

We implemented a coculture system of pDCs with HCV-infected hepatoma cell line, Huh7.5. We used three HCV derivatives in order to gain insight into pDCs’ behavior against HCV and associated antiviral mechanisms. The results with this cell coculture system support the capacity of pDCs to inhibit HCV replication and infectivity mainly via IFN-α, but also through additional mechanisms associated with pDC maturation. We provided evidence that TLR agonists can enhance antiviral pDCs’ function and can induce phenotypic changes that may facilitate the interplay with other immune cells. These findings suggest the possibility of including TLR agonists in the strategies of HCV vaccine development.
KEYWORDS: HCV, innate immunity, pDCs
ABSTRACT
Plasmacytoid dendritic cells (pDCs) are innate immune cells with high antiviral activity triggered by Toll-like receptor 7 (TLR-7) and TLR-9 stimulation. Moreover, they are important mediators between innate and adaptive immunity. Although nowadays there is available an effective therapeutic arsenal against hepatitis C virus (HCV), a protective vaccine is not available. We have analyzed the pDCs’ response to HCV infection in a hepatitis C virus (HCV)-Huh7.5 virus-cell system, which allows completion of the virus infectious cycle. pDCs were cocultured following human immunodeficiency virus (HIV) aldrithiol-2 (AT-2 [TLR-7 agonist]) inactivation and CpG (TLR-9 agonist) stimulation. We employed three virus derivatives—wild-type Jc1, interferon (IFN)-resistant virus IR, and high-replicative-fitness virus P100—in order to explore additional IFN-α-related virus inhibition mechanisms. pDCs inhibited HCV infectivity and replication and produced IFN-α. After TLR-7 and TLR-9 stimulation, inhibition of infectivity and IFN-α production by pDCs were enhanced. TLR-7 stimulation drove higher TNF-related apoptosis-inducing ligand (TRAIL) expression in pDCs. Additionally, TLR-7- and TLR-9-stimulated pDCs exhibited a mature phenotype, improving the antigen presentation and lymph node homing-related markers. In conclusion, pDCs could serve as a drug target against HCV in order to improve antiviral activity and as an enhancer of viral immunization.
IMPORTANCE We implemented a coculture system of pDCs with HCV-infected hepatoma cell line, Huh7.5. We used three HCV derivatives in order to gain insight into pDCs’ behavior against HCV and associated antiviral mechanisms. The results with this cell coculture system support the capacity of pDCs to inhibit HCV replication and infectivity mainly via IFN-α, but also through additional mechanisms associated with pDC maturation. We provided evidence that TLR agonists can enhance antiviral pDCs’ function and can induce phenotypic changes that may facilitate the interplay with other immune cells. These findings suggest the possibility of including TLR agonists in the strategies of HCV vaccine development.
INTRODUCTION
Hepatitis C virus (HCV) is an RNA virus. According to the 2017 World Health Organization report, around 71 million people around the world are living with HCV infection, and 1.75 million new HCV infections in 2015 were estimated (1). HCV infection is a major public health problem, as in the absence of treatment most of the infections become chronic, increasing the risk of hepatic disorders (e.g., hepatocellular carcinoma [2]) and extrahepatic diseases (e.g., cardiovascular diseases [3]). During the last few years, new direct-acting antiviral drugs (DAAs) demonstrating great efficacy have been included in the anti-HCV clinical arsenal (4). However, there are difficult-to-treat patients, drug resistance outcomes, and importantly, reinfections (5, 6). Thus, for large-scale control of HCV-associated diseases, it is extremely desirable to work toward the development of an effective anti-HCV vaccine. In this regard, despite advances in the knowledge of anti-HCV T- and B-cell functions, (7, 8), the understanding of how innate immunity responds to HCV infection could help in the development of immunotherapeutic strategies. Plasmacytoid dendritic cells (pDCs) are the main interferon alpha (IFN-α) producer cells in humans by Toll-like receptor 7 (TLR-7) and TLR-9 stimulation (9). Moreover, after TLR-7 stimulus, pDCs turns into TNF-related apoptosis-inducing ligand (TRAIL)-expressing induced killer pDCs (IKpDCs), which are able to lyse infected cells (10). pDCs are considered a link between innate and adaptive immunity not only through their antiviral activity but also as antigen-presenting cells, through regulatory T-cell induction, and through bystander cell activation function, among other activities (11). pDCs have been associated with spontaneous control of HIV infection through IFN-α production and HIV-infected CD4+ T-cell apoptosis induction (12, 13).pDCs have also been reported to be involved in HCV control. pDCs of HCV spontaneous resolvers exhibit higher sensitivity to single-stranded RNA (ssRNA) stimulation (TLR-7 agonist) than nonresolvers (14). pDCs from chronically HCV-infected patients showed resting-state levels of maturation markers, but maintained the responsiveness to TLR stimulation, suggesting that pDCs could be a viable drug target (15). pDCs sense in vitro HCV-infected cells, produce IFN-α, and inhibit HCV replication (16), and this ability is enhanced by interaction with other components of innate immunity (NK cells and monocytes) (17).
TLR-7 and TLR-9 agonists have been used recently in immunomodulatory strategies and demonstrated improvements in pDC functionality and antiviral adaptive responses (18–20). One study using a TLR-7 agonist has been tested in the setting of HIV vaccine development, revealing promising results (21). So, it was interesting to investigate how pDCs inhibit HCV replication and infectivity and whether TLR-7 or -9 agonists could serve as potential boosters of this pDC activity.
To this aim, we developed a pDC coculture system with three HCV cell culture (HCVcc) derivatives that infect Huh7.5 cells. A wild-type virus, Jc1FLAG(p7-nsGluc2A) (Jc1), described by Marukian et al. (22), was passaged 100 times either in the presence of increasing IFN-α concentrations, leading to a population that displayed higher fitness and partial IFN-α resistance (termed IR) (23), or in the absence of IFN-α, which resulted in a high-fitness population (termed P100) (24, 25). We have analyzed the phenotypic consequences of the pDCs’ response to the three HCV populations following stimulation of TLR-7 and TLR-9. The results show that pDCs are a potential new drug target to enhance the immune response against HCV, with implications for vaccine design.
RESULTS
Time course for coculture conditions of pDCs and HCV-infected Huh7.5 cells.
Preliminary time course experiments on virus progeny production indicated that infections at multiplicity of infection (MOI) of 0.1 50% tissue culture infective dose (TCID50)/cell and at 24 h to 48 h of pDC coculture were adequate to test the effect of coculture with pDCs (Fig. 1). Since the inhibition of HCV infectious progeny production was detectable under both the unstimulated and CpG-stimulated conditions at 48 h and not at 24 h, a 48-h coculture time was used for subsequent experiments.
FIG 1.

Time course of HCV infection of Huh7.5 cells and pDC coculture. (a) Kinetics of infectious HCV progeny production by the indicated viruses at an MOI of 0.1 TCID50/cell. (b) Infectious HCV progeny production (virus indicated in abscissa) following Huh7.5 infection and stimulated or unstimulated pDCs (left) and percentage of TCID50/ml inhibition relative to the yield of the infection without pDC coculture (“control” [right]). The experiments were performed with Jc1 virus, the HCV wild-type virus, with IR, the IFN-α-resistant virus, and with P100, the high-replication virus, as described in Materials and Methods. Results represent one of two independent experiments.
HCV replication and infectivity inhibition by pDCs.
HCV infectivity in the cell culture supernatants, as well as intracellular and extracellular viral RNA, was measured, and the results were expressed as percentage of inhibition with respect to infected Huh7.5 cells without pDC addition (Fig. 2). pDCs without any TLR-7 or -9 prestimulation (“unstimulated” pDCs) inhibited progeny production of HCV wild-type Jc1, the IFN-α-resistant IR virus, and the high-replicative-fitness P100 virus. Interestingly, the infectivity of IR and P100 was less inhibited than that of the wild-type virus Jc1 (P = 0.001 and P = 0.008, respectively, by Mann-Whitney U test) (Fig. 2a). These results suggest that resistance to inhibition of HCV infectious progeny production by pDCs might depend on IFN-α or replicative fitness, traits embodied in HCV IR (IFN-α resistance and fitness) and P100 (fitness). No differences were observed in the inhibition capacity of intracellular and extracellular HCV RNA copies among the three viruses (Fig. 2b and c).
FIG 2.

Inhibition of HCV replication by pDCs. (a) TCID50 per milliliter (infectivity) (left) and percentage of TCID50 per milliliter of inhibition (right). (b) Extracellular RNA production (left) and percentage of extracellular RNA inhibition (right). (c) Intracellular RNA production (left) and percentage of intracellular RNA inhibition (right). Percentages of inhibition were calculated with respect to the corresponding virus Huh7.5 infection without pDC addition (“control”). Lines that link the different conditions correspond to experiments made with pDCs from the same donor. Results are expressed as medians with ranges from 10 independent experiments. Continuous lines correspond to Mann-Whitney U tests, and dotted lines correspond to the Wilcoxon test. Statistical values are shown by asterisks as follows: *, P = 0.05 to 0.01; and **, P = 0.01 to 0.001.
The differences in inhibition of infectious progeny production were lost when the pDCs used in the coculture with Huh7.5 cells were prestimulated with AT-2 (TLR-7 stimulus) or CpG (TLR-9 stimulus), reaching high levels of inhibition of infectivity and viral RNA production, although the effect was more pronounced with CpG-stimulated pDCs (Fig. 2a to c).
IFN-α production by pDCs depending on type of virus.
IFN production has been proposed as the main mechanism of HCV infectivity inhibition mediated by pDCs (16, 17). No differences in the pDC response in terms of IFN-α production were observed among Jc1, IR, and P100. As expected, all IFN-α levels were higher with than without TLR stimulation (Fig. 3), suggesting that pDCs sense viruses equally, independently of the viral fitness or IFN-α resistance. There was a threshold of at least 3 log IFN-α pg/ml levels, which was high enough to reach top levels of infectivity inhibition with AT-2 or CpG (Fig. 2a).
FIG 3.
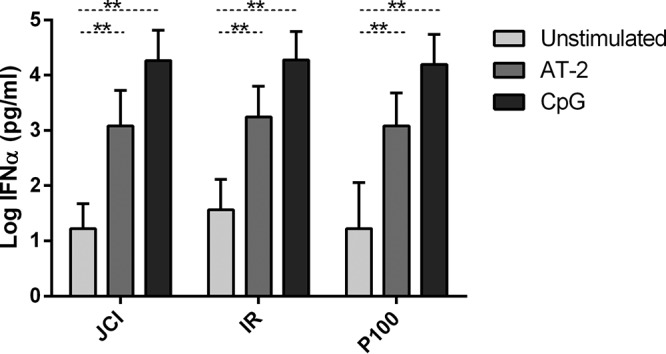
IFN-α production by pDCs. IFN-α was quantified by enzyme-linked immunosorbent assay (ELISA) in the supernatant of cocultured cells. Results are expressed as medians with ranges from 10 independent experiments. Dotted lines correspond to the Wilcoxon test. Statistical values are shown by asterisks as follows: **, P = 0.01 to 0.001.
Additional antiviral mechanisms of pDCs depending on stimuli.
As we did not observe differences in pDCs’ sensing of different types of HCV in terms of IFN-α production, some extracellular and intracellular molecules of pDCs were analyzed by flow cytometry. Higher IFN regulatory factor 7 (IRF-7) levels were observed in CpG-stimulated pDCs than in unstimulated or AT-2-stimulated pDCs (P = 0.002 in both cases, Mann-Whitney U test) (Fig. 4a). However, the opposite was noted with pDCs’ TRAIL expression. Unstimulated and AT-2-stimulated pDCs showed higher TRAIL levels than CpG-stimulated cells (P = 0.002 and P = 0.015, respectively, Mann-Whitney U test) (Fig. 4b), suggesting that pDCs could exert additional pDC antiviral mechanisms dependent on TLR stimulation. Furthermore, intracellular TNF-α production by pDCs was higher after CpG simulation compared with that of unstimulated and AT-2-stimulated pDCs (P = 0.002 in both cases, Mann-Whitney U test). The gating strategy is shown in Fig. 4d.
FIG 4.

Flow cytometry of pDCs. (a to c) Percentages of pDCs expressing (a) IRF-7, (b) TRAIL, and (c) TNF-α without any prestimulation and after AT-2 and CpG stimulation. (d) Gating strategy for pDC analysis. Results are expressed as medians with ranges from six independent experiments. Continuous lines correspond to Mann-Whitney U tests. Statistical values are shown by asterisks as follows: **, P = 0.01 to 0.001.
Huh7.5 cell apoptosis and TRAIL receptor expression.
Huh7.5 cell apoptosis was quantified by annexin V and Topro III staining. The gating strategy is shown in Fig. 5a. Following the coculture period, all viruses evoked similar level of apoptosis in Huh7.5 cells (Fig. 5b). However, there was a trend toward higher apoptosis sensitivity in terms of expression of TRAIL receptors DR4 and DR5 of Huh7.5 cells infected with IR and P100 than Jc1 (Fig. 5c and d), presumably associated with viral fitness. CpG-simulated pDCs evoked higher levels of apoptosis than unstimulated pDCs in Jc1 or IR infections (P = 0.018 in both cases, Wilcoxon test) and AT-2-stimulated pDCs in all three viral populations (P = 0.018, P = 0.018, and P = 0.028, respectively, Wilcoxon test) (Fig. 5e). These higher apoptosis levels correlated with levels of DR5 expression in infected Huh7.5 cells, being higher for CpG-stimulated pDCs than for unstimulated pDCs (P = 0.043 for Jc1 and P = 0.063 for IR, Wilcoxon test) or AT-2-stimulated pDCs (P = 0.063 for Jc1, P = 0.043 for IR, and P = 0.046 for P100, Wilcoxon test) (Fig. 5f).
FIG 5.

Huh7.5 apoptosis and TRAIL receptor expression. (a) Gating strategy for Huh7.5 cells. A flow cytometry dot plot and histograms of a representative experiment are presented. (b to d) Huh7.5-infected cells with each HCV population after 96 h plus 48 h, without pDC addition (control conditions). (b to d) Percentages of Huh7.5 cells expressing (b) annexin V and Topro III (apoptosis), (c) TRAIL receptor 2 (DR5), and (d) TRAIL receptor 1 (DR4). (e and f) Huh7.5 cells infected with each viral population after 96 h plus 48 h, with pDC addition. Also shown are the percentages of Huh7.5 cells expressing (e) annexin V and Topro III (apoptosis) and (f) annexin V, Topro III (apoptotic cells), and TRAIL receptor 2 (DR5). Results are expressed as medians with ranges from seven independent experiments. Continuous lines correspond to Mann-Whitney U tests. Dotted lines correspond to the Wilcoxon test. Statistical values are shown by asterisks as follows: *, P = 0.05 to 0.01.
Differences in pDC phenotypes among TLR simulations.
Phenotypic changes of pDCs were analyzed following the coculture step with the different types of TLR simulation (Fig. 6). Following AT-2 and CpG stimulation, pDCs exhibited a mature phenotype, which implied upregulation of HLA-DR, CCR7, CD40, and CD86 (Fig. 6a to d). Unstimulated and AT-2-stimulated pDCs showed lower CD4 mean fluorescence intensity (MFI), probably due to protein internalization. Regarding TLR-7 and TLR-9 pDC expression, we observed a lower MFI of TLR-7 in unstimulated and AT-2-stimulated pDCs than CpG-stimulated pDCs (Fig. 6f) and lower TLR-9 MFI in CpG-stimulated pDCs (Fig. 6g). Altogether, these results indicate that TLR stimulation, besides enhancing inhibition of HCV replication and infectivity of pDCs, induced the expression of molecules involved in the interplay with other components of the immune system.
FIG 6.

Phenotype of pDCs after coculture. Shown are flow cytometry data from pDCs after coculture, expressed as percentage or mean fluorescence intensity (MFI). Results are expressed as medians with ranges from six independent experiments. Histograms of a representative experiment are presented. Continuous lines correspond to Mann-Whitney U tests. Statistical values are shown by asterisks as follows: *, P = 0.05 to 0.01; **, P = 0.01 to 0.001; and ***, P < 0.001.
DISCUSSION
Herein we have described in an HCV-Huh7.5 virus-cell system that pDCs inhibit virus replication and infectivity in an IFN-α- and TRAIL-dependent manner. This virus control was enhanced after HIV attenuated AT-2 and CpG pDC stimulation (TLR-7 and TLR-9 agonists, respectively). Moreover, we used IFN-α-resistant and high-fitness HCV derivatives to evaluate a possible contribution of IFN-α in the antiviral state. Progeny production of all viruses was inhibited as a result of pDC TLR-7 or TLR-9 stimulation. Furthermore, after stimulation, pDCs showed a mature phenotype, thus, enabling potential interactions with other components of the immune system (11, 26).
Takahasi et al. described for the first time that pDCs produced IFN-α in the presence of HCV-infected cells via TLR-7 (16). It was further documented that pDC activation required cell-cell contact (16, 27). In that model, supernatants of pDCs and HCV-infected Huh7.5 cells contained large amounts of IFN-α, which inhibited HCV replication (16). In our study, we have confirmed and extended these findings by including the IR and P100 HCV populations and different TLR stimulations in the experimental protocol. pDCs inhibited replication and infectivity of the three HCV derivatives used (23, 24). With these tools, we observed a lower infectivity inhibition in the IFN-α-resistant virus IR and the high-fitness virus P100, probably due to their lower IFN sensitivity (23, 24). In addition, the lower inhibition of P100 compared to Jc1 production is in agreement with previous studies on HCV fitness as a drug resistance determinant (24, 28).
pDC stimulation with TLR-7 and TLR-9 triggered a high IFN-α production, responsible for the majority of virus replication and infectivity inhibition in all the HCV derivatives. This effect was also observed with HCV IR, probably because the amount of IFN-α released by pDCs exceeded the level required to manifest the IFN-α-resistant phenotype (24). We did not observe differences in IFN-α production by pDCs without TLR prestimulation after the coculture of Huh7.5-infected cells with the three viruses, suggesting that pDCs sensed equally all three HCV populations. Different responses were associated with different TLR-7 and TLR-9 stimulations. Interestingly, the amount of IFN-α produced by AT-2-stimulated pDCs was lower than that in CpG-stimulated pDCs, but the virus inhibition levels were similar. The IFN-α mechanisms could be complemented with induction of apoptosis, as evidenced by higher TRAIL expression with the AT-2 than with the CpG-A stimulus. Of note, similar levels of TRAIL expression were observed in pDCs with or without AT-2 prestimulation. This suggests that also unstimulated pDCs are being stimulated by HCV within the coculture system via TLR-7. It has been described that TLR-7-stimulated pDCs express high levels of TRAIL (10) and along with IFN-α production can provide a mechanism of viral control such as that described for HIV infection (13). pDCs in response to TLR stimulation not only produced IFN-α but also up to 19 type I IFN subtypes and multiple other cytokines (15, 29). However, it has been documented that direct stimulation by RNA viruses such as HCV, dengue virus, and chikungunya virus does not trigger the NF-κB pathway in pDCs (30, 31). This pathway is responsible for the production of IL-6 and TNF-α cytokines and of the expression of CD80 and CD86 (32). This could explain why we only observed TNF-α mainly in pDCs following CpG stimulation. Additionally, after TLR stimulation, pDCs showed upregulation of some key molecules involved in pDCs’ lymph node homing (CCR7) (33) and antigen presentation (HLA-DR, CD40) (25), as well as maturation markers that enable cross talk with other cells (e.g., CD86) (26). These phenotypic changes provide pDCs with additional mechanisms for HCV control apart from IFN-α production, thus, establishing a link between innate and adaptive immunity. A collaborative effect among innate cells (pDCs, NK cells, and monocytes) for HCV control has been previously documented by Kloss et al. (17).
With all these data, we propose that pDCs might be a potential target for immunomodulatory strategies. It should be considered, however, that TLR stimulation could be a double-edged sword, as pDC overactivation may lead to an antiviral T-cell response restriction (34) and to lymph node destruction (35, 36). So, the high IFN-α levels and high apoptosis sensitivity of Huh7.5 cells observed in CpG-stimulated pDCs could indicate that this stimulus might be too strong as a therapeutic target. The TLR-7 agonist AT-2 may induce a more balanced IFN-α production and maturation marker expression by pDCs. Recently, a new TLR-7 agonist has been developed demonstrating an improvement of pDC functionality (37), an increase of the anti-HIV adaptive immunity (18), and as an adjuvant, the improvement of response in HIV vaccine strategies (21). Since other TLR-9 agonists are under development (19, 20), it is suggested to include these strategies in the research of HCV vaccines.
In conclusion, activation of pDCs with a TLR agonist is a potential strategy to improve not only the antiviral activity of these cells but also their interplay with other cells involved in the immune response. This fact is of special interest as TLR agonists would be ideal candidates as adjuvants in the development of vaccines against HCV, as pDCs could act both as effectors and as antigen-presenting cells that may evoke an effective HCV-specific T-cell response.
MATERIALS AND METHODS
Cells and viruses.
The origin of Huh7.5 cell lines and procedures for cell growth have been described previously (22, 38–41). Briefly, infected and uninfected cells were cultured in Dulbecco’s modification of Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 2% glutamine, 1% nonessential amino acids, 0.1% penicillin–streptomycin, and 0.1% gentamicin at 37°C and 5% CO2.
The three HCV populations used (Jc1, IR, and P100) have been previously described (23, 24). Briefly, Huh7.5 cells were initially infected with Jc1FLAG(p7-nsGluc2A) (Jc1) virus. Then, Jc1 virus was passaged up to 100 times in the presence of a gradual amount of IFN-α (from 0.25 to 10 IU/ml), leading to a virus progeny with an IFN-α resistance phenotype, evidenced by an enhanced progeny production in an IFN-α environment (IR) (23). In parallel, Jc1 virus was passaged up to 100 times in the absence of IFN-α, leading to progeny with a high replicative fitness with a partial multidrug phenotype (P100) (24).
pDC isolation and stimulations.
Fresh pDCs were isolated from buffy coat from HIV-1- and hepatitis C virus (HCV)-seronegative blood bank donors obtained from the Regional blood Transfusion Center of Seville-Huelva (Seville, Spain) using the EasySep Human Plasmacytoid DC enrichment kit (StemCell) according to the manufacturer's instructions (purity of >90% [12]). After isolation, pDCs were cultured overnight (o/n) in DMEM without any stimuli (hereinafter called “unstimulated pDCs”) or with 1 µM CpG-A (ODN 2216; InvivoGen) or inactivated aldrithiol (AT-2) HIV-1MN at 20 ng/ml p24CA equivalents, kindly provided by J. D. Lifson (SAIC-National Cancer Institute, Frederick, Maryland).
Virus titration and RNA quantification using qRT-PCR.
Virus titration and quantitative real-time PCR (qRT-PCR) for RNA quantification were performed as previously described (23, 24). Briefly, for titration of HCV, supernatants of Huh7.5-infected cells were serially diluted and applied to Huh7.5 cells in 96-well plates (6,400 cells/well seeded 16 h earlier). Three days later, cell monolayers were washed with phosphate-buffered saline (PBS), fixed with ice-cold methanol, and stained for the presence of NS5A using anti-NS5A monoclonal antibody 9E10, as previously described (41, 42). All titrations were made in duplicate. The virus titer (infectivity) was expressed as 50% tissue culture infective dose (TCID50) per milliliter.
Intracellular and extracellular RNAs were extracted from infected cells using a Qiagen RNeasy microkit and from cell culture supernatants using the QiaAmp Viral RNA minikit (Qiagen, Valencia, CA), respectively, according to the manufacturer’s instructions. Quantitative real-time PCR (qRT-PCR) of HCV RNA was carried out using a Light Cycler RNA Master SYBR green I kit (Roche) using as primers oligonucleotides HCV-5UTR-F2 (5′-TGAGGAACTACTGTCTTCACGCAGAAAG [sense orientation] and HCV-5UTR-R2 (TGCTCATGGTGCACGGTCTACGAG [antisense orientation]). A standard curve was obtained with known amounts of HCV RNA synthesized by in vitro transcription of HCV cDNA (23).
HCV infection of Huh7.5 cells and pDC coculture.
For the coculture system (Fig. 7), Huh7.5 cells were infected with HCV Jc1, IR, or P100 at multiplicity of infection (MOI) of 0.1 TCID50/cell for 96 h (20,000 cells/well seeded 16 h earlier). Then, supernatants were replaced by pDCs with their corresponding supernatants, which had been previously stimulated with CpG-A or AT-2 or without stimulation at a 1:1 ratio of Huh7.5 cells to pDCs for 48 h. A condition without pDCs was included as control to analyze virus production without pDC inhibition. In this case, after 96 h of culture, supernatants were replaced only with DMEM and maintained for an additional 48 h. Depending on the amount of isolated pDCs, the experiments were performed in duplicate or triplicate.
FIG 7.
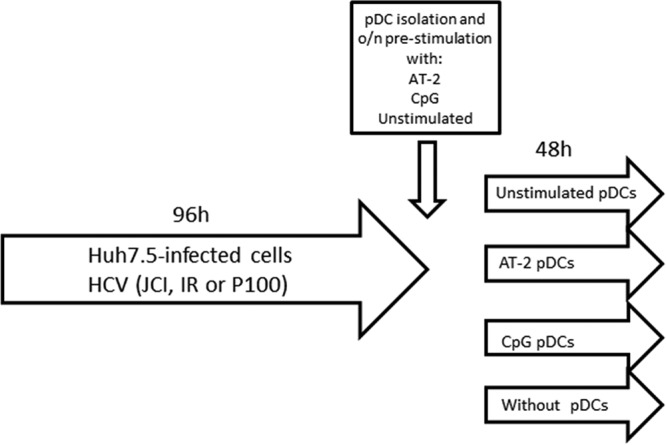
Scheme of HCV-infected Huh7.5 and pDC coculture. The origins of the cells and viruses and the different experimental steps are described in the introduction and Materials and Methods. o/n, overnight.
Flow cytometry.
Huh7.5 cells were washed with PBS, detached with trypsin-EDTA (0.25%), and resuspended in DMEM. Then they were washed and resuspended with annexin buffer (BD Biosciences), and stained for 20 min at room temperature with phycoerythrin (PE)-conjugated anti-TRAIL receptor 1 (DR4), peridinin chlorophyll protein (PerCP) anti-TRAIL receptor 2 (DR5) (R&D Systems), fluorescein isothiocyanate (FITC)-conjugated anti-annexin V (BD Biosciences), and allophycocyanin (APC)-conjugated Topro III (Invitrogen) antibodies. Control isotype antibodies were also added for DR4 and DR5 analyses. Flow cytometry analysis was performed on a Canto II flow cytometer using FACS Diva software (BD Biosciences). For pDC flow cytometry, supernatants were centrifuged and pDCs were pelleted and washed with PBS-fetal bovine serum (FBS [2%]) and stained for 20 min at room temperature with LIVE/DEAD fixable violet dead cell stain (Life Technologies), BV711 anti-HLA-DR, BV650 anti-CD86, BV786 anti-CCR7, PE-CF594 anti-CD123, BV605 anti-CD4, APC–anti-CD40 (BD Biosciences), and PE–anti-TRAIL (R&D Systems). Then pDCs were washed with BD Cytofix/Cytoperm buffer (BD Biosciences) and intracellularly strained with AF488 anti-IRF-7, PE-Cy7 anti-TNF-α (BD Biosciences), PerCP–anti-TLR-7 (R&D System), and DyLight 405 anti-TLR-9 (Novus Biological). pDCs were gated based on CD123+ HLA-DR+ expression. Flow cytometry analyses were performed on an LRS Fortessa flow cytometer using FACS Diva software (BD Biosciences). Data were analyzed using the FlowJo software (Treestar, Ashland, OR).
IFN-α production by pDCs.
The amount of IFN-α in cell culture supernatants was assessed by an IFN-α multisubtype enzyme-linked immunosorbent assay kit (PBL Interferon Source) according to the manufacturer’s instructions.
Statistical analysis.
Statistical analyses were performed by using Statistical Package for the Social Sciences software (SPSS 22.0; SPSS, Inc., Chicago, IL). Differences between conditions with different viruses were analyzed by two-tailed Mann-Whitney U tests. The Wilcoxon test was used to analyze related samples (i.e., experiments made with pDCs from the same donor). All differences with a P value of <0.05 were considered statistically significant. Graphs were generated with Prism, version 5.0 (GraphPad Software, Inc.).
ACKNOWLEDGMENTS
This work was supported by the Instituto de Salud Carlos III (research projects PI12/02283 and PI16/00684 and research contracts CPII014/00025 to E.R.-M. and FI14/00431 to L.T.-D.). E.R.-M. was supported by Consejería de Salud y Bienestar Social of Junta de Andalucía through the Nicolás Monardes program (C-0032/17). This work was also supported by Red Temática de Investigación Cooperativa en SIDA (RD12/0017/0029 and RD16/0025/0020), which is included in the Acción Estratégica en Salud, Plan Nacional de Investigación Científica, Desarrollo e Innovación Tecnológica 2008–2011, Instituto de Salud Carlos III, Fondos FEDER. B.D.-M. received a grant from The Spanish Ministry of Education (FPU13/02451). C.P. is supported by the Miguel Servet program of the Instituto de Salud Carlos III (CP14/00121), cofinanced by the European Regional Development Fund (ERDF). CIBERehd (Centro de Investigación en Red de Enfermedades Hepáticas y Digestivas) is funded by the Instituto de Salud Carlos III. The work in Madrid was supported by grants SAF2014-52400-R, SAF2017-87846-R, and S2013/ABI-2906 (PLATESA from Comunidad de Madrid/FEDER). Institutional grants from the Fundación Ramón Areces and Banco Santander to the CBMSO are also acknowledged.
We declare that no conflicts of interest exist.
E.R.-M. and M.L. designed the study. B.D.-M. performed experimental procedures and analyses and wrote the manuscript. K.M. and L.T.-D. collaborated on experimental procedures. C.P., J.L.S., I.G., and E.D. contributed with the elaboration of HCVcc derivatives used in this work and data management. All authors contributed to the writing and review of the manuscript.
This study would not have been possible without the collaboration of all of the volunteers, medical and nursery staff, and data managers who have taken part in the project.
REFERENCES
- 1.World Health Organization 2017. Global hepatitis report, 2017. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Petruzziello A. 2018. Epidemiology of hepatitis B virus (HBV) and hepatitis C virus (HCV) related hepatocellular carcinoma. Open Virol J 12:26–32. doi: 10.2174/1874357901812010026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Babiker A, Jeudy J, Kligerman S, Khambaty M, Shah A, Bagchi S. 2017. Risk of cardiovascular disease due to chronic hepatitis C infection: a review. J Clin Transl Hepatol 5:1–20. doi: 10.14218/JCTH.2017.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morozov VA, Lagaye S. 2018. Hepatitis C virus: morphogenesis, infection and therapy. World J Hepatol 10:186–212. doi: 10.4254/wjh.v10.i2.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sorbo MC, Cento V, Di Maio VC, Howe AYM, Garcia F, Perno CF, Ceccherini-Silberstein F. 2018. Hepatitis C virus drug resistance associated substitutions and their clinical relevance: update 2018. Drug Resist Updat 37:17–39. doi: 10.1016/j.drup.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 6.Taherkhani R, Farshadpour F. 2017. Global elimination of hepatitis C virus infection: progresses and the remaining challenges. World J Hepatol 9:1239–1252. doi: 10.4254/wjh.v9.i33.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swadling L, Capone S, Antrobus RD, Brown A, Richardson R, Newell EW, Halliday J, Kelly C, Bowen D, Fergusson J, Kurioka A, Ammendola V, Del Sorbo M, Grazioli F, Esposito ML, Siani L, Traboni C, Hill A, Colloca S, Davis M, Nicosia A, Cortese R, Folgori A, Klenerman P, Barnes E. 2014. A human vaccine strategy based on chimpanzee adenoviral and MVA vectors that primes, boosts, and sustains functional HCV-specific T cell memory. Sci Transl Med 6:261ra153. doi: 10.1126/scitranslmed.3009185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christiansen D, Earnest-Silveira L, Chua B, Meuleman P, Boo I, Grubor-Bauk B, Jackson DC, Keck ZY, Foung SKH, Drummer HE, Gowans EJ, Torresi J. 2018. Immunological responses following administration of a genotype 1a/1b/2/3a quadrivalent HCV VLP vaccine. Sci Rep 8:6483. doi: 10.1038/s41598-018-24762-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, Antonenko S, Liu YJ. 1999. The nature of the principal type 1 interferon-producing cells in human blood. Science 284:1835–1837. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 10.Hardy AW, Graham DR, Shearer GM, Herbeuval J-P. 2007. HIV turns plasmacytoid dendritic cells (pDC) into TRAIL-expressing killer pDC and down-regulates HIV coreceptors by Toll-like receptor 7-induced IFN-alpha. Proc Natl Acad Sci U S A 104:17453–17458. doi: 10.1073/pnas.0707244104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swiecki M, Colonna M. 2015. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol 15:471–485. doi: 10.1038/nri3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Machmach K, Leal M, Gras C, Viciana P, Genebat M, Franco E, Boufassa F, Lambotte O, Herbeuval JP, Ruiz-Mateos E. 2012. Plasmacytoid dendritic cells reduce HIV production in elite controllers. J Virol 86:4245–4252. doi: 10.1128/JVI.07114-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barblu L, Machmach K, Gras C, Delfraissy J-F, Boufassa F, Leal M, Ruiz-Mateos E, Lambotte O, Herbeuval J-P. 2012. Plasmacytoid dendritic cells (pDCs) from HIV controllers produce interferon-α and differentiate into functional killer pDCs under HIV activation. J Infect Dis 206:790–801. doi: 10.1093/infdis/jis384. [DOI] [PubMed] [Google Scholar]
- 14.Pelletier S, Said E, Ancuta P, Bruneau J, Shoukry NH. 2013. Sustained hyperresponsiveness of dendritic cells is associated with spontaneous resolution of acute hepatitis C. J Virol 87:e02445. doi: 10.1128/JVI.02445-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Decalf J, Fernandes S, Longman R, Ahloulay M, Audat F, Lefrerre F, Rice CM, Pol S, Albert ML. 2007. Plasmacytoid dendritic cells initiate a complex chemokine and cytokine network and are a viable drug target in chronic HCV patients. J Exp Med 204:2423–2437. doi: 10.1084/jem.20070814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takahashi K, Asabe S, Wieland S, Garaigorta U, Gastaminza P, Isogawa M, Chisari FV. 2010. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc Natl Acad Sci U S A 107:7431–7436. doi: 10.1073/pnas.1002301107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klöss V, Grünvogel O, Wabnitz G, Eigenbrod T, Ehrhardt S, Lasitschka F, Lohmann V, Dalpke AH. 2017. Interaction and mutual activation of different innate immune cells is necessary to kill and clear hepatitis C virus-infected cells. Front Immunol 8:1238. doi: 10.3389/fimmu.2017.01238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsai A, Irrinki A, Kaur J, Cihlar T, Kukolj G, Sloan DD, Murry JP. 2017. Toll-like receptor 7 agonist GS-9620 induces HIV expression and HIV-specific immunity in cells from HIV-infected individuals on suppressive antiretroviral therapy. J Virol 91:e02166-16. doi: 10.1128/JVI.02166-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vibholm L, Schleimann MH, Højen JF, Benfield T, Offersen R, Rasmussen K, Olesen R, Dige A, Agnholt J, Grau J, Buzon M, Wittig B, Lichterfeld M, Petersen AM, Deng X, Abdel-Mohsen M, Pillai SK, Rutsaert S, Trypsteen W, De Spiegelaere W, Vandekerchove L, Østergaard L, Rasmussen TA, Denton PW, Tolstrup M, Søgaard OS. 2017. Short-course Toll-like receptor 9 agonist treatment impacts innate immunity and plasma viremia in individuals with human immunodeficiency virus infection. Clin Infect Dis 64:1686–1695. doi: 10.1093/cid/cix201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krarup AR, Abdel-Mohsen M, Schleimann MH, Vibholm L, Engen PA, Dige A, Wittig B, Schmidt M, Green SJ, Naqib A, Keshavarzian A, Deng X, Olesen R, Petersen AM, Benfield T, Østergaard L, Rasmussen TA, Agnholt J, Nyengaard JR, Landay A, Søgaard OS, Pillai SK, Tolstrup M, Denton PW. 2018. The TLR9 agonist MGN1703 triggers a potent type I interferon response in the sigmoid colon. Mucosal Immunol 11:449–461. doi: 10.1038/mi.2017.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borducchi EN, Cabral C, Stephenson KE, Liu J, Abbink P, Ng’ang’a D, Nkolola JP, Brinkman AL, Peter L, Lee BC, Jimenez J, Jetton D, Mondesir J, Mojta S, Chandrashekar A, Molloy K, Alter G, Gerold JM, Hill AL, Lewis MG, Pau MG, Schuitemaker H, Hesselgesser J, Geleziunas R, Kim JH, Robb ML, Michael NL, Barouch DH. 2016. Ad26/MVA therapeutic vaccination with TLR7 stimulation in SIV-infected rhesus monkeys. Nature 540:284–287. doi: 10.1038/nature20583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marukian S, Jones CT, Andrus L, Evans MJ, Ritola KD, Charles ED, Rice CM, Dustin LB. 2008. Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology 48:1843–1850. doi: 10.1002/hep.22550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perales C, Beach NM, Gallego I, Soria ME, Quer J, Esteban JI, Rice C, Domingo E, Sheldon J. 2013. Response of hepatitis C virus to long-term passage in the presence of alpha interferon: multiple mutations and a common phenotype. J Virol 87:7593–7597. doi: 10.1128/JVI.02824-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheldon J, Beach NM, Moreno E, Gallego I, Pineiro D, Martinez-Salas E, Gregori J, Quer J, Esteban JI, Rice CM, Domingo E, Perales C. 2014. Increased replicative fitness can lead to decreased drug sensitivity of hepatitis C virus. J Virol 88:12098–12111. doi: 10.1128/JVI.01860-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moreno E, Gallego I, Gregori J, Lucía-Sanz A, Soria ME, Castro V, Beach NM, Manrubia S, Quer J, Esteban JI, Rice CM, Gómez J, Gastaminza P, Domingo E, Perales C. 2017. Internal disequilibria and phenotypic diversification during replication of hepatitis C virus in a noncoevolving cellular environment. J Virol 91:e02505-16. doi: 10.1128/JVI.02505-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanabuchi S, Liu Y-J. 2011. In vivo role of pDCs in regulating adaptive immunity. Immunity 35:851–853. doi: 10.1016/j.immuni.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 27.Webster B, Assil S, Dreux M. 2016. Cell-cell sensing of viral infection by plasmacytoid dendritic cells. J Virol 90:10050–10053. doi: 10.1128/JVI.01692-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gallego I, Sheldon J, Moreno E, Gregori J, Quer J, Esteban JI, Rice CM, Domingo E, Perales C. 2016. Barrier-independent, fitness-associated differences in sofosbuvir efficacy against hepatitis C virus. Antimicrob Agents Chemother 60:3786–3793. doi: 10.1128/AAC.00581-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y-J. 2005. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol 23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 30.Dental C, Florentin J, Aouar B, Gondois-Rey F, Durantel D, Baumert TF, Nunes JA, Olive D, Hirsch I, Stranska R. 2012. Hepatitis C virus fails to activate NF-κB signaling in plasmacytoid. J Virol 86:1090–1096. doi: 10.1128/JVI.05444-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Webster B, Werneke SW, Zafirova B, This S, Coléon S, Décembre E, Paidassi H, Bouvier I, Joubert P-E, Duffy D, Walzer T, Albert ML, Dreux M. 2018. Plasmacytoid dendritic cells control dengue and chikungunya virus infections via IRF7-regulated interferon responses. eLife 7:e34273. doi: 10.7554/eLife.34273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gilliet M, Cao W, Liu Y-J. 2008. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol 8:594–606. doi: 10.1038/nri2358. [DOI] [PubMed] [Google Scholar]
- 33.Lehmann C, Lafferty M, Garzino-Demo A, Jung N, Hartmann P, Fätkenheuer G, Wolf JS, van Lunzen J, Romerio F. 2010. Plasmacytoid dendritic cells accumulate and secrete interferon alpha in lymph nodes of HIV-1 patients. PLoS One 5:e11110. doi: 10.1371/journal.pone.0011110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boasso A, Royle CM, Doumazos S, Aquino VN, Biasin M, Piacentini L, Tavano B, Fuchs D, Mazzotta F, Lo Caputo S, Shearer GM, Clerici M, Graham DR. 2011. Over-activation of plasmacytoid dendritic cell inhibits anti-viral T cell responses: a model for HIV immunopathogenesis. Blood 118:5152–5163. doi: 10.1182/blood-2011-03-344218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baenziger S, Heikenwalder M, Johansen P, Schlaepfer E, Hofer U, Miller RC, Diemand S, Honda K, Kundig TM, Aguzzi A, Speck RF. 2009. Triggering TLR7 in mice induces immune activation and lymphoid system disruption, resembling HIV-mediated pathology. Blood 113:377–388. doi: 10.1182/blood-2008-04-151712. [DOI] [PubMed] [Google Scholar]
- 36.Heikenwalder M, Polymenidou M, Junt T, Sigurdson C, Wagner H, Akira S, Zinkernagel R, Aguzzi A. 2004. Lymphoid follicle destruction and immunosuppression after repeated CpG oligodeoxynucleotide administration. Nat Med 10:187–192. doi: 10.1038/nm987. [DOI] [PubMed] [Google Scholar]
- 37.Bam RA, Hansen D, Irrinki A, Mulato A, Jones GS, Hesselgesser J, Frey CR, Cihlar T, Yant SR. 2017. TLR7 agonist GS-9620 is a potent inhibitor of acute HIV-1 infection in human peripheral blood mononuclear cells. Antimicrob Agents Chemother 61:e01369-16. doi: 10.1128/AAC.01369-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones CT, Catanese MT, Law LMJ, Khetani SR, Syder AJ, Ploss A, Oh TS, Schoggins JW, MacDonald MR, Bhatia SN, Rice CM. 2010. Real-time imaging of hepatitis C virus infection using a fluorescent cell-based reporter system. Nat Biotechnol 28:167–171. doi: 10.1038/nbt.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koutsoudakis G, Herrmann E, Kallis S, Bartenschlager R, Pietschmann T. 2007. The level of CD81 cell surface expression is a key determinant for productive entry of hepatitis C virus into host cells. J Virol 81:588–598. doi: 10.1128/JVI.01534-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Friebe P, Boudet J, Simorre J-P, Bartenschlager R. 2005. Kissing-loop interaction in the 3′ end of the hepatitis C virus genome essential for RNA replication. J Virol 79:380–392. doi: 10.1128/JVI.79.1.380-392.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich H-G, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]