fnbpA | S. aureus Newman | Deletion of fnbpA gene | [18,19] |
| fnbpB | S. aureus Newman | Deletion of fnbpB gene | [18,19] |
| Srdcde | S. aureus Newman | Deletion of SdrCDE genes | [18,19] |
| sasB | S. aureus Newman | Deletion of sasB gene | [18,19] |
| sasC | S. aureus Newman | Deletion of sasC gene | [18,19] |
| L. lactis pIL253 | L. lactis subsp. cremoris 1363 | Empty vector expressing erythromycin resistance determinant | [11] |
| L. lactis FnBpA | L. lactis subsp. cremoris 1363 | Insertion of staphylococcal fnba gene | [11] |
| L. lactis FnBpB | L. lactis subsp. cremoris 1363 | Insertion of staphylococcal fnbb gene | [11] |
| L. lactis ClfA | L. lactis subsp. cremoris 1363 | Insertion of staphylococcal clfa gene | [20] |
Expression and purification of proteins
Recombinant His6-vWbp (rvWbp) without the signal sequence and lacking coagulase activity was cloned with plasmid pET15 and was purified from E. coli BL21 (DE3) using Ni-trilo-triacetic acid (Ni-NTA) chromatography as previously described [21]. Recombinant vWbp-Strep was cloned with pET22b and was purified from E. coli BL21 (DE3) using Strep-Tactin affinity chromatography as previously described [22]. Recombinant His6-ClfA1–520 was (ClfA, Clumping factor A) cloned with plasmid pET15 and purified from E. coli BL21 (DE3) using Ni-NTA chromatography as previously described [23]. Glutathione-S-transferase (GST)–VWF A1-domain proteins were produced as described before [24].
Cell wall and secreted protein extraction
S. aureus Newman (wild-type) and coa/vwb deletion mutant (Table 1) were grown overnight in Tryptic Soy Broth at 37 °C. Bacteria were washed, resuspended in 50 mL phosphate buffered saline (PBS) and incubated for 4 h at 37 °C. Where indicated, 20 µg mL−1 rvWbp was added to the S. aureus coa/vwb strain and incubated for 1 h at 37 °C. The bacterial pellet and supernatant were stored separately at −20 °C. To recover the cell wall proteins, the pellet was first washed in 0.05 M Tris buffer (pH 7.4) and resuspended in 0.05 M Tris buffer (pH 7.4) containing 0.002 M MgCl2. Bacterial cells were disrupted with a homogenizer for 10 min and the cell suspension was placed at 75 °C for 10 min to inactivate cell-wall autolytic enzymes. Supernatants were recovered and centrifuged at high speed to recover the cell wall. Cell walls were washed with 5 mL 0.05 M Tris buffer pH 7.4 + 1 M NaCl and extracted with 2% Triton at room temperature for 30 min, followed by incubating the pellet in 0.05 M Tris buffer (pH 7.4) + 0.145 M NaCl + 50 µg mL−1 lysostaphin (AMBI Products, New York, NY, USA) at 37 °C for 2 h. To recover secreted proteins, supernatant was filtered (Millex Filter Unit 0.22 lm, Merck Millipore, Overijse, Belgium) and concentrated using centrifugal filters with a 50 kD threshold (Centrifugal Filer Units, Merck Millipore).
Western blot analysis
Cell wall and secreted proteins were subjected to SDS-poly-acrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes in a Trans-Blot Turbo apparatus (Bio-Rad, Nazareth, Belgium). Membranes were incubated overnight with a house-made polyclonal anti-body against rvWbp (1:500). After adding horseradish per-oxidase-conjugated secondary antibodies, immunoreactive bands were visualized by Enhanced chemiluminescence (Amersham Biosciences, Diegem, Belgium).
In vitro perfusion experiments
In vitro perfusion experiments were performed as previously described [6,25]. Glass coverslips (24 × 50 mm, VWR International, Leuven, Belgium) were coated with 50 µg mL−1 VWF (Haemate P, CSL Behring, Mechelen, Belgium), 200 µg mL−1 Horm collagen (Takeda, Linz, Austria), 30 µg mL−1 rvWbp or 50 µg mL−1 VWF A1-domain in a humidified container at room temperature for 4 h. The coverslips were mounted in a micro-parallel flow chamber [26] and perfused for 10 min with a high-accuracy Harvard pump (PHD 2000 Infusion, Harvard Apparatus, Holliston, MA, USA) generating flow rates of 1000 s−1. Bacteria were labeled with 5(6)-carboxy-fluorescein N-hydroxysuccinimidyl ester (Sigma-Aldrich) (final concentration of 30 µg mL−1 for subsequent perfusion experiments) and diluted in PBS to an OD600 (optical density) of 0.65 or 1.2 (corresponding to approximately 3 × 108 and 6 × 109 colony forming units (CFU) mL−1). Coated coverslips were perfused with labeled bacteria, with or without soluble VWF (60 µg mL−1), rvWbp or bacterial supernatant added to the perfusate. Membranebound rvWbp was prepared by supplementing vwb (S. aureus strain lacking vWbp) with rvWbp and after an incubation period of 15 min, unbound rvWbp was removed by centrifugation. Bacterial supernatant was prepared by incubating washed bacteria for 4 h at 37 °C in PBS. Bacteria were then removed by centrifugation and filtering (Millex Filter Unit 0.22 lm, Merck Millipore). Live images were obtained using an inverted fluorescence microscope and video microscopy as reported [6]. Images were digitally stored and fluorescence was measured with ImageJ analysis software (National Institutes of Health, Bethesda, MD, USA). The intensity of fluo-rescence is reported as relative bacterial adhesion.
Bacterial adhesion to endothelial cells
Human umbilical vein endothelial cells (HUVECs) were isolated from fresh umbilical cords of healthy donors as described before [6,25]. HUVECs were seeded on 1% gelatin-coated (Sigma-Alderich) plastic coverslips (Sarstedt, Numbrecht,€ Germany) and grown to confluence. The coverslips were mounted in a micro-parallel flow chamber and the HUVECs were activated with 0.1 mM Ca2+-iono-phore A23187 (Sigma-Aldrich) for 10 min and perfused for 10 min with fluorescently labeled bacteria (OD600 of 1.2). Where indicated, 20 µg mL−1 rvWbp, 2.5 µg mL−1 rADAMTS-13 and/or 10 µg mL−1 anti-VWF A1 domain antibody were added to the bacterial perfusate. Bacterial adhesion was recorded as described above.
In vivo mesenteric perfusion model
Six- to eight-week-old C57Bl/6 mice were anesthetized with ketamine/xylazine and their right jugular veins were catheterized (Portex intravenous cannula, 2F). The peritoneal cavity was opened via midline abdominal incision and the mesenteric microcirculation was visualized with an inverted microscope (Axio-observer D1, Carl-Zeiss NV, Zaventem, Belgium). To activate the endothelium and cause VWF release, 5 µL of the Ca2+- ionophore A23187 (10 mM) was locally applied to the vascular bed. Subsequently, 100 µL of a suspension of fluorescently labeled bacteria (final concentration of 60 µg mL−1 carboxy-fluorescein and an OD600 of 1.8, corresponding to approximately 1 × 109 CFU mL−1) was injected through the catheter. Where indicated, 20 µg mL−1 of rvWbp was added to the bacterial inoculum. Live time-lapse images (1 image per second, 40 images) were acquired with an inverted fluorescence microscope, captured via a black and white camera and developed using image software. The fluorescent signal in the blood vessel was quantified manually for each frame and reported as described above. Animal experiments were approved by the Ethical Committee of the University of Leuven (P110/2014). For immunofluorescence staining, the blood vessels were dissected and fixed with paraformaldehyde 4% overnight and imbedded in parafine. A polyclonal anti-VWF rabbit antibody (31 µg mL−1) (Dako, Glostrup, Denmark) was used as primary antibody and a goat anti-rabbit Alexa Fluor-568 (20 µg mL−1) (Invitrogen, Carlsbad, CA, USA) as secondary antibody. Endothelial cells were counterstained with 4’,6-diamidino-2-phenylindole.
Surface plasmon resonance
Affinity and rates of association were measured on a BIA-core 3000 (GE Healthcare, Hillerod, Denmark). Buffers were sterile filtered and degassed. A nitrilotriacetic acid (NTA) chip (GE Healthcare, Diegem, Belgium) was used to capture histidine-tagged [27] ligands. The NTA chip was prepared for ligand capturing by injecting NiCl2 (0.5 M) (GE Healthcare, Diegem, Belgium) followed by injection of 20 lL His-ClfA1–520 (200 nM). His-ClfA1–520 and vWbp-Strep were diluted in running buffer (HBS-P buffer (20 mM HEPES [pH 7.4], 150 mM NaCl, 0.005% [vol/vol] surfactant P20) (GE Healthcare, Diegem, Belgium), 50 uM ethylenediaminetetraacetic acid (EDTA) (GE Healthcare, Diegem, Belgium) and 1 mM imidazole). To study the His-ClfA1–520–vWbp interaction, vWbp-Strep was injected at 6.25 nM, 12.5 nM and 25 nM for 180 s, followed by regeneration of the chip with 50 lM EDTA and 1 mM imidazole. All injections were performed at a flow rate of 10 lL min−1. Kinetic coefficients, KA and KD, were determined using the BiaEvaluation software (GE Healthcare, Diegem, Belgium) and best fit was determined with a 1 : 1 binding model with drifting baseline and local Rmax. All experiments were repeated in triplicate on at least three occasions.
Statistical analysis
All calculations were carried out with GraphPad Prism 5.0d (GraphPad Software, San Diego, CA USA). Groups were compared with the one-way ANOVA or a two-tailed Student’s t-test. All values are reported as mean standard error of the mean (SEM). A P-value of < 0.05 was considered significant (*P < 0.05; **P < 0.01; ***P < 0.001).
Results
Secreted vWbp interacts with both VWF and the S. aureus cell wall
We previously described that the shear-dependent adhesion of S. aureus to endothelial cells and subendothelial matrix is mediated by complex formation between VWF and staphylococcal vWbp [6]. To promote bacterial adhesion, secreted vWbp has to be able to interact with the S. aureus cell wall.
Western blotting confirmed that vWbp is secreted by the S. aureus Newman wild-type strain (WT), but it was also found attached to the cell wall (Fig. 1A). An S. aureus mutant strain lacking vWbp lacked both secreted and cell wall-bound vWbp. Added exogenous rvWbp was able to bind to the cell wall of the mutant strain lacking vWbp.
Fig. 1.
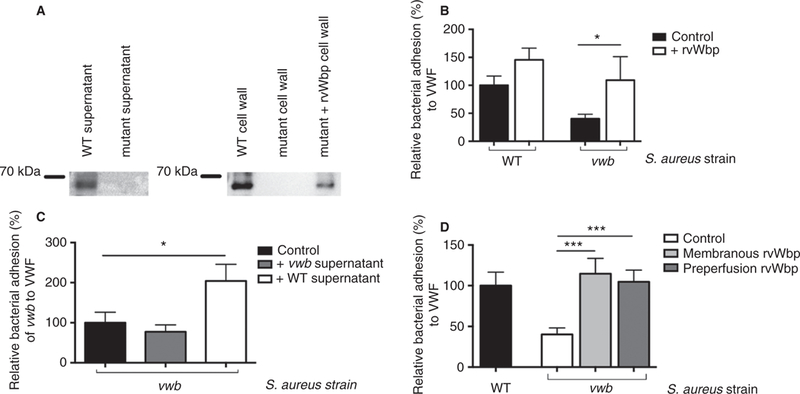
vWbp is a secreted staphylococcal protein that binds to S. aureus. (A) Western blot analysis on secreted proteins and cell wall proteins of S. aureus WT and mutant lacking vWbp. Where indicated, 30 µg mL−1 rvWbp was added to the mutant. (B) Micro-parallel plate flow chamber perfusion over coated VWF (50 µg mL−1) with fluorescently labeled WT and vwb strains at a shear rate of 1000 s−1 (n ≥ 8). Where indicated, 15 µg mL−1 rvWbp was added to the bacterial perfusate. All results are expressed as mean SEM. *P < 0.05. (C) Micro-parallel plate flow chamber perfusion over coated VWF (50 µg mL−1) with fluorescently labeled vwb strain at a shear rate of 1000 s−1 (n ≥ 8). Where indicated, supernatant of the WT strain (containing secreted vWbp) or vwb supernatant (lacking secreted vWbp) was added to the bacterial perfusate. All results are expressed as mean SEM. *P < 0.05. (D) Micro-parallel plate flow chamber perfusion over coated VWF (50 µg mL−1) with fluorescently labeled WT or vwb strains at a shear rate of 1000 s−1 (n ≥ 8). Where indicated, prior to perfusion, vwb was supplemented with 30 µg mL−1 rvWbp and after an incubation period of 15 minutes, unbound rvWbp was removed. Where indicated, VWF was pre-perfused with 30 µg mL−1 rvWbp. All results are expressed as mean SEM. ***P < 0.001. vWbp, von Willebrand factor-binding protein; WT, wild type; rvWbp, recombinant His6-vWbp; VWF, von Willebrand factor.
Compared with WT, perfusion of vwb over VWF resulted in reduced adhesion. Normal adhesion could be restored by either exogenous rvWbp (Fig. 1B) or by adding supernatant of the WT strain (Fig. 1C), but not by adding supernatant of the vwb strain. Exogenous vWbp (rvWbp or vWbp present in WT supernatant) (Fig. 1B, C) led to a 2-fold increase in the adhesion of vwb to VWF under flow (P = 0.021 and P = 0.048, respectively), indicating that vWbp interacts with S. aureus regardless of its ability to secrete vWbp.
This increase in adhesion was seen both when vwb was pre-incubated with rvWbp and when rvWbp was pre-perfused over VWF (Fig. 1D) (P = 0.0003 and P = 0.003, respectively).
These findings show that vWbp is a secreted protein capable of both binding to VWF under flow and sticking to S. aureus, thereby promoting the adhesion of S. aureus to the vessel wall under flow via a ternary interaction.
vWbp binds to S. aureus via an SrtA-dependent surface protein
We hypothesized that S. aureus interacts with vWbp via a staphylococcal cell wall-anchored surface protein processed by SrtA.
In contrast to WT, the srtA strain (deficient in SrtA, which lacks all sortase A-dependent cell wall-anchored proteins) was not able to adhere to coated rvWbp (Fig. 2A) (P < 0.0001). Binding of vwb to the rvWbp coating was similar to that of the WT strain (P = 0.56).
Fig. 2.
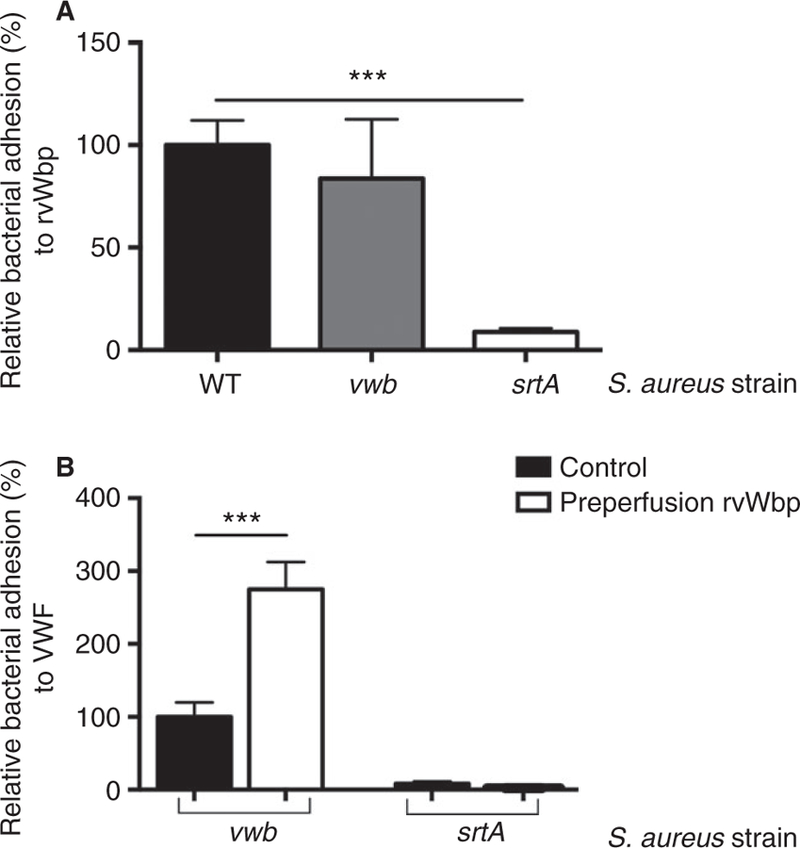
vWbp binds to S. aureus via an SrtA-dependent surface protein. (A) Micro-parallel plate flow chamber perfusion over coated rvWbp (30 µg mL−1) with fluorescently labeled WT, vwb and srtA strains at a shear rate of 1000 s−1 (n ≥ 8). All results are expressed as mean SEM. ***P < 0.001. (B) Micro-parallel plate flow chamber perfusion over coated VWF (50 µg mL−1) with fluorescently labeled S. aureus vwb and srtA strains at a shear rate of 1000 s−1 (n ≥ 8). Where indicated, pre-perfusion with rvWbp (30 µg mL−1) was performed. All results are expressed as mean SEM. ***P < 0.001. vWbp, von Willebrand factor-binding protein; SrtA, Sortase A; rvWbp, recombinant His6-vWbp; WT, wild type; VWF, von Willebrand factor.
Pre-perfusion of coated VWF with rvWbp increased the adhesion of vwb but had no effect on the adhesion of srtA (Fig. 2B) (P = 0.10), strengthening our interpretation that vWbp interacts with S. aureus through a sortase A-dependent cell wall-anchored protein.
vWbp forms a complex with VWF and ClfA, promoting bacterial adhesion to endothelial cells and subendothelial matrix
To identify which SrtA-dependent protein is crucial for vWbp binding, we screened a set of mutants deficient in one single SrtA-dependent cell wall-anchored surface protein for their adhesiveness to coated rvWbp under flow. When compared with the WT strain, two SrtA-dependent cell wall-anchored surface protein deletion mutants showed decreased adhesion to rvWbp (i.e. the mutant lacking Clumping factor A [clfA] and the mutant lacking staphylococcal protein A) [28] (Fig. 3A) (P = 0.0001 and P = 0.0092, respectively).
Fig. 3.
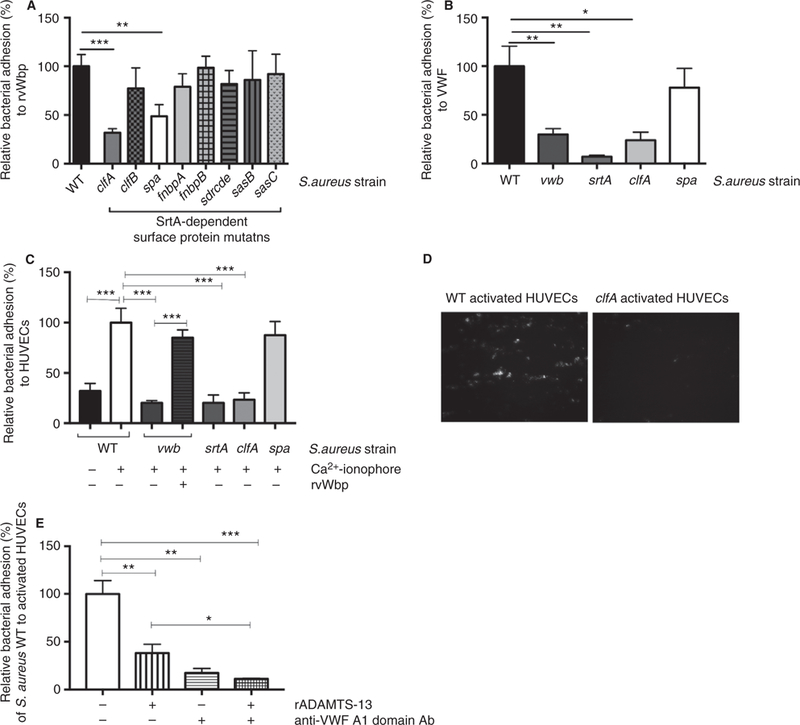
vWbp forms a complex with VWF and ClfA to promote bacterial adhesion to VWF and to endothelial cells. (A) Micro-parallel plate flow chamber perfusion over coated rvWbp (30 µg mL−1) with fluorescently labeled WT, vwb, srtA, clfA, clfB, spa, fnbpA, fnbpB, sdrcde, sasB and sasC strains at a shear rate of 1000 s−1 (n ≥ 8). All results are expressed as mean SEM. **P < 0.01 ***P < 0.001. (B) Micro-parallel plate flow chamber perfusion over coated VWF (50 µg mL−1) with fluorescently labeled WT, vwb, srtA, clfA and spa strains at a shear rate of 1000 s−1 (n ≥ 8). All results are expressed as mean SEM. *P < 0.05, **P < 0.01. (C) Micro-parallel plate flow chamber perfusion over HUVECs with fluorescently labeled WT, vwb, srtA, clfA and spa strains at a shear rate of 1000 s−1. Where indicated, HUVECs were activated by a 5-min perfusion with a Ca2+-ionophore. Where indicated, 20 µg mL−1 rvWbp was added to the perfusate (n ≥ 6). All results are expressed as mean SEM. ***P < 0.001. (D) Representative image of WT bacteria (left) and clfa bacteria [36] adhering to activated endothelial cells under flow. White bar is 100 micron. (E) Micro-parallel plate flow chamber perfusion over HUVECs with fluorescently labeled WT strain at a shear rate of 1000 s−1. HUVECs were activated by a 5-min perfusion with a Ca2+-ionophore. Where indicated, 2.5 µg mL−1 rADAMTS-13 and/or 10 µg mL−1 anti-VWF A1 domain antibody were added to the perfusate (n ≥ 6). All results are expressed as mean SEM. *P < 0.05, **P < 0.01, ***P < 0.001. vWbp, von Willebrand factor-binding protein; VWF, von Willebrand factor; ClfA, Clumping factor A; rvWbp, recombinant His6-vWbp; WT, wild type; HUVECs, human umbilical vein endothelial cells.
We further assessed the adhesion profile of srtA, clfA and spa to VWF (Fig. 3B). Compared with the WT strain, absence of SrtA and ClfA reduced adhesion by more than 75% (P = 0.0066 and P = 0.0066, respectively), whereas absence of SpA did not significantly reduce bacterial adhesion (P = 0.48).
Similarly, when compared with WT, srtA and clfA were unable to bind to collagen, the main component of the subendothelial matrix, regardless of the presence of VWF in the perfusate (Figure SI) (P = 0.0009 and P = 0.0069, respectively). The adhesion of spa to collagen in the presence of VWF was similar to that of WT (P = 0.49), suggesting that ClfA is the main bacterial surface binding partner for vWbp mediating S. aureus adhesion to VWF under flow.
Exogenous rvWbp increased the adhesion of vwb to VWF under flow; however, different concentrations of rvWbp did not affect the adhesion of clfA to VWF (Figure S2).
To validate whether this mechanism is capable of explaining bacterial adhesion to the endothelium, we examined the adhesion of WT, vwb, srtA, clfA and spa to resting and stimulated endothelial cells under flow.
Activation of endothelial cells facilitated the adhesion of the WT strain (Fig. 3C,D). In contrast, in the absence of vWbp, SrtA or ClfA, S. aureus was no longer able to bind to the VWF-strings and bacterial adhesion was low even to stimulated endothelial cells (Fig. 3C) (P = 0.0087, P = 0.0053 and P = 0.0077, respectively). However, addition of rvWbp increased the adhesion of vwb comparable to WT. Cleavage of VWF multimers by rADAMTS-13 decreased the adhesion of the WT strain by 60% (P = 0.0085) and combining rADAMTS-13 with an anti-VWF A1 domain antibody further decreased the WT adhesion to 10% (P = 0.0007) (Fig. 3E).
These data identify ClfA as a crucial factor in the VWF-mediated binding of S. aureus to endothelial cells in flow conditions by acting as a bacterial binding partner for vWbp.
vWbp forms a complex with the VWF A1-domain and ClfA
Presence of isolated VWF A1-domain on the coverslip was sufficient to trigger adhesion of the WT strain, but not of vwb, srtA and clfA, suggesting that vWbp binds to the A1-portion of VWF (Fig. 4A) (P = 0.0072, P = 0.0013 and P = 0.0045, respectively). This finding further provides an explanation for the previously recognized role of the VWF A1-domain in S. aureus binding to VWF [6].
Fig. 4.
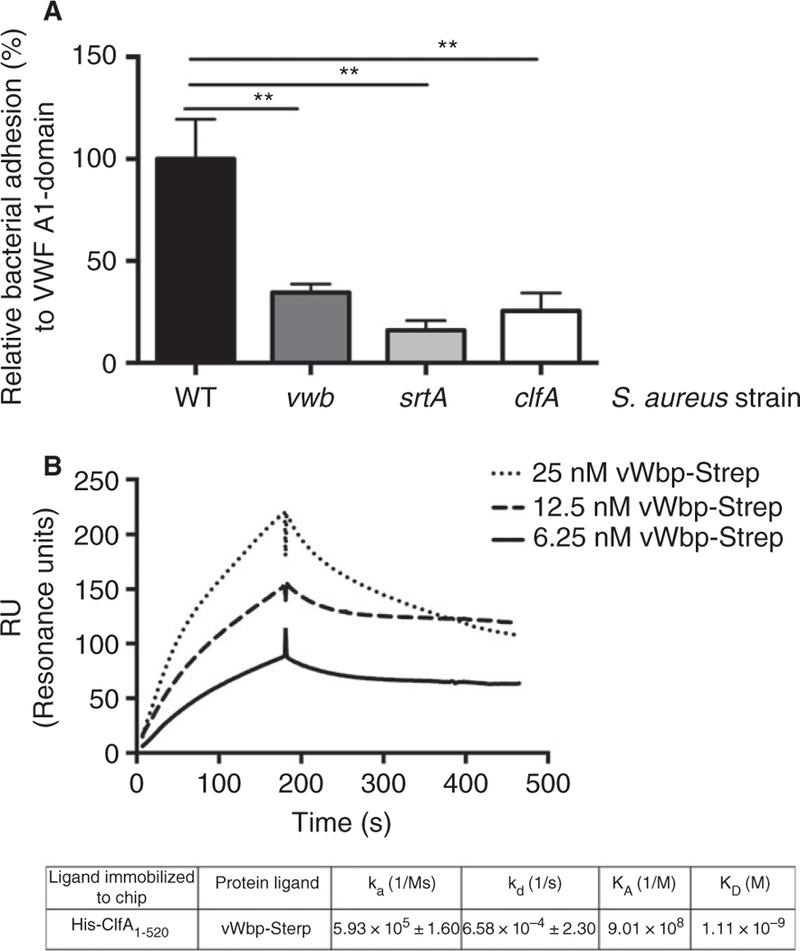
vWbp forms a complex with the VWF A1-domain and ClfA. (A) Micro-parallel plate flow chamber perfusion over coated VWF A1-domain (50 µg mL−1) with fluorescently labeled WT, vwb, srtA, clfA and spa at a shear rate of 1000 s−1 (n > 8). All results are expressed as mean SEM. **P < 0.05. (B) Protein–protein interaction study with surface plasmon resonance. 200 nM His-ClfA1–520 was captured with an NTA chip and perfused with different concentrations of recombinant vWbp-Strep. All injections were performed with a flow rate of 10 lL min−1. ka = association rate, kd = dissociation rate, KA = association constant, KD = dissociation constant. vWbp, von Willebrand factor-binding protein; VWF, von Willebrand factor; ClfA, Clumping factor A; WT, wild type; NTA, nitrilotri-acetic acid.
To extend the submolecular localization of vWbp binding to the VWF A1-domain to interactions with ClfA, we measured the association of His-ClfA1–520 with vWbp-Strep by surface plasmon resonance (Fig. 4B). Using a range of concentrations, we calculated the dissociation constant (KD) to be around 1 nM for the interaction between soluble vWbp-Strep and immobilized His-ClfA1–520, representative of a moderately high affinity (Fig. 4B).
These findings confirmed that vWbp interacts with the VWF A1-domain and with the S. aureus surface protein ClfA simultaneously.
L. lactis expressing staphylococcal ClfA binds to vWbp and to VWF in the presence of vWbp
We independently verified our findings using Lactococcus lactis (L. lactis) bacteria expressing single staphylococcal surface molecules on their cell walls. The control L. lactis pIL253 strain showed only minimal adhesion to rvWbp under flow. Expression of ClfA in L. lactis sufficed to allow adhesion to rvWbp under flow (Fig. 5A) (P = 0.0018). L. lactis expressing FnBPA (Fibronectin binding protein A) or FnBPB (Fibronectin binding protein B), two well-described staphylococcal SrtA-dependent surface proteins, was unable to adhere to rvWbp. No adhesion of the L. lactis strains was observed to coated VWF A1-domain. However, when rvWbp was present, L. lactis expressing ClfA showed a significantly increased adhesion to the VWF A1-domain under flow (Fig. 5B) (P = 0.0354). rvWbp had no effect on the adhesion of L. lactis pIL253 or the lactococci expressing FnBPA or FnBPB (data not shown). Next, we examined the adhesion of L. lactis to endothelial cells under flow. L. lactis expressing ClfA was not able to adhere to activated endothelial cells. Adding rvWbp to the perfusate remarkably increased adhesion of this strain to activated endothelial cells under flow (Fig. 5C) (P = 0.0013).
Fig. 5.
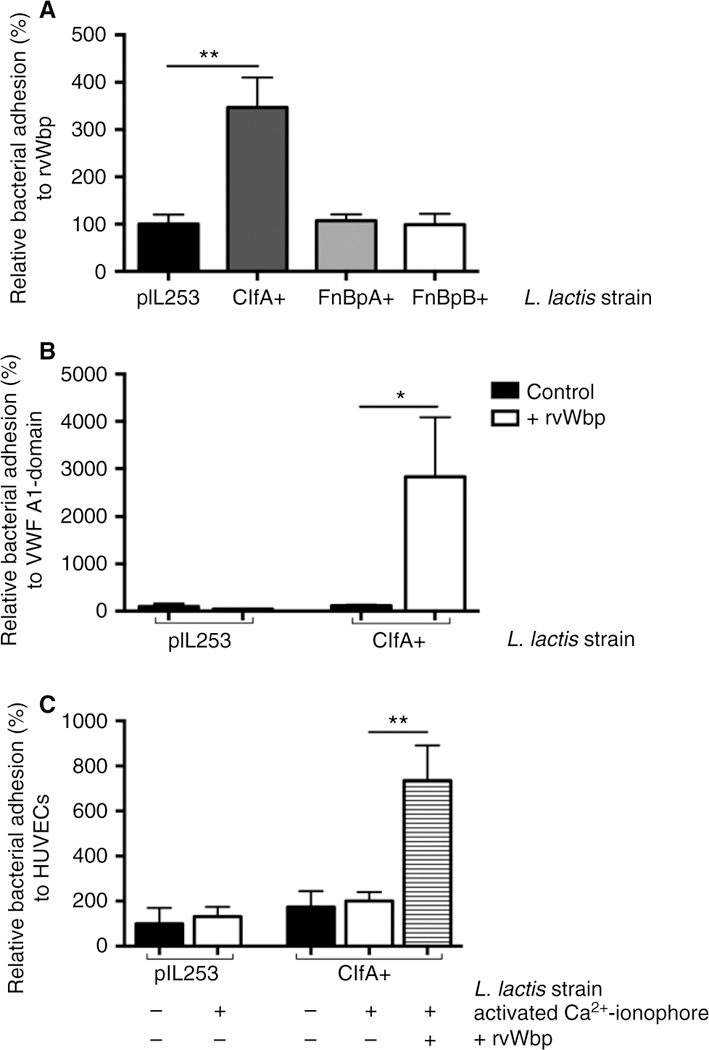
L. lactis expressing staphylococcal ClfA binds to vWbp. (A) Micro-parallel plate flow chamber perfusion over coated rvWbp (30 µg mL−1) with fluorescently labeled L. lactis pIL253, L. lactis ClfA+, L. lactis FnBpA+ and L. lactis FnBpB+ strains at a shear rate of 1000 s−1 (n = 8). All results are expressed as mean SEM. **P < 0.01. (B) Micro-parallel plate flow chamber perfusion over coated VWF A1-domain (50 µg mL−1) with fluorescently labeled L. lactis pIL253 and L. lactis ClfA+ strains at a shear rate of 1000 s−1 (n ≥ 9). Where indicated, rvWbp (20 µg mL−1) was added. All results are expressed as mean SEM. *P < 0.05. (C) Micro-parallel flow chamber perfusion over HUVECs with fluorescently labeled L. lactis pIL253 and L. lactis ClfA+, strains at a shear rate of 1000 s−1. Where indicated, HUVECs were activated by a 5-min perfusion with a Ca2+-ionophore. Where indicated, rvWbp (50 µg mL−1) was added (n ≥ 8). All results are expressed as mean SEM. **P < 0.01. ClfA, Clumping factor A; vWbp, von Willebrand factor-binding protein; rvWbp, recombinant His6-vWbp; VWF, von Willebrand factor; HUVECs, human umbilical vein endothelial cells.
Together, these data show that expression of ClfA is sufficient to bind to the VWF A1-domain, but only in the presence of vWbp.
Adhesion of S. aureus to the vessel wall in vivo is mediated by the VWF-vWbp-ClfA complex
We confirmed these findings using an in vivo intravital mesenteric perfusion model. The WT strain was able to roll over and adhere to the activated murine vessel wall (Fig. 6A/B, Video S1) at the site of locally stimulated VWF release. Similar to our previous observations, the absence of ClfA or vWbp mitigated bacterial adhesion to the vessel wall compared with the WT strain (P = 0.0033 and P = 0.0050, respectively). However, supplementing vwb with rvWbp restored its adhesion to the vessel wall (P = 0.0005) (Video S2 and S3).
Fig. 6.
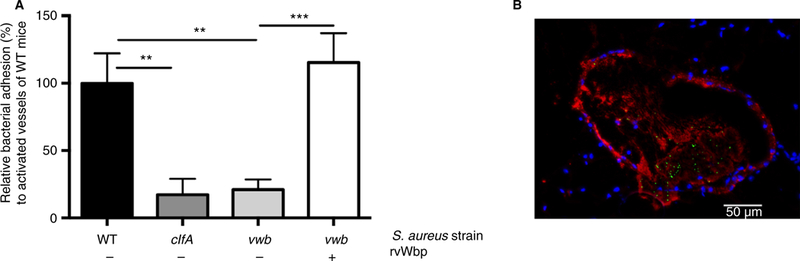
Adhesion of S. aureus to the vessel wall in vivo is mediated by the ternary complex VWF-vWbp-ClfA. (A) In vivo venous mesenteric perfusion model with WT mice. A total of 5 lL of the Ca2+ -ionophore A23187 (10 mM) was applied to the region of the visualized vascular bed to trigger endothelial cell activation and VWF release. A suspension of fluorescent-labeled WT, clfA and vwb strains was injected through the jugular catheter. Where indicated, 20 µg mL−1 rvWbp was added to the bacterial perfusate (n ≥ 14). All results are expressed as mean SEM. **P < 0.01, ***P < 0.001. (B) Fluorescence image (9 630) of S. aureus (green) adhering to activated murine vessel wall with immuno-staining for VWF (red) and 4’,6-diamidino-2-phenylindole-staining of the cell nucleus (blue). White bar is 50 lm. VWF, von Wille-brand factor; vWbp, von Willebrand factor-binding protein; ClfA, Clumping factor A; WT, wild type. [Color figure can be viewed at wileyonlinelibrary.com]
We conclude that vWbp interacts both with sheared VWF and with the staphylococcal surface protein ClfA. The ternary complex formed by endothelial VWF, secreted vWbp and bacterial ClfA mediates adhesion of S. aureus to the vascular endothelium with a high efficiency.
Discussion
Despite improvement in medical supportive care and the implementation of a more aggressive surgical approach, S. aureus infective endocarditis continues to have a very high mortality [29]. Furthermore, antibiotic resistance spreads at an alarming pace, urging new ways to prevent and treat this severe disease. The inability to improve out-come once infective endocarditis has been diagnosed underlines the importance of intervening at the early stages of the disease, preferably to prevent infective endocarditis from developing in the first place.
Patients with S. aureus bacteremia are at high risk of developing infective endocarditis [30]. Infection of cardiac valves requires the binding of S. aureus to the endothelium under the high shear stress of flowing blood. Identifying virulence factors that mediate this initial binding can help to develop strategies to prevent infective endocarditis.
In our previous work we showed that S. aureus exploits the VWF-mediated binding that localizes platelets to sites of vascular damage or inflammation [6]. Although we identified the bacterial protein vWbp as a crucial factor, the precise mechanisms remained unclear, because vWbp is a secreted protein that lacks a cell-wall anchoring sequence.
In this study, we unravel how vWbp, a secreted protein, can bind to the bacterial cell wall, thus allowing shear-resistant binding of S. aureus to the inflamed or damaged endothelium and to the subendothelial matrix. We identified ClfA, an SrtA-mediated surface protein, as the bacterial surface binding partner for vWbp. Both vWbp and ClfA were shown to be crucial factors in the initial adhesion of S. aureus to the vascular endothelium.
S. aureus has many surface proteins that enable its binding to host proteins, endothelial cells or to subendothelial matrix. These surface proteins or MSCRAMMs (e.g. ClfA, FnBPA/B and SpA) are covalently bound to the cell wall by a transpeptidase, SrtA. Absence of SrtA leads to the defective anchoring of about 20 staphylococcal surface proteins [13–15]. Our findings indicate that the srtA gene and the subsequent correct anchoring of staphylococcal surface proteins are vital for S. aureus binding to the vascular endothelium via VWF. However, the adhesive contribution of these proteins in a flow field remains uncertain. Whereas the surface proteins ClfA and SpA can bind to vWbp, only ClfA contributed to the adhesion of S. aureus to the vascular endothelium via VWF in flow. ClfA is known to adhere to endothelial cells via fibrinogen and fibronectin, but it has never been associated with VWF binding. SpA binding to VWF has been reported in static conditions; however, in flow, the recruitment of S. aureus to endothelial cells is SpA independent [5,6].
VWF circulates in a compact globular form, but is progressively unfolded in a flow field or when bound to collagen, thereby exposing VWF A1, A2 and A3-domain [7]. It has been described that bacteria can bind to endothelial cells via VWF [5,6,31]. Similar to our findings, Pappelbaum et al. showed that ADAMTS-13 decreased VWF-mediated S. aureus adhesion to endothelial cells by 50% [5]. We have previously shown that binding of S. aureus to VWF can be blocked by an A1 neutralizing antibody, an antibody that also blocks the binding of platelets to VWF [6]. We now show that S. aureus binds directly to the VWF A1-domain and does so via vWbp and ClfA.
L. lactis, non-pathogenic bacteria, process their surface proteins in a similar way to S. aureus via a LPXTG motif. L. lactis expressing single staphylococcal surface molecules on their cell walls are therefore widely used to study the adhesive properties of a single surface protein. In 2013, Veloso et al. demonstrated the relevance of these lactococci expressing single surface proteins using a low-grade bacteremia model [11,20,32]. Using an L. lactis strain expressing ClfA, we confirmed that the simple presence of ClfA was sufficient to confer adhesion to the VWF A1-domain, but only in the presence of vWbp.
S. aureus exploits a variety of mechanisms to interact with and bind to the host’s tissue. Therefore, targeting a single virulence factor may be insufficient to block clinically relevant bacterial adhesion. We confirmed the pathophysiological relevance of this adhesion mechanism by studying in vivo adhesion of S. aureus to the murine mesenteric circulation. Indeed, S. aureus binding to activated endothelium in vivo was also VWF, vWbp and ClfA dependent. Adding vWbp restored the adhesive phenotype of the vWbp mutant strain, again highlighting the importance of this protein in bacterial adhesion to the vessel wall. As absence of either ClfA or vWbp prevented bacterial adhesion in vivo, it is tempting to speculate about the therapeutic potential of a strategy targeting these factors in patients with S. aureus bacteremia [33– 35]. Interestingly, it was previously shown that a vaccine strategy against ClfA reduced the incidence of experimental infective endocarditis in bacteremic mice [34]. Similarly, vaccinating against ClfA also reduced binding of S. aureus to an aortic patch in mice [29].
In summary, our work identifies ClfA as a novel bacterial binding partner for staphylococcal vWbp. Together, these two proteins promote the adhesion of S. aureus to vascular endothelium. Further unraveling of the interactions between VWF, secreted vWbp and bacterial ClfA may lead to novel preventive or therapeutic strategies that reduce the high mortality of S. aureus infective endocarditis.
Supplementary Material
Essentials.
Staphylococcus aureus (S. aureus) binds to endothelium via von Willebrand factor (VWF).
Secreted VWF-binding protein (vWbp) mediates S. aureus adhesion to VWF under shear stress.
vWbp interacts with VWF and the Sortase A-dependent surface protein Clumping factor A (ClfA).
VWF-vWbp-ClfA anchor S. aureus to vascular endothelium under shear stress.
Acknowledgements
We thank A. Gils from the Department of Pharmaceutical and Pharmacological Sciences at the University of Leuven for the use of equipment and help with the surface plasmon resonance experiments. We thank K. Vanhoorelbeke from the Laboratory for Thrombosis Research, KULAK, for the kind gift of the anti-VWF A1 domain antibody 6D1 and rADAMTS-13. We thank K. Cludts, M. Lox, S. Van Kerckhoven and G. Compernolle for their skillful technical assistance.
This work was supported by the University of Leuven (OT 14/097) and by the Fonds voor Wetenschappelijk Onderzoek (FWO) Vlaanderen.
M. Peetermans reports grants from Research Foundation Flanders (FWO-Vlaanderen, grant number 11I0113N) during the conduct of the study and non-financial support from Pfizer outside the submitted work. P. Verhamme reports grants and personal fees from Boehringer-Ingelheim, Bayer, and Daiichi Sankyo, as well as personal fees from Pfizer, outside the submitted work.
Footnotes
Disclosure of Conflict of Interests
The other authors state that they have no conflict of interest.
References
- 1.Petti CA, Fowler VG Jr. Staphylococcus aureus bacteremia and endocarditis. Infect Dis Clin North Am 2002; 16: 413–35. [DOI] [PubMed] [Google Scholar]
- 2.Lowy FD. Staphylococcus aureus infections. N Eng J Med 1998; 339: 520–32. [DOI] [PubMed] [Google Scholar]
- 3.Moreillon P, Que YA. Infective endocarditis. Lancet 2004; 363: 139–49. [DOI] [PubMed] [Google Scholar]
- 4.Prendergast BD, Tornos P. Surgery for infective endocarditis: who and when? Circulation 2010; 121: 1141–52. [DOI] [PubMed] [Google Scholar]
- 5.Pappelbaum KI, Gorzelanny C, Grassle S, Suckau J, Laschke MW, Bischoff M, Bauer C, Schorpp-Kistner M, Weidenmaier C, Schneppenheim R, Obser T, Sinha B, Schneider SW. Ultralarge von Willebrand factor fibers mediate luminal Staphylococcus aureus adhesion to an intact endothelial cell layer under shear stress. Circulation 2013; 128: 50–9. [DOI] [PubMed] [Google Scholar]
- 6.Claes J, Vanassche T, Peetermans M, Liesenborghs L, Vanden-briele C, Vanhoorelbeke K, Missiakas D, Schneewind O, Hoylaerts MF, Heying R, Verhamme P. Adhesion of Staphylococcus aureus to the vessel wall under flow is mediated by von Wille-brand factor-binding protein. Blood 2014; 124: 1669–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem 1998; 67: 395–424. [DOI] [PubMed] [Google Scholar]
- 8.Dong JF, Moake JL, Nolasco L, Bernardo A, Arceneaux W, Shrimpton CN, Schade AJ, McIntire LV, Fujikawa K, Lopez JA. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood 2002; 100: 4033–9. [DOI] [PubMed] [Google Scholar]
- 9.Heying R, van de Gevel J, Que YA, Moreillon P, Beekhuizen H. Fibronectin-binding proteins and clumping factor A in Staphylo-coccus aureus experimental endocarditis: FnBPA is sufficient to activate human endothelial cells. Thromb Haemost 2007; 97: 617–26. [PubMed] [Google Scholar]
- 10.Patti JM, Allen BL, McGavin MJ, Hook M. MSCRAMM-mediated adherence of microorganisms to host tissues. Annu Rev Microbiol 1994; 48: 585–617. [DOI] [PubMed] [Google Scholar]
- 11.Que YA, Francois P, Haefliger JA, Entenza JM, Vaudaux P, Moreillon P. Reassessing the role of Staphylococcus aureus clumping factor and fibronectin-binding protein by expression in Lactococcus lactis. Infect Immun 2001; 69: 6296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mazmanian SK, Liu G, Ton-That H, Schneewind O. Staphylo-coccus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science 1999; 285: 760–3. [DOI] [PubMed] [Google Scholar]
- 13.Roche FM, Massey R, Peacock SJ, Day NP, Visai L, Speziale P, Lam A, Pallen M, Foster TJ. Characterization of novel LPXTG-containing proteins of Staphylococcus aureus identified from genome sequences. Microbiology 2003; 149: 643–54. [DOI] [PubMed] [Google Scholar]
- 14.Mazmanian SK, Liu G, Jensen ER, Lenoy E, Schneewind O. Staphylococcus aureus sortase mutants defective in the display of surface proteins and in the pathogenesis of animal infections. Proc Natl Acad Sci USA 2000; 97: 5510–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsompanidou E, Denham EL, Sibbald MJ, Yang XM, Seinen J, Friedrich AW, Buist G, van Dijl JM. The sortase A substrates FnbpA, FnbpB, ClfA and ClfB antagonize colony spreading of Staphylococcus aureus. PLoS One 2012; 7: e44646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baba T, Bae T, Schneewind O, Takeuchi F, Hiramatsu K. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J Bacteriol 2008; 190: 300–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng AG, McAdow M, Kim HK, Bae T, Missiakas DM, Schneewind O. Contribution of coagulases towards Staphylococcus aureus disease and protective immunity. PLoS Pathog 2010; 6: e1001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bae T, Banger AK, Wallace A, Glass EM, Aslund F, Schneewind O, Missiakas DM. Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proc Natl Acad Sci USA 2004; 101: 12312–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng AG, Kim HK, Burts ML, Krausz T, Schneewind O, Missiakas DM. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J 2009; 23: 3393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Que YA, Haefliger JA, Francioli P, Moreillon P. Expression of Staphylococcus aureus clumping factor A in Lactococcus lactis subsp. cremoris using a new shuttle vector. Infect Immun 2000; 68: 3516–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vanassche T, Verhaegen J, Peetermans WE, Van Ryn J, Cheng A, Schneewind O, Hoylaerts MF, Verhamme P. Inhibition of staphylothrombin by dabigatran reduces Staphylococcus aureus virulence. J Thromb Haemost 2011; 9: 2436–46. [DOI] [PubMed] [Google Scholar]
- 22.Thomer L, Schneewind O, Missiakas D. Multiple ligands of von Willebrand Factor binding protein (vWbp) promote Staphylococcus aureus clot formation in human plasma. J Biol Chem 2013; 288: 28283–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McAdow M, Kim HK, Dedent AC, Hendrickx AP, Schneewind O, Missiakas DM. Preventing Staphylococcus aureus sepsis through the inhibition of its agglutination in blood. PLoS Pathog 2011; 7: e1002307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonnefoy A, Yamamoto H, Thys C, Kito M, Vermylen J, Hoylaerts MF. Shielding the front-strand beta 3 of the von Wille-brand factor A1 domain inhibits its binding to platelet glycoprotein Ibalpha. Blood 2003; 101: 1375–83. [DOI] [PubMed] [Google Scholar]
- 25.Claes J, Liesenborghs L, Lox M, Verhamme P, Vanassche T, Peetermans M. In Vitro and In Vivo Model to Study Bacterial Adhesion to the Vessel Wall Under Flow Conditions. J Vis Exp 2015; 11: e52862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Theilmeier G, Lenaerts T, Remacle C, Collen D, Vermylen J, Hoylaerts MF. Circulating activated platelets assist THP-1 monocytoid/endothelial cell interaction under shear stress. Blood 1999; 94: 2725–34. [PubMed] [Google Scholar]
- 27.Nassar T, Akkawi S, Shina A, Haj-Yehia A, Bdeir K, Tarshis M, Heyman SN, Higazi AA. In vitro and in vivo effects of tPA and PAI-1 on blood vessel tone. Blood 2004; 103: 897–902. [DOI] [PubMed] [Google Scholar]
- 28.Salgado-Pabon W, Breshears L, Spaulding AR, Merriman JA, Stach CS, Horswill AR, Peterson ML, Schlievert PM. Superantigens are critical for Staphylococcus aureus Infective endocarditis, sepsis, and acute kidney injury. MBio 2013; 4: e00494–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG Jr. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 2015; 28: 603–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fowler VG Jr, Li J, Corey GR, Boley J, Marr KA, Gopal AK, Kong LK, Gottlieb G, Donovan CL, Sexton DJ, Ryan T. Role of echocardiography in evaluation of patients with Staphylococcus aureus bacteremia: experience in 103 patients. J Am Coll Cardiol 1997; 30: 1072–8. [DOI] [PubMed] [Google Scholar]
- 31.Liesenborghs L, Peetermans M, Claes J, Veloso TR, Vanden-briele C, Criel M, Lox M, Peetermans WE, Heilbronner S, de Groot PG, Vanassche T, Hoylaerts MF, Verhamme P. Shear-Resistant Binding to von Willebrand Factor Allows Staphylococcus lugdunensis to Adhere to the Cardiac Valves and Initiate Endocarditis. J Infect Dis 2016; 213: 1148–56. [DOI] [PubMed] [Google Scholar]
- 32.Veloso TR, Chaouch A, Roger T, Giddey M, Vouillamoz J, Majcherczyk P, Que YA, Rousson V, Moreillon P, Entenza JM. Use of a human-like low-grade bacteremia model of experimental endocarditis to study the role of Staphylococcus aureus adhesins and platelet aggregation in early endocarditis. Infect Immun 2013; 81: 697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peetermans M, Verhamme P, Vanassche T. Coagulase Activity by Staphylococcus aureus: a potential target for therapy? Semin Thromb Hemost 2015; 41: 433–44. [DOI] [PubMed] [Google Scholar]
- 34.Veloso TR, Mancini S, Giddey M, Vouillamoz J, Que YA, Moreillon P, Entenza JM. Vaccination against Staphylococcus aureus experimental endocarditis using recombinant Lactococcus lactis expressing ClfA or FnbpA. Vaccine 2015; 33: 3512–7. [DOI] [PubMed] [Google Scholar]
- 35.McAdow M, DeDent AC, Emolo C, Cheng AG, Kreiswirth BN, Missiakas DM, Schneewind O. Coagulases as determinants of protective immune responses against Staphylococcus aureus. Infect Immun 2012; 80: 3389–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ellison-Wright Z, Heyman I, Frampton I, Rubia K, Chitnis X, Ellison-Wright I, Williams SC, Suckling J, Simmons A, Bullmore E. Heterozygous PAX6 mutation, adult brain structure and fronto-striato-thalamic function in a human family. Eur J Neuorsci 2004; 19: 1505–12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.