

Viral encephalitis is a serious and often life-threatening inflammation of the brain. Mammalian orthoreoviruses are promising oncolytic therapeutics for humans but establish virulent, serotype-dependent disease in the central nervous system (CNS) of many young mammals. Serotype 1 reoviruses infect ependymal cells and produce hydrocephalus, whereas serotype 3 reoviruses infect neurons and cause encephalitis. Reovirus neurotropism is hypothesized to be dictated by the filamentous σ1 viral attachment protein. However, it is not apparent how this protein mediates disease. We discovered that sequences forming the most virion-distal domain of T1 and T3 σ1 coordinate infection of either ependyma or neurons, respectively, leading to mutually exclusive patterns of tropism and disease in the CNS. These studies contribute new knowledge about how reoviruses target cells for infection in the brain and inform the rational design of improved oncolytic therapies to mitigate difficult-to-treat tumors of the CNS.
KEYWORDS: central nervous system infections, encephalitis, hydrocephalus, neurovirulence, reovirus
ABSTRACT
Viral capsid components that bind cellular receptors mediate critical functions in viral tropism and disease pathogenesis. Mammalian orthoreoviruses (reoviruses) spread systemically in newborn mice to cause serotype-specific disease in the central nervous system (CNS). Serotype 1 (T1) reovirus infects ependymal cells to cause nonlethal hydrocephalus, whereas serotype 3 (T3) reovirus infects neurons to cause fulminant and lethal encephalitis. This serotype-dependent difference in tropism and concomitant disease is attributed to the σ1 viral attachment protein, which is composed of head, body, and tail domains. To identify σ1 sequences that contribute to tropism for specific cell types in the CNS, we engineered a panel of viruses expressing chimeric σ1 proteins in which discrete σ1 domains have been reciprocally exchanged. Parental and chimeric σ1 viruses were compared for replication, tropism, and disease induction following intracranial inoculation of newborn mice. Viruses expressing T1 σ1 head sequences infect the ependyma, produce relatively lower titers in the brain, and do not cause significant disease. In contrast, viruses expressing T3 σ1 head sequences efficiently infect neurons, replicate to relatively higher titers in the brain, and cause a lethal encephalitis. Additionally, T3 σ1 head-expressing viruses display enhanced infectivity of cultured primary cortical neurons compared with T1 σ1 head-expressing viruses. These results indicate that T3 σ1 head domain sequences promote infection of neurons, likely by interaction with a neuron-specific receptor, and dictate tropism in the CNS and induction of encephalitis.
IMPORTANCE Viral encephalitis is a serious and often life-threatening inflammation of the brain. Mammalian orthoreoviruses are promising oncolytic therapeutics for humans but establish virulent, serotype-dependent disease in the central nervous system (CNS) of many young mammals. Serotype 1 reoviruses infect ependymal cells and produce hydrocephalus, whereas serotype 3 reoviruses infect neurons and cause encephalitis. Reovirus neurotropism is hypothesized to be dictated by the filamentous σ1 viral attachment protein. However, it is not apparent how this protein mediates disease. We discovered that sequences forming the most virion-distal domain of T1 and T3 σ1 coordinate infection of either ependyma or neurons, respectively, leading to mutually exclusive patterns of tropism and disease in the CNS. These studies contribute new knowledge about how reoviruses target cells for infection in the brain and inform the rational design of improved oncolytic therapies to mitigate difficult-to-treat tumors of the CNS.
INTRODUCTION
Viral invasion of the central nervous system (CNS) is a significant cause of morbidity and mortality worldwide, particularly in young children (1). The nervous system presents a challenging site for viruses to access, with multiple physical and immunological barriers that limit pathogen invasion. To penetrate the CNS, viruses must access cell surface receptors to mediate binding and entry events. Virus-receptor interactions also govern tropism and often control disease type and severity. For many viruses, the identities of receptors and other cellular determinants of viral tropism remain elusive. Understanding where and how viral capsid components engage neural receptors and the effect of these interactions on tropism and disease may illuminate targets to prevent viral neuroinvasion.
Mammalian orthoreoviruses (reoviruses) provide a highly tractable and well-established system to study mechanisms of viral neuroinvasion. Reoviruses are nonenveloped particles containing a 10-segmented, double-stranded RNA genome that replicate well in culture and can be modified via reverse genetics (2, 3). While reovirus causes similar age-restricted disease in many young mammals (4–6), most studies employ newborn mice. Following peroral or intracranial inoculation of newborn mice, reovirus displays serotype-specific patterns of tropism in the brain and concomitant disease. Serotype 1 (T1) strains infect ependymal cells lining the ventricles of the brain and cause a nonlethal hydrocephalus (7). In contrast, serotype 3 (T3) strains infect specific neuronal populations in the CNS and produce a fulminant, and often lethal, encephalitis (8). These differences in tropism and disease have been genetically mapped to the reovirus S1 gene using single-gene reassortant viruses (9).
The S1 gene encodes two protein products, σ1s and σ1, in alternate but overlapping reading frames. The nonstructural σ1s protein is dispensable for both T1 (10) and T3 (11) replication in the brain, suggesting that this protein does not mediate serotype-dependent CNS infection. In contrast, the σ1 viral attachment protein is well suited to mediate tropism differences. A long, filamentous trimer composed of multifunctional head, body, and tail domains (Fig. 1A), σ1 extends from the virion surface and engages cellular receptors. While the T1 and T3 σ1 proteins are structurally similar (12, 13), they share less than 30% amino acid identity (14) and are hypothesized to mediate tropism differences in the CNS by binding distinct receptors on the surfaces of ependyma and neurons.
FIG 1.
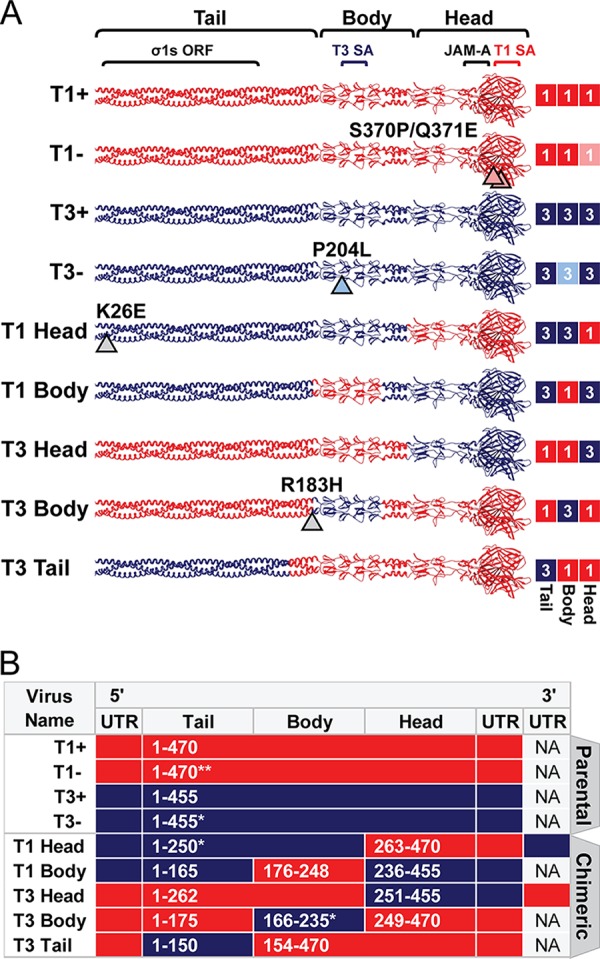
Chimeric σ1-encoding viruses used in this study. (A) Schematic of engineered σ1 proteins. A model of the σ1 trimer (adapted from reference 12) is shown as a ribbon diagram (left) or a simplified box schematic (right). T1 sequences (red; 1) and T3 sequences (blue; 3) are indicated for the tail, body, and head domains. T1 and T3 parental strains bind glycans (+) or contain mutations that abrogate glycan binding (−). Mutations that differ from T1+ and T3+ are indicated by arrowheads and noted. Approximate sites of the overlapping σ1s ORF and receptor binding domains for T1 sialic acid (SA), T3 SA, and JAM-A are annotated. The schematic on the right indicates sequence origin of σ1 domains for each virus (red, T1; blue, T3). (B) T1 (red) or T3 (blue) S1 gene elements are depicted for each virus. The approximate σ1 head, body, and tail domains are noted. Native untranslated regions (UTR) are shown for 5′ and 3′ gene termini. S1 genes of T1 Head and T3 Head viruses contain additional UTR sequences appended to the 3′ termini. Amino acid boundaries are provided for chimeras and numbered by parental strain origin (1 to 470 of T1 and 1 to 455 of T3). Asterisks indicate the number of amino acids that differ from either T1+ or T3+ parental strains in the corresponding domain. NA, not applicable.
Although multiple attachment moieties have been identified for reovirus, cellular receptors engaged in the CNS by T1 or T3 reovirus have not been identified. Moderately conserved pockets at the base of both the T1 and T3 σ1 head domains engage junctional adhesion molecule A (JAM-A) (15, 16) to mediate hematogenous dissemination in mice (17). JAM-A expression is dispensable for reovirus replication in the brain following intracranial inoculation (17). Another reovirus receptor, Nogo receptor 1 (NgR1), is predominantly expressed in CNS neurons with a distribution that overlaps with sites of T3 neurotropism and represents an attractive candidate for a T3-specific receptor (18). However, both T1 and T3 strains can use NgR1 to infect nonneuronal cell types (18), suggesting it is not the sole cellular determinant of reovirus neuronal tropism.
The only reovirus receptors known to be engaged in a serotype-specific manner are sialylated glycans. Glycans are not thought to directly mediate reovirus entry. Instead, glycans facilitate adhesion strengthening to cells, whereby reovirus transiently binds sialic acid with low affinity until a higher-affinity internalization receptor like JAM-A is encountered (19). The T1 σ1 head domain engages a specific α2,3-sialylated glycan found on various proteins and the GM2 ganglioside (13), whereas the T3 σ1 body domain can engage a variety of α2,3-, α2,6-, and α2,8-sialylated glycans (20). Compared with glycan-binding strains, T1 and T3 reoviruses engineered with mutations that specifically abrogate sialic-acid-binding capacity infect similar sites in the brain and produce similar yields (21, 22), leaving the viral and cellular determinants of reovirus neurotropism unclear.
The entry of reovirus into host cells has been elucidated primarily using nonpolarized, cultured cells. Following receptor-mediated endocytosis, reovirus virions undergo a stepwise, cathepsin-mediated, proteolytic disassembly of the outer capsid to form infectious subvirion particles (ISVPs) (23, 24). ISVPs are characterized by loss of major outer-capsid protein σ3, cleavage of μ1 to the δ and φ fragments, and conformational rearrangement of the σ1 protein (24, 25). Following further conformational changes and σ1 loss, viral particles penetrate endosomal membranes to initiate replication in the cytosol (26). The functions of σ1 conformational changes in reovirus infection are not known. However, these changes may modify receptor-binding capacity (27, 28).
In this study, we used reverse genetics to engineer a panel of viruses expressing T1-T3 chimeric σ1 proteins to identify S1 gene sequences and coordinating protein domains that dictate reovirus neurotropism and virulence in mice. We validated that chimeric σ1 proteins were expressed, encapsidated, folded, and functional to bind receptors and facilitate efficient viral replication in nonneuronal cells. We found that neurotropism and neurovirulence correlate with sequences in the T3 σ1 head domain. The reciprocal sequences of T1 σ1 track with infection of ependymal cells. Together, these findings indicate that homologous domains of the reovirus attachment protein coordinate distinct patterns of tropism in the CNS and suggest that the σ1 head domain engages unknown receptors that target virus to distinct cell populations.
RESULTS
Construction of chimeric σ1 reoviruses to identify protein domains that dictate serotype-specific patterns of neurovirulence.
To elucidate determinants of reovirus neurotropism, we designed a panel of viruses that express different S1 genes in an otherwise isogenic T1 background. Four parental, or control, S1 genes were used to establish well-characterized σ1-specific differences of the two reovirus serotypes and their respective glycan-binding capacities. T1+ and T3+ parental S1 genes (Fig. 1B) encode σ1 proteins (Fig. 1A) that efficiently bind glycan (13, 19) and dictate divergent patterns of tropism in the brain. Viruses with specific mutations introduced into the glycan-binding domains of T1+ (22) and T3+ (19) display diminished glycan-binding capacity and are called T1− and T3−, respectively.
To define domains of T3 σ1 that mediate infection of neurons, we designed gain-of-function constructs in which a T1 σ1 domain was exchanged with homologous T3 sequences and, concordantly, loss-of-function constructs in which a T3 σ1 domain was exchanged with homologous T1 sequences. We hypothesized that sequence exchange between serotypes at the two interdomain regions (located at the σ1 tail-body and body-head junctions) would yield replication-competent viruses. Furthermore, the head, body, and tail domains of T1 and T3 σ1 proteins demonstrate comparable amino acid identity by sequence alignment (approximately 26%, 24%, and 25% identity, respectively). Therefore, we used primary sequences and available crystal structures of T1 σ1 and T3 σ1 to identify sequences neighboring the interdomain regions that contain sequence or structural conservation. Using this information, S1 genes were designed to encode the approximate σ1 head, body, or tail domain of one strain exchanged with homologous sequences of the reciprocal strain (Fig. 1B). Virus and S1 gene nomenclature are interchangeable and reflect the domain that was acquired. For example, T3 Head virus contains sequences of the T3 σ1 head domain appended to sequences of the T1 body and tail domains. The σ1s open reading frame remains intact within the overlapping σ1 tail domain of all S1 genes used in this study. Within a reovirus gene, terminal 5′ and 3′ sequences are predicted to bind with complementarity and function to promote efficient replication or packaging (29–32). Chimeric genes containing discordant termini were either modified at the 3′ end to include extended 3′ untranslated sequences of the parental gene (Fig. 1B) or modified at the 5′ end to express the native 5′ untranslated region of the parental gene (Fig. 1B). Following design, chimeric S1 genes were engineered using a combination of molecular cloning and de novo gene synthesis.
Chimeric σ1 reoviruses can be recovered by reverse genetics.
Plasmids encoding parental or chimeric S1 genes were transfected together with the nine remaining T1 genes into BHK-T7 cells to produce recombinant virus strains as described previously (3, 33). BHK-T7 cell lysates yielded plaques on L929 cell monolayers for all viruses shown (Fig. 1A and B). However, despite several attempts, T1 Tail virus was not recovered. For other constructs, individual virus plaques were isolated and amplified in L929 cells for two passages before extraction of total RNA and sequence confirmation of the S1 gene.
The S1 genes of most viruses were genetically identical to the input plasmid sequences. However, approximately one-half of the T3 Body virus clones contained a point mutation (T3 σ1-R183H) near the 5′ junction of T1 and T3 sequences (Fig. 1A and B). This mutation coincided with higher viral titers, larger plaques, and enhanced replicative capacity (data not shown). Therefore, the S1 gene of T3 Body was modified to encode a histidine at T3-σ1 position 183 and recovered by reverse genetics. A point mutation also was identified in all propagated clones of T1 Head virus (T3 σ1-K26E) in a region of σ1 that is predicted to be embedded into the viral capsid (34, 35). Before introduction into mice, these chimeric strains were evaluated using several in vitro correlates of viral fitness.
Chimeric σ1 proteins are incorporated into purified reovirus particles.
To determine whether σ1-chimeric reoviruses faithfully recapitulate properties of parental reoviruses and efficiently encapsidate chimeric σ1 proteins, equivalent particle numbers of purified virions of each strain were resolved by SDS-PAGE. Proteins were visualized following colloidal blue staining or immunoblot detection of μ1/δ, and the optical densities (ODs) of bands corresponding to specific viral proteins were quantified. All viruses contain comparable levels of major large (λ), medium (μ), and small (σ) structural proteins (Fig. 2A, top panel). Moreover, σ1-chimeric viruses do not demonstrate significant ISVP contamination, a correlate of particle instability that is discerned by the appearance of the δ fragment of μ1 and loss of σ3 (Fig. 2A, bottom panel). However, T1 Head virus incorporates significantly less σ1 protein onto virions (Fig. 2A and B), perhaps because of the T3-K26E mutation identified in the σ1 tail domain of this strain. Combined, these data demonstrate that reovirus particles can assemble and encapsidate chimeric σ1 trimers, and most viruses do so with an efficiency comparable to that of the parental strains.
FIG 2.

Fitness correlates of chimeric σ1-containing viruses. (A, B) Incorporation of σ1 and δ in virions. Purified virions (5 × 1010) or ISVPs (5 × 109) were resolved by SDS-polyacrylamide gel electrophoresis. Proteins were visualized following colloidal blue staining (top panel) or immunoblotting to detect μ1/δ (bottom panel) and imaged using an Odyssey fluorescence scanner. (A) Representative images are shown. Bands corresponding to viral λ, μ, and σ proteins and molecular weight markers (in kilodaltons) are indicated. (B) Optical density of bands corresponding to σ1 and μ structural proteins were quantified. Results from three independent experiments are expressed as the mean relative optical density of σ1 bands normalized to μ bands. Error bars indicate standard deviations (SD). The schematic on the left indicates the sequence origin of σ1 domains for each virus (red, T1; blue, T3). (C, D) Conformation-specific antibody neutralization of viral infectivity. L929 fibroblasts were inoculated with the virus strains shown preincubated with vehicle or 4-fold dilutions of MAbs 5C6 (C) or 9BG5 (D). Infected cells were enumerated 24 h postinoculation by indirect immunofluorescence. Antibodies 5C6 and 9BG5 recognize epitopes in the σ1 head domain of T1 or T3, respectively. Symbol color reflects the sequences present in the σ1 head domain (red, T1; blue, T3). Results are expressed as the mean percentages of infected cells in treated samples relative to those in vehicle-treated samples for four fields of view per well in duplicate wells for three independent experiments. Error bars indicate standard errors of the means (SEM). (E, F) Viral replication efficiency. L929 fibroblasts were inoculated with the virus strains shown at an MOI of 0.5 PFU/cell. At the times shown postinoculation, viral titer in cell lysates was determined by plaque assay. Results are expressed as the mean viral yield from duplicate wells for three independent experiments. Viruses are displayed with appropriate parental strain controls; panel E and F assays were performed simultaneously and results can be compared directly. Error bars indicate SEM. Viral titers that differ significantly from those of T1+ (E) or T3+ (F) at 60 h postinoculation by one-way analysis of variance (ANOVA) and Dunnett's test are indicated (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Virion-associated parental and chimeric σ1 proteins are recognized comparably by conformation-specific σ1 antibodies.
To determine whether chimeric σ1 proteins are correctly folded and displayed on the surface of virions, we assessed the capacity of conformationally specific σ1 antibodies to neutralize reovirus infectivity using a fluorescent focus unit (FFU) assay. Monoclonal antibodies (MAbs) 5C6 and 9BG5 recognize distinct epitopes that span subunits in the trimeric σ1 head domain of T1 and T3 reoviruses, respectively (36). Parental and σ1-chimeric reoviruses were preincubated with MAb or vehicle and adsorbed to L929 cells. Cells were fixed at 24 h postadsorption, stained with a reovirus-specific antiserum, and imaged to determine the percentage of infected cells. Viruses containing T1 σ1-head sequences (T1+, T1−, T1 Head, T3 Body, and T3 Tail) were efficiently neutralized by T1-specific 5C6 antibody, whereas viruses containing T3 σ1-head sequences (T3+, T3−, T1 Body, and T3 Head) were unaffected by increasing concentrations of 5C6 (Fig. 2C). Similarly, the T3-specific antibody 9BG5 reduced infectivity of viruses containing T3 σ1-head sequences (Fig. 2D), but even at high concentrations, 9BG5 did not diminish infectivity of viruses containing T1 σ1-head sequences. Notably, the efficiencies with which chimeric strains are neutralized by the 5C6 and 9BG5 antibodies are comparable to those of parental strains expressing the same σ1 antibody epitope. Combined, these data suggest that chimeric σ1 trimers are natively folded and displayed on the virion surface.
Chimeric σ1 reoviruses replicate efficiently.
Reovirus strains are capable of efficiently infecting a broad range of cell types. However, they often do so in a strain-specific manner. In particular, reovirus infectivity of many cultured cell lines is dependent on serotype-specific engagement of sialylated cell surface glycans (13, 19, 22). Unlike with many other cell lines, reovirus infection of L929 cells is largely glycan and serotype independent (19). To determine whether viruses with chimeric σ1 proteins replicate efficiently, L929 cells were adsorbed with equivalent infectious units, and viral titer was determined at multiple intervals postinoculation by plaque assay. All viruses tested replicated with comparable kinetics and reached similar peak viral titers (Fig. 2E and F). T3 Head and T3 Tail display replication kinetics similar to those of parental strains T1+ and T1−, and T3 Head virus even reached modestly, but significantly, higher titers than did T1+ at 60 h postadsorption. T3 Body virus produced lower mean yields at early time points than its parental strain, T1+, but reached comparable yields at 60 h postinoculation (Fig. 2E). Similarly, T1 Head and T1 Body viruses produced slightly lower yields at early time points than their parental strain, T3+, but both chimeric viruses reached yields comparable to T3+ at 60 h postinoculation (Fig. 2F). Notably, T3− produced significantly higher titers than did T3+ at late time points in this assay, which may be a consequence of diminished cell viability following infection with T3+ (37). Together, these data demonstrate that σ1-chimeric viruses are capable of replicating with similar efficiency and producing equivalent or enhanced peak titers relative to parental strains.
Chimeric σ1 proteins display functional glycan-binding domains.
Reovirus strains display serotype-specific patterns of hemagglutination (HA) as a consequence of binding to different sialic acid moieties on the surface of erythrocytes (22, 38–41). The T1 glycan-binding site in the σ1 head domain mediates agglutination of human erythrocytes (13), whereas sequences in the T3 σ1 head domain do not contribute to hemagglutination. Instead, the glycan-binding site in the T3 σ1 body domain mediates less-efficient agglutination of human erythrocytes and a serotype-specific agglutination of bovine erythrocytes (42).
To evaluate the functionality of sialic acid-binding sequences conferred to chimeric σ1 proteins, we assessed hemagglutination capacity of reovirus particles using human and bovine erythrocytes. T1+ agglutinated human erythrocytes efficiently (mean HA titer = 431), while disruption of the T1 glycan-binding pocket in the σ1 head domain of T1− virus resulted in a 33-fold reduction in HA titer (Fig. 3A). Residual hemagglutination capacity of T1− is thought to be mediated by σ1 cross-linking non-sialic acid moieties on the cell surface (22). T3+ agglutinated human erythrocytes with a mean HA titer of 38, which is 11-fold less than that of T1+. Disruption of the T3 glycan-binding domain in T3− virus abolished agglutination of these cells. Two σ1-chimeric viruses (T1 Body and T3 Head) do not contain any predicted glycan-binding sequences, and as expected, these strains did not agglutinate human erythrocytes. T3 Tail virus contains a predicted T1 glycan-binding domain and agglutinated human erythrocytes with an HA titer identical to that of T1+, suggesting that the T1 σ1 head domain functions efficiently to engage sialic acid. Two σ1-chimeric viruses (T1 Head and T3 Body) incorporate sequences for both the T1 and T3 glycan-binding domains and were predicted to have identical hemagglutination profiles. However, T1 Head agglutinated human erythrocytes poorly (38-fold less than T1+ and 3-fold less than T3+), indicating an impairment in binding to human erythrocyte glycans. In contrast, the mean HA titer of T3 Body was 7-fold greater than that of T1+ and 76-fold greater than that of T3+, suggesting a synergistic effect of the two glycan-binding domains (Fig. 3A).
FIG 3.

Chimeric viruses display distinct sialic acid-binding profiles. (A, B) Hemagglutination capacity of parental and σ1-chimeric viruses. Schematics on left indicate the predicted hemagglutination (HA) capacity of σ1 head, body, and tail domains for each virus. Filled boxes (red, T1+; blue, T3+) indicate domains with predicted hemagglutination capacity. Gray boxes with central buttons indicate that the domain is not hypothesized to contribute to hemagglutination (red, T1+; pink, T1−; dark blue, T3+; light blue, T3−). Purified reovirus virions (1011) were serially diluted 2-fold in PBS. Human (A) or bovine (B) erythrocytes were resuspended in PBS at a concentration of 1% (vol/vol). Equal volumes of virus and erythrocyte mixtures were combined and incubated at 4°C for 4 h, and hemagglutination was assessed. Results are expressed as mean log2-transformed HA titers from three independent experiments. Error bars indicate SD.
Of the viruses tested, only T3+, T1 Head, and T3 Body contain T3 glycan-binding sequences, and therefore, these strains were hypothesized to specifically agglutinate bovine erythrocytes. T3+ and T3 Body viruses agglutinated bovine cells equivalently (Fig. 3B), suggesting that the conferred T3 σ1 body domain is sufficient to mediate sialic acid binding. However, T1 Head virus failed to agglutinate bovine erythrocytes. The combined impairment of T1 Head in agglutination of human and bovine erythrocytes is likely attributable to the decreased encapsidation of σ1 onto virions (Fig. 2A and B). Together, these data indicate that four of the five chimeric σ1 proteins bind glycan receptors as predicted and provide validated tools to identify σ1 domains required for reovirus neurovirulence.
Sequences encoding the σ1 head domain dictate reovirus neurovirulence and replication in the brain.
To define reovirus sequences that govern neurovirulence, we inoculated newborn mice intracranially with ∼100 PFU of reovirus and monitored infected animals for symptoms of disease. Moribund animals were euthanized. T1− and T3− viruses were excluded from these studies to reduce the number of mice used, as these viruses display lethality at this dose comparable to that of their glycan-binding counterparts, T1+ (22) and T3+ (43), respectively. Inoculation with T1+ or T3 Tail virus caused no detectable disease, and all inoculated mice survived infection (Fig. 4A). Following inoculation with T1 Head or T3 Body, one mouse in each cohort was euthanized because of significantly impaired weight gain over the course of infection. However, lethality following inoculation with these strains did not differ statistically compared with T1+. In contrast, most mice inoculated with T3+, T1 Body, or T3 Head displayed weight loss, lethargy, and neurological impairment. Neurological signs included agitated behavior, repetitive movements such as scratching or circling locomotion, seizures, and ataxia, which are all consistent with reovirus-induced meningoencephalitis (44, 45). Some mice inoculated with T1 Body or T3 Head either did not display detectable signs of disease or recovered from illness. Viruses that did not initiate statistically significant lethal disease in newborn mice all express sequences corresponding to the T1 σ1 head domain, whereas viruses causing lethal disease express sequences corresponding to the T3 σ1 head domain.
FIG 4.
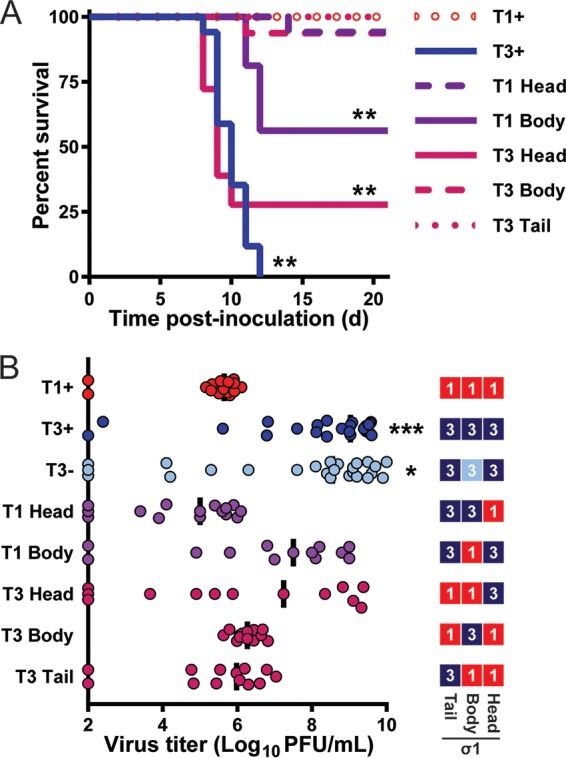
The σ1 head domain dictates reovirus neurovirulence and viral replication capacity in the brain. (A) Survival following intracranial inoculation. Newborn C57BL/6 mice (n = 16 to 18 for each virus strain) were inoculated intracranially with 100 PFU of purified virions of the strains shown. Mice were monitored for illness for 21 days and euthanized when moribund. Values that differ significantly from T1+ by log rank test are indicated (*, P < 0.05; **, P < 0.01). (B) Viral titers in the brain following intracranial inoculation. Schematic on the right indicates sequence origin of σ1 domains for each virus (red, T1; blue, T3). An independent cohort of identically inoculated mice (n = 13 to 26 for each virus strain) were euthanized 8 days postinoculation, and viral titers in the homogenized right brain hemispheres were determined by plaque assay. Each symbol represents the viral titer of a single mouse. Data are log transformed and displayed with a linear x axis scale. The median viral titer is indicated by a vertical bar. Values that differ significantly from T1+ by one-way ANOVA and Dunnett's test are indicated (*, P < 0.05; ***, P < 0.001).
To determine the replicative capacity of σ1-chimeric viruses in the brain, we inoculated mice intracranially with ∼100 PFU of reovirus and euthanized infected animals 8 days postinoculation to quantify viral titers in the right brain hemisphere by plaque assay. T1− was excluded from these studies to minimize the number of mice used but was previously shown to produce slightly lower titers than T1+ at this dose and time point (22). Median and peak virus titers in the brain for all other strains were compared to those of T1+ to identify viruses that have enhanced replicative capacity at that site. For viruses expressing T1 σ1 head sequences (T1+, T1 Head, T3 Body, and T3 Tail), maximum titers ranged from 6.1 to 7.0 log10 PFU/brain, and median titers ranged from 5.0 to 6.3 log10 PFU/brain (Fig. 4B). In contrast, viruses expressing T3 σ1 head sequences (T3+, T3−, T1 Body, and T3 Head) displayed maximum titers 770- to 12,000-fold higher than that for T1+ and ranged from 9.0 to 10.2 log10 PFU/brain, while median titers ranged from 7.2 to 9.0 log10 PFU/brain (Fig. 4B). The broader distribution of viral titers in brain tissue observed for higher-replicating viruses may reflect differences in receptor utilization or sites targeted within the brain. Thus, viruses that express T1 σ1 head sequences produce lower viral titers in the brain and do not initiate significant disease, whereas viruses that express T3 σ1 head sequences produce higher viral titers in the brain and induce lethal disease (Fig. 4A and B), further implicating the σ1 head domain as the major determinant of reovirus neurovirulence.
Sequences in the σ1 head domain dictate sites of reovirus infection in the brain.
To test whether the σ1 head domain influences sites of viral replication in the brain, mice were inoculated with ∼100 PFU of parental or chimeric reovirus strains and euthanized 8 days postinoculation. The left brain hemisphere was sectioned and stained with a reovirus-specific antiserum. Except for glial cells (identified by their dendritic morphology), which were infected by all strains, viral tropism was mutually exclusive to either ependyma or neurons for all tissue sections assessed (Fig. 5). Following inoculation with T1+, T1 Head, T3 Body, or T3 Tail (all viruses that express T1 σ1 head sequences), reovirus antigen was detected in cells adjacent to the lumen of the lateral ventricle that lack extended processes (Fig. 5). This staining pattern is consistent with both the location and morphology of ependymal cells, as is expected for T1+ tropism (7, 22). Following inoculation with T3+, T3−, T1 Body, or T3 Head (all viruses that express T3 σ1 head sequences), reovirus antigen was detected in specific neuronal subsets throughout the brain (Fig. 5), as described previously for T3+ (21). All neuronal subsets infected by T3+ also were infected by T3−, T1 Body, and T3 Head. In particular, pyramidal neurons of the cortex, neurons of the thalamus and hypothalamus, and Purkinje neurons of the cerebellum were targeted by viruses expressing a T3 σ1 head domain. These findings indicate that the σ1 head domain dictates the tropism of both T1 and T3 reoviruses in the brain.
FIG 5.

Reovirus neurotropism is dictated by sequences in the σ1 head domain. Newborn C57BL/6 mice were inoculated intracranially with ∼100 PFU of purified virions of the strains shown. Mice were euthanized 8 days postinoculation, and brains were resected and hemisected. Right brain hemispheres were homogenized for determination of viral titer by plaque assay. Left brain hemispheres were fixed in formalin and embedded in paraffin. Coronal sections of the left brain hemisphere were stained with reovirus-specific antiserum and hematoxylin. Low-magnification overview images at the depth of the hippocampus are shown. Regions corresponding to high-magnification insets of the lateral ventricle and lateral thalamus are indicated in the overview micrographs by red or blue boxes, respectively. Representative sections are shown. Viral titers from the paired right brain hemispheres are displayed above the micrographs. Reovirus-infected ependymal cells (open red triangles), neurons (filled blue triangles), and glia (black arrowhead), all identified using morphological criteria, are indicated.
Sialic acid-independent infectivity of primary cultured neurons phenocopies in vivo neuronal tropism.
Results obtained thus far demonstrate that the σ1 head domain of T3 reovirus is required for infection of neurons in the CNS and that glycan engagement by the T3 body domain is dispensable for infection of these cells. To test this hypothesis directly, we quantified reovirus infection of cultured primary neurons with or without neuraminidase pretreatment to remove cell surface sialic acid. Vehicle-treated neurons were poorly infected by T1+, T1−, T1 Head, and T3 Tail, moderately infected by T3−, T1 Body, T3 Head, and T3 Body, and most heavily infected by T3+ (Fig. 6A and C). These results demonstrate serotype-dependent infection of neurons (when comparing T1+ and T1− with T3+ and T3−) and sialic acid-enhanced infection of neurons (when comparing T3+ with T3−), as shown previously for the parental strains (17). Similar to viral replication efficiency in the brain, T1 Body and T3 Head infected neurons in culture as efficiently as T3−, which also lacks a functional T3 glycan-binding domain. Infectivity of cultured neurons with T1 Head and T3 Tail also mirrored in vivo replication capacity, with low-level infectivity like T1+ and T1−. Surprisingly, a virus that did not efficiently infect neurons in vivo, T3 Body, demonstrated enhanced infectivity in cultured neurons relative to the T1 parental strains. This trend of enhanced infectivity cannot be entirely explained by the presence of T3 body domain sequences, as T1 Head virus does not show this trend.
FIG 6.

Infection of cultured cortical neurons is primarily dependent on sequences in the T3 σ1 head domain. Cultured primary rat cortical neurons were treated with a vehicle control (A, C) or 40 mU/ml of neuraminidase (B, D), inoculated with reovirus at an MOI of 500 PFU/cell, fixed at 24 h postinoculation, and stained with reovirus-specific antiserum, an antibody to detect Tuj1, and DAPI. (A, B) Representative micrographs of reovirus-infected neurons (white staining) are shown. (C, D) Infected neurons were enumerated using indirect immunofluorescence. Results are expressed as mean numbers of infected neurons per field of view for six images per well in quadruplicate wells for five independent experiments. Error bars indicate SEM. Values that differ significantly from T1+ by one-way ANOVA and Dunnett's test are indicated (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
To determine the influence of sialic acid on infection of primary neuronal cultures, neurons were treated with Arthrobacter ureafaciens neuraminidase, which removes α2,3-, α2,6-, and α2,8-linked terminal sialic acid residues, prior to infection. In contrast to vehicle-treated neurons, infectivity trends of neuraminidase-treated neurons precisely mimicked those observed from in vivo studies. Viruses containing T1 σ1 head sequences infected neuraminidase-treated neurons poorly (Fig. 6B and D). Of note, the diminished infectivity of neuraminidase-treated neurons by T3 Body virus suggests that infection of cultured neurons by this strain is dependent on binding to cell surface sialic acid. Viruses containing T3 σ1 head sequences (T3+, T3−, T1 Body, and T3 Head) infected neuraminidase-treated neurons robustly and comparably (Fig. 6B and D). We also noted that T3−, T1 Body, and T3 Head viruses displayed approximately 2-fold-enhanced infectivity of neuraminidase-treated neurons compared with vehicle-treated neurons. Collectively, these data demonstrate that the σ1 head domain controls serotype-dependent infection of neurons and provide additional evidence that glycan engagement is not required for infection of neurons in vivo.
DISCUSSION
The reovirus S1 gene dictates serotype-dependent differences in neurotropism and disease, a finding first reported nearly 40 years ago (9). However, even as new functions are described for S1-encoded proteins, the mechanism by which S1 gene sequences determine these differences in pathogenesis has remained elusive. In this study, we used a reverse genetics platform to engineer a panel of reoviruses that encode chimeric S1 genes to identify σ1 sequences that mediate serotype-specific differences in reovirus neurologic disease. We found that infection of neurons does not correlate with T3 sequences encoding the σ1s protein, the σ1 tail domain, or serotype-specific glycan engagement in the σ1 body domain. Instead, we discovered that sequences encoding the T3 σ1 head domain are required for infection of neurons in the murine brain (Fig. 5). These same sequences also influence encephalitis induction and survival outcome (Fig. 4A). Viruses expressing T3 σ1 head domain sequences replicate to high titers in the brain, likely because of the greater number of cells targeted than with viruses expressing T1 σ1 head domain sequences (Fig. 4B and 5). Using cultured neurons, we further demonstrate that this infection of neurons is strictly dependent on the T3 σ1 head domain when cell surface sialic acid is removed (Fig. 6B). Furthermore, we found that reciprocal sequences in T1 σ1 mediate infection of ependyma (Fig. 5), lower viral brain titers (Fig. 4B), and survival (Fig. 4A) following intracranial inoculation of mice. These data establish that homologous sequences in the reovirus σ1 head domain coordinate infection at discrete sites in the CNS (Fig. 7A and B).
FIG 7.
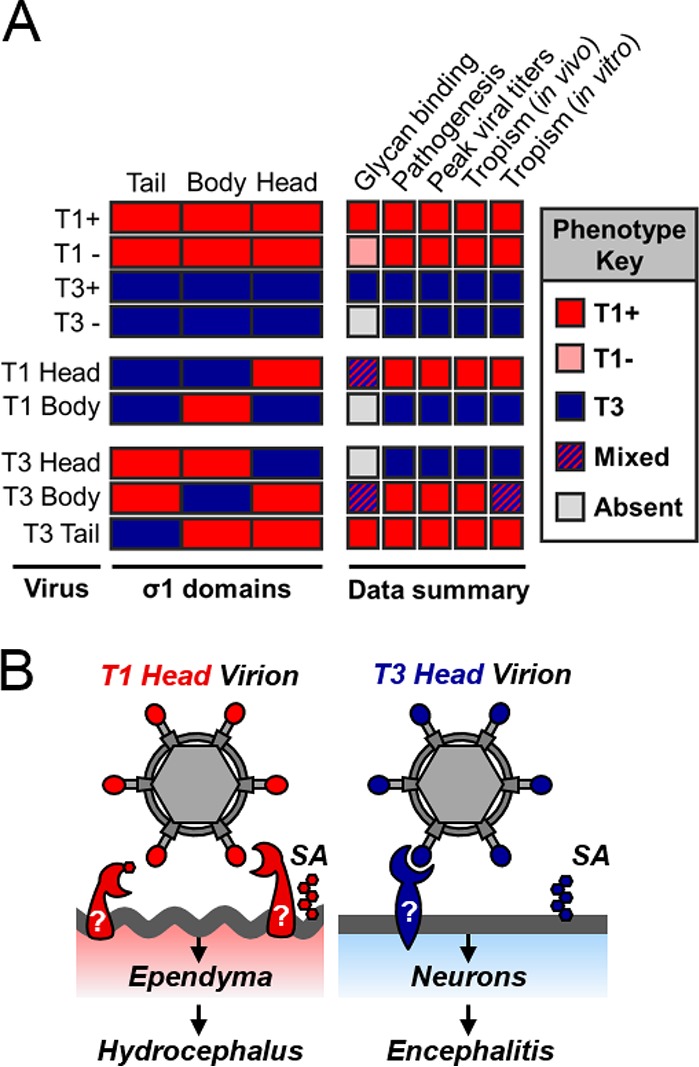
Sequences in the σ1 head domain dictate serotype-dependent patterns of viral tropism and neurologic disease in a glycan-independent manner. (A) Summary of key findings. The σ1 protein domain for each virus strain indicated is presented as a rectangle (red, T1; blue, T3). The phenotype key on the right displays results as more similar to T1 (red box), more similar to T3 (blue box), a mix between T1 and T3 (hatched box), or absent (gray box) in the case of hemagglutination. (B) Model of serotype-dependent neurotropism. Viruses expressing T1 σ1 head domains infect ependyma, and we predict they would cause hydrocephalus with a higher inoculation dose. Viruses expressing T3 σ1 head domains infect neurons and initiate a lethal, fulminant encephalitis. Sialic acid (SA) and other glycans are depicted by hexagons. Predicted T1- and T3-specific proteinaceous receptors are indicated with question marks.
It was not known whether reoviruses expressing chimeric genes derived from different serotypes could be recovered. Chimeric σ1 proteins have been expressed and purified from insect cell lysates (46), and the S1 gene of replication-competent viruses has allowed insertion of transgenes, although transgene sequences are variably stable (47–49). Our study demonstrates that replication-competent reoviruses expressing chimeric S1 genes and gene products can be recovered and are genetically maintained over multiple generations. We were not able to recover a virus expressing the T1 σ1 tail domain appended to the T3 σ1 body and head domains (T1 Tail). It is possible that the sequences chosen for T1 Tail construction do not allow proper protein folding. Alternatively, the viral RNA may contain an incompatibility between T1 and T3 sequences (29). Recently available structures of the T1 and T3 σ1 tail domains (12) should allow improved design of future σ1-chimeric proteins.
Similar to other glycan-binding viruses, reoviruses use an adhesion-strengthening mechanism of attachment to cells, in which low-affinity engagement of glycans adheres virus to the cell surface prior to ligation of a higher-affinity receptor capable of promoting internalization (19, 50, 51). Reovirus binding to sialic acid is not thought to directly initiate viral entry, although there is some evidence that glycan engagement may influence postbinding signaling events (37). Our data support an adhesion-strengthening model of reovirus neurotropism. Viral titers in the brain and lethality are highest for T3+, a virus that engages glycans using the σ1 body domain. However, viruses with limited or absent T3 glycan-binding affinity (T3−, T1 Body, and T3 Head) retain the capacity to target neurons for infection (Fig. 5 and 6A) and initiate lethal encephalitis (Fig. 4A), albeit less efficiently than T3+.
T3 Body virus does not exhibit neurotropic capacity in vivo (Fig. 5). However, this virus is capable of infecting cultured neurons using a sialic acid-dependent mechanism (Fig. 5A and B). It is possible that T3 glycan engagement is sufficient to initiate infection of neurons in vivo at a level that was not detected or that in vitro-cultured neurons do not faithfully recapitulate in vivo receptor requirements. Interestingly, purified virion preparations of T3 Body may contain more ISVPs (indicated by the δ cleavage product of μ1 protein) than other contemporaneously purified viruses (Fig. 2A). Cultured neurons are significantly more susceptible to ISVPs than virions (52), and while ISVPs are presumed to use receptors comparably to virions, ISVPs hemagglutinate more efficiently than virions, and ISVP infectivity is significantly more sensitive to neuraminidase treatment than virion infectivity (28). These data indicate that receptor requirements differ for reovirus virions and in vitro-prepared ISVPs. Thus, we hypothesize that T3 Body ISVPs enter neurons in the absence of a receptor engaged by the T3 σ1 head domain.
Treatment of cultured neurons with A. ureafaciens neuraminidase diminishes T3+ virus infectivity to that of T3− virus and does not alter T3− infectivity (18, 21). Surprisingly, we observed that T3−, T1 Body, and T3 Head viruses demonstrate a 2-fold increase in infectivity of cultured neurons following neuraminidase treatment (Fig. 6A and B), despite the prediction that these viruses do not bind sialic acid and should be unaffected by neuraminidase treatment. A nearly identical finding has been reported for HIV-1 in studies using both primary and cultured cells (53). It is possible that removal of sialic acid from the cell surface in certain settings enhances the accessibility of σ1 to a T3 head-specific receptor.
How do sequences in the σ1 head domain mediate tropism? No currently known functions of σ1 account for the CNS tropism observed in our study. However, data reported here raise new questions. To efficiently infect either ependymal cells or neurons, reovirus must be able to traffic to, bind, and replicate within these cells. Because T1 and T3 tropism in the CNS is manifested following multiple different routes of inoculation, and cultured neurons recapitulate serotype-dependent differences in neurotropism (54) (Fig. 6A), we do not think that trafficking to affected CNS cell types accounts for differences in tropism. While the σ1 head domain may function in postbinding/preentry replication steps to promote tropism differences, serotype-dependent binding to primary ependyma (55) and neurons (54) supports a receptor-dependent mechanism of target cell selection.
In addition to JAM-A, GM2 glycan is bound by the T1 σ1 head domain (13). T1 strains deficient in glycan engagement are altered in virulence but not in tropism within the CNS (22). While the GM2 glycan bound by T1 reovirus is appended to both proteins and lipids, the full array of host components expressing this glycan is not known. We hypothesize that T1 σ1 head sequences coordinate binding to a proteinaceous receptor, which may be engaged independent of, or overlapping with, the GM2-glycan-binding site (Fig. 7B). In the case of T3 σ1, the serotype-specific, glycan-binding domain is distant from the neurotropism-determining σ1 head domain (Fig. 7B). Both T1 and T3 reoviruses can spread by hematogenous routes to infect multiple organs, but T3 strains also spread via neural routes (11, 56, 57). We hypothesize that T3 σ1 head sequences engage a neurally specific receptor to initiate infection of neurons, which also may mediate neural dissemination from sites of initial infection.
If the σ1 head domain binds a T1- or T3-specific proteinaceous receptor, as we anticipate, then reovirus would have evolved two proximal binding sites at the virion-distal end of a long filamentous protein, one site for JAM-A and one that determines CNS tropism. We hypothesize that this location might be advantageous to either provide a larger surface area for binding and concordantly promote a more affine interaction (58) or serve as a flexible probe to interrogate molecular contacts at the cell surface. Other viruses also display receptor-binding domains at the termini of an extended trimeric protein (59, 60), further highlighting a shared design for viral adhesion to cells.
Our studies contribute to an overall understanding of mechanisms of neuroinvasion and highlight the evolutionary pressure for RNA viruses to co-opt viral proteins to serve multiple, critical functions in infection. By dissecting those functions, we have uncovered an important, serotype-specific determinant of reovirus neurotropism. We also consider this panel of viruses to be a useful toolbox to answer questions about the function of reovirus attachment in replication, dissemination, and tropism outside the CNS. While reovirus does not cause disease in humans, it preferentially infects and kills cancer cells (61–63). A prototype T3 reovirus strain, licensed as Reolysin, has been used in more than 30 clinical trials to date and shows both safety and potential efficacy in treatment of several different cancers, including those of the CNS (64). The reovirus σ1 protein shares a striking resemblance to the multifunctional attachment protein of adenovirus (65), another promising oncolytic virus. Adenoviruses expressing reovirus σ1 are viable and display reovirus-predicted tropism (60). By combining reovirus and adenovirus receptor-binding domains, a new class of tailored therapeutics can be envisioned. Such strategies to genetically manipulate the attachment functions of reovirus could improve cancer targeting and cell killing. Thus, understanding molecular determinants of reovirus tropism informs mechanisms of viral neuroinvasion and may contribute to tailored and improved oncolytic therapies.
MATERIALS AND METHODS
Cells and antibodies.
Spinner-adapted L929 fibroblasts were maintained in Joklik's minimal essential medium (JMEM) supplemented to contain 5% fetal bovine serum, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B. BHK-T7 cells were maintained as previously described (33). Reovirus-neutralizing antibodies 5C6 (66), 9BG5 (67), and 8H6 (66) were purified from hybridoma cell supernatants using protein G chromatography (GE Healthcare) (36). Hybridoma cell lines are deposited at the Developmental Studies Hybridoma Bank (Iowa City, IA, USA). Reovirus polyclonal serum was collected from rabbits immunized and boosted with reovirus strain T1L or T3D. Sera from T1L- and T3D-inoculated rabbits were mixed 1:1 (vol/vol) and cross-adsorbed on L929 cells to deplete nonspecific antibodies.
Viruses.
All viruses were recovered using plasmid-based reverse genetics (3, 33) to contain nine gene segments from reovirus strain T1L (33) and a unique S1 gene. The S1 gene of T1+ is derived from strain T1L. T1− differs from T1+ by two point mutations (S370P and Q371E) (22). The S1 gene of T3+ expresses the primary amino acid sequence of strain T3C44-MA and was engineered by site-directed mutagenesis of cDNA carrying the S1 gene of strain T3D (accession no. EF494441) to incorporate five σ1 polymorphisms (V22I, I88T, I246T, T249I, and T408A) (42) and one σ1s polymorphism (P70S). T3− differs from strain T3+ by a single point mutation (P204L).
T1 Head, T1 Tail, and T3 Body S1 genes were synthesized with a 5′ T7 promoter site and 3′ sequences encoding an HDZ ribozyme (GenScript) and initially introduced into a pUC57 vector with Xba1 flanking sequences. Genes and flanking sequences were excised by restriction-enzyme digest and subcloned into existing Xba1 sites of rescue plasmid pBacPak8 (Clontech).
T3 Head and T3 Tail S1 genes were engineered by cloning T3 S1 sequences into modified T1L-S1-pBacPak plasmids (modified from reference 33). In the T1L-S1-pBacPak plasmid (33), NcoI or NheI sites were engineered by site-directed mutagenesis at sites flanking T1 head or T1 tail sequences. Primers encoding terminal NcoI or NheI sequences were used to amplify T3+ S1 head or tail sequences. Sequences corresponding to the σ1 head or tail regions of the T1L S1 gene were excised by restriction enzyme digestion and replaced with PCR-amplified and NcoI/NheI-digested cDNA fragments of the T3+ σ1 head or tail. NcoI and NheI restriction sites in the newly constructed chimeric gene were converted to T1 or T3 sequences by site-directed mutagenesis.
The T1 Body S1 gene was engineered similar to the T3 Head and T3 Tail S1 genes. NcoI and NheI sites were introduced in the T3+-pBacPak plasmid, and T3+ sequences corresponding to the body domain were excised by restriction enzyme digest and replaced with T1 Body sequences. T1 Body sequences were synthesized (GenScript). NcoI and NheI sites of the chimeric gene were removed by site-directed mutagenesis.
All S1 plasmid sequences were confirmed using primers complementary to the pBakPak8 plasmid (Bac1, 5′AACCATCTCGCAAATAAATA; Bac2, 5′ACGCACAGAATCTAGCGCTT). Virus was plaque purified from BHK-T7 cell lysates and propagated for 3 or 4 passages in L929 cells. Virus was purified from infected L929 cells by cesium chloride gradient centrifugation (34), and viral titers were determined by plaque assay (68) or FFU assay using L929 cells. The S1 gene sequences of purified viruses were confirmed using applicable primers (T1 Fwd, 5′GCTATTCGCGCCTATGGATG; T1 Rev, 5′GATGATTGACCCCTTGTGCC; T3 Fwd, 5′GCTATTGGTCGGATGGATCC; T3 Rev, 5′GATGAAATGCCCCAGTGCCG). Virus particle number was estimated by spectral absorbance at 260 nm (1 OD260 = 2.1 × 1012 particles/ml).
Quantification of σ1 abundance in virions by colloidal blue-stained acrylamide gels.
Purified reovirus particles (5 × 1010) were incubated 1:1 (vol/vol) with 2× Laemmli sample buffer containing 5% β-mercaptoethanol at 95°C for 5 min and electrophoresed in 16-cm-long 10% polyacrylamide gels under Laemmli buffer using the Protean II Xi system (Bio-Rad) at room temperature for 6 h at 37 mA. Gels were stained with colloidal blue (Invitrogen) and scanned using an Odyssey CLx imager (Li-Cor) at 700 nm λ. Optical densities of bands corresponding to σ1 and μ (includes μ1, μ1C, and μ2) proteins were quantified using ImageStudio software (Li-Cor). The relative OD of σ1 was calculated per gel using the following equation: Relative ODσ1 = [(σ1x − σ1blank)/(σ1max − σ1blank)] × 100, where raw OD values are reported for experimental lanes (σ1x), for lanes containing the maximum σ1 intensity measured per gel (σ1max), and for lanes measured at the same relative electrophoretic mobility in the gel where virus was not loaded (σ1blank). Relative OD for μ (ODμ) was calculated similarly. Normalized σ1 per virus = relative ODσ1/relative ODμ.
Quantification of δ abundance in virions by immunoblotting.
ISVPs were prepared by treating reovirus particles with α-chymotrypsin as described previously (69). Purified reovirus virions (5 × 1010 particles) or ISVPs (5 × 109 particles) were incubated 3:1 (vol/vol) with 4× Laemmli sample buffer containing 10% β-mercaptoethanol at 95°C for 5 min and electrophoresed under Laemmli buffer in 7.5% polyacrylamide gels at room temperature. Proteins were transferred to nitrocellulose at 100 V at 4°C for 1 h and immunoblotted using μ1C-specific 8H6 mouse MAb (1 μg/ml) and IRDye800CW (Li-Cor) (1:20,000). Membranes were scanned using an Odyssey CLx imaging system (Li-Cor).
Assessment of reovirus replication by plaque assay.
L929 cells (2 × 105) were adsorbed with reovirus in Dulbecco's phosphate-buffered saline (PBS) at a multiplicity of infection (MOI) of 0.5 PFU/cell at 37°C for 1 h, washed with PBS, and incubated in 1 ml of fresh medium. At various intervals, cells were frozen and thawed twice before determination of viral titer by plaque assay using L929 cells. Viral yields were calculated using the equation log10 (PFU/ml)tx − log10 (PFU/ml)tx=0, where tx is the time postinfection.
FFU neutralization with conformation-specific antibodies to assess σ1 folding.
L929 cells were adsorbed with serial 2-fold dilutions of virus in complete JMEM at 37°C for 1 h, washed with PBS, incubated in fresh medium at 37° for 24 h, washed with PBS, and fixed with cold methanol. Fixed cells were incubated with reovirus polyclonal serum diluted 1:1,000 in 0.5% Triton X-100, followed by incubation with Alexa Fluor 488-labeled secondary IgG. Cells were counterstained with DAPI (4′,6-diamidino-2-phenylindole) and imaged using a Lionheart FX imaging system (BioTek). The percentage of infected cells was quantified using Gen5 software (BioTek), and the concentration of virus required to infect approximately 60% of cells (TCID60) was determined.
Antibody neutralization efficiency was determined by incubating 1 TCID60 of virus with an equal volume of complete JMEM or 4-fold serial dilutions of 9BG5 or 5C6 antibody at room temperature for 1 h, followed by virus adsorption to cells at 37°C for 1 h. Cells were washed, fixed, imaged, and enumerated as described above. The percent neutralization was determined using the following formula: percent infection[MAb]/mean percent infectionJMEM.
Hemagglutination assay to determine viral glycan-binding capacity.
Purified reovirus virions were serially diluted 2-fold in 0.05 ml of PBS. The viral concentrations used ranged from 5.0 × 1010 particles in 0.05 ml to 2.4 × 107 particles in 0.05 ml. Bovine erythrocytes (Innovative Research) or human type O− erythrocytes (University of Pittsburgh Medical Center Blood Bank) were washed with PBS and diluted to 1% in PBS (vol/vol). Equal volumes (0.05 ml) of virus and erythrocyte mixtures were gently mixed in U-bottom assay plates and incubated at 4°C for 4 h. Hemagglutination (HA) was defined as a partial or complete shield of erythrocytes within the well. The lowest number of reovirus particles required to produce HA, termed HALow, was used to calculate the HA titer using the equation HA titer = (5.0 × 1010 reovirus particles)/(HALow).
Mice and rats.
C57BL/6J mice (Jackson) were used to establish breeding colonies in specific-pathogen-free facilities at Vanderbilt University and the University of Pittsburgh, and experiments were performed using animal biosafety level 2 (ABSL2) facilities and guidelines. Timed pregnant Sprague-Dawley rats (Charles River) were housed in specific-pathogen-free facilities at the University of Pittsburgh before euthanasia and embryo resection. Experiments using mice and rats included approximately equivalent numbers of males and females.
All animal husbandry and experimental procedures were performed in accordance with U.S. Public Health Service policy and approved by the Institutional Animal Care and Use Committees at Vanderbilt University and the University of Pittsburgh.
Infection of mice and histology.
Two or three litters of 2- to 3-day-old C57BL/6J mice weighing between 1.4 and 2.2 g were inoculated intracranially in the right cerebral hemisphere with 100 PFU of virus (±3-fold) diluted in PBS using a 30-gauge needle and syringe (Hamilton). Viral titers of inocula were confirmed by plaque assay. For analyses of virulence, inoculated mice were monitored daily for symptoms of disease. Death was not used as an endpoint in these studies; moribund mice were euthanized. Qualifications for moribundity included severe lethargy or seizures, paralysis, or 25% body weight loss.
For analyses of viral replication and immunohistopathology, mice were euthanized 8 days postinoculation, and brains were excised and hemisected along the longitudinal fissure. The right brain hemisphere was collected into 1 ml of PBS and frozen and thawed twice prior to homogenization using a TissueLyser (Qiagen). Viral titers were determined by plaque assay using L929 cells. The left brain hemisphere was fixed in 10% neutral buffered formalin for at least 24 h, imbedded in paraffin wax, and cut into 5-μm-thick sections. Using the Bond-Max automated system (Leica), tissue was deparaffinized, incubated with Epitope Retrieval Solution 2 (Bond) at 100°C for 20 min (followed by gradual, stepwise temperature reduction), blocked with serum-free protein block (Dako) at room temperature for 10 min, and incubated with reovirus polyclonal serum diluted 1:45,000 in primary antibody diluent (Bond). Polymer Refine detection reagents (Bond) were used to visualize reovirus antigen and nuclei using 3,3′-diaminobenzidine-conjugated rabbit secondary IgG and a hematoxylin counterstain, respectively. Stained slides were scanned using a Lionheart FX imager.
Cortical neuron culture.
Surfaces of 48-well tissue culture grade plates were treated with Neurobasal medium (Gibco) containing 10 μg/ml poly-d-lysine (BD Biosciences) and 1 μg/ml laminin (BD Biosciences) at 4°C overnight and washed three times with PBS. Plating medium (Neurobasal medium supplemented to contain 10% fetal bovine serum, 50 U/ml penicillin, 50 μg/ml streptomycin, and 0.6 mM GlutaMAX [Gibco]) was added to treated wells, and plates were incubated at 37°C until use.
Cortices of embryonic day 17.5 Sprague-Dawley rats were isolated, separated from meninges, and collected in Hanks' balanced salt solution without calcium and magnesium (HBSS) on ice. Cortices were incubated with 130 U/ml trypsin (Worthington) in HBSS at room temperature for 30 min, washed twice with HBSS, and dissociated in 5 ml of plating medium by trituration with fire-polished borosilicate glass Pasteur pipettes. Debris was allowed to settle for 2 min, and the supernatant was transferred to 5 ml of plating medium for additional trituration. Cell viability was assessed by trypan blue exclusion, and 105 viable cells were suspended in plating medium per well and incubated overnight. Plating medium was removed, and cells were maintained in Neurobasal medium supplemented to contain 1× B27 supplement (Gibco), 50 U/ml penicillin, 50 μg/ml streptomycin, and 0.6 mM GlutaMAX. Half of the medium was replaced every 4 days, and neurons were cultivated in vitro for 10 to 12 days prior to infection.
Defining sialic acid-dependent and -independent infectivity of cultured neurons.
Cultured cortical neurons were incubated with either vehicle control or 40 mU/ml of Arthrobacter ureafaciens neuraminidase (MP Biomedicals, LLC) diluted in complete Neurobasal medium at 37°C for 1 h. Neurons were washed with PBS and adsorbed with reovirus diluted in PBS with added calcium and magnesium at an MOI of 500 PFU/cell at 37°C for 1 h. The inoculum was removed, warm complete medium was added to cells, and cells were incubated for 24 h prior to fixation with 4% paraformaldehyde and 0.2% glutaraldehyde in PBS at 37°C for 15 min. Fixation was quenched with 100 mM glycine in PBS, and cells were blocked with 5% bovine serum albumin in PBS.
Fixed cells were incubated with reovirus polyclonal serum diluted 1:1,000 in 0.5% Triton X-100, followed by incubation with Alexa Fluor 488-labeled secondary IgG. Cells were counterstained with DAPI, and neurons were identified by indirect immunofluorescence using a mouse anti-tubulin beta 3 (Tuj1) antibody (BioLegend) diluted 1:1,000, followed by incubation with Alexa Fluor 546-labeled secondary IgG. Cells were imaged at a magnification of ×4 with 6 images/well in quadruplicate wells per virus per experiment using a LionHeart FX imager. DAPI-positive nuclei were enumerated using Gen5 software, and reovirus-positive neurons were manually counted.
Statistical analysis.
All statistical tests were conducted using Prism 7 (GraphPad Software). P values of less than 0.05 were considered to be statistically significant. Descriptions of the specific tests used are found in the figure legends, and differences were calculated relative to T1+ virus unless otherwise noted.
Accession number(s).
Reovirus S1 gene sequences, previously described (T1+) and those designed for this study (T3+, T1 Head, T1 Body, T3 Head, T3 Body, and T3 Tail), are available under NCBI GenBank accession numbers as follows: T1+, accession no. EF494445.1 (33); T3+, accession no. MH822896 (see Acknowledgments); T1 Head, accession no. MH822897; T1 Body, accession no. MH822898; T3 Head, accession no. MH822899; T3 Body, accession no. MH822900; T3 Tail, accession no. MH822901.
ACKNOWLEDGMENTS
We thank Kerstin Reiss for recommendations about early chimeric constructs. We are grateful to Mini Ikizler for engineering the T3+ plasmid used here. We acknowledge the staff of the Vanderbilt Translational Pathology Shared Resource (with special thanks to Cindy Lowe and Sherry Smith) and the Research Pathology/Health Sciences Tissue Bank at UPMC Hillman Cancer Center. We thank Sarah Katen and the Vanderbilt Antibody and Protein Resource for 5C6 and 9BG5 hybridoma subcloning and antibody production. We appreciate Gwen Taylor for technical assistance with histology. We thank Anthony Lentscher, Laurie Silva, and Gwen Taylor for critical review of the manuscript.
D.M.S. designed and performed the experiments, analyzed the results, and wrote the manuscript. P.A. designed and performed the experiments and analyzed the results. M.H.D. and T.S. designed the experiments and analyzed the results. T.S.D. designed the experiments, analyzed the results, and wrote the manuscript.
This work was supported by the U.S. Public Health Service awards R01 AI038296 (D.M.S., P.A., and T.S.D.), R01 AI118887 (D.M.S., T.S., and T.S.D.), and T32 AI007281 (D.M.S.) and the Elizabeth B. Lamb Center for Pediatric Research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Dahm T, Rudolph H, Schwerk C, Schroten H, Tenenbaum T. 2016. Neuroinvasion and inflammation in viral central nervous system infections. Mediators Inflamm 2016:8562805. doi: 10.1155/2016/8562805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kanai Y, Komoto S, Kawagishi T, Nouda R, Nagasawa N, Onishi M, Matsuura Y, Taniguchi K, Kobayashi T. 2017. Entirely plasmid-based reverse genetics system for rotaviruses. Proc Natl Acad Sci U S A 114:2349–2354. doi: 10.1073/pnas.1618424114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kobayashi T, Ooms LS, Ikizler M, Chappell JD, Dermody TS. 2010. An improved reverse genetics system for mammalian orthoreoviruses. Virology 398:194–200. doi: 10.1016/j.virol.2009.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Margolis G, Kilham L. 1969. Hydrocephalus in hamsters, ferrets, rats, and mice following inoculations with reovirus type I. II. Pathologic studies. Lab Invest 21:189–198. [PubMed] [Google Scholar]
- 5.Raine CS, Fields BN. 1973. Reovirus type 3 encephalitis—a virologic and ultrastructural study. J Neuropathol Exp Neurol 32:19–33. doi: 10.1097/00005072-197301000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Wu AG, Pruijssers AJ, Brown JJ, Stencel-Baerenwald JE, Sutherland DM, Iskarpatyoti JA, Dermody TS. 2018. Age-dependent susceptibility to reovirus encephalitis in mice is influenced by maturation of the type-I interferon response. Pediatr Res 83:1057–1066. doi: 10.1038/pr.2018.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masters C, Alpers M, Kakulas B. 1977. Pathogenesis of reovirus type 1 hydrocephalus in mice. Significance of aqueductal changes. Arch Neurol 34:18–28. doi: 10.1001/archneur.1977.00500130038008. [DOI] [PubMed] [Google Scholar]
- 8.Weiner HL, Drayna D, Averill DR Jr, Fields BN. 1977. Molecular basis of reovirus virulence: role of the S1 gene. Proc Natl Acad Sci U S A 74:5744–5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weiner HL, Powers ML, Fields BN. 1980. Absolute linkage of virulence and central nervous system cell tropism of reoviruses to viral hemagglutinin. J Infect Dis 141:609–616. doi: 10.1093/infdis/141.5.609. [DOI] [PubMed] [Google Scholar]
- 10.Boehme KW, Guglielmi KM, Dermody TS. 2009. Reovirus nonstructural protein sigma1s is required for establishment of viremia and systemic dissemination. Proc Natl Acad Sci U S A 106:19986–19991. doi: 10.1073/pnas.0907412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boehme KW, Frierson JM, Konopka JL, Kobayashi T, Dermody TS. 2011. The reovirus sigma1s protein is a determinant of hematogenous but not neural virus dissemination in mice. J Virol 85:11781–11790. doi: 10.1128/JVI.02289-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dietrich MH, Ogden KM, Long JM, Ebenhoch R, Thor A, Dermody TS, Stehle T. 2018. Structural and functional features of the reovirus sigma1 tail. J Virol doi: 10.1128/JVI.00336-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reiss K, Stencel JE, Liu Y, Blaum BS, Reiter DM, Feizi T, Dermody TS, Stehle T. 2012. The GM2 glycan serves as a functional co-receptor for serotype 1 reovirus. PLoS Pathog 8:e1003078. doi: 10.1371/journal.ppat.1003078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nibert ML, Dermody TS, Fields BN. 1990. Structure of the reovirus cell-attachment protein: a model for the domain organization of s1. J Virol 64:2976–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirchner E, Guglielmi KM, Strauss HM, Dermody TS, Stehle T. 2008. Structure of reovirus sigma1 in complex with its receptor junctional adhesion molecule-A. PLoS Pathog 4:e1000235. doi: 10.1371/journal.ppat.1000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stettner E, Dietrich MH, Reiss K, Dermody TS, Stehle T. 2015. Structure of serotype 1 reovirus attachment protein sigma1 in complex with junctional adhesion molecule A reveals a conserved serotype-independent binding epitope. J Virol 89:6136–6140. doi: 10.1128/JVI.00433-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Antar AAR, Konopka JL, Campbell JA, Henry RA, Perdigoto AL, Carter BD, Pozzi A, Abel TW, Dermody TS. 2009. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 5:59–71. doi: 10.1016/j.chom.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Konopka-Anstadt JL, Mainou BA, Sutherland DM, Sekine Y, Strittmatter SM, Dermody TS. 2014. The Nogo receptor NgR1 mediates infection by mammalian reovirus. Cell Host Microbe 15:681–691. doi: 10.1016/j.chom.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. 2001. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J Biol Chem 276:2200–2211. doi: 10.1074/jbc.M004680200. [DOI] [PubMed] [Google Scholar]
- 20.Reiter DM, Frierson JM, Halvorson EE, Kobayashi T, Dermody TS, Stehle T. 2011. Crystal structure of reovirus attachment protein sigma1 in complex with sialylated oligosaccharides. PLoS Pathog 7:e1002166. doi: 10.1371/journal.ppat.1002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frierson JM, Pruijssers AJ, Konopka JL, Reiter DM, Abel TW, Stehle T, Dermod TS. 2012. Utilization of sialylated glycans as coreceptors enhances the neurovirulence of serotype 3 reovirus. J Virol 86:13164–13173. doi: 10.1128/JVI.01822-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stencel-Baerenwald J, Reiss K, Blaum BS, Colvin D, Li XN, Abel T, Boyd K, Stehle T, Dermody TS. 2015. Glycan engagement dictates hydrocephalus induction by serotype 1 reovirus. mBio 6:e02356-14. doi: 10.1128/mBio.02356-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ebert DH, Deussing J, Peters C, Dermody TS. 2002. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J Biol Chem 277:24609–24617. doi: 10.1074/jbc.M201107200. [DOI] [PubMed] [Google Scholar]
- 24.Sturzenbecker LJ, Nibert ML, Furlong DB, Fields BN. 1987. Intracellular digestion of reovirus particles requires a low pH and is an essential step in the viral infectious cycle. J Virol 61:2351–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dryden KA, Wang G, Yeager M, Nibert ML, Coombs KM, Furlong DB, Fields BN, Baker TS. 1993. Early steps in reovirus infection are associated with dramatic changes in supramolecular structure and protein conformation: analysis of virions and subviral particles by cryoelectron microscopy and image reconstruction. J Cell Biol 122:1023–1041. doi: 10.1083/jcb.122.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dermody TS, Parker JS, Sherry B. 2013. Orthoreoviruses, p 1304–1346. In Knipe DM, Howley PM (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 27.Borsa J, Morash BD, Sargent MD, Copps TP, Lievaart PA, Szekely JG. 1979. Two modes of entry of reovirus particles into L cells. J Gen Virol 45:161–170. doi: 10.1099/0022-1317-45-1-161. [DOI] [PubMed] [Google Scholar]
- 28.Nibert ML, Chappell JD, Dermody TS. 1995. Infectious subvirion particles of reovirus type 3 Dearing exhibit a loss in infectivity and contain a cleaved s1 protein. J Virol 69:5057–5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chappell JD, Goral MI, Rodgers SE, Depamphilis CW, Dermody TS. 1994. Sequence diversity within the reovirus S2 gene: reovirus genes reassort in nature, and their termini are predicted to form a panhandle motif. J Virol 68:750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roner MR, Roehr J. 2006. The 3′ sequences required for incorporation of an engineered ssRNA into the Reovirus genome. Virol J 3:1. doi: 10.1186/1743-422X-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roner MR, Steele BG. 2007. Localizing the reovirus packaging signals using an engineered m1 and s2 ssRNA. Virology 358:89–97. doi: 10.1016/j.virol.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 32.Zou S, Brown EG. 1992. Identification of sequence elements containing signals for replication and encapsidation of the reovirus M1 genome segment. Virology 186:377–388. doi: 10.1016/0042-6822(92)90003-8. [DOI] [PubMed] [Google Scholar]
- 33.Kobayashi T, Antar AA, Boehme KW, Danthi P, Eby EA, Guglielmi KM, Holm GH, Johnson EM, Maginnis MS, Naik S, Skelton WB, Wetzel JD, Wilson GJ, Chappell JD, Dermody TS. 2007. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 1:147–157. doi: 10.1016/j.chom.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Furlong DB, Nibert ML, Fields BN. 1988. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol 62:246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leone G, Mah DCW, Lee PWK. 1991. The incorporation of reovirus cell attachment protein s1 into virions requires the amino-terminal hydrophobic tail and the adjacent heptad repeat region. Virology 182:346–350. doi: 10.1016/0042-6822(91)90678-5. [DOI] [PubMed] [Google Scholar]
- 36.Dietrich MH, Ogden KM, Katen SP, Reiss K, Sutherland DM, Carnahan RH, Goff M, Cooper T, Dermody TS, Stehle T. 2017. Structural insights into reovirus σ1 interactions with two neutralizing antibodies. J Virol 91:e01621-16. doi: 10.1128/JVI.01621-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Connolly JL, Barton ES, Dermody TS. 2001. Reovirus binding to cell surface sialic acid potentiates virus-induced apoptosis. J Virol 75:4029–4039. doi: 10.1128/JVI.75.9.4029-4039.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brubaker MM, West B, Eillis RJ. 1964. Human blood group influence on reovirus hemagglutination titers. Proc Soc Exp Biol Med 115:1118–1120. doi: 10.3181/00379727-115-29130. [DOI] [PubMed] [Google Scholar]
- 39.Eggers HJ, Gomatos PJ, Tamm I. 1962. Agglutination of bovine erythrocytes: a general characteristic of reovirus type 3. Proc Soc Exp Biol Med 110:879–881. doi: 10.3181/00379727-110-27679. [DOI] [PubMed] [Google Scholar]
- 40.Gentsch JR, Pacitti AF. 1987. Differential interaction of reovirus type 3 with sialylated receptor components on animal cells. Virology 161:245–248. doi: 10.1016/0042-6822(87)90192-9. [DOI] [PubMed] [Google Scholar]
- 41.Paul RW, Lee PWK. 1987. Glycophorin is the reovirus receptor on human-erythrocytes. Virology 159:94–101. doi: 10.1016/0042-6822(87)90351-5. [DOI] [PubMed] [Google Scholar]
- 42.Chappell JD, Gunn VL, Wetzel JD, Baer GS, Dermody TS. 1997. Mutations in type 3 reovirus that determine binding to sialic acid are contained in the fibrous tail domain of viral attachment protein s1. J Virol 71:1834–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barton ES, Youree BE, Ebert DH, Forrest JC, Connolly JL, Valyi-Nagy T, Washington K, Wetzel JD, Dermody TS. 2003. Utilization of sialic acid as a coreceptor is required for reovirus-induced biliary disease. J Clin Invest 111:1823–1833. doi: 10.1172/JCI16303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stanley NF, Dorman DC, Ponsford J. 1953. Studies on the pathogenesis of a hitherto undescribed virus (hepato-encephalomyelitis) producing unusual symptoms in suckling mice. Aust J Exp Biol Med Sci 31:147–159. doi: 10.1038/icb.1953.18. [DOI] [PubMed] [Google Scholar]
- 45.Tyler KL. 1998. Pathogenesis of reovirus infections of the central nervous system. Curr Top Microbiol Immunol 233:93–124. [DOI] [PubMed] [Google Scholar]
- 46.Chappell JD, Duong JL, Wright BW, Dermody TS. 2000. Identification of carbohydrate-binding domains in the attachment proteins of type 1 and type 3 reoviruses. J Virol 74:8472–8479. doi: 10.1128/JVI.74.18.8472-8479.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Demidenko AA, Blattman JN, Blattman NN, Greenberg PD, Nibert ML. 2013. Engineering recombinant reoviruses with tandem repeats and a tetravirus 2A-like element for exogenous polypeptide expression. Proc Natl Acad Sci U S A 110:E1867–E1876. doi: 10.1073/pnas.1220107110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eaton HE, Kobayashi T, Dermody TS, Johnston RN, Jais PH, Shmulevitz M. 2017. African swine fever virus NP868R capping enzyme promotes reovirus rescue during reverse genetics by promoting reovirus protein expression, virion assembly, and RNA incorporation into infectious virions. J Virol 91:e02416-16. doi: 10.1128/JVI.02416-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van den Wollenberg DJ, Dautzenberg IJ, Ros W, Lipinska AD, van den Hengel SK, Hoeben RC. 2015. Replicating reoviruses with a transgene replacing the codons for the head domain of the viral spike. Gene Ther 22:267–279. doi: 10.1038/gt.2014.126. [DOI] [PubMed] [Google Scholar]
- 50.Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell FJ, Nusrat A, Parkos CA, Dermody TS. 2001. Junction adhesion molecule is a receptor for reovirus. Cell 104:441–451. doi: 10.1016/S0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- 51.Haywood AM. 1994. Virus receptors: binding, adhesion strengthening, and changes in viral structure. J Virol 68:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pruijssers AJ, Hengel H, Abel TW, Dermody TS. 2013. Apoptosis induction influences reovirus replication and virulence in newborn mice. J Virol 87:12980–12989. doi: 10.1128/JVI.01931-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun J, Barbeau B, Sato S, Tremblay MJ. 2001. Neuraminidase from a bacterial source enhances both HIV-1-mediated syncytium formation and the virus binding/entry process. Virology 284:26–36. doi: 10.1006/viro.2001.0889. [DOI] [PubMed] [Google Scholar]
- 54.Dichter MA, Weiner HL. 1984. Infection of neuronal cell cultures with reovirus mimics in vitro patterns of neurotropism. Ann Neurol 16:603–610. doi: 10.1002/ana.410160512. [DOI] [PubMed] [Google Scholar]
- 55.Tardieu M, Weiner HL. 1982. Viral receptors on isolated murine and human ependymal cells. Science 215:419–421. doi: 10.1126/science.6276976. [DOI] [PubMed] [Google Scholar]
- 56.Morrison LA, Sidman RL, Fields BN. 1991. Direct spread of reovirus from the intestinal lumen to the central nervous system through vagal autonomic nerve fibers. Proc Natl Acad Sci U S A 88:3852–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tyler KL, McPhee DA, Fields BN. 1986. Distinct pathways of viral spread in the host determined by reovirus S1 gene segment. Science 233:770–774. doi: 10.1126/science.3016895. [DOI] [PubMed] [Google Scholar]
- 58.Chen J, Sawyer N, Regan L. 2013. Protein-protein interactions: general trends in the relationship between binding affinity and interfacial buried surface area. Protein Sci 22:510–515. doi: 10.1002/pro.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bewley MC, Springer K, Zhang YB, Freimuth P, Flanagan JM. 1999. Structural analysis of the mechanism of adenovirus binding to its human cellular receptor, CAR. Science 286:1579–1583. doi: 10.1126/science.286.5444.1579. [DOI] [PubMed] [Google Scholar]
- 60.Mercier GT, Campbell JA, Chappell JD, Stehle T, Dermody TS, Barry MA. 2004. A chimeric adenovirus vector encoding reovirus attachment protein s1 targets cells expressing junctional adhesion molecule 1. Proc Natl Acad Sci U S A 101:6188–6193. doi: 10.1073/pnas.0400542101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Duncan MR, Stanish SM, Cox DC. 1978. Differential sensitivity of normal and transformed human cells to reovirus infection. J Virol 28:444–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Strong JE, Lee PW. 1996. The v-erbB oncogene confers enhanced cellular susceptibility to reovirus infection. J Virol 70:612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. 1998. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J 17:3351–3362. doi: 10.1093/emboj/17.12.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clements D, Helson E, Gujar SA, Lee PW. 2014. Reovirus in cancer therapy: an evidence-based review. Oncolytic Virother 3:69–82. doi: 10.2147/OV.S51321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chappell JD, Prota A, Dermody TS, Stehle T. 2002. Crystal structure of reovirus attachment protein s1 reveals evolutionary relationship to adenovirus fiber. EMBO J 21:1–11. doi: 10.1093/emboj/21.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Virgin HW IV, Mann MA, Fields BN, Tyler KL. 1991. Monoclonal antibodies to reovirus reveal structure/function relationships between capsid proteins and genetics of susceptibility to antibody action. J Virol 65:6772–6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burstin SJ, Spriggs DR, Fields BN. 1982. Evidence for functional domains on the reovirus type 3 hemagglutinin. Virology 117:146–155. doi: 10.1016/0042-6822(82)90514-1. [DOI] [PubMed] [Google Scholar]
- 68.Virgin HW, Bassel-Duby R, Fields BN, Tyler KL. 1988. Antibody protects against lethal infection with the neurally spreading reovirus type 3 (Dearing). J Virol 62:4594–4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mainou BA, Dermody TS. 2011. Src kinase mediates productive endocytic sorting of reovirus during cell entry. J Virol 85:3203–3213. doi: 10.1128/JVI.02056-10. [DOI] [PMC free article] [PubMed] [Google Scholar]