

A dynamic relationship exists between viruses and their hosts in which each ostensibly attempts to exploit the other’s vulnerabilities. A window is opened into the established condition, which evolved over millennia, if loss-of-function mutations occur in either the virus or host. Thus, the inability of viral host range mutants to replicate in specific cells can be overcome by identifying and inactivating the opposing cellular gene. Here, we investigated a C7/K1 host range mutant of vaccinia virus in which the cellular gene SAMD9 serves as the principal host restriction factor. Host restriction was triggered early in infection and manifested as a block in translation of viral mRNAs. Features of the block include inhibition of cap-dependent and internal ribosome entry site-mediated translation, sequestration of viral RNA, and inability to overcome the inhibition by inactivation of protein kinase R, ribonuclease L, or G3 binding proteins, suggesting a novel mechanism of host restriction.
KEYWORDS: host range, posttranscriptional control mechanisms, poxvirus, regulation of gene expression, translational control, vaccinia virus
ABSTRACT
Replication of vaccinia virus in human cells depends on the viral C7 or K1 protein. A previous human genome-wide short interfering RNA (siRNA) screen with a C7/K1 double deletion mutant revealed SAMD9 as a principal host range restriction factor along with additional candidates, including WDR6 and FTSJ1. To compare their abilities to restrict replication, the cellular genes were individually inactivated by CRISPR/Cas9 mutagenesis. The C7/K1 deletion mutant exhibited enhanced replication in each knockout (KO) cell line but reached wild-type levels only in SAMD9 KO cells. SAMD9 was not depleted in either WDR6 or FTSJ1 KO cells, suggesting less efficient alternative rescue mechanisms. Using the SAMD9 KO cells as controls, we verified a specific block in host and viral intermediate and late protein synthesis in HeLa cells and demonstrated that the inhibition could be triggered by events preceding viral DNA replication. Inhibition of cap-dependent and -independent protein synthesis occurred primarily at the translational level, as supported by DNA and mRNA transfection experiments. Concurrent with collapse of polyribosomes, viral mRNA was predominantly in 80S and lighter ribonucleoprotein fractions. We confirmed the accumulation of cytoplasmic granules in HeLa cells infected with the C7/K1 deletion mutant and further showed that viral mRNA was sequestered with SAMD9. RNA granules were still detected in G3BP KO U2OS cells, which remained nonpermissive for the C7/K1 deletion mutant. Inhibition of cap-dependent and internal ribosome entry site-mediated translation, sequestration of viral mRNA, and failure of PKR, RNase L, or G3BP KO cells to restore protein synthesis support an unusual mechanism of host restriction.
IMPORTANCE A dynamic relationship exists between viruses and their hosts in which each ostensibly attempts to exploit the other’s vulnerabilities. A window is opened into the established condition, which evolved over millennia, if loss-of-function mutations occur in either the virus or host. Thus, the inability of viral host range mutants to replicate in specific cells can be overcome by identifying and inactivating the opposing cellular gene. Here, we investigated a C7/K1 host range mutant of vaccinia virus in which the cellular gene SAMD9 serves as the principal host restriction factor. Host restriction was triggered early in infection and manifested as a block in translation of viral mRNAs. Features of the block include inhibition of cap-dependent and internal ribosome entry site-mediated translation, sequestration of viral RNA, and inability to overcome the inhibition by inactivation of protein kinase R, ribonuclease L, or G3 binding proteins, suggesting a novel mechanism of host restriction.
INTRODUCTION
Chordopoxviruses have nearly 100 conserved genes that enable essential functions, such as cell entry, genome replication, transcription, and assembly of infectious particles, as well as a similar number of less well-conserved genes, some of which counteract cellular immune responses (1–4). A subset of the latter genes affect replication in specific cell types, and their absence produces host range defects (5). Unlike the situation with many other viruses, the poxvirus host range genes typically affect postentry steps in replication. Examples of vaccinia virus (VACV) host range genes include those encoding a double-stranded RNA binding protein, an inhibitor of cellular protein kinase R (PKR), a serine protease inhibitor, and C7 and K1, which together are required for replication in human cells (4). More information regarding the host range defect of VACV C7/K1 double deletion mutants, which is the focus of the present investigation, is provided below.
Following nitrous acid mutagenesis, Drillien and coworkers (6) isolated a VACV mutant with a large deletion near the left end of the genome that cannot replicate in human cells. Subsequent studies revealed that the host range defect depends on the loss of both K1 (7, 8) and C7 (9) genes, which are unrelated in sequence and have no nonpoxvirus homologs. Replication in human cells occurs if either of the two genes is present, while K1 but not C7 is essential in rabbit kidney cells (9, 10). Furthermore, a third unrelated gene from cowpox virus can substitute for C7 and K1 in some cells (9, 11, 12). The original studies, as well as later ones, concurred that C7/K1 mutants are defective in postreplicative protein synthesis (13–16). Although enhanced PKR and eIF2α phosphorylation has been described in the absence of C7/K1, this is thought to be a secondary effect, since protein synthesis is not restored by knockdown or inactivation of PKR (13, 16, 17). K1 was also reported to prevent the degradation of IkBα and inhibit NF-κB activation in RK-13 cells (18) and to bind ACAP2 (19), but the latter does not appear related to the host range function of K1 (20).
The interferon sensitivity of C7/K1 mutant replication in human Huh7 cells suggests a role for one or more interferon-stimulated genes (ISGs) (13). Screening of a library of more than 350 ISGs identified interferon-regulated factor 1 (IRF1) as an inhibitor of the C7/K1 deletion mutant but not wild-type VACV (21). However, IRF1 induces expression of other ISGs that may be more direct inhibitors. Independent proteomic (22, 23) and genome-wide short interfering RNA (siRNA) (24) screens identified SAMD9 as a human host factor responsible for inhibiting replication of myxoma virus C7 homolog MO62 and VACV C7/K1 deletion mutants. The siRNA screen detected additional candidate host factors, namely, WDR6, FTSJ1, and CDC37. SAMD9 binds VACV C7 (24, 25) and K1 (24) as well as MO62 (23). Liu and coworkers (22) described the formation of SAMD9-containing granules, which appear related but not identical to typical stress granules, in the absence of C7/K1 of VACV or MO62 of myxoma virus. A mutagenesis study indicated that the N-terminal 385 amino acids of SAMD9 retain the ability to interact with MO62 but are insufficient to prevent replication of the host range mutants (26). SAMD9L, a paralog of SAMD9, also inhibits the C7/K1 mutant (27).
Aside from an antiviral role, there is evidence for a function of SAMD9 in human health (28). Mutations of SAMD9 are responsible for a familial disease characterized by calcified skin tumors (29, 30) and for a form of adrenal hypoplasia (31). SAMD9 has been identified as a possible myeloid tumor suppressor gene, and deletion of the related SAMD9L gene in mice causes myeloid disorders (32). SAMD9 has been shown to interact with RGL2, a probable guanine nucleotide exchange factor, and knockdown of RGL2 increased expression of the transcription factor EGR1 (33). There is also evidence that SAMD9 binds to the protein EEA1 and is involved in homotypic fusion of endosomes and degradation of cytokine receptors (32).
Although there is general agreement that the host range defect of C7/K1 mutants involves inhibition of postreplicative protein synthesis, the mechanism remains elusive. The aims of the present study were to use CRISPR/Cas9 mutagenesis to confirm the effect of knockdown of WDR6 and FTSJ1 in addition to SAMD9 on replication of the VACV C7/K1 mutant, determine the stage of viral replication in which the translational defect is triggered in nonpermissive cells, and evaluate the roles of translation initiation factors and mRNA sequestration. CRISPR/Cas9-modified cell lines unable to express PKR, RNase L, or G3BP provided tools for this analysis.
RESULTS
Comparison of SAMD9, WDR6, and FTSJ1 inactivation on replication of ΔC7K1.
We previously identified SAMD9 as a principal restriction factor for inhibition of a VACV C7/K1 deletion mutant (ΔC7K1) from a high-throughput human genome-wide siRNA library screen and confirmed this result by CRISPR/Cas9 mutagenesis of HeLa cells (24). However, knockdown of additional genes, including WDR6 and FTSJ1, also enhanced spread of ΔC7K1. CRISPR/Cas9 mutagenesis of the genes encoding WDR6 and FTSJ1 was carried out to further compare their relative roles in restricting replication of ΔC7K1. Genome sequencing of PCR products confirmed frameshift mutations within the first exons of SAMD9, WDR6, and FTSJ1, and the sequences of cloned DNAs with out-of-frame mutations in the first exon are shown in Fig. 1A.
FIG 1.

Comparison of ΔC7K1 replication in SAMD9, WDR6, and FTSJ1 CRISPR/Cas9 KO cells. (A) Genome sequences of CRISPR/Cas9 modifications in SAMD9, WDR6, and FTSJ1 genes. The SAMD9, WDR6, and FTSJ1 genes of HeLa cells were modified by Cas9 using guide RNAs that targeted their first exons. Representative cell clones that exhibited enhanced replication of ΔC7K1 were expanded and further analyzed by genome sequencing. Primers that anneal to DNA flanking the Cas9 guide RNA target sites were used for PCR amplification. The PCR products were cloned in plasmids and subjected to Sanger sequencing. Unmodified and CRISPR/Cas9-modified sequences are shown with amino acid codons below the nucleotide sequences. Dashes and asterisks indicate deleted nucleotides and stop codons, respectively. (B) Infection and spread of the C7/K1 deletion mutant. HeLa, SAMD9−/−, WDR6−/−, and FTSJ1−/− cells were infected in triplicate at a multiplicity of infection of 0.03 PFU/cell with ΔC7K1 encoding GFP regulated by the P11 late promoter. After 18 h, GFP-positive cells were counted by flow cytometry. (C) HeLa and SAMD9−/− cells were infected with WT or ΔC7K1 VACV and analyzed as described above. Error bars represent standard errors of the means (SEM). ****, P < 0.0001; **, P < 0.004; *, P < 0.025.
To assess the biological effects of inactivating these genes, unmodified HeLa and SAMD9, WDR6, and FTSJ1 KO cells were inoculated with a low multiplicity of infection of ΔC7K1, which expresses green fluorescent protein (GFP) regulated by a late promoter, to allow infection and spread. After 18 h, GFP-expressing cells were quantified by flow cytometry. Spread of ΔC7K1 was enhanced in all three KO cell lines compared to that of HeLa cells (P < 0.0001) but was greater in the SAMD9−/− cells than in the WDR6−/− (P < 0.025) and FTSJ1−/− (P < 0.004) cells (Fig. 1B). The much higher replication of ΔC7K1 in SAMD9−/− cells than HeLa cells is also shown in Fig. 1C. Whereas there was an enormous difference between the replication of WT virus and ΔC7K1 in HeLa cells (P < 0.0001), their replication was equivalent in SAMD9−/− cells (P > 0.9999) (Fig. 1C). Interestingly, even though WT VACV replicates well in HeLa cells, the yield was higher in the SAMD9−/− cells (P < 0.0001), suggesting a partial inhibitory effect of SAMD9 despite the presence of C7 and K1 (Fig. 1C).
To further compare the permissiveness of the KO cell lines, each was infected with 5 PFU/cell of WT or ΔC7K1 KO viruses to provide synchronous infections. After 8 h, Western blotting was carried out with antibodies to the early I3 and the postreplicative D13 and A3 proteins. In HeLa cells, similar amounts of I3 were detected after infection with WT and ΔC7K1, but both D13 and A3 were severely diminished after infection with the mutant virus (Fig. 2A). I3 was similarly expressed in each of the KO cells infected with WT and ΔC7K1, whereas expression of D13 and A3 was fully restored in SAMD9−/− cells but only modestly increased in WDR6−/− and FTSJ1−/− cells infected with ΔC7K1 (Fig. 2A).
FIG 2.

Protein synthesis in HeLa and KO cell lines. (A) Western blot. HeLa, SAMD9−/−, WDR6−/−, and FTSJ1−/− cells were infected with WT VACV or ΔC7K1 at a multiplicity of infection of 5 PFU/cell. At 8 h, lysates were prepared, and proteins were resolved by SDS-PAGE and then transferred to membranes. The blots were probed with primary antibodies to I3, D13, and A3, followed by secondary antibodies. Protein bands were visualized with an infrared imager. Inter, intermediate. (B) Expression of SAMD9. HeLa, SAMD9−/−, WDR6−/−, and FTSJ1−/− cells were infected as described for panel A and analyzed by SDS-PAGE. Blots were probed with antibody to SAMD9. (C) Detection of newly synthesized proteins by incorporation of [35S]methionine and cysteine. HeLa and SAMD9−/− cells were infected with ΔC7K1 at a multiplicity of 3 PFU/cell and incubated with [35S]methionine and cysteine for 15 min at the indicated times. The cells were then washed, lysed, and analyzed by SDS-PAGE. Radiolabeled proteins were detected with a biomolecular imager. HPI, hours postinfection.
Since SAMD9−/− cells were most permissive for ΔC7K1, we considered the possibility that WDR6 and FTSJ1 act by regulating SAMD9 levels. SAMD9 was not detected in SAMD9−/− cells, consistent with other evidence for a disruption of the gene (Fig. 2B). However, Western blotting showed that SAMD9 was undiminished in WDR6−/− or FTSJ1−/− cells (Fig. 2B). Thus, WDR6 and FTSJ1 may act at different steps than SAMD9. In subsequent experiments, SAMD9−/− cells were used as a control to ensure that host range defects in ΔC7K1 replication were specific.
Metabolic labeling was carried out for a more global assessment of protein synthesis in HeLa and SAMD9−/− cells infected with ΔC7K1. In SAMD9−/− cells, bands corresponding to abundant postreplicative proteins became apparent at 4 h after infection (Fig. 2C). In contrast, there was a total diminution of all bands between 4 and 8 h in infected HeLa cells, indicating that host protein synthesis was also shut down, in addition to a failure to synthesize viral proteins (Fig. 2C).
Inhibition of cap-dependent and IRES-mediated protein synthesis in cells infected with ΔC7K1.
VACV and cellular mRNAs have cap structures at their 5′ ends (34) that are pivotal for translation efficacy through the recruitment and positioning of the ribosomes. In contrast, some viruses have an internal ribosome entry site (IRES) within the mRNA 5′ leader that enables cap-independent translation in the absence of some or, as in the case of the cricket paralysis virus (CrPV) IRES, all translation initiation factors (35, 36). We considered that insights into the possible mechanisms of translation inhibition can be obtained by comparing cap-dependent and IRES-mediated translation. Specific features of poxvirus replication enabled the strategy outlined in Fig. 3A. Poxvirus early mRNAs are synthesized within the virus core and extruded into the cytoplasm almost immediately after infection, and neither de novo protein synthesis nor DNA replication are needed for this to occur. Synthesis of early proteins, which include RNA polymerase subunits and specific intermediate transcription factors, are necessary for intermediate gene expression. During a normal infection, viral DNA replication is also required to provide the template for intermediate transcription. Therefore, if DNA replication is inhibited by AraC, only early genes are expressed. Importantly for our purposes, genes regulated by intermediate promoters can be transcribed from a transfected plasmid template even in the absence of viral genome replication (37, 38). Thus, our plan was to infect HeLa and SAMD9−/− cells with ΔC7K1 in the presence of AraC and transfect a plasmid carrying a reporter gene. The reporter plasmid contained a bicistronic gene comprised of the firefly luciferase (FLuc) open reading frame (ORF) regulated by the intermediate G8 promoter preceding the mRNA start site, followed by the renilla luciferase (RLuc) ORF regulated by the CrPV IRES. FLuc and RLuc have different substrates and were assayed independently.
FIG 3.

Host restriction of cap-dependent and IRES-mediated translation. (A) Experimental plan. Features of VACV transcription, including the inhibition by AraC of intermediate and late gene expression, are indicated on the left. The plan to assess expression in VACV-infected cells from a transfected plasmid with a bicistronic gene encoding FLuc regulated by an intermediate promoter, followed by RLuc regulated by the CrPV IRES, is illustrated on the right. (B) HeLa and SAMD9−/− cells were infected with 3 PFU/cell of ΔC7K1 and transfected 1 h later. Expression of FLuc was determined at 2, 4, 6, and 8 h after infection. (C) HeLa cells were infected in triplicate, as described for panel B, with ΔC7K1 or WT virus in the absence or presence of 40 µg/ml AraC and transfected with the bicistronic reporter plasmid. At 6 h after infection, the cells were lysed and FLuc and RLuc activities determined. Error bars represent SEM.
The first step in implementing the scheme was to determine whether intermediate promoter-regulated gene expression from a plasmid was impaired in nonpermissive cells. HeLa and SAMD9−/− cells were infected with ΔC7K1 and transfected with the reporter plasmid. FLuc activity was detected at 4 h and plateaued between 6 and 8 h (Fig. 3B). The activity was about 5-fold higher in SAMD9−/− cells than in unmodified HeLa cells, indicating gene expression from the plasmid was reduced in the latter cells. We next infected HeLa cells with WT and mutant ΔC7K1 viruses in the absence and presence of AraC and transfected the FLuc/RLuc reporter plasmid. The low expression of FLuc and RLuc in cells infected with ΔC7K1 indicated that the restriction affected translation initiation factor-dependent and -independent protein synthesis mediated by cap and IRES elements, respectively (Fig. 3C). Furthermore, the inhibition occurred in the presence of AraC, indicating that neither viral DNA replication nor the onset of viral intermediate protein synthesis was necessary to trigger the shutdown. AraC increased FLuc and RLuc expression by WT virus, possibly by suppressing competition with viral intermediate and late mRNAs or by enhancing mRNA stability by preventing expression of the D10 decapping enzyme (39).
Relative amounts of VACV pre- and postreplicative mRNAs in HeLa and SAMD9−/− cells infected with ΔC7K1.
We considered that reduced amounts of viral mRNAs might contribute to decreased cap- and IRES-mediated protein synthesis in nonpermissive cells infected with ΔC7K1. To evaluate this possibility, the mRNAs encoding the I3, D13, and A3 proteins were quantified by reverse transcription and Droplet Digital PCR (ddPCR). By also determining the amount of 18S rRNA in each sample and knowing that HeLa cells contain 9.5 × 106 ribosomes (40), we calculated the number of copies of each mRNA per cell. At 2 h after infection, I3 was the predominant mRNA and was even higher in the cells infected with ΔC7K1 than the control virus, although the amounts were similar at 8 h (Fig. 4A). Due to the sensitivity of ddPCR, small amounts of D13 were also detected at 2 h but increased dramatically at 8 h. At the latter time, there were approximately 175,000 copies of D13 mRNA and 150,000 copies of A3 mRNA in HeLa cells and slightly larger amounts in SAMD9−/− cells (Fig. 3A). Although these copy numbers might be inflated to some extent because of readthrough from upstream genes, they indicated highly reiterative transcription of the DNA templates. In HeLa cells infected with ΔC7K1, the copies of D13 and A3 mRNAs were reduced approximately 3- and 6-fold, respectively, compared to infection with the control virus, whereas the control and mutant viruses had similar amounts of D13 and A3 mRNAs in SAMD9−/− cells (Fig. 4A). Although the D13 and A3 genes both have intermediate and late promoter elements, D13 is predominantly intermediate and A3 predominantly late (41, 42). This difference might account for the greater inhibition of A3 compared to D13, as intermediate transcription factor proteins are products of early genes and late transcription factor proteins are products of intermediate genes. Nevertheless, it was uncertain whether the reductions in D13 and A3 mRNAs in HeLa cells infected with ΔC7K1 were sufficient to entirely account for the inhibition of synthesis of viral proteins, and therefore experiments were carried out to distinguish between transcriptional and translational blocks.
FIG 4.

Quantification of mRNAs synthesized by ΔC7K1 and WT virus and translation of transfected mRNAs. (A) mRNA quantification. HeLa and SAMD9−/− cells were infected in triplicate with ΔC7K1 or WT virus at a multiplicity of infection of 3 PFU/cell, and RNA was extracted at 2 and 8 h, reversed transcribed, and quantified by ddPCR using primers specific for I3, D13, and A3 transcripts and rRNA. The absolute amounts of viral mRNA in each sample were calculated using the 18S rRNA concentration and the number of ribosomes in HeLa cells. (B) Translation of mRNA synthesized in vitro. Control HeLa and SAMD9−/− cells were infected in triplicate with ΔC7K1 at a multiplicity of infection of 3 PFU/cell in the absence or presence of AraC and then transfected with capped, methylated, and polyadenylated mRNA with a 5′ poly(A) or non-poly(A) leader preceding the FLuc ORF. FLuc was measured after 6 h. Error bars represent SEM.
Evidence for a translational block.
To more directly investigate the possibility of a translational block, we synthesized mRNAs in vitro and transfected them into infected cells. HeLa and SAMD9−/− cells were infected with ΔC7K1 in the presence or absence of AraC and transfected with capped mRNAs synthesized in vitro containing a 3′ poly(A) tail and with a 5′ poly(A) or a non-poly(A) leader preceding the FLuc ORF. The 5′ poly(A) leader is characteristic of VACV intermediate and late mRNAs and has been reported to enhance translation under certain conditions (43), whereas this feature is absent from most early and cellular mRNAs. With both leader sequences, FLuc expression was greatly reduced in HeLa cells compared to the level in SAMD9−/− cells in the absence or presence of AraC (Fig. 4B). AraC increased the amount of FLuc in SAMD9−/− cells, similar to the result observed with transfected plasmids. Taken together, the strong effect on viral protein synthesis, incomplete effect on viral mRNA abundance, and the inhibition of expression of transfected mRNA are consistent with a defect in mRNA translation. Furthermore, the block occurred in the presence of AraC, again showing that neither DNA replication nor postreplicative protein synthesis were necessary to trigger the translational defect.
Localization of mRNA in cells infected with ΔC7K1.
We next investigated the association of viral mRNAs with ribosomes in cells infected with ΔC7K1. The number and size of polyribosomes, comprised of ribosomes with associated mRNA and nascent peptides, can be determined by sucrose gradient sedimentation. Polyribosomes following VACV infection are smaller than those of uninfected cells (44), due at least in part to the smaller size of VACV mRNAs. In accordance, we found fewer large polyribosomes after infection of HeLa cells with wild-type virus than in uninfected cells. More importantly, there was a near total collapse of polyribosomes in cells infected with ΔC7K1 (Fig. 5A). Analysis of gradient fractions from SAMD9−/− cells infected with ΔC7K1 showed substantial amounts of D13 mRNA associated with polyribosomes and in fractions lighter than 80S ribosomes (Fig. 5B). In contrast, there was less D13 mRNA associated with polyribosomes of HeLa cells infected with ΔC7K1, and most was in the 80S ribosome and lighter fractions. Both the polyribosome collapse and the distribution of mRNA support a reduction in translation initiation.
FIG 5.

Localization of viral mRNA on sucrose gradients. (A) Polyribosome profiles. HeLa cells were not infected (NI) or were infected with WT or ΔC7K1 virus for 6 h, after which their cytosols were centrifuged on sucrose gradients. The gradients were collected from the top while we continuously recorded the absorbance (A260) to monitor ribosomes and polyribosomes. The 80S ribosome position is indicated. (B) Distribution of D13 mRNA. HeLa and SAMD9−/− cells were infected with ΔC7K1, and the cytosols were centrifuged on sucrose gradients as described for panel A. RNA was extracted from the resulting fractions and reversed transcribed, and D13 mRNA was quantified using ddPCR. Fraction 1 is the top of the gradient.
We were curious as to the intracellular location of viral mRNAs in HeLa cells infected with ΔC7K1, as they were not being actively translated. The nucleoside analog 5′ethynyl uridine (EU) followed by the attachment of a fluor-azide with click chemistry can be used to localize nascent RNA (45), although to our knowledge this has not been reported for a VACV infection. HeLa and SAMD9−/− cells were infected with ΔC7K1 and 5 h later were incubated with EU for 1 h. In HeLa cells infected with ΔC7K1, the nascent RNA formed clustered punctae in and around 4′,6-diamidino-2-phenylindole (DAPI)-stained viral factories (Fig. 6A). In contrast, cytoplasmic EU staining was more diffuse in SAMD9−/− cells infected with ΔC7K1 (Fig. 6A). The drug triptolide, an inhibitor of cellular RNA polymerase I and II that covalently binds to a subunit of TFIIH and inhibits DNA-dependent ATPase activity (46–48), was used to help differentiate cellular and viral RNAs. HeLa cells and SAMD9−/− cells were treated with triptolide from 4 to 6 h after infection with ΔC7K1 and then incubated with EU for 1 h before performing click chemistry. There was less overall nuclear labeling with EU, which was mostly concentrated in nucleoli, consistent with the known activity of triptolide (Fig. 6B). However, the nascent cytoplasmic RNA still localized in punctae in HeLa cells and was diffuse in SAMD9−/− cells. Taken together, the data indicated that in HeLa cells infected with ΔC7K1, the viral mRNA did not sediment with polyribosomes and was localized in discrete structures associated with cytoplasmic viral factories.
FIG 6.
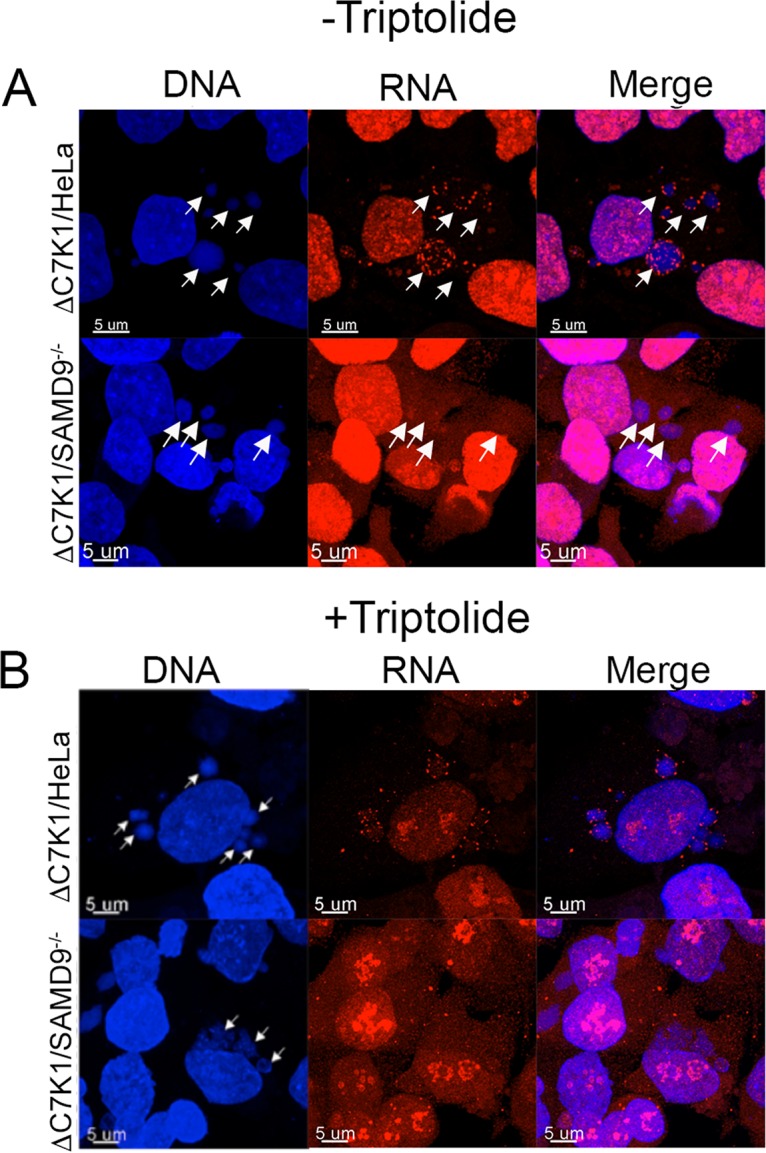
Localization of EU-labeled nascent RNA. (A) HeLa and SAMD9−/− cells were grown on coverslips, infected with ΔC7K1, and incubated with 1 mM EU from 5 to 6 h after infection. Cells were washed, fixed, and permeabilized, and EU-labeled RNA was detected by conjugating Alexa Fluor 568 azide using click chemistry. DNA was detected by staining with DAPI. Arrows pointing to the positions of the viral factories visualized with DAPI are shown in all panels. Magnification is indicated by size bars. (B) HeLa and SAMD9−/− cells were infected with ΔC7K1, as described for panel A, except that triptolide was added at 4 h and 1 mM EU at 5 h. At 6 h, the cells were fixed and treated as described for panel A. Arrows pointing to viral factories visualized with DAPI are shown only in the DNA panels.
Association of RNA with stress granule markers.
We suspected that the RNA punctae detected with EU corresponded to the SAMD9 granules previously described in nonpermissive cells infected with myxoma virus MO62 and VACV C7/K1 deletion mutants (23). In that study, SAMD9 colocalized with the stress granule markers as well as the translation factor eIF4G and double-stranded RNA, but the presence of nascent mRNA was not investigated. We infected HeLa cells and SAMD9−/− cells with ΔC7K1 in the presence of triptolide. G3BP was associated with granules in and around viral factories in HeLa cells infected with ΔC7K1, whereas G3BP was more diffuse in infected SAMD9−/− cells (Fig. 7A). As shown in the merge, EU-labeled RNA punctae in HeLa cells colocalized with many of the G3BP granules. Liu and McFadden (23) reported that SAMD9 and G3BP colocalize in granules that form in HeLa cells infected with the MO62 and C7K1L deletion mutants. We found that SAMD9 also colocalized at sites of EU labeling in cells infected with ΔC7K1 (Fig. 7B).
FIG 7.
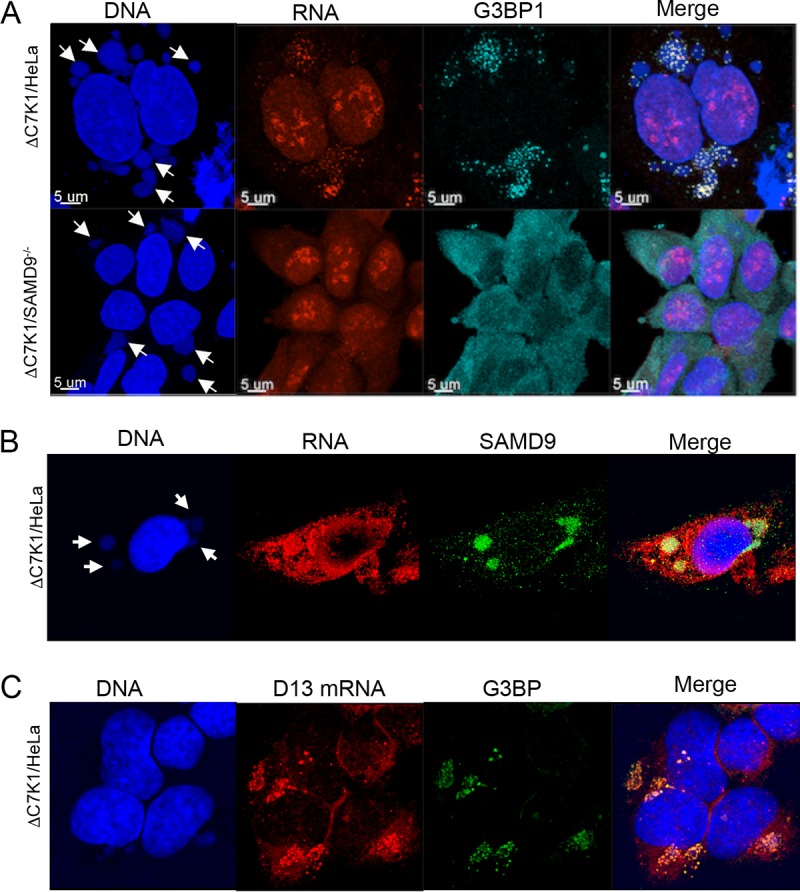
Association of viral RNA with the G3BP stress granule marker and SAMD9. (A) Colocalization of EU-labeled cytoplasmic RNA and G3BP1 at viral factories. HeLa and SAMD9−/− cells were infected with ΔC7K1. At 4 h after infection, cells were treated with 1 µM triptolide for 1 h and then 1 mM EU in the continued presence of triptolide for an additional 1 h. EU-labeled RNA was detected as described in the legend to Fig. 6A, and G3BP was visualized with a rabbit polyclonal antibody followed by a fluorescent secondary antibody. Arrows point to DAPI-labeled DNA factories. Magnification is indicated by size bars. (B) Colocalization of EU-labeled RNA and SAMD9. HeLa cells were infected and analyzed as described for panel A, except that the antibody was to SAMD9 instead of G3BP1. (C) Colocalization of viral D13 mRNA with G3BP1. HeLa cells were infected as described for panel A except for the absence of triptolide. Following fixation and permeabilization, D13 mRNA was detected by FISH using probes to D13 mRNA. G3BP1 was detected with specific antibody.
Although the cytoplasmic RNA labeled with EU at late times after infection was almost certainly viral, fluorescent in situ hybridization (FISH) was employed to prove this. A D13 antisense probe partially colocalized with G3BP granules in HeLa cells infected with ΔC7K1 (Fig. 7C). Thus, we confirmed the formation of SAMD9-containing stress-like granules in the cytoplasm during nonpermissive infection with ΔC7K1 and importantly provided evidence for their colocalization with viral mRNA.
Host range defect of ΔC7K1 is not overcome by inactivation of G3BP, PKR, or RNase L.
Mammalian stress granules contain stalled translation preinitiation complexes that are assembled into discrete granules by specific RNA-binding proteins, such as G3BP. Kedersha and coworkers (49) reported that CRISPR/Cas9-modified human U2OS cells unable to synthesize G3BP1 and G3BP2 (ΔΔG3BP1/2) do not form stress granules in response to eIF2α phosphorylation or eIF4A inhibition, although they still form after severe heat or osmotic stress. The ΔΔG3BP1/2 cells therefore provided a means to determine whether G3BP was required to form the ΔC7K1-induced RNA granules and mediate host restriction. Prominent granules were detected with antibodies to G3BP and eIF3A in unmodified U2OS cells treated with arsenite, a strong inducer of stress granules, or infected with ΔC7K1 (Fig. 8A). As expected, the granular pattern detected with antibody to eIF3A was greatly diminished in arsenite-treated ΔΔG3BP1/2 cells. However, in ΔΔG3BP1/2 cells infected with ΔC7K1, the granular pattern was more persistent (Fig. 7A).
FIG 8.

Effect of G3BP1/2 deletion on granule formation and virus spread. (A) Effect of G3BP1/2 deletion on granule formation. U2OS cells (left) and ΔΔG3BP1/2 U2OS cells (right) were grown on coverslips and left untreated or incubated for 1 h with 100 µM arsenite to induce stress granule formation or inoculated with ΔC7K1 virus at a multiplicity of infection of 3 PFU/cell for 6 h. Cells were fixed and granules were detected with antibodies to G3BP and eIF3A and fluorescent secondary antibodies. Large images on the left represent a merge of DAPI and antibody staining; areas within the boxes are shown at higher magnification on the right as a composite of DAPI and anti-G3BP (upper) and anti-eIF3A (lower). Magnification is indicated by size bars. (B) Effect of G3BP1/2 deletion on spread of ΔC7K1. HeLa, SAMD9−/−, U2OS, and ΔΔG3BP1/2 cells were infected in triplicate with ΔC7K1 at a multiplicity of 0.05 PFU/cell for 18 h. GFP-positive cells were determined by flow cytometry. (C) Effect of PKR and RNase L deletion on spread of ΔC7K1. The procedure was similar to that described for panel B, except that A549 and PKR/RNase L knockout cells were used. Error bars represent SEM.
An important question was whether inactivation of G3BP1/2 enhanced the replication of ΔC7K1. Whereas KO of SAMD9 from HeLa cells greatly increased the spread of ΔC7K1, KO of G3BP1/2 did not improve the spread (Fig. 8B). Similarly, PKR/RNase L double KO A549 cells were unable to support replication of ΔC7K1 (Fig. 8C), unlike the situation with E3 and decapping enzyme mutants (50). We concluded that the restriction of ΔC7K1 was not mediated by G3BP, PKR, or RNase L.
DISCUSSION
In a previous human genome-wide siRNA screen, SAMD9 was the most prominent hit based on enhancement of ΔC7K1 spread in HeLa cells (24). Nevertheless, additional potential host restriction factors were identified and confirmed by repeat siRNA assays. Because the extent of knockdown can vary due to protein stability and other causes, we used CRISPR/Cas9 to introduce out-of-frame mutations in the first exons of two of the genes encoding additional potential restriction factors, namely, WDR6 and FTSJ1. The new KO cell lines enhanced replication and late protein synthesis of ΔC7K1 but to a lesser extent than SAMD9 KO cells. Although the possibility that WDR6 and FTSJ1 regulated SAMD9 abundance would be a parsimonious explanation for the existence of multiple host factors, SAMD9 was not reduced in either WDR6 or FTSJ1 KO cells, suggesting alternative but less efficient restriction mechanisms. However, the limited information available regarding the latter host proteins does not suggest a mechanism for host restriction. WDR6, a member of the 40-amino-acid WD repeat family, has a mass of approximately 122 kDa, is located in the cytoplasm, and is reported to interact with serine/threonine kinase 11 and contribute to cell growth arrest (51). FTSJ1, a member of the methyltransferase superfamily, is a 36-kDa protein located in the cytoplasm and nucleolus, can methylate the anticodon loop of some tRNAs, and is implicated in cognitive disabilities (52). Intriguingly, there is evidence for a physical interaction between WDR6 and FTSJ1 (and also CDC37, another positive hit in the siRNA screen but not further evaluated here) (53, 54). It would be interesting to construct double WDR6/FTSJ1 KO cell lines to see if replication of ΔC7K1 is further enhanced.
The SAMD9−/− HeLa cells provided a useful control for further studies of the C7/K1 deletion mutant, which comprise the major portion of this paper. Previous studies had shown that the host range defect of C7/K1 mutants is manifested as a block in viral postreplicative protein synthesis. VACV mRNAs are capped by viral enzymes and use the cellular translation machinery, which includes ribosomes and a multitude of translation initiation factors. One hypothesis was that the cell responds to infection by C7/K1 mutants by inactivating one or more initiation factors. We tested this idea by taking advantage of the CrPV IRES element, which allows translation in the absence of any known translation initiation factor. By means of transfection experiments, we found that CrPV IRES-mediated as well as cap-dependent translation were restricted in HeLa cells infected with the C7/K1 deletion mutant. This suggested that translation initiation factors are not the target of host restriction. In this context, we also determined that KO of both PKR, which prevents phosphorylation of eIF2α in A549 cells, and RNase L did not alleviate the host range restriction.
Reduced amounts of viral mRNAs would impact both translation initiation factor-dependent and -independent protein synthesis. Using quantitative ddPCR, we found that synthesis of a representative early mRNA was unimpeded, whereas the two postreplicative intermediate/late mRNAs analyzed were reduced 3- to 6-fold in ΔC7K1-infected HeLa cells compared to the level in SAMD9−/− cells, which seemed insufficient to account entirely for the decrease in viral protein synthesis. Moreover, a decrease in translation of intermediate mRNAs and the consequent reduced levels of transcription factors could lead to a secondary decrease in late mRNA synthesis. In order to untangle the relationship between viral protein and mRNA synthesis, we transfected mRNAs made in vitro and found diminished translation in cells infected with ΔC7K1. Similar results were obtained with mRNA containing a 5′ poly(A) untranslated leader, which resembles intermediate and late mRNAs, and with a non-poly(A) leader, which resembles viral early and host cell mRNAs. A block at or before the initiation stage of translation was supported by the analysis of polyribosome profiles, which exhibited near total collapse at 8 h after infection of HeLa cells with ΔC7K1. Moreover, viral mRNA was largely in the 80S and lighter fractions. A similar collapse of polyribosomes and accumulation of cellular mRNA on ribosomes and ribosome subunits occurs in uninfected cells treated with arsenite, which causes eIF2a phosphorylation and stress granule formation (55).
The absence of viral mRNA in polyribosomes brought up the question of its intracellular location. We first examined this by incorporation of the clickable nucleoside analog EU at times at which nearly all newly synthesized cytoplasmic mRNA is viral during a normal infection. The EU-labeled cytoplasmic RNA in HeLa cells infected with ΔC7K1 was present in punctae that localized in and around viral factories, whereas the EU-labeled RNA was more diffusely cytoplasmic in permissive SAMD9−/− cells. Similar results were obtained when triptolide, a specific inhibitor of cellular RNA polymerase I and II, was used, supporting the viral nature of the EU-labeled RNA. More definitive evidence was obtained by FISH using a probe to a specific viral intermediate mRNA. The stress granule marker G3BP, as well as SAMD9, colocalized with the viral mRNA. The latter result fit with a prior report that SAMD9 localized in stress-like granules in cells infected with deletions of the myxoma virus MO62 gene and the VACV C7/K1 deletion mutant (23). Stress-like granules have also been detected in cells infected with a VACV E3 deletion mutant and were called antiviral because of the associated replication block (56). However, whether antiviral granule formation in that system is a cause or a result of the translation block, which involves phosphorylation of PKR and eIF2α, remains unknown. We were therefore interested in this question in relation to the C7/K1 deletion mutant.
Mammalian stress granules form under conditions associated with disassembly of polyribosomes and contain stalled translation preinitiation complexes containing 40S ribosomes and mRNA bound to RNA binding proteins, including G3BP (57–61). The phosphorylations of PKR and eIF2α are often associated with stress granule formation. Kedersha and coworkers (49) recently showed that human U2OS cells lacking G3BP1 and G3BP2 are unable to form stress granules in response to phosphorylation of eIF2α induced by arsenite or inhibition of eIF4A but still form stress granules in response to heat or osmotic shock. We confirmed the requirement for G3BPs for arsenite-induced granules but found that RNA granules still formed in G3BP1/2 KO U2OS cells that were infected with ΔC7K1 virus. Furthermore, the G3BP1/2 KO cells remained nonpermissive for ΔC7K1, indicating that G3BP was not directly involved in the inhibition of replication.
Since SAMD9 is constitutively expressed in HeLa and other cell lines, it seems that VACV must trigger an activation step that is suppressed or counteracted by C7 or K1 binding. We showed that the triggering event does not require viral DNA replication, even though the major effect appears to be on intermediate and late protein synthesis. Although intermediate and late mRNAs are distinguished from the majority of cellular and viral early mRNAs by the presence of a 5′ poly(A) leader, the presence or absence of this feature did not affect translatability in HeLa cells infected with ΔC7K1. A more likely explanation for the lesser effect on early gene expression is that it takes time to establish the inhibitory state. In support of this idea, Meng et al. (13) reported that C7 is required for the sustained synthesis of E3, an early protein.
The possibility that SAMD9 inhibits protein synthesis following a direct interaction with mRNA instead of translation factors could explain the effect on both cap-dependent and IRES-mediated translation, the failure of the inhibition to be alleviated by KO of PKR or RNase L, and the collapse of polyribosomes. Moreover, such an interaction would be consistent with the secondary colocalization of viral mRNA and SAMD9 in stress-like granules independent of G3BP. A recent bioinformatic analysis revealed that SAMD9 has a complex architecture with domains predicted to exhibit DNA/RNA binding, Sir2-like activity, P-loop ATPase, and an OB-fold with RNA binding properties (62). To test the above-described model, it will be necessary to directly investigate mRNA binding by SAMD9 and determine the effects of site-specific mutations, particularly in the putative RNA binding site of SAMD9, on host range restriction.
MATERIALS AND METHODS
Cells and viruses.
BS-C-1 cells were grown in minimum essential medium with Earle's salts; HeLa, U2OS, and A549 cells were grown in Dulbecco’s minimum essential medium. In both cases the media were supplemented with 10% fetal bovine serum (FBS), 100 U of penicillin, and 100 μg of streptomycin per ml (Quality Biologicals, Gaithersburg, MD). HeLa SAMD9−/− cells (24) and A549 PKR/RNase L−/− cells (50) were described previously. U2OS ΔΔG3BP1/2 cells (49) were a kind gift of Nancy Kedersha and Paul Anderson (Brigham and Women's Hospital, Boston, MA). Recombinant VACV vTF7-3 (63) expressing p11-GFP was previously described (64) and used here as the wild-type (WT) virus. vTF7-3 with deletions of the C7L and K1L genes and expressing p11-GFP (15) was a gift of Yan Xiang (University of Texas, San Antonio).
CRISPR/Cas9 mutagenesis of HeLa cells.
Guide sequences for WDR6 (CACCACTGGATTTGGGATGCACGCGTTT) and FTSJ1 (CACCGCGGGATGTCTACTACCGCCGTTT) were inserted into the CAS9 plasmid pSpCas9(BB)-2A-GFP (PX458) (48138; deposited by Feng Zheng; Addgene). Plasmids were transfected into HeLa cells grown in a 6-well plate, and fluorescence-activated cell sorting was used to select for GFP+ cells, which were distributed into 96-well plates with 1 to 3 GFP+ cells/well. Individual colonies were randomly selected and tested for the ability to support the spread of ΔC7K1. Cells were grown in 6-well plates, and DNA was extracted using a Qiagen DNeasy blood and tissue kit. The DNA was then PCR amplified using primers flanking the CRISPR target. The PCR products were gel purified, cloned into pCR-Blunt II TOPO plasmid vector, and sequenced at Eurofins, using M13 F and R primers.
Expression plasmids and in vitro mRNA synthesis.
A plasmid was constructed that contained the VACV G8 intermediate promoter regulating the FLuc ORF, which was followed by the CrPV IRES and the RLuc ORF. Infected cells were transfected using Lipofectamine 2000 (Thermo Fisher).
A PCR product with a T7 promoter followed by 12 adenylate residues or the sequence GTAGTAGTAGTA preceding the FLuc ORF was used as the template for in vitro transcription using an mMessage mMachine kit by Ambion. The poly(A) tail was added to the 3′ end of the mRNA using the poly(A) tailing kit (Thermo Fisher). mRNAs were purified by lithium chloride precipitation. For expression, 750 ng of mRNA was transfected into cells using Lipofectamine 2000 (Thermo Fisher).
Antibodies.
Polyclonal antibodies specific for hSAMD9 and eIF3B were purchased from Sigma and Bethyl Laboratories, respectively. The rabbit polyclonal antibody recognizing G3BP1 was previously described (65).
Western blot analysis.
Proteins of whole-cell lysates and purified virions were separated in 4 to 12% Novex NuPAGE acrylamide gels with 2-(N-morpholino)ethanesulfonic acid buffer and were transferred to nitrocellulose membranes using the iBlot system (Invitrogen). Membranes were blocked with 5% nonfat milk in phosphate-buffered saline (PBS) with 0.05% Tween 20 and then incubated for 1 h at room temperature or overnight at 4°C in the same solution with primary antibodies at appropriate dilutions. Excess antibodies were removed by washing with PBS containing Tween 20, followed by PBS without detergent. IRDye 800- or 700-conjugated secondary antibodies against mouse and rabbit antibodies were added, and the mixture was incubated for 1 h at room temperature, washed, and developed using an Odyssey infrared imager (LI-COR Biosciences, Lincoln, NE). Images were acquired with Image Studio Software (LI-COR Biosciences, Lincoln, NE) and prepared with Photoshop.
Luciferase assays.
HeLa or SAMD9−/− cells in 24-well plates were infected with the indicated viruses for 1 h at 4°C. Cells were then placed in a 37°C incubator and transfected with plasmids encoding the luciferase gene using Lipofectamine 2000 (Thermo-Fisher). At 6 h posttransfection, cells were lysed with 100 µl of passive cell lysis buffer X1 and diluted 1:100, and luciferase activity was assayed using a Dual-Glo Luciferase assay system (Promega).
Virus infectivity and replication assays.
Plaque assays were performed in BS-C-1 cell monolayers in 12-well culture plates with duplicate 10-fold serial dilutions of virus. The virus was adsorbed for 1 h at room temperature, after which unbound virus was removed, and the cells were washed and incubated with medium containing 0.5% (wt/vol) methylcellulose. To measure virus spread, cells in 24-well plates were infected in triplicate with viruses expressing GFP at a multiplicity of 0.01 PFU/cell for 1 h at 37°C. The cells were washed, incubated with medium for 18 h at 37°C, trypsin treated, and fixed with 2% paraformaldehyde. Cells expressing GFP were detected by flow cytometry and analyzed with FlowJo software (FlowJo, Ashland, OR). P values were calculated by one-way analysis of variance and postreplicative test with Bonferroni corrections (Prism).
Metabolic labeling of nascent proteins.
HeLa cells grown in 24-well plates were infected with 3 PFU/cell of the indicated viruses for 1 h at room temperature. The inoculum was removed, and the cells were incubated with complete Dulbecco’s minimum essential medium for the indicated times. At 30 min before labeling, the cells were incubated with cysteine- and methionine-free medium at 37°C and then pulse labeled for 15 min with 100 μCi of [35S]methionine-cysteine (Perkin-Elmer). The cells were washed with cold PBS and lysed in 0.5% Nonidet P-40 containing cOmplete Protease Inhibitor Cocktail (Roche Applied Science). Proteins in the clarified cell lysate were resolved by SDS-PAGE and the gels imaged with a Typhoon imager FL7000 (GE Life Sciences).
Polysomal profile analysis.
Polysomal profiles were performed according to Johannes and Sarnow (66), with modifications. Specifically, cells were grown in T150 flasks, incubated for 5 min with 100 μg of cycloheximide (CHX), harvested, and stored at −70°C. Prior to analysis, the cells were resuspended in 0.4 ml of LBA buffer (18 mM Tris, pH 7.5, 50 mM KCl, 10 mM MgCl, 10 mM NaF, 10 mM α-glycerolphosphate, 1.4 μg/ml pepstatin, 2 μg/ml leupeptin, EDTA-free cOmplete Protease Inhibitor Cocktail [Roche], 70 μg/ml CHX, 1.25 mM dithiothreitol, and 200 μg/ml heparin), and Triton X-100 and deoxycholate were added to a final concentration of 1.2% each for lysis over a 5-min period on ice. Twenty optical density units (at 260 nm) were loaded on each sucrose gradient. Gradients were centrifuged for 100 min at 39,000 rpm (SW-41 rotor) and then resolved using a Teledyne ISCO (model UA-6). RNA absorbance was continuously monitored using 260-nM UV light, and 0.5-ml fractions were collected. For RNA extraction, 1 ml of cold ethanol was added to each fraction, which was then incubated for 12 h at −20°C and centrifuged for 15 min at 20,000 × g. RNA was extracted from each pellet with TRIzol reagent (ThermoFisher) according to the manufacturer’s instructions. Experiments were carried out twice, and a representative is shown.
mRNA quantification by ddPCR.
Total RNA was purified using TRIzol reagent (Life Technologies, Carlsbad, CA) according to the manufacturer’s protocols. A portion of each RNA sample was treated with DNase (Life Technologies) for 1 h at 37°C prior to reverse transcription with oligo(dT) and random hexamer primers using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Serial dilutions of the cDNA were added to a reaction mix containing QX200 EvaGreen supermix (Bio-Rad), and droplets were generated using an automated droplet generator (Bio-Rad). Oligonucleotides generating short amplicons within cellular 18S rRNA and viral I3, D13, and A3 genes were used for the PCR. Upon completion, droplets were analyzed in a QX200 Droplet Digital PCR system (Bio-Rad).
Confocal microscopy.
Cells grown on coverslips were infected for specified times, fixed with 4% paraformaldehyde, and permeabilized with 0.1% Triton X-100 in PBS. The samples were blocked with 5% bovine serum albumin and 10% FBS, followed by incubation with primary antibodies in 10% serum for 2 h. Cells were washed with PBS and incubated with appropriate secondary antibodies conjugated to fluorescent dyes (Molecular Probes, Eugene, OR) for an additional 1 h with DAPI. Coverslips were washed and mounted on a glass slide using ProLong Gold (Invitrogen). The slides were examined with a Leica SP5 inverted four-channel confocal microscope, and images were prepared using IMARIS software, version 8.2 (Bitplane Scientific Software, St. Paul, MN). Colocalization was determined using IMARIS on individual focal planes prior to z-stack superimposition. For RNA labeling with EU (Thermo Fisher), cells were grown on coverslips and infected with VACV. In specific experiments, cells were treated with 1 μM triptolide (Sigma) to inhibit cellular polymerase I and II transcription at 1 to 2 h before labeling with EU. Cells were pulse labeled with 1 mM EU for 1 h, washed, fixed with 4% paraformaldehyde, and reacted with Alexa Fluor 594 azide (Click-iT RNA Alexa Fluor 594 imaging kit; ThermoFisher). Cells were washed with the kit’s wash buffer and were processed further for confocal microscopy as described above.
FISH.
HeLa cells were plated on 12-mm glass circular coverslips in a 24-well dish and the next day were infected with 3 PFU/cell of ΔC7K1. At 8 h postinfection, cells were washed 3× with Dulbecco’s phosphate-buffered saline (DPBS) containing calcium and magnesium. Cells were then fixed for 10 min in 3.7% formaldehyde in 1× DPBS. Fixed cells were then washed 4× in DPBS and placed in 70% ethanol overnight. Ethanol was removed and cells were washed 4× at 5-min intervals with wash buffer containing 10% formamide in 2× SSC buffer (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate). Fluorescently labeled VACV D13 antisense deoxyoligonucleotides (Bioresearch Technologies, Petaluma, CA) were resuspended in TE (10 mM Tris-HCl, 1 mM sodium EDTA, pH 8.0) to give a final solution of 25 μM. The probe was diluted 1:100 with hybridization buffer (100 mg/ml dextran sulfate, 10% formamide in 2× SSC), put on cells, and incubated in a humidified incubator at 37˚C overnight. Cells were washed 4× at 15-min intervals with wash buffer at 37˚C. Cells were then incubated for 3 h at 37˚C with rabbit polyclonal antibody to G3BP1 (65) diluted 1:100 in 2× SSC, 8% formamide, 2 mM vanadyl-ribonucleoside complex, and 0.2% bovine serum albumin. Cells were washed 3× at 10-min intervals with wash buffer at 37˚C. Secondary antibody diluted 1:100 in the same buffer as the primary antibody (Thermo Fisher Scientific) was added for 3 h at 37˚C. Cells were washed as described above and incubated with 5 ng/ml DAPI in wash buffer for 30 min at 37˚C. After washing, 3× coverslips were mounted on a glass slide with ProLong gold (Thermo Fisher Scientific). Analysis was done using a Leica SP5 confocal microscope.
ACKNOWLEDGMENTS
We thank Catherine Cotter for maintaining cells. The C7/K1 KO virus was kindly provided by Yan Xiang and the U2OS ΔΔG3BP1/2 cells by Nancy Kedersha and Paul Anderson. The Division of Intramural Research, NIAID, NIH, supported the study.
REFERENCES
- 1.Upton C, Slack S, Hunter AL, Ehlers A, Roper RL. 2003. Poxvirus orthologous clusters: toward defining the minimum essential poxvirus genome. J Virol 77:7590–7600. doi: 10.1128/JVI.77.13.7590-7600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moss B. 2013. Poxviridae, p 2129–2159. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, vol 2 Lippincott Williams & Wilkins, Pennsylvania, PA. [Google Scholar]
- 3.Smith GL, Benfield CTO, de Motes CM, Mazzon M, Ember SWJ, Ferguson BJ, Sumner RP. 2013. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J Gen Virol 94:2367–2392. doi: 10.1099/vir.0.055921-0. [DOI] [PubMed] [Google Scholar]
- 4.Haller SL, Peng C, McFadden G, Rothenburg S. 2014. Poxviruses and the evolution of host range and virulence. Infect Genet Evol 21:15–40. doi: 10.1016/j.meegid.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bratke KA, McLysaght A, Rothenburg S. 2013. A survey of host range genes in poxvirus genomes. Infect Genet Evol 14:406–425. doi: 10.1016/j.meegid.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drillien R, Koehren F, Kirn A. 1981. Host range deletion mutant of vaccinia virus defective in human cells. Virology 111:488–499. doi: 10.1016/0042-6822(81)90351-2. [DOI] [PubMed] [Google Scholar]
- 7.Gillard S, Spehner D, Drillien R. 1985. Mapping of a vaccinia virus host range sequence by insertion into the viral thymidine kinase gene. J Virol 53:316–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gillard S, Spehner D, Drillien R, Kirn A. 1986. Localization and sequence of a vaccinia virus gene required for multiplication in human cells. Proc Natl Acad Sci U S A 83:5573–5577. doi: 10.1073/pnas.83.15.5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perkus ME, Goebel SJ, Davis SW, Johnson GP, Limbach K, Norton EK, Paoletti E. 1990. Vaccinia virus host range genes. Virology 179:276–286. doi: 10.1016/0042-6822(90)90296-4. [DOI] [PubMed] [Google Scholar]
- 10.Oguiura N, Spehner D, Drillien R. 1993. Detection of a protein encoded by the vaccinia virus C7L open reading frame and study of its effect on virus multiplication in different cell lines. J Gen Virol 74:1409–1413. doi: 10.1099/0022-1317-74-7-1409. [DOI] [PubMed] [Google Scholar]
- 11.Ramsey-Ewing AL, Moss B. 1996. Complementation of a vaccinia virus host range K1L gene deletion by the non-homologous CP77 gene. Virology 222:75–86. doi: 10.1006/viro.1996.0399. [DOI] [PubMed] [Google Scholar]
- 12.Hsiao JC, Chung CS, Drillien R, Chang W. 2004. The cowpox virus host range gene, CP77, affects phosphorylation of eIF2 alpha and vaccinia viral translation in apoptotic HeLa cells. Virology 329:199–212. doi: 10.1016/j.virol.2004.07.032. [DOI] [PubMed] [Google Scholar]
- 13.Meng XZ, Chao J, Xiang Y. 2008. Identification from diverse mammalian poxviruses of host-range regulatory genes functioning equivalently to vaccinia virus C7L. Virology 372:372–383. doi: 10.1016/j.virol.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willis KL, Patel S, Xiang Y, Shisler JL. 2009. The effect of the vaccinia K1 protein on the PKR-eIF2 alpha pathway in RK13 and HeLa cells. Virology 394:73–81. doi: 10.1016/j.virol.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meng X, Jiang C, Arsenio J, Dick K, Cao J, Xiang Y. 2009. Vaccinia virus K1L and C7L inhibit antiviral activities induced by type I interferons. J Virol 83:10627–10636. doi: 10.1128/JVI.01260-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Backes S, Sperling KM, Zwilling J, Gasteiger G, Ludwig H, Kremmer E, Schwantes A, Staib C, Sutter G. 2010. Viral host-range factor C7 or K1 is essential for modified vaccinia virus Ankara late gene expression in human and murine cells, irrespective of their capacity to inhibit protein kinase R-mediated phosphorylation of eukaryotic translation initiation factor 2 alpha. J Gen Virol 91:470–482. doi: 10.1099/vir.0.015347-0. [DOI] [PubMed] [Google Scholar]
- 17.Najera JL, Gomez CE, Domingo-Gil E, Gherardi MM, Esteban M. 2006. Cellular and biochemical differences between two attenuated poxvirus vaccine candidates (MVA and NYVAC) and role of the C7L gene. J Virol 80:6033–6047. doi: 10.1128/JVI.02108-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shisler JL, Jin XL. 2004. The vaccinia virus K1L gene product inhibits host NF-kB activation by preventing IkBa degradation. J Virol 78:3553–3560. doi: 10.1128/JVI.78.7.3553-3560.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bradley RR, Terajima M. 2005. Vaccinia virus K1L protein mediates host-range function in RK-13 cells via ankyrin repeat and may interact with a cellular GTPase-activating protein. Virus Res 114:104–112. doi: 10.1016/j.virusres.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 20.Meng XZ, Xiang Y. 2006. Vaccinia virus K1L protein supports viral replication in human and rabbit cells through a cell-type-specific set of its ankyrin repeat residues that are distinct from its binding site for ACAP2. Virology 353:220–233. doi: 10.1016/j.virol.2006.05.032. [DOI] [PubMed] [Google Scholar]
- 21.Meng XZ, Schoggins J, Rose L, Cao JX, Ploss A, Rice CM, Xiang Y. 2012. C7L family of poxvirus host range genes inhibits antiviral activities induced by type I interferons and interferon regulatory factor 1. J Virol 86:4538–4547. doi: 10.1128/JVI.06140-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu J, Wennier S, Zhang LL, McFadden G. 2011. M062 Is a host range factor essential for myxoma virus pathogenesis and functions as an antagonist of host SAMD9 in human cells. J Virol 85:3270–3282. doi: 10.1128/JVI.02243-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J, McFadden G. 2015. SAMD9 is an innate antiviral host factor with stress response properties that can be antagonized by poxviruses. J Virol 89:1925–1931. doi: 10.1128/JVI.02262-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sivan G, Ormanoglu P, Buehler EC, Martin SE, Moss B. 2015. Identification of restriction factors by human genome-wide RNA interference screening of viral host range mutants exemplified by discovery of SAMD9 and WDR6 as inhibitors of the vaccinia virus K1L-C7L-mutant. mBio 6:e01122. doi: 10.1128/mBio.01122-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meng XZ, Krumm B, Li YC, Deng JP, Xiang Y. 2015. Structural basis for antagonizing a host restriction factor by C7 family of poxvirus host-range proteins. Proc Natl Acad Sci U S A 112:14858–14863. doi: 10.1073/pnas.1515354112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nounamo B, Li Y, O’Byrne P, Kearney AM, Khan A, Liu J. 2017. An interaction domain in human SAMD9 is essential for myxoma virus host-range determinant M062 antagonism of host anti-viral function. Virology 503:94–102. doi: 10.1016/j.virol.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meng XZ, Zhang FS, Yan B, Si CP, Honda H, Nagamachi A, Sun LZ, Xiang Y. 2018. A paralogous pair of mammalian host restriction factors form a critical host barrier against poxvirus infection. PLoS Pathog 14:e1006884. doi: 10.1371/journal.ppat.1006884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lemos de Matos A, Liu J, McFadden G, Esteves PJ. 2013. Evolution and divergence of the mammalian SAMD9/SAMD9L gene family. BMC Evol Biol 13:121. doi: 10.1186/1471-2148-13-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Topaz O, Indelman M, Chefetz I, Geiger D, Metzker A, Altschuler Y, Choder M, Bercovich D, Uitto J, Bergman R, Richard G, Sprecher E. 2006. A deleterious mutation in SAMD9 causes normophosphatemic familial tumoral calcinosis. Am J Hum Genet 79:759–764. doi: 10.1086/508069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chefetz I, Ben Amitai D, Browning S, Skorecki K, Adir N, Thomas MG, Kogleck L, Topaz O, Indelman M, Uitto J, Richard G, Bradman N, Sprecher E. 2008. Normophosphatemic familial tumoral calcinosis is caused by deleterious mutations in SAMD9, encoding a TNF-alpha responsive protein. J Investig Dermatol 128:1423–1429. doi: 10.1038/sj.jid.5701203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Narumi S, Amano N, Ishii T, Katsumata N, Muroya K, Adachi M, Toyoshima K, Tanaka Y, Fukuzawa R, Miyako K, Kinjo S, Ohga S, Ihara K, Inoue H, Kinjo T, Hara T, Kohno M, Yamada S, Urano H, Kitagawa Y, Tsugawa K, Higa A, Miyawaki M, Okutani T, Kizaki Z, Hamada H, Kihara M, Shiga K, Yamaguchi T, Kenmochi M, Kitajima H, Fukami M, Shimizu A, Kudoh J, Shibata S, Okano H, Miyake N, Matsumoto N, Hasegawa T. 2016. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet 48:792–797. doi: 10.1038/ng.3569. [DOI] [PubMed] [Google Scholar]
- 32.Nagamachi A, Matsui H, Asou H, Ozaki Y, Aki D, Kanai A, Takubo K, Suda T, Nakamura T, Wolff L, Honda H, Inaba T. 2013. Haploinsufficiency of SAMD9L, an endosome fusion facilitator, causes myeloid malignancies in mice mimicking human diseases with monosomy 7. Cancer Cell 24:305–317. doi: 10.1016/j.ccr.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 33.Hershkovitz D, Gross Y, Nahum S, Yehezkel S, Sarig O, Uitto J, Sprecher E. 2011. Functional characterization of SAMD9, a protein deficient in normophosphatemic familial tumoral calcinosis. J InvestDermatol 131:662–669. doi: 10.1038/jid.2010.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei CM, Moss B. 1975. Methylated nucleotides block 5′-terminus of vaccinia virus mRNA. Proc Natl Acad Sci U S A 72:318–322. doi: 10.1073/pnas.72.1.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilson JE, Pestova TV, Hellen CU, Sarnow P. 2000. Initiation of protein synthesis from the A site of the ribosome. Cell 102:511–520. doi: 10.1016/S0092-8674(00)00055-6. [DOI] [PubMed] [Google Scholar]
- 36.Sasaki J, Nakashima N. 2000. Methionine-independent initiation of translation in the capsid protein of an insect RNA virus. Proc Natl Acad Sci U S A 97:1512–1515. doi: 10.1073/pnas.010426997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vos JC, Stunnenberg HG. 1988. Derepression of a novel class of vaccinia virus genes upon DNA replication. EMBO J 7:3487–3492. doi: 10.1002/j.1460-2075.1988.tb03224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baldick CJ, Keck JG, Moss B. 1992. Mutational analysis of the core, spacer and initiator regions of vaccinia virus intermediate class promoters. J Virol 66:4710–4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parrish S, Resch W, Moss B. 2007. Vaccinia virus D10 protein has mRNA decapping activity, providing a mechanism for control of host and viral gene expression. Proc Natl Acad Sci U S A 104:2139–2144. doi: 10.1073/pnas.0611685104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolf SF, Schlessinger D. 1977. Nuclear metabolism of ribosomal RNA in growing, methionine-limited, and ethionine-treated HeLa cells. Biochemistry 16:2783–2791. doi: 10.1021/bi00631a031. [DOI] [PubMed] [Google Scholar]
- 41.Yang Z, Reynolds SE, Martens CA, Bruno DP, Porcella SF, Moss B. 2011. Expression profiling of the intermediate and late stages of poxvirus replication. J Virol 85:9899–9908. doi: 10.1128/JVI.05446-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Z, Maruri-Avidal L, Sisler J, Stuart C, Moss B. 2013. Cascade regulation of vaccinia virus gene expression is modulated by multistage promoters. Virology 447:213–220. doi: 10.1016/j.virol.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dhungel P, Cao S, Yang Z. 2017. The 5′-poly(A) leader of poxvirus mRNA confers a translational advantage that can be achieved in cells with impaired cap-dependent translation. PLoS Pathog 13:e1006602. doi: 10.1371/journal.ppat.1006602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Becker Y, Joklik WK. 1964. Messenger RNA in cells infected with vaccinia virus. Proc Natl Acad Sci U S A 51:577–584. doi: 10.1073/pnas.51.4.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jao CY, Salic A. 2008. Exploring RNA transcription and turnover in vivo by using click chemistry. Proc Natl Acad Sci U S A 105:15779–15784. doi: 10.1073/pnas.0808480105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pan J. 2010. RNA polymerase–an important molecular target of triptolide in cancer cells. Cancer Lett 292:149–152. doi: 10.1016/j.canlet.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 47.Vispe S, DeVries L, Creancier L, Besse J, Breand S, Hobson DJ, Svejstrup JQ, Annereau JP, Cussac D, Dumontet C, Guilbaud N, Barret JM, Bailly C. 2009. Triptolide is an inhibitor of RNA polymerase I and II-dependent transcription leading predominantly to down-regulation of short-lived mRNA. Mol Cancer Ther 8:2780–2790. doi: 10.1158/1535-7163.MCT-09-0549. [DOI] [PubMed] [Google Scholar]
- 48.Titov DV, Gilman B, He QL, Bhat S, Low WK, Dang Y, Smeaton M, Demain AL, Miller PS, Kugel JF, Goodrich JA, Liu JO. 2011. XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat Chem Biol 7:182–188. doi: 10.1038/nchembio.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kedersha N, Panas MD, Achorn CA, Lyons S, Tisdale S, Hickman T, Thomas M, Lieberman J, McInerney GM, Ivanov P, Anderson P. 2016. G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J Cell Physiol 212:845–860. doi: 10.1083/jcb.201508028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu R, Moss B. 2016. Opposing roles of double-stranded RNA effector pathways and viral defense proteins revealed with CRISPR/Cas9 knock-out cell lines and vaccinia virus mutants. J Virol 90:7864–7879. doi: 10.1128/JVI.00869-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xie X, Wang Z, Chen Y. 2007. Association of LKB1 with a WD-repeat protein WDR6 is implicated in cell growth arrest and p27(Kip1) induction. Mol Cell Biochem 301:115–122. doi: 10.1007/s11010-006-9402-5. [DOI] [PubMed] [Google Scholar]
- 52.Guy MP, Shaw M, Weiner CL, Hobson L, Stark Z, Rose K, Kalscheuer VM, Gecz J, Phizicky EM. 2015. Defects in tRNA anticodon loop 2′-O-methylation are implicated in nonsyndromic X-linked intellectual disability due to mutations in FTSJ1. Hum Mutat 36:1176–1187. doi: 10.1002/humu.22897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wan C, Borgeson B, Phanse S, Tu F, Drew K, Clark G, Xiong X, Kagan O, Kwan J, Bezginov A, Chessman K, Pal S, Cromar G, Papoulas O, Ni Z, Boutz DR, Stoilova S, Havugimana PC, Guo X, Malty RH, Sarov M, Greenblatt J, Babu M, Derry WB, Tillier ER, Wallingford JB, Parkinson J, Marcotte EM, Emili A. 2015. Panorama of ancient metazoan macromolecular complexes. Nature 525:339–344. doi: 10.1038/nature14877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, Hyman AA, Mann M. 2015. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163:712–723. doi: 10.1016/j.cell.2015.09.053. [DOI] [PubMed] [Google Scholar]
- 55.Brostrom CO, Prostko CR, Kaufman RJ, Brostrom MA. 1996. Inhibition of translational initiation by activators of the glucose-regulated stress protein and heat shock protein stress response systems. Role of the interferon-inducible double-stranded RNA-activated eukaryotic initiation factor 2alpha kinase. J Biol Chem 271:24995–25002. doi: 10.1074/jbc.271.40.24995. [DOI] [PubMed] [Google Scholar]
- 56.Simpson-Holley M, Kedersha N, Dower K, Rubins KH, Anderson P, Hensley LE, Connor JH. 2011. Formation of antiviral cytoplasmic granules during orthopoxvirus infection. J Virol 85:1581–1593. doi: 10.1128/JVI.02247-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kedersha NL, Gupta M, Li W, Miller I, Anderson P. 1999. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Physiol 147:1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bordeleau ME, Matthews J, Wojnar JM, Lindqvist L, Novac O, Jankowsky E, Sonenberg N, Northcote P, Teesdale-Spittle P, Pelletier J. 2005. Stimulation of mammalian translation initiation factor eIF4A activity by a small molecule inhibitor of eukaryotic translation. Proc Natl Acad Sci U S A 102:10460–10465. doi: 10.1073/pnas.0504249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dang Y, Kedersha N, Low WK, Romo D, Gorospe M, Kaufman R, Anderson P, Liu JO. 2006. Eukaryotic initiation factor 2alpha-independent pathway of stress granule induction by the natural product pateamine A. J Biol Chem 281:32870–32878. doi: 10.1074/jbc.M606149200. [DOI] [PubMed] [Google Scholar]
- 60.Bounedjah O, Desforges B, Wu T-D, Pioche-Durieu C, Marco S, Hamon L, Curmi PA, Guerquin-Kern J-L, Piétrement O, Pastré D. 2014. Free mRNA in excess upon polysome dissociation is a scaffold for protein multimerization to form stress granules. Nucleic Acids Res 42:8678–8691. doi: 10.1093/nar/gku582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anderson P, Kedersha N. 2006. RNA granules. J Cell Physiol 172:803–838. doi: 10.1083/jcb.200512082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mekhedov SL, Makarova KS, Koonin EV. 2017. The complex domain architecture of SAMD9 family proteins, predicted STAND-like NTPases, suggests new links to inflammation and apoptosis. Biol Direct 12:13. doi: 10.1186/s13062-017-0185-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fuerst TR, Niles EG, Studier FW, Moss B. 1986. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci U S A 83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bengali Z, Townsley AC, Moss B. 2009. Vaccinia virus strain differences in cell attachment and entry. Virology 389:132–140. doi: 10.1016/j.virol.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Katsafanas GC, Moss B. 2004. Vaccinia virus intermediate stage transcription is complemented by Ras-GTPase-activating protein SH3 domain-binding protein (G3BP) and cytoplasmic activation/proliferation-associated protein (p137) individually or as a heterodimer. J Biol Chem 279:52210–55217. doi: 10.1074/jbc.M411033200. [DOI] [PubMed] [Google Scholar]
- 66.Johannes G, Sarnow P. 1998. Cap-independent polysomal association of natural mRNAs encoding c-myc, BiP, and eIF4G conferred by internal ribosome entry sites. RNA 4:1500–1513. doi: 10.1017/S1355838298981080. [DOI] [PMC free article] [PubMed] [Google Scholar]