

cccDNA elimination is a major goal in future curative regimens for chronic HBV patients. However, PCR-based assays for cccDNA quantification show a principally constrained specificity when high levels of input virus or replicative intermediates are present. Here, we characterized T5 exonuclease as a suitable enzyme for medium-throughput in vitro assays that preserves cccDNA but efficiently removes rcDNA prior to PCR-based quantification. We compared T5 exonuclease with the previously described exonuclease III and showed that both nucleases are suitable for reliable quantification of cccDNA by PCR. We substantiated the applicability of our method through examination of early cccDNA formation and stable accumulation in several in vitro infection models and analyzed cccDNA stability after administration of anti-HBV drugs. Our results support the use of T5 exonuclease for fast and convenient rcDNA removal, especially for early cccDNA quantification and rapid drug testing in in vitro studies.
KEYWORDS: covalently closed circular DNA, Myrcludex B, T5 exonuclease, cccDNA, hepatitis B virus
ABSTRACT
Chronic infection with the human hepatitis B virus (HBV) is a major health problem. Virus persistence requires the establishment and maintenance of covalently closed circular DNA (cccDNA), the episomal virus template in the nucleus of infected hepatocytes. Compared to replicative DNA intermediates (relaxed circular DNA [rcDNA]), copy numbers of cccDNA in infected hepatocytes are low. Accordingly, accurate analyses of cccDNA require enrichment of nuclear fractions and Southern blotting or selective quantitative PCR (qPCR) methods allowing discrimination of cccDNA and rcDNA. In this report, we analyzed cccDNA-specific primer pairs for their ability to amplify cccDNA selectively. Using mixtures of defined forms of HBV and genomic DNA, we determined the potential of different nucleases for targeted digestion of the open/relaxed circular DNA forms in the absence and presence of genomic DNA without affecting cccDNA. We found that the combination of T5 exonuclease with a primer set amplifying an approximately 1-kb fragment permits reliable quantification of cccDNA without the requirement of prior nucleus enrichment or Hirt extraction. We tested this method in four different in vitro infection systems and quantified cccDNA copy numbers at increasing multiplicities of inoculated genome equivalents. We further analyzed the kinetics of cccDNA formation and the effect of drugs (interferon, entry inhibitors, and capsid inhibitors) on cccDNA. Our method allows reliable cccDNA quantification at early stages of infection in the presence of a high excess of input virus and replicative intermediates and is thereby suitable for drug screening and investigation of cccDNA formation and maintenance.
IMPORTANCE cccDNA elimination is a major goal in future curative regimens for chronic HBV patients. However, PCR-based assays for cccDNA quantification show a principally constrained specificity when high levels of input virus or replicative intermediates are present. Here, we characterized T5 exonuclease as a suitable enzyme for medium-throughput in vitro assays that preserves cccDNA but efficiently removes rcDNA prior to PCR-based quantification. We compared T5 exonuclease with the previously described exonuclease III and showed that both nucleases are suitable for reliable quantification of cccDNA by PCR. We substantiated the applicability of our method through examination of early cccDNA formation and stable accumulation in several in vitro infection models and analyzed cccDNA stability after administration of anti-HBV drugs. Our results support the use of T5 exonuclease for fast and convenient rcDNA removal, especially for early cccDNA quantification and rapid drug testing in in vitro studies.
INTRODUCTION
Chronic hepatitis B virus (HBV) infection is a major infectious disease, leading to liver cirrhosis and hepatocellular carcinoma (1). Presently, about 240 million people worldwide are chronic HBV carriers (2). Although vaccines are currently accessible in most countries of the world, the number of chronically infected patients is not expected to decrease in the near future due to the growing world population (3). Current treatment options for chronic HBV patients are limited to alpha interferon (IFN-α) and nucleos(t)ide analogs, which modulate immune responses and suppress viral replication. However, they fail to eliminate the episomal template of virus replication, covalently closed circular DNA (cccDNA), in the majority of patients (4, 5). cccDNA is formed in the nucleus of infected hepatocytes, where it serves as the template for all HBV transcripts. cccDNA is supposed to persist within hepatocytes with half-lives comparable to those of its host cell. For still-unknown reasons, the copy numbers of HBV cccDNA in human hepatocytes are very low, although a very high number of replicative intermediates are present, which could replenish and amplify the intracellular “cccDNA pool” (6). This contrasts with the findings in other hepadnaviruses (e.g., duck hepatitis B virus [DHBV]), where numbers of 50 cccDNA molecules and higher have been reported, especially when the envelope protein is depleted (7–10). During viral replication, pregenomic RNA (pgRNA) is transcribed from cccDNA, transported to the cytoplasm, encapsidated into nucleocapsids, and reverse transcribed within the capsid by the viral polymerase to form a polymerase-bound relaxed circular DNA (rcDNA) complex. HBV rcDNA is partially double stranded and contains a complete minus strand but an incomplete plus strand. In an infected cell, encapsidated cytosolic rcDNA molecules are present in a ∼100-fold excess (11) over the nuclear cccDNA (6, 12, 13). Moreover, released virions and released naked nucleocapsids contain rcDNA.
Due to the longtime lack of convenient infection systems, which became available only in 2012 after identification of human sodium taurocholate cotransporting polypeptide (hNTCP) as the major receptor (14, 15), many aspects of HBV cccDNA molecular biology were unknown. Accordingly, many studies have been performed in the DHBV system showing that cccDNA is rapidly formed from incoming virus and becomes “amplified” upon import of newly formed rcDNA-containing nucleocapsids (7). However, we lack information on whether such amplification occurs in HBV-infected cells (16). Particularly, it is unclear whether and to what extent cccDNA turnover in infected livers occurs (e.g., via intracellular replenishment or via an extracellular route involving de novo infection of hepatocytes). With now-available infection systems, a major (not trivial) problem is the reliable early detection of cccDNA in the presence of a high excess of rcDNA coming from virus input or replicative intermediates (6).
Southern blotting is the gold standard for cccDNA detection and quantification; however, the limited number of samples that can be analyzed and the large amounts of DNA needed are not compatible with, e.g., drug screening approaches for cccDNA inhibitors (17). To overcome this limitation, quantitative PCR (qPCR) methods are desirable but require selective detection and/or biochemical separation of cccDNA from other HBV DNA (18–20). One approach to gain specificity is the use of cccDNA-specific primers spanning the gap region of rcDNA (19, 20). However, this approach is limited in its specificity since primer extensions of these oligonucleotides during the initial PCR cycles generate overlapping annealing products that serve as the templates similar to cccDNA for subsequent cycles (6).
To solve that problem, several approaches to purify cccDNA before selective qPCRs have been applied: (i) enrichment of nuclei, (ii) Hirt extraction to remove polymerase-bound rcDNA (21), and (iii) Plasmid-Safe DNase (PSD) digestion aiming at hydrolyzation of rcDNA (22). However, these methods either incompletely remove rcDNA (first and second methods) or are not selective enough regarding the hydrolysis of rcDNA (third method) (13, 23). Particularly, PSD, an enzyme originally applied for removing bacterial chromosomal DNA from plasmid preparations, preferentially hydrolyzes double-stranded linear DNA (dslDNA) and, with a lower efficiency, linear and closed circular single-stranded DNAs (ssDNAs) (Lucigen, Middleton, WI). PSD shows negligible enzymatic activity against closed circular supercoiled DNA and is also not active against nicked circular double-stranded DNA (23). Nevertheless, PSD-based protocols have been developed to remove replicative HBV intermediates prior to cccDNA quantification, especially for samples obtained from livers of chronic HBV-infected patients in which such intermediates were suppressed by inhibitors (22). Recently, a combination of exonuclease I (Exo I) and exonuclease III (Exo III) was successfully used to degrade DNA strands with free 3′ ends prior to cccDNA quantification (24).
Here, we aimed at systematically investigating the effect of different nucleases, including PSD and the recently described Exo I and Exo III, on selective removal of rcDNA from cccDNA and established a reliable protocol for HBV cccDNA quantification from total-cell lysates from in vitro-infected cells. We confirmed Exo III, a combination of Exo I and Exo III, and—as characterized in more detail here—T5 exonuclease (T5 Exo) (25) as suitable enzymes that efficiently remove HBV rcDNA in the presence of host genomic DNA without degrading cccDNA. Combining T5 Exo treatment with optimized TaqMan qPCR allowed us to scale down cccDNA quantification to a 96-well format. We analyzed the kinetics of cccDNA formation in primary human hepatocytes (PHH) and HepaRG and HepG2hNTCP cells and describe the effects of selected anti-HBV drugs (alpha interferon [IFN-α], Myrcludex B, tenofovir, and the capsid inhibitor GLS4) on cccDNA formation and maintenance.
RESULTS
Residual rcDNA amplification by qPCR using cccDNA-specific primers.
To establish a cccDNA-specific qPCR assay, we compared five different cccDNA primer sets (972-1995, 1558-1958, 1548-1886, 1578-1867, and 1583-2301) described in the literature (14, 18, 26–28) with a newly designed set (1040-1996, probe p1085) (Table 1). All the cccDNA-specific primer pairs (pp’s) spanning the gap region of rcDNA were compared with pp466-541, which does not discriminate between rcDNA and cccDNA forms (Fig. 1A). We compared cytosolic fractions (rcDNA without any cccDNA) with nuclear lysates (cccDNA and low copy numbers of protein-free rcDNA) using all different primer sets under the respective optimized conditions (see legend to Fig. 1B). While the nonselective pp466-541 showed a 36.8-fold-higher signal in the cytosolic fraction than the nuclear fraction, pp1040-1996 as well as five published pp’s resulted in comparable signals for the two fractions, verifying the specificity of pp1040-1996 and other pp’s under the respective conditions for cccDNA (Fig. 1B). However, none of the cccDNA primers showed absolute specificity since cytosolic DNA provided a signal comparable with that of nuclear DNA. A closer comparison between the two primers pp1040-1996 and pp972-1995, leading to a 1-kb amplicon, indicated a higher sensitivity of pp1040-1996 (Fig. 1C). We therefore used pp1040-1996 in all subsequent experiments.
TABLE 1.
Primers in the study
| Pair namea | Sequence information | Product size, bp (reference) |
|---|---|---|
| pp1040-1996 | 1040: 5′-GTGGTTATCCTGCGTTGAT-3′ | 974 |
| 1996: 5′-GAGCTGAGGCGGTATCT-3′ | ||
| p1085: 5′-FAM-AGTTGGCGAGAAAGTGAAAGCCTGC-TAMRA-3′b | ||
| pp972-1995 | 972: 5′-GCCTATTGATTGGAAAGTATGT-3′ | 1,040 (18) |
| 1995: 5′-AGCTGAGGCGGTATCTA-3′ | ||
| pp1558-1958 | 1558: 5′-GTGCCTTCTCATCTGCCGG-3′ | 420 (28) |
| 1958: 5′-GGAAAGAAGTCAGAAGGCAA-3′ | ||
| pp1548-1886 | 1548: 5′-CTCCCCGTCTGTGCCTTCT-3′ | 356 (26) |
| 1886: 5′-GCCCCAAAGCCACCCAAG-3′ | ||
| pp1578-1867 | 1578: 5′-CCGTGTGCACTTCGCTTCA-3′ | 308 (27) |
| 1867: 5′-GCACAGCTTGGAGGCTTGA-3′ | ||
| pp1583-2301 | 1583: 5′-TGCACTTCGCTTCACCT-3′ | 735 (14) |
| 2301: 5′-AGGGGCATTTGGTGGTC-3′ | ||
| pp466-541 | 466: 5′-GTTGCCCGTTTGTCCTCTAATTC-3′ | 125 (18) |
| 541: 5′-GGAGGGATACATAGAGGTTCCTTGA-3′ | ||
| β-Globin | Forward: 5′-AGGTACGGCTGTCATCACTTAGA-3′ | 161 (57) |
| Reverse: 5′-CATGGTGTCTGTTTGAGGTTGCTA-3′ |
EcoRI site in HBV genome: nucleotide numbers 1 to 6.
FAM, 6-carboxyfluorescein; TAMRA, 6-carboxytetramethylrhodamine.
FIG 1.

Excessive rcDNA provides false-positive signals when using cccDNA-specific primers for PCR amplification. (A) cccDNA pp’s (pp1040-1996 with TaqMan probe p1085) were designed to span the nick and gap region of the HBV genome. pp972-1995 was used as a control (18). Other cccDNA-specific primers (pp1558-1958, pp1548-1886, pp1578-1867, and pp1583-2301) selected from previous studies were also implemented (Table 1). Nonselective DNA primers (pp466-541) amplified all HBV DNA species. (B) Cytosolic and nuclear DNA samples from the same infected HepG2hNTCP cells were analyzed using pp’s under the respective programs in the original literature as follows: pp1040-1996, 95°C for 15 min followed by 50 cycles of 95°C for 10 s and then 63°C for 70 s; pp972-1995, 95°C for 10 min followed by 45 cycles of 95°C for 15 s, 60°C for 5 s, and 72°C for 45 s; pp1558-1958, 95°C for 10 min followed by 45 cycles of 95°C for 10 s, 58°C for 5 s, 63°C for 10 s, and 72°C for 20 s; pp1548-1886, 95°C for 10 min followed by 45 cycles of 95°C for 10 s, 62°C for 10 s, and 72°C for 20 s; pp1578-1867, 95°C for 10 min followed by 45 cycles of 95°C for 15 s, 60°C for 1 min, and 72°C for 10 s; pp1583-2301, 95°C for 5 min followed by 45 cycles of 95°C for 30 s, 62°C for 25 s, and 72°C for 45 s; pp466-541, 95°C for 10 min followed by 40 cycles of 95°C for 10 s and 60°C for 30 s. RFU, relative fluorescence units. (C) Copies (103, 102, and 101) of the pSHH2.1 plasmid template were subjected to a TaqMan reaction using pp1040-1996 or pp972-1995 with the p1085 probe. Amplification curves (left) and products (right) are shown. (D) DNA samples were collected from infected HepG2hNTCP cells at indicated times (days 1, 3, 7, 10, and 14 p.i. and day 0 without inoculum). Using pp1040-1996 and pp466-541, cccDNA (left) and total DNA copies (right) were quantified in total lysates or nuclear fractions, without any enzyme predigestion.
We further analyzed the potential of pp466-541 and pp1040-1996 to discriminate between rcDNA and cccDNA from in vitro-infected cells. Thus, we infected HepG2hNTCP cells with cell culture-derived HBV at a multiplicity of genomic equivalents (mge) of 300 per cell; prepared total-cell lysates and nuclear fractions at days 1, 3, 7, 10, and 14 postinfection (p.i.); and performed qPCR using pp1040-1996 and pp466-541 (Fig. 1D). Both primer pairs gave rise to strong signals at day 1 p.i., which decreased at day 3 p.i and increased again at later time points. Since cccDNA synthesis is not completed within 24 h p.i., we assumed that input virion rcDNA associated with the nucleus-enriched fraction provided false-positive signals. At later points in time, the purported cccDNA copy numbers in total-lysate samples appeared 3-fold higher than in the respective nuclear lysates, indicating that progeny rcDNA contributes to the determined cccDNA signal.
More extensive analyses using susceptible and nonsusceptible cell lines, which were inoculated with HBV in the presence or absence of Myrcludex B and/or polyethylene glycol (PEG), supported this assumption. Observed signals of input rcDNA in HeLahNTCP and two mousehNTCP cells excluded human- or hepatic tissue-specific effects (Fig. 2A). Especially, the addition of PEG resulted in enhanced input-derived signals, which is in line with enhanced attachment of HBV to heparin sulfate proteoglycans (HSPG) (Fig. 2B and C) (29, 30).
FIG 2.

Early rcDNA signals are mostly caused by PEG addition in inoculum but not Myrcludex B treatment or hNTCP expression. (A) Total DNA kinetics from inoculum-incubated mouse Hepa1-6hNTCP, mouse Hepa56DhNTCP, human HeLahNTCP, and infected HepG2hNTCP cells (days 1, 2, 3, 6, and 9 p.i. and day 0 without inoculum) were determined by pp466-541. (B and C) Samples from HepG2hNTCP and HepG2 cells infected with HBV inoculum in the presence or absence of PEG (4%) (B) or Myrcludex B (1 μM) (C) during the infection were analyzed by pp466-541.
T5 exonuclease specifically hydrolyzes HBV rcDNA.
We next analyzed the effect of different nucleases with distinct enzymatic activities on rcDNA and dslDNA on purified virus inoculum used for infection (Fig. 3A). As shown by Southern blot analysis, PSD and exonuclease V efficiently hydrolyzed dslDNA but only partially removed rcDNA. In contrast, T5 Exo and BAL-31 nuclease completely hydrolyzed all forms of viral DNA, including rcDNA (Fig. 3B). Since BAL-31 requires high salt concentrations, which block the Taq polymerase activity in the subsequent qPCR analysis, we focused on T5 Exo for further analysis.
FIG 3.
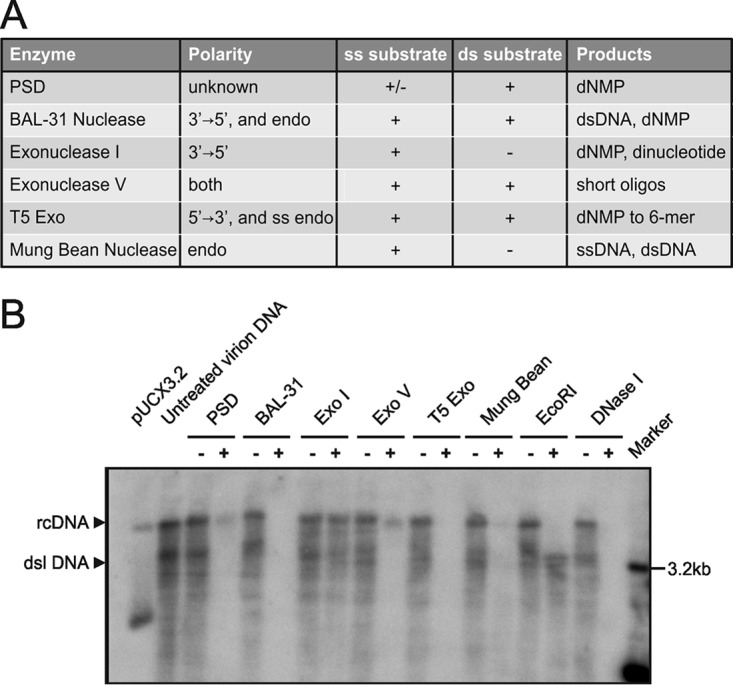
Identification of exonucleases selectively digesting rcDNA. (A) Properties of exonucleases tested in this study. +, strong activity; -, no significant activity; +/-, reduced activity; ss, single stranded; ds, double stranded; endo, endonuclease activity; dNMP, deoxyribonucleoside monophosphate; oligos, oligonucleotides. (B) Copies (3 × 108) of cell culture-derived viral DNA containing rcDNA and dslDNA were incubated for 1 h at 37°C with PSD (5 U), BAL-31 (5 U), Exo I (5 U), Exo V (5 U), and T5 Exo (5 U). Mung bean nuclease (5 U), EcoRI (5 U), and DNase I (5 U) were included as controls. After heat inactivation, the products were subjected to Southern blotting. The plasmid pUCX3.2 served as a marker for indicating the expected sizes of rcDNA and cccDNA.
Since PSD has been previously used for HBV rcDNA digestion (14, 22), we directly compared the two enzymes. As depicted in Fig. 4A, T5 Exo (5 U, 60 min) hydrolyzed rcDNA extracted from purified virus more efficiently than PSD (5 U, 60 min). As controls, we used EcoRI and DNase I (both 5 U for 60 min) that linearized rcDNA or completely hydrolyzed it. The same held true when a cloned HBV genome (pSHH2.1) was used (Fig. 4B). In its linearized form (top panel), it showed sensitivity to T5 Exo (5 U, 60 min) but was hydrolyzed by PSD only at the highest concentration (10 U, 60 min). The circular forms (middle panel) showed resistance to PSD, while T5 Exo selectively hydrolyzed the open circular (oc) form, leaving the closed circular form intact. Genomic DNA from HepG2hNTCP cells was only partially hydrolyzed by PSD (10 U, 60 min) but was sensitive to digestion with T5 Exo already at 1 U for 60 min (bottom panel). As expected, DNase I efficiently hydrolyzed all types of DNA. To mimic the situation during HBV infection, we mixed HBV DNA, extracted from cell culture-derived virus preparations (rcV) or the HBV-containing plasmid pUCX3.2 (representing cccDNA), with cellular genomic DNA and subjected the mixture to hydrolyzation with PSD or T5 Exo (Fig. 4C). PSD (5 U, 60 min) partially removed rcDNA in the absence of genomic DNA but showed reduced activity in its presence, indicating substrate competition by genomic DNA. In contrast, T5 Exo (5 U, 60 min) removed viral rcDNA (left) as well as the open circular DNA from pUCX3.2 (right) in addition to the genomic DNA with high efficiency (Fig. 4C). Next, viral DNA (rcV) or plasmid (pSHH2.1) was hydrolyzed with 10 U of PSD versus 5 U of T5 Exo. Subsequently, we performed qPCR using the nonselective pp466-541 and the cccDNA-selective pp1040-1996 (Fig. 4D). While T5 Exo induced a 2.5-log reduction in rcDNA signals, PSD reduced rcDNA only by 0.9 log. Combination of T5 Exo treatment with qPCR using pp1040-1996 achieved an 8.0-fold log10 reduction. This indicates that T5 Exo hydrolyzed all rcDNA (Fig. 4D). We further incubated genomic DNA or pSHH2.1 plasmid with different levels of T5 Exo and PSD for different times. Titration analysis showed that 5 U of T5 Exo for 60 min hydrolyzed all genomic DNA without affecting supercoiled DNA. However, when T5 Exo was incubated for 16 h, it decreased 60% of supercoiled DNA. Thus, to avoid T5-mediated hydrolyzation of cccDNA, enzyme amounts and incubation time should be carefully adjusted (Fig. 5A and B).
FIG 4.

T5 Exo efficiently removes rcDNA and genomic DNA from DNA preparation. (A) Copies (3 × 108) of virion DNA from purified HBV virions were incubated with PSD (5 U), T5 Exo (5 U), EcoRI (5 U), or DNase I (5 U) at 37°C for 1 h and further subjected to Southern blotting. pUCX3.2 plasmid (3.2 kb) was loaded as well to indicate the positions of rcDNA and cccDNA. (B) (Top) Two micrograms of purified 3.2-kb linear HBV monomer released from the pSHH2.1 plasmid by EcoRI digestion was incubated with indicated units of T5 Exo or PSD at 37°C for 1 h. (Middle) A mixture of 3.2-kb open circular DNA (2 μg) that was artificially nicked by Nb.BtsI endonuclease and 3.2-kb supercoiled pUCX3.2 plasmid (2 μg) was subjected to T5 Exo or PSD digestion at 37°C for 1 h. (Bottom) Two micrograms of genomic DNA from uninfected HepG2hNTCP cells was similarly treated with T5 Exo or PSD. All digestion products are shown on agarose gels, and for relative quantification, band density of untreated samples is set as 100%. (C) Copies (108) of virion DNA or pUCX3.2 plasmid were digested with T5 Exo (5 U) or PSD (5 U) in the absence (0 μg) or presence (2 μg) of genomic DNA (as shown above; 1% agarose gel) at 37°C for 1 h, and the products were loaded for Southern blotting (bottom). (D) Virion DNA (rcV) or pSHH2.1 plasmid was incubated with T5 Exo (5 U) or PSD (10 U) at 37°C for 1 h, and products were further analyzed by pp466-541 (left) or pp1040-1996 (right), respectively. ns, no significance. (E) Total DNA samples from HBV-infected HepG2hNTCP cells (days 1, 2, 3, 6, and 9 p.i. and day 0 without inocula) were incubated with T5 Exo (5 U) or PSD (10 U) as described above, and cccDNA (left) and total DNA (right) copies were quantified by respective primers.
FIG 5.

Titration analysis of T5 Exo and PSD. (A) Two micrograms of genomic DNA samples from HBV-free HepG2hNTCP cells was incubated with T5 Exo or PSD in time-dependent (1, 2, 4, and 16 h) and dose-dependent (1 and 5 U) manners. After digestion, products were visualized on an agarose gel, and the expression level of the human β-globin gene was measured as a representative readout to show digestion degree of genomic DNA. (B) Two micrograms of pSHH2.1 plasmid was incubated with T5 Exo or PSD similarly, and the remaining plasmid in products was determined by pp466-541 or directly visualized on an agarose gel.
To test the applicability of T5 Exo (5 U, 60 min) in comparison to PSD (10 U, 60 min) under standard in vitro infection conditions, we inoculated HepG2hNTCP cells with HBV (mge/cell of 300), washed cells extensively, and extracted total DNA at days 1, 2, 3, 6, and 9 p.i. Using pp1040-1996 (Fig. 4E, left panel), we detected 2 × 106 copies of HBV DNA already at day 1 p.i., which did not significantly decline until day 9 p.i. Hydrolysis with PSD reduced the signal 10-fold; however, at day 1 p.i. (when cccDNA is not fully established), 1.5 × 105 genome equivalents could still be detected. Remarkably, treatment with T5 Exo eliminated this early signal. At later points in time, the signal tended to reach comparable levels of the PSD-treated samples. Using pp466-541 (Fig. 4E, right panel), we detected very high levels of HBV DNA early after inoculation in untreated and even PSD-treated samples which correlate with input virus associated with the cells. T5 Exo eliminated these signals, indicating that it is HBV DNA derived from the inoculum (at least at the early points in time). After decline of the signal, an increase of total HBV DNA was observed (days 6 to 9), which is consistent with the described kinetics of HBeAg and HBsAg formation (15). Taken together, these results indicate that PSD, in contrast to T5 Exo, only partially removes HBV rcDNA from total DNA extractions of infected cells, and they identify T5 Exo as the preferred nuclease to enrich cccDNA for PCR analysis.
T5 Exo and Exo III efficiently remove HBV rcDNA.
A combination of Exo I and Exo III has been recently reported to efficiently hydrolyze all HBV DNA besides cccDNA (24). Since Exo I alone had only limited activity on virion DNA (Fig. 3B), we performed a direct comparison between T5 Exo, Exo I, Exo III, Exo I plus III, and PSD, using DNA samples from in vitro-infected cells, and performed Southern blotting and qPCR. As shown in Fig. 6A, T5 Exo (5 U, 60 min) and Exo III (25 U, 60 min) comparably removed cytosolic HBV DNA replicative intermediates. The same samples quantified side by side by real-time PCR showed that Exo III removed 98%, T5 Exo removed 97%, and Exo I plus III removed 99% of total HBV DNA. In contrast, Exo I (5 U, 60 min) was less efficient and matched PSD (10 U, 60 min), although it slightly enhanced the activity of Exo III. After Hirt extraction, T5 Exo and Exo III digestion (alone or in combination with Exo I) resulted in a single cccDNA band while Exo I alone and PSD still showed rcDNA remnants (Fig. 6B). This strongly supports the application of T5 Exo or Exo III for purification of cccDNA.
FIG 6.

T5 Exo and Exo III remove HBV replicative intermediates without affecting cccDNA. HepG2hNTCP cells were seeded in a 6-well plate and infected at an mge/cell of 3,000. To block entry, Myrcludex B (2 μM) was used as a control. (A) On day 7 p.i., cytosolic DNA samples were extracted as described in Materials and Methods and hydrolyzed by Exo I (5 U, 60 min), Exo III (25 U, 60 min), Exo I and III (5 U plus 25 U, 60 min), T5 Exo (5 U, 60 min), PSD (10 U, 60 min), and EcoRI (10 U, 60 min) at 37°C for 1 h, and later on, all enzymes were heat denatured at 70°C. Samples were analyzed by Southern blotting (left) and PCR with pp466-541 (right). (B) HepG2hNTCP cells were infected in a 6-well plate format for 7 days, and the DNA samples were Hirt extracted and hydrolyzed by the respective enzymes prior to Southern blotting (left) and cccDNA-specific PCR using pp1040-1996 (right).
Kinetics of cccDNA establishment in in vitro-infected hepatocytes.
Taking advantage of T5 Exo digestion of total DNA followed by qPCR, we analyzed cccDNA formation in in vitro-infected HepG2hNTCP cells at different mge’s. Using mge’s of 30 to 3,000 per cell, secreted HBsAg confirmed productive infection (Fig. 7B). At an mge of >1,000, the entry inhibitor Myrcludex B still profoundly reduced HBcAg expression, indicating that HBV replication even under these conditions is exclusively initiated by NTCP-mediated entry of virions and is cccDNA dependent (Fig. 7C). The rise of mge per cell from 30 to 1,000 resulted in an increase of infected cells and a corresponding increase of cccDNA copies per well. Consistently, total DNA also increased. The correlation of mge with HBsAg secretion and an increased number of infected cells was observed previously in HepaRG cells and PHH (30). Interestingly, at an mge of >1,000 no further proportional increase of cccDNA copy numbers and total DNA was observed (Fig. 7A). Thus, cccDNA copy numbers show a saturation when a higher mge of virus is applied under conditions where the majority of cells are already infected.
FIG 7.

cccDNA profiles in infections with increasing mge. (A) HepG2hNTCP cells were infected with different amounts of virus inoculum (mge/cell of 30, 100, 300, 1,000, and 3,000) in parallel, and total DNA samples were prepared on day 10 p.i. Samples were hydrolyzed by T5 Exo (5 U, 60 min) at 37°C for 1 h, and cccDNA was determined using pp1040-1996. Total DNA copy numbers were also determined in undigested samples using pp466-541. (B) Within the same infections, secreted HBsAg values from day 7 to 10 p.i. were detected. (C) On day 10 p.i., intracellular HBcAg expression levels (red) were visualized. As a control to verify NTCP-mediated entry of the virus, Myrcludex B (1 μM) was administered during the infection.
To achieve reasonable infection rates, we further used an mge value of 300 and analyzed the kinetics of cccDNA formation in HepG2hNTCP, HepaRGhNTCP, and HepaRG cells and PHH. Infection rates were 24.0%, 8.6%, 5.3%, and 16.7%, respectively (determined by counting HBcAg-positive cells at day 7 p.i.). We also quantified secreted HBsAg and HBeAg (Fig. 8B and C). In PHH, cccDNA was almost undetectable at day 1 p.i. but accumulated rapidly to an average of 5.8 copies per infected cell at day 10 p.i. (Fig. 9A). Mean cccDNA copy numbers in PHH from three different donors ranged between 2.6 and 10.5 (data not shown). cccDNA formation was blocked by Myrcludex B but not affected by tenofovir. Total HBV DNA (rcDNA plus cccDNA) rapidly increased at time points after day 4, indicating induction of pgRNA synthesis followed by reverse transcription. As expected, total DNA synthesis was sensitive to tenofovir (Fig. 9A). In HepaRG and HepaRGhNTCP cells, cccDNA was already detectable on day 1 p.i. Copy numbers increased until day 3 and remained at a level of 1 to 4 cccDNA copies per infected cell until day 14. This indicates that most of the cccDNA molecules form simultaneously and do not amplify significantly (Fig. 9B and C). Remarkably, cccDNA formation in infected HepG2hNTCP cells behaved similarly to PHH with a moderate linear increase from 0.5 copy/cell at day 3 p.i. to about 1 copy/cell at day 14 (Fig. 9D). Consistent with previous results, high copy numbers of total DNA representing input virions were detected in all cell types on day 1 p.i. Later on, a slow but steady increase of total DNA levels could be observed in all cell types (Fig. 9). Although the kinetics of cccDNA formation differ in PHH and NTCP-transduced cell lines, mean cccDNA copy numbers remain generally low (ca. 5 copies/cell), although mge values of >1,000 have been used for inoculation. Moreover, we did not observe a profound increase of copy number per infected cell within the following 2 weeks, indicating that massive amplification of cccDNA after initial establishment does not occur.
FIG 8.

Elution efficacy in DNA extraction and infectivity in in vitro-infected hepatocytes. (A) Elution efficiency reflecting the in-column loss during DNA extraction was calculated by coloading a series of the diluted pSHH2.1 plasmid with HBV-free genomic carrier DNA onto columns. Eluted DNA (percentage of input) was quantified by pp466-541. (B) Infectivity ratios (day 7 p.i., mge/cell of 300) in indicated cultures were determined by counting HBcAg-positive and Hoechst stain-positive cell numbers from 20 independent views under microscopy. (C) Correlated with Fig. 9, productive infection in all in vitro infections was validated by detecting secreted HBeAg and HBsAg values during days 7 to 10 p.i.
FIG 9.
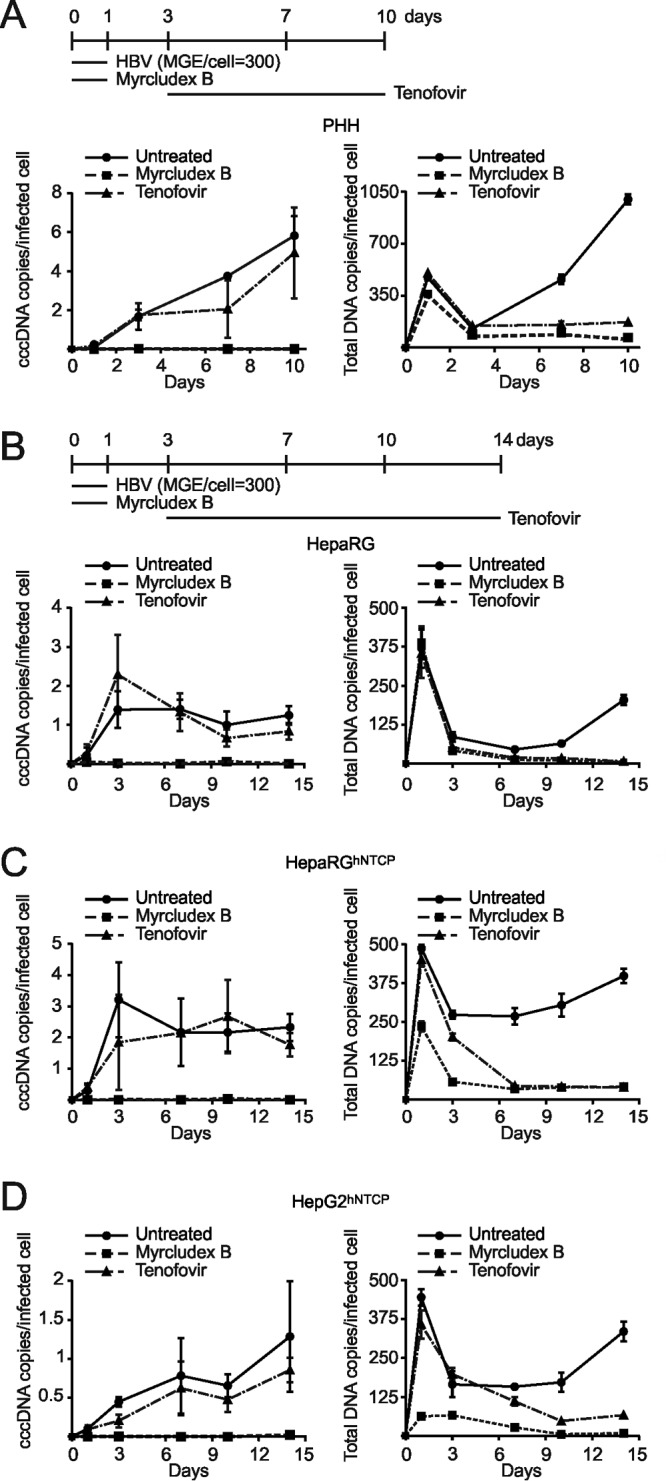
Comparability of cccDNA formation and accumulation in different in vitro infection systems. In vitro infections with a moderate mge (mge/cell of 300) were performed in PHH (A), differentiated HepaRG cells (B), differentiated HepaRGhNTCP cells (C), and HepG2hNTCP cells (D). Myrcludex B (1 μM) and tenofovir (15 μM) were implemented as controls as shown in the timelines. For PHH, total DNA was extracted on days 1, 3, 7, and 10 p.i.; for the other cell lines, samples were collected until day 14 p.i. To measure the respective infection rates, cell counting of HBcAg-positive cells was performed (Fig. 8B). Undigested DNA samples were directly used for total HBV DNA as well as human β-globin quantification. T5 Exo treatment (37°C, 5 U, 60 min) was performed prior to cccDNA quantification. cccDNA copy number was normalized by a factor that was derived from β-globin level to eliminate variation from inputs and further calculated as number of cccDNA copies/cell. Finally, the value of cccDNA copies per infected cell was determined by using number of cccDNA copies/cell to divide elution ratio and infectivity ratio (Fig. 8).
Effect of different drugs on cccDNA formation.
We evaluated our method on a small number of representative drugs, acting at different steps of the HBV replication cycle. We included IFN-α-2b, the two nucleos(t)ide analogs lamivudine and tenofovir, the entry inhibitors Myrcludex B (31) and cyclosporine (32), and the two capsid inhibitors BAY41-4109 (33) and GLS4 (34) at two concentrations and applied them during or after infection (Fig. 10A). In the first set of experiments, IFN-α, Myrcludex B, and cyclosporine were coadministered with the virus while nucleos(t)ide analogs and capsid inhibitors were applied postinoculation (Fig. 10A). We excluded cytotoxicity of the drugs by a lactate dehydrogenase (LDH) assay (Fig. 11B). Consistent with previous reports (18, 35), IFN-α slightly inhibited cccDNA formation and HBeAg secretion in a dose-dependent manner when administered at 1,000 IU/ml (Fig. 10B and C). As expected, Myrcludex B completely blocked de novo cccDNA formation and HBeAg secretion when coadministered with the virus (Fig. 10B and C) but not postinfection (Fig. 10D). Antiviral effects of IFN-α and Myrcludex B were confirmed by immunofluorescence (IF) staining for HBcAg (Fig. 11A). A similar but much weaker effect was observed for cyclosporine, which also prevents entry by inhibiting NTCP (50% inhibitory concentration [IC50] of 8 µM) (32, 36). Lamivudine and tenofovir did not affect cccDNA levels either when applied at day 1 p.i. (Fig. 10C) or when administered with the virus (Fig. 10D). At 1 µM (which is 20-fold/100-fold the respective IC50s for capsid assembly inhibition [34, 37]), neither BAY41-4109 nor GLS4 affected cccDNA copy numbers when administered at day 1 p.i. However, a slight reduction of cccDNA copy numbers (by ca. 30%) was observed at 10 μM (Fig. 10C). Coadministration of GLS4 with HBV at 2 μM affected early cccDNA formation, which is consistent with the observation that GLS4 can act on assembled capsids and functionally inactivate them (Fig. 10D) (38). However, the concentrations required for this activity are much higher than required for interference with assembly.
FIG 10.

Effect of antiviral drugs on cccDNA establishment and maintenance. (A) IFN-α-2b, the entry inhibitor Myrcludex B, and cyclosporine (which shows entry inhibition activity through NTCP binding) were coadministered with HBV inocula to initiate infection in HepG2hNTCP cells. The drugs were maintained in cell culture medium for 1 week. Lamivudine, tenofovir, BAY41-4109, and GLS4 were added from days 1 to 7 p.i. (B and C) Secreted HBeAg values (B) and calculated cccDNA (left) and total DNA copies (right) (C) per infected HepG2hNTCP cell are shown. (D) Myrcludex B, GLS4, and tenofovir were coadministered during infection (cotreatment) or from days 1 to 7 p.i. (posttreatment). cccDNA expression levels were measured on day 7.
FIG 11.

Effect of antiviral drugs on HBcAg expression. (A) Correlated with Fig. 10, infected HepG2hNTCP cells were treated with IFN-α-2b (100 or 1,000 IU/ml), lamivudine (1 or 10 μM), tenofovir (1 or 10 μM), Myrcludex B (0.5 or 5 μM), and cyclosporine (1 or 10 μM) for 1 week. Cells were fixed, and HBcAg staining was performed by an IF assay. Hoechst dye indicates nuclei. (B) Cell viability in Fig. 10 is shown here. Viability, survival ratio (percent) compared to the untreated cells (100%). Dashed line, 80%.
To determine if the method was suitable for medium-throughput screening, we infected HepG2hNTCP cells in 96-well plates (Fig. 12A and B) and tested three drugs (Myrcludex B, IFN-α, and tenofovir) at eight concentrations in triplicates. While Myrcludex B profoundly reduced cccDNA levels and HBeAg (IC50, ca. 10 nM; IC90, ca. 100 nM) (Fig. 12C), IFN-α-2a showed only slight effects on the cccDNA level but stronger inhibition of HBeAg. Tenofovir had no effect on cccDNA formation and HBeAg secretion. Taken together, combined T5 Exo treatment with cccDNA-specific PCR allows reliable screening of cccDNA-affecting drugs in a 96-well format.
FIG 12.

Detection of cccDNA and validation of Myrcludex B in 96-well plate format. (A) HepG2hNTCP cells seeded in a 96-well plate were infected at an mge/cell of 1,000. Myrcludex B was coadministered, tenofovir was added postinoculation, and IFN-α-2a was applied during and after infection. (B) Cells were treated with each antiviral at eight doses (1:3.2 serial dilutions) in triplicates. (C) On day 7 p.i., DNA samples were extracted together using a vacuum-based system. Crude DNA in 100 μl of elute was ethanol precipitated and resuspended with 10 μl of water. T5 Exo digestion was performed prior to cccDNA quantification by PCR. HBeAg levels during days 5 to 7 p.i. in the supernatant in all wells were measured.
DISCUSSION
Although cccDNA-specific primer pairs span the gap and nick region in rcDNA of replicative intermediates and virus genomes to avoid direct amplification, they are limited in selective amplification of cccDNA (Fig. 1A). The reason for this is the ability of the linear single-strand products formed by primer extensions to anneal within the short overlapping region (between DR1 and DR2). This generates a linear template for further exponential amplification, leading to a PCR product indistinguishable from cccDNA-derived products. Our study confirms this by demonstrating that all tested primers amplify cytosolic DNA extracted from infected HepG2hNTCP cells. Unlike nonselective primers, cccDNA primers in the optimized PCR program indeed showed limited specificity. However, this cannot guarantee absolute specificity in amplifying cccDNA when it coexists with rcDNA. Taken together, the use of cccDNA-specific primers allows specific amplification of ccc forms of HBV DNA; however, the primer-dependent selectivity for cccDNA is—in the best case—about 36-fold (Fig. 1B).
The limited specificity for amplifying cccDNA demands elimination of other HBV DNA species when exceeding this discrimination threshold. Selective enzymatic hydrolysis, Hirt extraction, or both can achieve this (14, 17). Regarding the efficiency of such a separation, e.g., when high sample numbers have to be analyzed, selective enzymatic digestion of non-cccDNA intermediates would be superior. Previous attempts have shown that PSD is suitable to decrease virion-associated DNA extracted from the serum of a viremic patient or from livers of HBV-infected humanized liver-chimeric mice (22, 39). However, incubation of total DNA extracted from woodchuck hepatitis B virus (WHV)-infected hepatocytes with PSD did not result in complete rcDNA digestion, as shown by Southern blotting (22). This might be due to increased competition of chromosomal DNA as a substrate for PSD, as we have shown in Fig. 3B, 4B, and 6A. PSD hydrolyzes short dslDNA and, with lesser efficacy, genomic DNA and oc/rcDNA and prefers genomic DNA with higher efficacy than rcDNA (Fig. 4B and C). This implies that total (genomic) DNA, which is present in samples, limits the application of PSD. Exo I plus Exo III were recently described to facilitate specific and sensitive detection of cccDNA by PCR (24). We confirmed this result and further noticed that Exo III alone is sufficient and comparable to T5 Exo for removing most non-cccDNA intermediates (Fig. 6A). Unlike Exo III preserving the intact strand of nicked DNA, T5 Exo acts on nicks in duplex DNA. Thus, special caution should be used during Hirt preparation, which may introduce nicks into cccDNA (40, 41).
Efficient in vitro infection of susceptible hepatic cells with HBV requires inoculation with high mge’s for extended times (4 to 24 h) in the presence of PEG (30, 42). This unusual infection characteristic is probably due to a very slow transition of nonmature virions to an NTCP-binding competent state (required for bona fide infection), while profound cellular association via HSPG occurs. The consequence is a profound association of virions and subviral particles with even nonsusceptible cells (Fig. 2 and unpublished data). Moreover, virus preparations from cell culture supernatants (e.g., HepAD38 cells) contain rcDNA-containing naked nucleocapsids if not prepared by heparin affinity chromatography (43). These naked nucleocapsids also show a strong tendency to attach to different cells even in the absence of NTCP (Y. Ni and S. Urban, unpublished results). Accordingly, early after virus inoculation (when cccDNA is just about being formed), the rc/cccDNA ratio is extraordinarily high compared to in vivo samples of patients or mouse livers or in vitro samples harvested at later time points after infection. PCR analyses of purified virions (Fig. 4D) following nuclease treatment showed that PSD only partially hydrolyzed rcDNA from the inoculum while T5 Exo completely removed rcDNA. This supports previous findings by Werle-Lapostolle et al. (22). However, the presence of chromosomal DNA competed for rcDNA hydrolysis by PSD, which is in line with its substrate specificity (44) (Lucigen, Middleton, WI). In contrast, T5 Exo efficiently removed rcDNA in the presence of artificially added genomic DNA (Fig. 4C) or from infected cell culture DNA extracts (Fig. 4E). This favors T5 Exo for decomposing rcDNA from the inoculum, a prerequisite for quantitative studies of in vitro cccDNA formation early during infection. Yet, as shown in Fig. 5B, incubation of the cccDNA-like plasmid pSHH2.1 with 5 units of T5 Exo for >4 h resulted in a significant degradation (60% after a 16-hour incubation). Thus, incubation conditions should be restricted to a maximum of 5 U for 60 min. Since we currently do not know to what extent transcription of cccDNA underlies a dynamic and regulated process of strand opening and ligation (e.g., through the action of topoisomerases and ligases), we cannot exclude the possibility that T5 Exo treatment of cellular extracts from HBV replicating cells discriminates between a transcriptionally active and silenced state. This has to be clarified in future studies.
A major focus in current drug discovery is the identification of molecules that directly or indirectly affect cccDNA levels in infected cells. Figure 12 exemplifies the suitability of our method for a 96-well plate format. Using improved methods (e.g., digital droplet PCR), this may be even further improved for high-throughput screening approaches. Thus, the direct cccDNA assay complements a recently developed cell culture system with HepDE19 and HepBHAe82 cells in which HBeAg serves as a surrogate marker for cccDNA transcriptional activity (45–47).
Our study also answered some principal questions related to HBV cccDNA biology. (i) It compares the kinetics of cccDNA formation in different HBV-susceptible cells (PHH, HepaRG, and HepG2hNTCP cells), demonstrating that copy numbers are low (1 to 5/cell) in all cell lines (Fig. 9). The numbers fit with previous reports on PHH and HepaRG cells (48, 49) and contrast with earlier findings in DHBV-infected duck livers and primary duck hepatocytes or WHV-infected woodchuck hepatocytes, which are in the range of 10 to 100 (50, 51). (ii) We further demonstrate that cccDNA copy numbers cannot be increased by raising the mge when 100% of cells are infected. This indicates the presence of an intracellular restriction for unlimited conversion of rcDNA to cccDNA and is in line with earlier observations which describe a saturation of HBV antigen production at very high mge’s per cell (30). (iii) We describe effects of selected inhibitors of infection on cccDNA formation and maintenance. Once established, cccDNA copy numbers do not further accumulate during late days of infection. This is in agreement with the observed kinetics of cccDNA formation leading to early saturation with no further increase of copy numbers (Fig. 9B and C) and the fact that very high mge’s do not promote an increase in the numbers of cccDNA molecules per cell (Fig. 7A).
Taken together, our data confirm the importance of limited primer specificity to obtain cccDNA-specific PCR products. Moreover, PCR-based cccDNA quantification from whole-cell lysates of in vitro-infected hepatocytes requires removal of rcDNA. Conventional PSD leads to uncompleted digestion of rcDNA within coexisting chromosomal DNA, while similarly to Exo III, T5 Exo treatment exhibits reliable quantification of cccDNA numbers. Applying this method, we showed that in in vitro models, HepaRGhNTCP and also the recently developed HepG2hNTCP cell line are suitable systems to study cccDNA early formation and accumulation. Finally, we demonstrate that entry inhibitors like Myrcludex B are able to block cccDNA formation completely. The protocol combining optimized TaqMan qPCR and T5 Exo purification will be a suitable tool for rapid and reliable cccDNA quantification in in vitro assays.
MATERIALS AND METHODS
Cells and enzymes.
HepG2, HepaRG, Hepa1-6, Hepa56D, and HeLa cells stably expressing hNTCP were cultivated as described previously (52–54). PHH were isolated from liver specimens obtained after partial hepatectomy and following written informed consent of the patients (approved by the ethics commission of Hannover Medical School/Ethik-Kommission der MHH, no. 252-2008). PHH and HepaRG and HepaRGhNTCP cells were maintained as previously described (53). HepG2hNTCP, Hepa1-6hNTCP, Hepa56DhNTCP, and HeLahNTCP cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 1 mM nonessential amino acids. A stable HepG2hNTCP-A3 cell clone established from limiting dilution of parental cells was validated with the best susceptibility and used for the experiments.
T5 Exo (M0363), BAL-31 nuclease (M0213), exonuclease I (M0293), exonuclease III (M0206), exonuclease V (RecBCD, M0345), mung bean nuclease (M0250), EcoRI (R0101), and Nb.BtsI (R0707) were purchased from New England Biolabs. PSD (E3105K) was provided by Epicentre; DNase I (grade II, catalog no. 10104159001) was from Roche.
HBV infection.
Virus stocks were prepared from culture supernatant from doxycycline-depleted HepAD38 cells (55) and concentrated by HiTrap Heparin HP affinity chromatography (catalog no. 10227781; GE Healthcare), and the virus titer (mge per milliliter) was determined by the RealTime HBV amplification reagent kit (Abbott) and in-house qPCR. Cells with 90% to 95% confluence were inoculated with HBV for 16 h at 37°C in the presence of 4% PEG 8000 (Sigma-Aldrich).
After washing, 2% (HepG2hNTCP) or 1.5% (HepaRG, HepaRGhNTCP, and PHH) dimethyl sulfoxide (DMSO) was added to the culture medium. Intron A was a product for injection use (Schering-Plough). IFN-α-2a was provided from PeproTech. Tenofovir (Sigma-Aldrich), BAY41-4109 (Bayer), and GLS4 (HEC Pharm) were purchased, and Myrcludex B was synthesized by Bachem (Bubendorf, Switzerland) (31). HBeAg values were quantified by the Advia Centaur XP immunoassay system (Siemens), and HBsAg values were quantified by an Architect assay (Abbott).
HBV DNA extraction.
DNA was extracted from purified virus stocks using the High Pure viral nucleic acid kit (catalog no. 11858874001; Roche) according to the manufacturer’s instructions. For real-time PCR, HBV DNA from infected hepatocytes was extracted using a NucleoSpin tissue kit (catalog no. 740952; Macherey-Nagel) according to the manufacturer’s manual with minor modifications. Briefly, cells were washed with PBS three times and immediately lysed in buffer T1 to obtain total lysates or in TNN buffer (50 mM Tris, 150 mM NaCl, and 0.5% NP-40 detergent, pH 8.0) for separation of cytosolic and nuclear fractions. Enriched nuclei were washed three times with TNN (400 rpm for 5 min) and lysed in buffer T1. Lysates were further incubated with proteinase K and buffer B3 at 70°C for 1 h and further treated according to the manufacturer’s protocol. DNA extraction from the 96-well plate was done using a NucleoSpin 8 tissue kit (catalog no. 740740; Macherey-Nagel) and a NucleoVac 96 vacuum manifold (catalog numbers 740681 and 740682; Macherey-Nagel). DNA samples were precipitated in ethanol and resuspended in 1/10 volume of water.
For cytosolic DNA detection by Southern blotting, cells were lysed in TENN buffer (50 mM Tris, 10 mM EDTA, 10 mM NaCl, 1% NP-40, pH 7.4) for 30 min at room temperature (RT). Nuclei were centrifuged down at 4,000 rpm for 5 min. Supernatant containing cytosolic DNA was carefully transferred and digested with proteinase K (0.6 mg/ml) and SDS (0.5%) at 65°C overnight. The next day, RNase A (0.05 mg/ml) was treated at 37°C for 30 min. Lysates were then extracted by phenol three times and phenol-chloroform (1:1 [vol/vol]) once, the aqueous phase was transferred, and DNA was precipitated by adding 2 volumes of ethanol. For cccDNA detection by Southern blotting, Hirt extraction was performed (17).
Real-time quantitative PCR.
SYBR Green Supermix (catalog no. 172-5121; Bio-Rad) and PerfeCTa qPCR ToughMix (catalog no. 95112-012; Quanta Biosciences) were used in the Bio-Rad CFX96 Touch real-time PCR detection system. The PerfeCTa qPCR ToughMix was identified as the best master mix for cccDNA qPCR. An optimized two-step qPCR program was established: 95°C for 15 min, followed by 95°C for 5 s (denaturation) and 63°C for 70 s (annealing and extension) for 50 cycles. A pSHH2.1 plasmid carrying two head-to-tail HBV monomers was used for the standard curve (56). For low-copy-number standards (103, 102, and 101 copies), 10-fold serial dilutions were performed in a diluent solution (10 mM Tris, 0.1 mM EDTA, 10 ng/ml yeast tRNA, and 0.01% Tween 20) instead of water (Quanta Biosciences, personal communication). DNA samples digested with T5 Exo (reaction mixture containing 5 μl eluted DNA, 1 μl 10× reaction buffer, 0.5 μl T5 Exo, and 3.5 μl water) at 37°C for 1 h and 70°C for 20 min were subjected to cccDNA qPCR. Undigested samples were directly used for human β-globin and HBV total DNA quantifications. β-Globin expression levels were used to normalize the inputs from different groups. Sequences of all the primers and probes used are summarized in Table 1.
Southern blotting.
DNA samples extracted as described above were separated on a 1.2% agarose gel. In-gel DNA was denatured (0.5 M NaOH, 1.5 M NaCl), neutralized (1 M Tris, 1.5 M NaCl, pH 7.4), and transferred (20× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate]) upward or downward onto a nylon membrane overnight as previously described (17). After UV cross-linking and prehybridization for 1 h, hybridization was performed by rotating and incubating the blots with α-[32P]dCTP randomly incorporated HBV-specific DNA probe (catalog no. 9661519; GE Healthcare) in the QuickHyb solution (Agilent; catalog no. 201221-21) at 60°C overnight. After two 30-minute washes in wash buffer (0.5% SDS, 1× SSC) at 60°C, radioactive signals were read by a phosphorimager (Bio-Rad) and analyzed by Quantity One software (Bio-Rad). pUCX3.2 plasmid (3.2 kb) containing a pUC18 backbone with a full-length HBV X protein coding frame was used to indicate the positions of 3.2-kb supercoiled and open circular DNA.
Immunofluorescence.
Infected cells were washed twice with PBS, fixed with 4% paraformaldehyde for 30 min, and permeabilized with 0.25% Triton X-100 for 30 min at RT. HBcAg staining was performed overnight at 4°C using the polyclonal anti-HBcAg antibody (catalog no. B0586; Dako; 1:3,000 dilution). After three 5-min washes with PBS, cells were incubated with Alexa Fluor 546-conjugated goat anti-rabbit IgG (heavy-plus-light chain [H + L]) antibody (catalog no. 1387801; Invitrogen; 1:1,000 dilution) and 1 μg/ml Hoechst dye at room temperature for 1.5 h. After three washes, images were collected under a DFC350 FX microscope (Leica). To calculate infectivity ratio, both HBcAg-positive and Hoechst stain-positive cell numbers were counted by eye in 20 random views, and HBcAg-positive cell number in every 100 Hoechst stain-positive cells was shown.
Cytotoxicity assay.
Released lactate dehydrogenase (LDH) was measured using a CytoTox96 nonradioactive cytotoxicity kit (G1780; Promega). Briefly, collected supernatant was centrifuged at 400 × g for 5 min to remove cell debris. Fifty microliters of the supernatant was incubated with 50 μl of CytoTox96 reagent for 30 min with protection from light. Then, 50 μl of stop solution was added, and the colorimetric absorbance at 490 nm was measured. The untreated group was set up as 100%, and viability for all the groups was normalized.
Statistical analysis.
A two-tailed Student t test was used to evaluate the data by Microsoft Excel software. χ2 analyses were used to calculate the data, with a probability value less than 0.05 considered a significant deviation.
ACKNOWLEDGMENTS
This work was funded by Deutsche Forschungsgemeinschaft (DFG) SFB/TRR179 (S.U. and B.Q.) and German Center for Infection Research (DZIF) TTU Hepatitis projects 05.704 and 05.807 (S.U., Y.N., and F.A.L.).
We acknowledge Franziska Schlund for her excellent skills in HepaRG and HepaRGhNTCP differentiation, Thomas Tu for fruitful discussions, Christina Kaufman for Myrcludex B synthesis, Jessica Sonnabend for the pUCX3.2 construct, and Christa Kuhn for the pSHH2.1 plasmid. We are grateful to Ralf Bartenschlager for his continuous support.
S.U. holds patents and intellectual property rights for Myrcludex B, the entry inhibitor used in this study.
B.Q., Y.N., and S.U. designed the study. B.Q., Y.N., and F.A.L. performed experiments. B.Q., Y.N., and S.U. analyzed the data. F.W.R.V. prepared primary human hepatocytes. B.Q., F.A.L., and S.U. wrote and revised the manuscript. S.U. supervised the study.
REFERENCES
- 1.Trepo C, Chan HL, Lok A. 2014. Hepatitis B virus infection. Lancet 384:2053–2063. doi: 10.1016/S0140-6736(14)60220-8. [DOI] [PubMed] [Google Scholar]
- 2.Wong GL, Wong VW. 2016. Eliminating hepatitis B virus as a global health threat. Lancet Infect Dis 16:1313–1314. doi: 10.1016/S1473-3099(16)30214-6. [DOI] [PubMed] [Google Scholar]
- 3.Nelson NP, Easterbrook PJ, McMahon BJ. 2016. Epidemiology of hepatitis B virus infection and impact of vaccination on disease. Clin Liver Dis 20:607–628. doi: 10.1016/j.cld.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lai CL, Yuen MF. 2013. Prevention of hepatitis B virus-related hepatocellular carcinoma with antiviral therapy. Hepatology 57:399–408. doi: 10.1002/hep.25937. [DOI] [PubMed] [Google Scholar]
- 5.Schinazi RF, Asselah T. 2017. From HCV to HBV cure. Liver Int 37:73–80. doi: 10.1111/liv.13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nassal M. 2015. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 64:1972–1984. doi: 10.1136/gutjnl-2015-309809. [DOI] [PubMed] [Google Scholar]
- 7.Wu TT, Coates L, Aldrich CE, Summers J, Mason WS. 1990. In hepatocytes infected with duck hepatitis B virus, the template for viral RNA synthesis is amplified by an intracellular pathway. Virology 175:255–261. doi: 10.1016/0042-6822(90)90206-7. [DOI] [PubMed] [Google Scholar]
- 8.Kajino K, Jilbert AR, Saputelli J, Aldrich CE, Cullen J, Mason WS. 1994. Woodchuck hepatitis virus infections: very rapid recovery after a prolonged viremia and infection of virtually every hepatocyte. J Virol 68:5792–5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu Y, Yamamoto T, Cullen J, Saputelli J, Aldrich CE, Miller DS, Litwin S, Furman PA, Jilbert AR, Mason WS. 2001. Kinetics of hepadnavirus loss from the liver during inhibition of viral DNA synthesis. J Virol 75:311–322. doi: 10.1128/JVI.75.1.311-322.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Summers J, Smith PM, Horwich AL. 1990. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J Virol 64:2819–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Volz T, Lutgehetmann M, Wachtler P, Jacob A, Quaas A, Murray JM, Dandri M, Petersen J. 2007. Impaired intrahepatic hepatitis B virus productivity contributes to low viremia in most HBeAg-negative patients. Gastroenterology 133:843–852. doi: 10.1053/j.gastro.2007.06.057. [DOI] [PubMed] [Google Scholar]
- 12.Thomas D, Zoulim F. 2012. New challenges in viral hepatitis. Gut 61:i1–i5. doi: 10.1136/gutjnl-2012-302122. [DOI] [PubMed] [Google Scholar]
- 13.Kock J, Rosler C, Zhang JJ, Blum HE, Nassal M, Thoma C. 2010. Generation of covalently closed circular DNA of hepatitis B viruses via intracellular recycling is regulated in a virus specific manner. PLoS Pathog 6:e1001082. doi: 10.1371/journal.ppat.1001082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, Li W. 2014. Correction: sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 3 doi: 10.7554/eLife.05570. [DOI] [PubMed] [Google Scholar]
- 15.Ni Y, Lempp FA, Mehrle S, Nkongolo S, Kaufman C, Falth M, Stindt J, Koniger C, Nassal M, Kubitz R, Sultmann H, Urban S. 2014. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 146:1070–1083. doi: 10.1053/j.gastro.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 16.Allweiss L, Dandri M. 2017. The role of cccDNA in HBV maintenance. Viruses 9:156. doi: 10.3390/v9060156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cai D, Nie H, Yan R, Guo JT, Block TM, Guo H. 2013. A Southern blot assay for detection of hepatitis B virus covalently closed circular DNA from cell cultures. Methods Mol Biol 1030:151–161. doi: 10.1007/978-1-62703-484-5_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, Sprinzl MF, Koppensteiner H, Makowska Z, Volz T, Remouchamps C, Chou WM, Thasler WE, Huser N, Durantel D, Liang TJ, Munk C, Heim MH, Browning JL, Dejardin E, Dandri M, Schindler M, Heikenwalder M, Protzer U. 2014. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 343:1221–1228. doi: 10.1126/science.1243462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chisari FV, Mason WS, Seeger C. 2014. Comment on “Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA.” Science 344:1237. doi: 10.1126/science.1254082. [DOI] [PubMed] [Google Scholar]
- 20.Xia Y, Lucifora J, Reisinger F, Heikenwalder M, Protzer U. 2014. Response to comment on “Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA.” Science 344:1237. doi: 10.1126/science.1254083. [DOI] [PubMed] [Google Scholar]
- 21.Hirt B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 22.Werle-Lapostolle B, Bowden S, Locarnini S, Wursthorn K, Petersen J, Lau G, Trepo C, Marcellin P, Goodman Z, Delaney WE IV, Xiong S, Brosgart CL, Chen SS, Gibbs CS, Zoulim F. 2004. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology 126:1750–1758. doi: 10.1053/j.gastro.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 23.Schnepp BC, Clark KR, Klemanski DL, Pacak CA, Johnson PR. 2003. Genetic fate of recombinant adeno-associated virus vector genomes in muscle. J Virol 77:3495–3504. doi: 10.1128/JVI.77.6.3495-3504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo J, Cui X, Gao L, Hu J. 2017. Identification of an intermediate in hepatitis B virus covalently closed circular (CCC) DNA formation and sensitive and selective CCC DNA detection. J Virol 91:e00539-17. doi: 10.1128/JVI.00539-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sayers JR, Eckstein F. 1991. A single-strand specific endonuclease activity copurifies with overexpressed T5 D15 exonuclease. Nucleic Acids Res 19:4127–4132. doi: 10.1093/nar/19.15.4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belloni L, Pollicino T, De Nicola F, Guerrieri F, Raffa G, Fanciulli M, Raimondo G, Levrero M. 2009. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc Natl Acad Sci U S A 106:19975–19979. doi: 10.1073/pnas.0908365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malmstrom S, Larsson SB, Hannoun C, Lindh M. 2012. Hepatitis B viral DNA decline at loss of HBeAg is mainly explained by reduced cccDNA load—down-regulated transcription of PgRNA has limited impact. PLoS One 7:e36349. doi: 10.1371/journal.pone.0036349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang D, Zuo C, Wang X, Meng X, Xue B, Liu N, Yu R, Qin Y, Gao Y, Wang Q, Hu J, Wang L, Zhou Z, Liu B, Tan D, Guan Y, Zhu H. 2014. Complete replication of hepatitis B virus and hepatitis C virus in a newly developed hepatoma cell line. Proc Natl Acad Sci U S A 111:E1264–E1273. doi: 10.1073/pnas.1320071111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schulze A, Gripon P, Urban S. 2007. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 46:1759–1768. doi: 10.1002/hep.21896. [DOI] [PubMed] [Google Scholar]
- 30.Schulze A, Mills K, Weiss TS, Urban S. 2012. Hepatocyte polarization is essential for the productive entry of the hepatitis B virus. Hepatology 55:373–383. doi: 10.1002/hep.24707. [DOI] [PubMed] [Google Scholar]
- 31.Petersen J, Dandri M, Mier W, Lutgehetmann M, Volz T, von Weizsacker F, Haberkorn U, Fischer L, Pollok JM, Erbes B, Seitz S, Urban S. 2008. Prevention of hepatitis B virus infection in vivo by entry inhibitors derived from the large envelope protein. Nat Biotechnol 26:335–341. doi: 10.1038/nbt1389. [DOI] [PubMed] [Google Scholar]
- 32.Nkongolo S, Ni Y, Lempp FA, Kaufman C, Lindner T, Esser-Nobis K, Lohmann V, Mier W, Mehrle S, Urban S. 2014. Cyclosporin A inhibits hepatitis B and hepatitis D virus entry by cyclophilin-independent interference with the NTCP receptor. J Hepatol 60:723–731. doi: 10.1016/j.jhep.2013.11.022. [DOI] [PubMed] [Google Scholar]
- 33.Stray SJ, Zlotnick A. 2006. BAY 41-4109 has multiple effects on hepatitis B virus capsid assembly. J Mol Recognit 19:542–548. doi: 10.1002/jmr.801. [DOI] [PubMed] [Google Scholar]
- 34.Wu G, Liu B, Zhang Y, Li J, Arzumanyan A, Clayton MM, Schinazi RF, Wang Z, Goldmann S, Ren Q, Zhang F, Feitelson MA. 2013. Preclinical characterization of GLS4, an inhibitor of hepatitis B virus core particle assembly. Antimicrob Agents Chemother 57:5344–5354. doi: 10.1128/AAC.01091-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mutz P, Metz P, Lempp FA, Bender S, Qu B, Schoneweis K, Seitz S, Tu T, Restuccia A, Frankish J, Dachert C, Schusser B, Koschny R, Polychronidis G, Schemmer P, Hoffmann K, Baumert TF, Binder M, Urban S, Bartenschlager R. 2018. HBV bypasses the innate immune response and does not protect HCV from antiviral activity of interferon. Gastroenterology 154:1791–1804.e22. doi: 10.1053/j.gastro.2018.01.044. [DOI] [PubMed] [Google Scholar]
- 36.Watashi K, Sluder A, Daito T, Matsunaga S, Ryo A, Nagamori S, Iwamoto M, Nakajima S, Tsukuda S, Borroto-Esoda K, Sugiyama M, Tanaka Y, Kanai Y, Kusuhara H, Mizokami M, Wakita T. 2014. Cyclosporin A and its analogs inhibit hepatitis B virus entry into cultured hepatocytes through targeting a membrane transporter, sodium taurocholate cotransporting polypeptide (NTCP). Hepatology 59:1726–1737. doi: 10.1002/hep.26982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deres K, Schroder CH, Paessens A, Goldmann S, Hacker HJ, Weber O, Kramer T, Niewohner U, Pleiss U, Stoltefuss J, Graef E, Koletzki D, Masantschek RN, Reimann A, Jaeger R, Gross R, Beckermann B, Schlemmer KH, Haebich D, Rubsamen-Waigmann H. 2003. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science 299:893–896. doi: 10.1126/science.1077215. [DOI] [PubMed] [Google Scholar]
- 38.Guo F, Zhao Q, Sheraz M, Cheng J, Qi Y, Su Q, Cuconati A, Wei L, Du Y, Li W, Chang J, Guo JT. 2017. HBV core protein allosteric modulators differentially alter cccDNA biosynthesis from de novo infection and intracellular amplification pathways. PLoS Pathog 13:e1006658. doi: 10.1371/journal.ppat.1006658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Belloni L, Allweiss L, Guerrieri F, Pediconi N, Volz T, Pollicino T, Petersen J, Raimondo G, Dandri M, Levrero M. 2012. IFN-alpha inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J Clin Invest 122:529–537. doi: 10.1172/JCI58847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Masamune Y, Fleischman RA, Richardson CC. 1971. Enzymatic removal and replacement of nucleotides at single strand breaks in deoxyribonucleic acid. J Biol Chem 246:2680–2691. [PubMed] [Google Scholar]
- 41.Smith AJ. 1979. The use of exonuclease III for preparing single stranded DNA for use as a template in the chain terminator sequencing method. Nucleic Acids Res 6:831–848. doi: 10.1093/nar/6.3.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gripon P, Diot C, Guguen-Guillouzo C. 1993. Reproducible high level infection of cultured adult human hepatocytes by hepatitis B virus: effect of polyethylene glycol on adsorption and penetration. Virology 192:534–540. doi: 10.1006/viro.1993.1069. [DOI] [PubMed] [Google Scholar]
- 43.Seitz S, Iancu C, Volz T, Mier W, Dandri M, Urban S, Bartenschlager R. 2016. A slow maturation process renders hepatitis B virus infectious. Cell Host Microbe 20:25–35. doi: 10.1016/j.chom.2016.05.013. [DOI] [PubMed] [Google Scholar]
- 44.Crosa JH, Brenner DJ, Falkow S. 1973. Use of a single-strand specific nuclease for analysis of bacterial and plasmid deoxyribonucleic acid homo- and heteroduplexes. J Bacteriol 115:904–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou T, Guo H, Guo JT, Cuconati A, Mehta A, Block TM. 2006. Hepatitis B virus e antigen production is dependent upon covalently closed circular (ccc) DNA in HepAD38 cell cultures and may serve as a cccDNA surrogate in antiviral screening assays. Antiviral Res 72:116–124. doi: 10.1016/j.antiviral.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 46.Cai D, Mills C, Yu W, Yan R, Aldrich CE, Saputelli JR, Mason WS, Xu X, Guo JT, Block TM, Cuconati A, Guo H. 2012. Identification of disubstituted sulfonamide compounds as specific inhibitors of hepatitis B virus covalently closed circular DNA formation. Antimicrob Agents Chemother 56:4277–4288. doi: 10.1128/AAC.00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cai D, Wang X, Yan R, Mao R, Liu Y, Ji C, Cuconati A, Guo H. 2016. Establishment of an inducible HBV stable cell line that expresses cccDNA-dependent epitope-tagged HBeAg for screening of cccDNA modulators. Antiviral Res 132:26–37. doi: 10.1016/j.antiviral.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller RH, Robinson WS. 1984. Hepatitis B virus DNA forms in nuclear and cytoplasmic fractions of infected human liver. Virology 137:390–399. doi: 10.1016/0042-6822(84)90231-9. [DOI] [PubMed] [Google Scholar]
- 49.Laras A, Koskinas J, Dimou E, Kostamena A, Hadziyannis SJ. 2006. Intrahepatic levels and replicative activity of covalently closed circular hepatitis B virus DNA in chronically infected patients. Hepatology 44:694–702. doi: 10.1002/hep.21299. [DOI] [PubMed] [Google Scholar]
- 50.Mason WS, Aldrich C, Summers J, Taylor JM. 1982. Asymmetric replication of duck hepatitis B virus DNA in liver cells: free minus-strand DNA. Proc Natl Acad Sci U S A 79:3997–4001. doi: 10.1073/pnas.79.13.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang YY, Zhang BH, Theele D, Litwin S, Toll E, Summers J. 2003. Single-cell analysis of covalently closed circular DNA copy numbers in a hepadnavirus-infected liver. Proc Natl Acad Sci U S A 100:12372–12377. doi: 10.1073/pnas.2033898100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, Guyomard C, Lucas J, Trepo C, Guguen-Guillouzo C. 2002. Infection of a human hepatoma cell line by hepatitis B virus. Proc Natl Acad Sci U S A 99:15655–15660. doi: 10.1073/pnas.232137699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ni Y, Urban S. 2017. Hepatitis B virus infection of HepaRG cells, HepaRG-hNTCP cells, and primary human hepatocytes. Methods Mol Biol 1540:15–25. doi: 10.1007/978-1-4939-6700-1_2. [DOI] [PubMed] [Google Scholar]
- 54.Lempp FA, Mutz P, Lipps C, Wirth D, Bartenschlager R, Urban S. 2016. Evidence that hepatitis B virus replication in mouse cells is limited by the lack of a host cell dependency factor. J Hepatol 64:556–564. doi: 10.1016/j.jhep.2015.10.030. [DOI] [PubMed] [Google Scholar]
- 55.Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother 41:1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cattaneo R, Will H, Darai G, Pfaff E, Schaller H. 1983. Detection of an element of the SV40 late promoter in vectors used for expression studies in COS cells. EMBO J 2:511–514. doi: 10.1002/j.1460-2075.1983.tb01455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Capovilla A, Arbuthnot P. 2002. Nucleotide excision repair by extracts of human fetal hepatocytes. FEBS Lett 518:144–148. doi: 10.1016/S0014-5793(02)02686-8. [DOI] [PubMed] [Google Scholar]