

Abstract
Metazoan replication-dependent histone mRNAs are the only known cellular mRNAs that are not polyadenylated. Histone mRNAs are present in large amounts only in S-phase cells, and their levels are coordinately regulated with the rate of DNA replication. In mammals, the stemloop at the 3′ end of histone mRNA is bound to stemloop binding protein, a protein required for both synthesis and degradation of histone mRNA, and an exonuclease, 3′hExo (ERI1). Histone mRNAs are rapidly degraded when DNA synthesis is inhibited in S-phase cells and at the end of S-phase. Upf1 is also required for rapid degradation of histone mRNA as is the S-phase checkpoint. We report that Smg1 is required for histone mRNA degradation when DNA replication is inhibited, suggesting it is the PI-like kinase that activates Upf1 for histone mRNA degradation. We also show that some mutant Upf1 proteins are recruited to histone mRNAs when DNA replication is inhibited and act as dominant negative factors in histone mRNA degradation. We report that the pathway of rapid histone mRNA degradation when DNA replication is inhibited in S-phase cells that are activating the S-phase checkpoint is similar to the pathway of rapid degradation of histone mRNA at the end of S-phase.
This article is part of the theme issue ‘5′ and 3′ modifications controlling RNA degradation’.
Keywords: histone mRNA, uridylation, RNA degradation, cell cycle
1. Introduction
Animal replication-dependent histone mRNAs are the only cellular mRNAs that are not polyadenylated, ending instead in a conserved stemloop sequence. Histone genes do not contain introns, and the only processing reaction necessary for histone mRNA formation is cleavage to form the 3′ end. The stemloop at the 3′ end of histone mRNA binds to the stemloop binding protein (SLBP), which is necessary for histone pre-mRNA processing. SLBP remains bound to histone mRNA as part of the cytoplasmic histone mRNP and is essential for translation. Histone mRNAs are tightly regulated during the cell cycle and are present in large amounts only in S-phase. In mammalian cells, the biosynthesis of histone mRNAs increases dramatically as cells enter S-phase, primarily owing to the large increase of SLBP, resulting in the activation of histone pre-mRNA processing [1]. At the end of S-phase, histone mRNAs are rapidly degraded, returning to the low levels present in G1 cells by mitosis [2]. Thus, most of the regulation of histone mRNA levels is posttranscriptional and is mediated through the stemloop at the 3′ end of histone mRNA.
The histone mRNA formed in the nucleus is cleaved 5 nts after the stemloop [3], in a reaction that requires holo-U7 snRNP, which contains a subset of polyadenylation factors, including CPSF73, which catalyses the cleavage of pre-mRNA [4]. This is the only processing reaction, other than co-transcriptional capping, required for histone mRNA maturation. However, the cytoplasmic histone mRNA has only 3 nts after the stemloop as a result of the removal of 2 nts by 3′hExo, a 3′–5′ exonuclease that binds to the stemloop together with SLBP [5,6]. The cytoplasmic histone mRNA is bound to both SLBP and 3′hExo (figure 1a). In the presence of SLBP, 3′hExo is blocked from continuing the 3′–5′ degradation of the histone mRNA.
Figure 1.

Cytoplasmic histone mRNAs. (a) A schematic of a mammalian histone mRNA showing the cap on the 5′ end, the short 5′ and 3′ UTRs and the stemloop at the 3′ end, which is bound by SLBP and 3′hExo. (b) Histone mRNA regulation during the cell cycle. Histone mRNAs increase just before entry into S-phase and decrease at the end of S-phase [2]. The levels of SLBP protein are also cell cycle regulated [1], while there is very little change in SLBP mRNA levels. Inhibition of DNA replication during S-phase (dotted line) results in rapid degradation of histone mRNA but not of SLBP. (c) The 3′ ends of the HIST2H2AA3 mRNAs from HCT116 S-phase cells, determined by high-throughput sequencing using the End-Seq approach [7]. The x-axis displays the nts at the 3′ end of the mRNA, with numbers referring to the drawing of the stemloop. The bars indicate the number of RNAs (y-axis) that have their last templated nt at that position. Blue indicates the RNA ends with the last templated nt, green indicates there was a single U added at that site, orange indicates there were two Us added at that site, and red indicates that there were more than 2 nts added at that site.
Histone mRNAs are present in high concentrations only in S-phase and are rapidly degraded at the end of S-phase (figure 1b). SLBP, which is essential for both histone pre-mRNA processing and translation of histone mRNA, is also cell cycle regulated [1]. As a result, the 3′ end of histone mRNA is a major cis-element regulating both histone mRNA synthesis and degradation during the cell cycle. Inhibition of DNA replication in S-phase cells results in rapid degradation of histone mRNA [8] (figure 1b). However, SLBP is not degraded when DNA replication is inhibited in S-phase but is only degraded at the end of S-phase [9].
In all mammalian histone mRNAs (approximately 50 different genes for the 5 histone proteins (10–15 copies of each core histone gene and 5 histone H1 genes), the conserved 26 nt region containing the stemloop (figure 1c) starts 24–75 nts after the stop codon [10]. A critical step in histone mRNA regulation is the rapid degradation of the mRNA when DNA synthesis is inhibited in S-phase or at the end of S-phase. The position of the stemloop is a critical parameter for regulation of histone mRNA degradation. It must be far enough from the stop codon to allow SLBP to remain bound during translation; if it is closer than 20 nts from the stop codon, then the histone mRNAs do not accumulate and are rapidly degraded [11]. It also must be close enough to the stop codon to allow communication between the stem loop and terminating ribosome to activate histone mRNA decay. If the stemloop is moved 200–300 nts after the stop codon, histone mRNAs are no longer degraded when DNA replication is inhibited [11,12]. We directly demonstrated that translation of histone mRNA is essential for degradation by using histone mRNAs with an IRE in the 5′ UTR to regulate the translation of the mRNA [11].
Supporting the idea that degradation of histone mRNA is tightly coupled to translation was the finding that Upf1 is required for histone mRNA degradation [13]. Upf1, which functions in nonsense-mediated decay (NMD) [14], as well as staufen-mediated decay [15], interacts with the terminating ribosome when termination is inefficient. In NMD, Upf1 is recruited to the stop codon, is activated by phosphorylation by Smg1, and subsequently recruits the NMD cofactors Upf2, Upf3 and SMG5, 6 and 7. The requirement for Upf1, as well as the necessity for the stop codon to be close to the stemloop, led us to propose that termination of histone mRNA translation is impaired as part of the signal that activates histone mRNA degradation [13].
In the 1980s, Jeff Ross and coworkers showed that histone mRNA degradation was likely initiated 3′–5′ [16,17] and identified a histone-specific exonuclease activity associated with polyribosomes that might be responsible for the initial steps in histone mRNA degradation [18,19]. This activity was almost certainly the 3′–5′ exonuclease, 3′hExo, which we cloned as a protein that specifically bound the stem-loop RNA [20]. In vitro in the presence of SLBP, 3′hExo can remove 2–3 nts from the 3′ end of histone mRNA, but further 3′–5′ degradation is blocked by SLBP [6]. Although our extensive attempts to affect histone mRNA degradation by RNAi knockdown of 3′hExo did not alter the rapid degradation of histone mRNA [20,21], when the 3′hExo gene was knocked out in mice, histone mRNAs were stabilized and were no longer rapidly degraded when DNA replication was inhibited [22].
Using RNA interference against known factors involved in mRNA degradation, we found that the largest effects on histone mRNA degradation resulted from knockdown of the exosome, Lsm1 or Upf1 [21]. Knockdown of the decapping enzyme, Dcp2, or the 5′–3′ exonuclease, Xrn1, had a smaller effect on degradation [21], and knockdown of Dis3L2 had no detectable effect [23].
In our initial attempts to determine the intermediates in histone mRNA degradation, we used circularization of putative degradation intermediates. We found that individual histone mRNA molecules could be simultaneously degraded 5′–3′ and 3′–5′. We cannot determine the relative importance of the 5′–3′ and 3′–5′ degradation from our studies, and it is very possible that most molecules undergo both 5′–3′ and 3′–5′ degradation. We also found that some histone mRNAs were oligouridylated in the stemloop [21]. Since we were not able to identify enough degradation intermediates to deduce the likely pathway of histone mRNA degradation, we subsequently developed a high-throughput sequencing strategy to identify the 3′ ends of histone mRNAs and their degradation intermediates, which also allowed us to identify any non-templated nts added to the degradation intermediates [23].
Two of the major questions that remain are: (1) How does Upf1 function to initiate histone mRNA degradation? (2) Since the S-phase checkpoint signal, which is involved in triggering histone mRNA degradation in S-phase cells when DNA synthesis is inhibited, is not activated at the end of S-phase, is the pathway of histone mRNA degradation similar at the end of S-phase? Here, we present additional evidence that the initial step in degradation is likely recruitment of Upf1 to the histone mRNP as a result of inefficient translation termination, followed by the activation of Upf1 by Smg1. We also show that the same degradation pathway is activated at the end of S-phase, as when DNA replication is inhibited in mid-S-phase, based on the similar pattern of uridylation and degradation intermediates that accumulate as cells exit S-phase.
2. Material and methods
(a). Plasmid construction, cell lines and antibodies
All primers used in this study are available upon request. The SLBP coding region was amplified by polymerase chain reaction (PCR) using primers that added a NotI site at its N-terminus and an AscI site at its C-terminus, and the digested amplicon was ligated into the pGLUE-C vector [24] (Ben Major, UNC) that had been digested with the same enzymes. pGLUE-SLBP was then stably transfected into HeLa cells to produce the SLBP–streptavidin-binding peptide (SBP) cell line. pcDNA3-HA-UPF1, pcDNA3-UPF1 (K498A) and pcDNA3-UPF1 (R843C) plasmids [6] were stably transfected into HeLa cells to form the corresponding cell lines. All HA-UPF1 truncation plasmids were constructed by amplifying the regions of interest from pcDNA3-HA-UPF1 with primers that added a HindIII site at the 5′ end and XbaI site at the 3′ end of the amplicon. The digested amplicons were ligated into pCDNA3-HA-UPF1 that had been digested with the same enzymes, which removed the entire UPF1 coding region, and the final plasmids were either transiently or stably transfected into HeLa cells or the SLBP–SBP cell line.
The anti-SLBP antibody has been described [1]; the anti-UPF1 antibody was kindly provided by L. Maquat, and the anti-Smg1 antibody was from Abcam.
(b). RNA interference
The SMG1 siRNA was obtained from Thermo-Scientific (Pennsylvania, USA) and was transfected into HeLa cells using Lipofectamine 2000 (Invitrogen; New York, USA). The siRNAs against Upf1 have been previously described [6].
(c). Protein binding assays
For all transient transfection experiments, approximately 1 × 106 SLBP–SBP cells were transfected with the indicated plasmid for 72 h. On the day of analysis, fresh media was placed on the cells for 5 h to promote active growth so that as many cells as possible would be in S-phase. The cells were treated with 5 mM hydroxyurea (HU) for 20 min, lysed using a hypotonic buffer and homogenized with a Dounce homogenizer. The lysates were cleared by centrifugation and used in binding assays. For stable cell lines, the cells were synchronized by double thymidine block and then released into S-phase for 3 h before being treated with 5 mM HU for 20 min. Proteins were isolated as above. For all assays, equal amounts of lysate (between 3 and 9 mg), depending on the expression of the Upf1 protein, were rotated overnight at 4°C with 30 µl of Streptavidin-Magnetic Sepharose beads (GE Healthcare; New Jersey, USA). The following day, the beads were washed with hypotonic buffer, boiled in 1× SDS-PAGE buffer, separated in an SDS-PAGE gel and transferred to PVDF membrane. The membranes were then subjected to immunoblot analysis using the appropriate antibodies.
(d). Histone mRNA decay analysis
For all decay assays, HeLa cells were plated so that they were at approximately 50% confluency on the day of HU treatment. For stable cell lines, the cells were synchronized by double thymidine block and then released into S-phase for 3 h before HU treatment. Otherwise, cells were fed fresh media for 3 h to ensure active growth before HU treatment. On the day of HU treatment, a final concentration of 5 mM HU was added to the plates, and RNA was isolated. One plate was not treated with HU and served as the t = 0 sample. Equal amounts of RNA (5 µg) from each time point were loaded onto a 6% acrylamide/8M urea-polyacrylamide gel, separated, transferred to nylon membrane and submitted to Northern analysis using probes specific for the H2a and 7SK RNAs [17]. Each experiment was repeated at least three times and quantified using a PhosphorImager.
(e). Cell cycle analysis
HCT116 cells were synchronized by double thymidine block using 12 h for the first block followed by a 9-h release and a second 12-h block, which we determined was optimal for this cell line. For cell cycle analysis, cells were labelled with 100 µM EdU for 30 min prior to collection. Cells were treated with ALEXA FLUOR 647 Azide to detect the EdU, stained with DAPI and analysed by flow cytometry in the UNC Flow Cytometry Facility.
(f). High-throughput sequencing analysis
Total cell RNA was prepared and treated with DNase I to remove any contaminating DNA. Libraries were prepared exactly as described [7]. The libraries were sequenced on an Illumina MiSeq and about 1 million reads were obtained for each library. Mapping of the data to the genome and subsequent analysis of the data were done as described [7]. At least two biological replicates of each sequencing experiment were done.
3. Results
The substrate for the initiation of histone mRNA is the translating histone mRNP associated with ribosomes. This mRNP contains the histone mRNA with the stemloop bound to SLBP and 3′hExo (figure 1a). To initiate histone mRNA degradation, we treat cells with DNA synthesis inhibitors to block DNA replication in S-phase cells, which contain the bulk of the histone mRNA. In RNAi experiments, we use exponentially growing cells 3 days after initiating RNAi treatment. For other experiments, we use either HeLa cells or HTC116 cells synchronized by double thymidine block and inhibit DNA synthesis 3 h after release into S-phase to activate histone mRNA degradation (figure 1b).
(a). The requirement of Upf1 and Smg1 for histone mRNA degradation
The role of Upf1 in histone mRNA degradation is almost certainly to bind to the terminating ribosome, as it does in NMD, as a result of translation termination becoming inefficient for an unknown reason. Upf1 is capable of binding to the 3′ UTR of histone mRNAs, as shown by the study of Muhlemann and coworkers who used iCLIP to compare binding of Upf1 to mRNAs that are being translated and binding of Upf1 to mRNAs when the mRNAs are released from polyribosomes by puromycin [25]. Upf1 was bound in low levels to the actively translated histone mRNA before it was released from polyribosomes. After releasing the mRNA from polyribosomes, Upf1 binding was substantially increased at the 3′ UTR of all the replication-dependent histone mRNAs, with the binding extending from the stop codon to the start of the stem loop [26]. Since the histone mRNA released from polyribosomes contains SLBP at the 3′ end, this result is consistent with SLBP being able to promote binding of Upf1 to the 3′ UTR of histone mRNA, after it is recruited to the terminating ribosome.
Treatment of cells with inhibitors of DNA replication results in activation of ATR and ATM, PI-like kinases that are activated as a result of DNA damage that occurs as a result of inhibiting DNA replication in S-phase cells [27]. These kinases then phosphorylate targets that result in the arrest of cells in S-phase, activating the S-phase checkpoint. These kinases can also phosphorylate Upf1 [28]. In addition to being present in the cytoplasm where NMD occurs, Upf1 is also present in the nucleus at replication forks and plays an unknown role in the nucleus [28,29]. In NMD, Upf1 is activated by phosphorylation by Smg1 [30]. To determine which of these PI kinases is critical for activation of Upf1 during histone mRNA degradation, we examined whether Smg1 is required for histone mRNA degradation and determined which domains of Upf1 are required.
We constructed an SLBP transgene in the pGLUE-C vector that adds a CBP (calmodulin binding peptide), SBP (Streptavidin-binding peptide) and HA tag at the C-terminus of the protein [24] (figure 2a) and created a stable cell line expressing the tagged SLBP–SBP, which could be detected with either antibodies to SLBP, SBP or the HA tag. Using RNAi to knockdown Smg1, we observed a similar reduction in the rate of histone mRNA degradation as when knocking down Upf1 (figure 2b). Knockdown of ATR, another member of the PI kinase family also required for histone mRNA degradation, did not affect the levels of Smg1 (figure 2b). Using the SLBP–SBP-expressing cell line, we showed that Upf1 associates with SLBP–SBP only after treatment with HU (figure 2c(i)). Smg1 also associates with SLBP–SBP only after HU treatment, consistent with a Upf1/Smg1 complex being formed on the 3′ end of the histone mRNA to activate Upf1 (figure 2c(ii)). As a control for these experiments, we tested whether the anti-HA antibody used to precipitate the SLBP–SBP precipitated any Smg1 from HeLa cells not expressing this protein. The amount of Smg1 recruited was clearly over background in multiple experiments, compared to the controls, although the recruitment of Smg1 was not as dramatic as the recruitment of Upf1. Note that these experiments were done by analysing the endogenous Upf1 and Smg1 proteins.
Figure 2.

Upf1/Smg1 are required for histone mRNA degradation. (a) Diagram of the C-terminal-tagged SLBP containing a CBP, a streptavidin-binding peptide and an HA tag (i). A stable HeLa cell line expressing the SLBP–SBP was generated, and SLBP and SLBP–SBP were detected by western blotting with the SLBP antibody (ii). (b) Knockdown of Smg1 or Upf1 slows the degradation of histone mRNA after inhibiting DNA replication. Cells treated with the indicated siRNAs were treated with HU for the indicated times, and the levels of histone mRNA were determined by Northern blotting and quantified with a PhosphorImager. The western blots below the graph show the effectiveness of the knockdown. (c) Lysates of synchronized SLBP–SBP-expressing cells were prepared 3 h after release into S-phase, before and 20 min after treating the cells with 5 mM HU. (i) The lysates were bound to streptavidin beads, and bound proteins were resolved by SDS gel electrophoresis and detected with the anti-HA antibody (bottom) or the anti-Upf1 antibody (top). (ii) The lysates were treated the same way, and the bound proteins were detected with anti-HA or with anti-Smg1 antibody. 5% of the input was analysed in the lane next to the antibody precipitate. Smg1 was analysed using a 4% polyacrylamide gel, while Upf1 was detected with a 6% polyacrylamide gel. IN, input. (d) Cells expressing SLBP–SBP were treated with control (C2) siRNA or SMG1 siRNA for 72 h. Lysates were prepared from C2 siRNA cells (lanes 1 and 2) or SMG siRNA-treated cells (lanes 3 and 4) 20 min after treatment with 5 mM HU, and lysates were prepared from the cells. The lysates were immunoprecipitated with anti-HA antibody. 5% of the input and the immunoprecipitate (lanes AP) were resolved by gel electrophoresis. SLBP–SBP and endogenous SLBP were detected by western blotting with anti-SLBP antibody and Upf1 was detected with an anti-Upf1 antibody. Lanes 1 and 3 are 5% of the input compared to the antibody precipitate in lanes 2 and 4. (Online version in colour.)
When we knocked down Smg1, we found that SLBP is still associated with Upf1 after HU treatment (figure 2d). Thus, recruitment of Upf1, in an inactive form, to histone mRNA may occur independently from phosphorylation by Smg1. These data are consistent with Upf1 recruitment and subsequent activation being an early step in histone mRNA degradation.
To further determine the requirements of Upf1 for histone mRNA degradation, we created deletion mutants of Upf1 that removed the C-terminal (which is phosphorylated by Smg1) or N-terminal region. A schematic of Upf1 is shown in figure 3a. Both deletion mutants had an HA-TAG at the N-terminus and had both the helicase and RNA binding activity intact. We had previously shown that expression of two HA-tagged point mutants, K498A and R843C, each of which inactivates helicase activity, reduces the degradation of histone mRNA when ectopically expressed in HeLa cells [13]. Expression of the deletion mutants of the N-terminus (215–1129) or C-terminus (1–984) also reduced the rate of histone mRNA degradation after HU treatment, in the SLBP–SBP-expressing cells, while expression of HA-tagged Upf1 had no effect (figure 3b,c).
Figure 3.

Requirements for Upf1 for histone mRNA degradation. (a) A schematic of the Upf1 protein. (b) Effect of deleting the N- or C-terminal domains of Upf1 on histone mRNA degradation. The SLBP–SBP-expressing HeLa cells (figure 2a) were transfected with the indicated HA-tagged Upf1 proteins. After 72 h, cells were treated with HU, and RNA samples analysed by Northern blotting to detect histone H2a mRNA and 7SK mRNA as a control. (c) The results of three independent biological replicates of the HU treatment are plotted, with s.d. shown. The * indicates that there is a significant retardation of histone mRNA degradation (p < 0.05). (d) Lysates were prepared from SLBP–SBP cells expressing the indicated HA-tagged Upf1 proteins, K498A, R843C or HA-Upf1, 20 min after HU treatment. The proteins bound to SLBP–SBP (lanes 2,4,6,8) were isolated as in figure 2 and detected by western blotting with SLBP antibody and with anti-HA to detect the transfected Upf1. 5% of the input is shown in lanes 1,3,5,7. (e) Lysates were prepared from SLBP–SBP cells expressing the indicated HA-tagged Upf1 deletion proteins, 215–1148 and 1–984, or HA-Upf1, 20 min after HU treatment. The proteins bound to SLBP–SBP (lanes 2,4,6) were isolated as in figure 2 and detected by western blotting with SLBP antibody and with anti-HA to detect the transfected Upf1. 5% of the input is shown in lanes 1,3,5. IN, input. (f) Western blot showing the expression of the HA-tagged Upf1 proteins 72 h after transfection. PTB (polypyrimidine tract binding protein) was used as a control. (Online version in colour.)
We tested whether the mutant Upf1 proteins were recruited to the 3′ end of histone mRNA. The SLBP–SBP cells were transfected with the four mutant HA-tagged proteins, as well as the wild-type HA-Upf1. Cells were treated with 5 mM HU for 20 min, and SLBP–SBP was isolated by binding to streptavidin beads. The tagged SLBP bound efficiently to the streptavidin beads, while the endogenous SLBP was not bound (figure 3d,e). The two mutant helicase proteins were bound to a similar extent as HA-Upf1 (figure 3d). Note that expression of the HA-tagged exogenous proteins results in overexpression of total Upf1 [13]. The amount of the UPF1 215–1129 deletion, which was expressed at lower levels than the other mutant proteins (figure 3f), bound to SLBP–SBP (figure 3e) was similar to that bound to HA-tagged Upf1. The proportion of Upf1 recruited to histone mRNA in these experiments was lower than when the experiments were done in cells expressing only endogenous Upf1 and SLBP (figure 2), because only a fraction of the total SLBP was precipitated with SLBP–SBP and the total amount of UPF1 was increased as result of expression of the tagged Upf1 proteins. The ability of all these mutant proteins to be recruited to the 3′ end of histone mRNA suggests that they may interfere with degradation by binding to the mRNA, but were not capable of initiating degradation of the histone mRNA. The 1–984 deletion, which also acted as a dominant negative protein, coprecipitated in low levels with SLBP, and may form a less stable complex with SLBP bound to the histone mRNP. This could be owing to the fact that Smg1 forms a less stable complex with SLBP or does not interact directly with SLBP–SBP or the histone mRNA, and hence may partially dissociate during the immunoprecipitation.
(b). The pathway of histone mRNA degradation is similar when DNA synthesis is inhibited and at the end of S-phase
The ability to isolate cells actively degrading histone mRNA by inhibiting DNA replication in S-phase, or by selecting cells at the end of S-phase, makes it possible to study the changes that take place as the relatively stable histone mRNAs are rapidly degraded either when DNA synthesis is inhibited or at the end of S-phase after completion of DNA replication. We can treat S-phase cells (3 h after release from double thymidine block) with HU to stop DNA replication and activate histone mRNA degradation (figure 4a). To determine whether the same pathway of histone mRNA degradation occurs in the unperturbed cell cycle at the end of S-phase, we allowed the synchronized cells to progress through the cell cycle and analysed histone mRNA levels during the cell cycle by Northern blotting (figure 4b). We also determined the phase of the cell cycle in the same samples by flow cytometry, using EdU pulse-labelling to determine the percentage of cells in S-phase (figure 4c). Based on this analysis, we selected cells 6 h after release into S-phase, when 20–25% of the cells had entered G2, and histone mRNA has been reduced by 25%, for analysis of histone mRNA.
Figure 4.

Histone mRNA is degraded by similar pathways after HU treatment and at the end of S-phase. (a) HCT116 cells were synchronized and released into S-phase. At 3 h, cell samples were harvested before treatment with 5 mM HU and 20 or 40 min after treatment with HU. Total RNA was prepared and analysed by Northern blotting for histone H2a mRNAs and 7SK RNA. (b) Cells were harvested at the indicated times after release into S-phase, and total cell RNA was harvested and analysed as in (a). The values for histone mRNA were determined relative to 7SK RNA. Note that the 7SK probe in (a) was of much lower specific activity than the probe used in (b). Below the gel, the amount of histone mRNA present relative to 3 h after release is indicated. (c) Cells from the experiment in (b) were analysed by flow cytometry to determine the percentage of cells in S-phase. The red dots indicate EdU-stained cells (intensity of staining on the y-axis). The cells in the lower left corner are cells that never entered S-phase. The percentage of cells in S-phase and the ratio of S-phase:G2-M cells are indicated under the time. (d,e) Libraries were prepared for analysis of histone mRNA 3′ ends from the cells in mid-phase (3 h), HU treated cells and cells near the end of S-phase (6 h), and the histone mRNAs were sequenced. The RNAs that had some degradation into the stemloop were plotted (ranging from nt 5 to nt 20, which is the end of the stem). The height of the bar indicating the percentage of total reads is indicated on each y-axis. (e) The 3′ ends of intermediates that extended up to 100 nts from the 3′ end are plotted. Note that these start at position 7. The position of the stop codon is indicated by the red octagon. The peak at about nt 75 is 15 nts after the stop codon. Note that there are many fewer degradation intermediates present in the mid-phase (3 h) sample. (f) A schematic of the 3′ end of the histone mRNA with the degradation intermediate after the stop codon indicated.
(c). Mapping the 3′ ends of histone mRNAs and mRNA degradation intermediates
We developed a method, End-SEQ, for determining the 3′ ends of histone mRNAs using a high-throughput sequencing strategy that allows the precise identification of the 3′ end of the RNA, including the identification of any untemplated nucleotides. The method, which has been described in detail [7], takes advantage of efficient ligation of a preadenylated linker to the 3′ ends of all cellular RNAs, followed by reverse transcription primed by a primer complementary to the linker. To enrich for histone mRNAs, primers complementary to the histone cDNAs together with the primer complementary to the linker were used to amplify the cDNA and attach the necessary Illumina linkers and barcodes. Since there are 10–15 different non-allelic genes for each core histone [31], we simultaneously get data on a large number of different histone mRNAs. The method allows unambiguous detection of a single non-templated nt, as well as longer stretches of non-templated nts, and the precise site in the histone mRNA where non-templated nts were added.
Using this approach, we found that the 3′ ends of histone mRNAs in S-phase are heterogeneous, ending 3 nts after the stemloop, but that in addition to the encoded ACC, there are also 3′ ends ending in ACU and AUU. These result from 3′hExo, which normally removes 2 nts from the 3′ end of histone mRNA, occasionally removing one or two additional nucleotides from the 3′ end (figure 1a,c). When this happens, a terminal uridyl transferase, primarily TUT7, restores the proper length of the 3′ end to 3 nts after the stemloop [32]. The result is that over 90% of the cytoplasmic histone mRNAs end in ACC, ACU or AUU during S-phase when they are relatively stable (figure 1c). The position of the last templated nucleotide is plotted on the x-axis, and non-templated nts are indicated by different coloured bars as no tails (blue), 1 nt tail (green), 2 nt tails (orange) or more than 2 nt tails (red). More than 95% of the non-templated nts are uridines. In some histone mRNAs, up to 70% of the mature molecules are uridylated, although this varies for individual histone mRNAs [32]. Thus, one function of uridylation is to maintain the proper 3′ end of histone mRNA during S-phase.
(d). Identifying mRNA degradation intermediates
After activating histone mRNA degradation by inhibiting DNA replication, we can profile the 3′ ends of the degradation intermediates by high-throughput sequencing, allowing us to deduce the 3′–5′ pathway of histone mRNA degradation. It is important to understand that when histone mRNAs (or any mRNAs) are rapidly degraded, entry of individual molecules into the degradation pathway occurs stochastically. Once a molecule is recruited into the degradation pathway, it is degraded very rapidly, while the other molecules remain unchanged. Thus, after one-half-life, 50% of the molecules have been degraded and the rest still are identical to the initial histone mRNAs present. When the half-life of histone mRNA is short, many more molecules are entering the degradation pathway, and a ‘steady-state’ distribution of degradation intermediates is formed. We have observed that the pattern of degradation intermediates is similar at all the time points we have analysed. In each experiment, we determine the amount of degradation with a goal of obtaining samples with about 50% of the molecules degraded; we have observed a similar degradation pattern of intermediates when less than 10% of the molecules have been degraded, as when 60–70% of the molecules have been degraded.
The pattern of degradation intermediates allows us to detect ‘barriers’ to degradation, spots at which intermediates are present in much higher amounts. The first kinetic intermediates we see are intermediates resulting from exonucleolytic degradation by 3′hExo, which stalls, resulting in the accumulation of a large number of intermediates into the stemloop. We can display these best by displaying the sequence of the stemloop (figure 4d). These intermediates are heavily uridylated, with many U-tails more than 2 nts, extending in length to 10–15 nts [23].
The second kinetic intermediate we see is 15 nts 3′ of the termination codon, which we attribute to the presence of the stalled terminating ribosome. There are very few intermediates between the stemloop and a site 15 nts 3′ of the stop codon (figure 4e), where there is an accumulation of large numbers of intermediates. The lack of intermediates between the stemloop and this site indicates that degradation occurs very rapidly through the 3′ UTR, and we have previously shown this is likely mediated by the exosome [23]. Degradation probably stalls because the exosome encounters the terminating ribosome. There are also intermediates in the coding region that likely represent interactions with other ribosomes on the mRNA, as the 3′–5′ degradation initiates and continues on translating mRNA [23]. These intermediates at the stalled ribosome and in the coding region are also uridylated.
Degradation does not initiate immediately after the addition of the DNA synthesis inhibitor during S-phase. There is a short lag (5–10 min, likely reflecting the time it takes to deplete the dNTP pools), followed by rapid degradation of the histone mRNA [33]. Our previous studies have all been done by inhibiting DNA replication, typically when cells are in mid-S-phase, 3 h after release from a double thymidine block or in exponentially growing cell cultures. In figure 4a, we show the analysis of the RNA in an experiment where less than 10% of the mRNA has been degraded 20 min after the addition of HU, and 64% has been degraded after 40 min. We analysed the sample 20 min after HU when degradation of histone mRNA has been initiated. Once degradation starts, the half-life of histone mRNA in HeLa cells after HU treatment is about 15 min [13], while the half-life of histone mRNA in S-phase cells is about 60 min [2,34].
We compared the amount and distribution of potential degradation intermediates in untreated cells (figure 4d(i),e(i)) with the cells treated for 20 min with HU (figure 4d(ii),e(ii)). We plotted the percentage of total reads for untreated cells and cells treated for 20 min with HU. Since in this experiment only about 6% of the total histone H2a mRNA has been degraded, the amount of full-length RNA was similar in both samples. In the cells treated with HU, there was a large increase in the number and a change in the distribution of these intermediates. This pattern of degradation intermediates in the stemloop is essentially identical to those previously reported in HeLa cells [23,32]. In the S-phase cells, there were a small amount of mRNAs ending at positions 5–7 in the stem, most of which had long U-tails. In the S-phase cells, there were also a small number of degradation intermediates 5′ of the stemloop with a peak of about 15 nts downstream of the stop codon. Many of these intermediates were uridylated. There was about a 20-fold increase in the amounts of these intermediates, with a clear lack of degradation intermediates between the stemloop and a prominent peak located 15 nts after the stop codon (figure 4e(ii)).
We analysed the 3′ ends of histone mRNAs in the sample taken at the end of S-phase (figure 4d(iii),e(iii)) and compared them with the histone mRNAs in the cells treated with HU for 20 min in mid-S-phase (figure 4d(ii),e(ii)). The same pattern of degradation intermediates was observed in the cells exiting S-phase as in the cells treated with HU, both in the intermediates present in the stemloop (figure 4d) and in the intermediates observed 5′ of the stemloop, with a prominent peak 15 nts 3′ of the stop codon (figure 4e,f). There was a similar quantitative difference (large increase) in the number of intermediates observed in the cells entering G2 compared to cells in S-phase cells (figure 4d(i),e(i)). Thus, the steady-state pattern of intermediates is similar in both samples, consistent with the same pathway of histone mRNA degradation occurring at the end of S-phase as when DNA replication is inhibited in S-phase.
4. Discussion
It is critical that the cell be able to rapidly adjust the synthesis of histone proteins to meet, but not exceed, the demands for histones as a result of DNA replication. In mammalian cells, the stemloop at the 3′ end of histone mRNAs is a critical cis-element for cell cycle regulation, participating in both the biosynthesis of histone mRNA, as well as its rapid degradation. Animal cells have developed a novel pathway for degrading histone mRNAs. The initial signal leading to degradation of histone mRNA is a defect in translation termination, which results in recruitment and subsequent activation of Upf1. The initial removal of nucleotides from the 3′ end involves the activation of 3′–5′ exonucleolytic decay by 3′hExo, an exonuclease that binds specifically to the 3′ end of histone mRNA and forms a ternary complex with SLBP and the stemloop on the RNA.
(a). The initial degradation intermediate
What are the components of the complex on the initial intermediate in the stemloop? It likely contains SLBP still bound to the 3′ end. Note that SLBP interacts primarily with the 5′ side of the stem, with no direct interactions with the 3′ side, and a specific interaction with the G at position 2 of the stem [5]. The base pair at this position is not essential for binding as long as the G at position 2 is present [35]. 3′hExo is present because it is degrading the RNA into the stemloop; since it is a distributive enzyme, it likely pauses in the stem, and uridylation may be required for it to continue degradation. Lsm 1–7 is bound to the U-tails on the stemloop, many of which at positions 7 and 9 are long enough to bind Lsm 1–7 [23]. Lsm4 also can bind directly with 3′hExo and SLBP [36]. Upf1 and Smg1 are bound to the 3′ UTR, with Upf1 activated by phosphorylation by Smg1. This model is consistent with all these factors coimmunoprecipitating with SLBP after initiating degradation, at which time substantial amounts of these intermediates have accumulated.
We postulate that the initial step in the degradation of histone mRNA is partial disruption of the SLBP/SL complex, either by activated Upf1 or a modification of SLBP, or both, resulting in the dissociation of the base of the stem (figure 5). Note that in the crystal structure of 3′hExo bound to the SL, the bottom three base pairs in the stem are broken, while in the SLBP/SL/3′hExo complex, they are intact [5], suggesting that partially disrupting SLBP binding could open up the stem to degradation by 3′hExo. A candidate for causing this disruption is the helicase activity of Upf1, which would allow 3′hExo to degrade into the stemloop, and is essential for rapid histone mRNA degradation [13]. The stalling of the 3′hExo in the stem results in TUT7 adding long U-tails. TUT7 is associated with the mature mRNP, because it is also primarily responsible for adding short U-tails to the 3′ end to maintain the proper length of the histone mRNA during S-phase [32].
Figure 5.
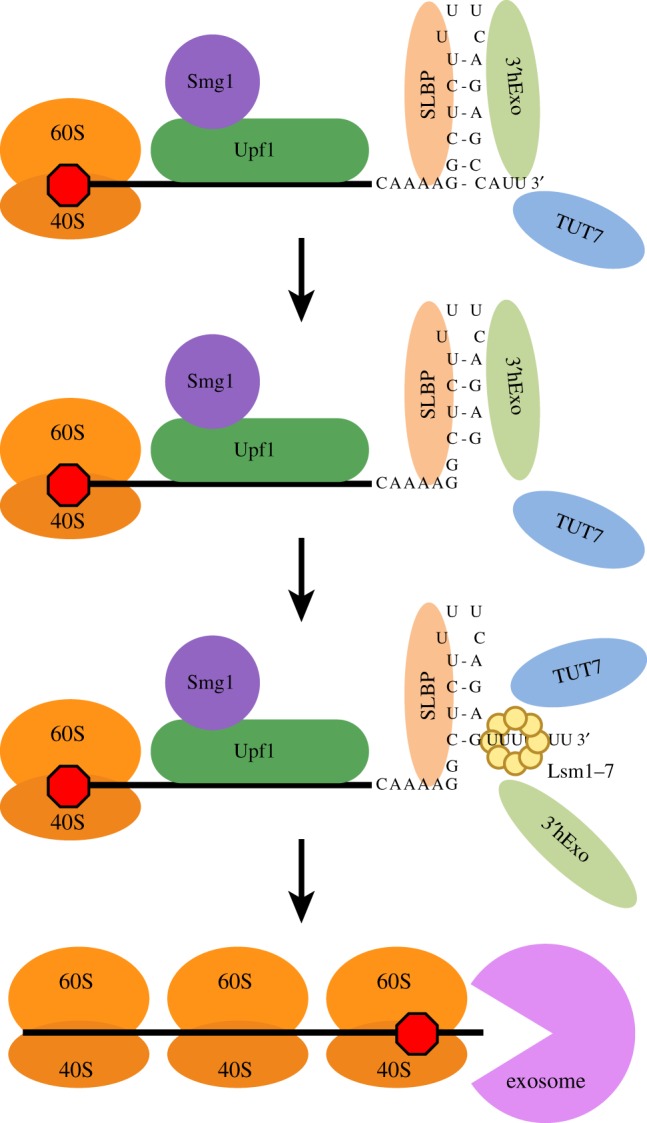
Intermediates in 3′–5′ degradation of histone mRNA. The histone mRNP associated with SLBP, 3′hExo and TUT7 (figure 1a) is the actively translating form of histone mRNA. The proposed initial step in histone mRNA degradation is loss of efficient translation termination (possibly by an unknown modification of SLBP). This results in binding of Upf1 and Smg1, phosphorylation of Upf1 and subsequent weakening of the SLBP/SL interaction, allowing 3′hExo to initiate degradation into the stemloop. The U-tails added in the stem bind Lsm1–7, which can also bind directly to SLBP and 3′hExo through the C-terminal tail of Lsm4. This intermediate accumulates but is resolved by loss of SLBP and 3′hExo, followed by loss of Upf1, resulting in the recruitment of the exosome and rapid degradation until the exosome encounters the ribosome bound to the stop codon, resulting in the formation of a uridylated intermediate. Subsequent degradation can occur either (or both) 5′–3′ as a result of continued exonucleolytic degradation and/or decapping of the mRNA and 5′–3′ degradation.
The intermediate is likely resolved by the loss of SLBP and 3′hExo from the 3′ end, probably facilitated by the helicase activity of Upf1 and possibly also by the interaction of Lsm4 with the SLBP/SL/3′hExo complex [36]. Once this happens, Upf1 dissociates from the complex (following the loss of SLBP), and the exosome can bind and processively degrade the 3′UTR until it reaches a terminating ribosome.
We do not detect significant numbers of long U-tails added to the single-stranded tail after the stemloop. When we primed cDNA synthesis with a primer ending in three As (so we would only detect U-tails of at least 3 nts long in the HTS analysis), we observed long U-tails predominantly on the Gs at position 7 and 9, suggesting that these may be the major site of Lsm1–7 binding, and few long tails after the stemloop [7]. Thus, our initial proposal that uridylation of the very 3′ end of the mRNA was critical for initiating degradation is not correct [21]. Long U-tails are only added once degradation has proceeded into the stem.
(b). Is 5′–3′ degradation an important component of the pathway of histone mRNA degradation?
Lsm1–7 bound to either oligo(A) or oligo(U) can promote decapping [37–39], and this is the major pathway of mRNA degradation in yeast. Our first approach to detect degradation intermediates was to circularize intermediates (which required that they be decapped). We observed a number of intermediates that were uridylated near the 3′ end, in the same region as the major degradation intermediate. Thus, clearly some of these intermediates are decapped, and these were being degraded both 5′–3′ and 3′–5′ on the same molecule [21]. It is impossible to tell how many of the intermediates we are detecting in our high-throughput sequencing experiments have been decapped, although a substantial proportion of them are still capped, based on immunoprecipitation with anti-cap antibody [23]. It is certainly possible that decapping ultimately occurs on most molecules, resulting in both rapid 5′–3′ degradation by Xrn1 as well as continued degradation 3′–5′ on the same molecule.
The complex on the 5′ end of histone mRNA has not been well characterized. There is a report that the nuclear cap binding complex, CBP80/20 is bound to much of the histone mRNAs [40] and that this complex contributes to rapid histone mRNA degradation. Since histone mRNAs have relatively short half-lives even in S-phase, and the mRNA is exported from the nucleus bound to CBP80/20, it is certainly possible that a substantial fraction of the histone mRNA may be bound to CBP80/20. The nuclear cap binding protein also promotes NMD initiated as a result of premature termination codons [41], consistent with a potential role for CBP80/20 in histone mRNA degradation.
(c). Remaining questions about histone mRNA degradation
There are several important questions still to be answered about histone mRNA degradation. In addition to the question of the relative contributions of 3′–5′ degradation and 5′–3′ degradation discussed above, we do not understand several other critical components of this process. It is not known what change occurs in the histone mRNP that results in inefficient termination leading to the activation of histone mRNA degradation. The polyA tail–PABP complex promotes termination of translation and recycling of ribosomes on polyadenylated mRNAs [42]. We hypothesize that SLBP also participates in the termination of translation of histone mRNAs. We think that termination of histone mRNA translation is compromised, possibly through modification of SLBP, leading to inefficient termination that then results in the recruitment of Upf1. The molecular details of the initial change in the translation of histone mRNA remain unknown.
(d). Other specific issues that need to be resolved
How does Upf1 act to allow 3′hExo to degrade into the stem of histone mRNA?
The mechanism that results in initiation of degradation by 3′hExo is not clear, but probably the interaction of SLBP with the stemloop must be altered, either by SLBP modification or activity of the Upf1 helicase, or both.
How is TUT7 recruited specifically to the histone mRNP? As TUT7 modifies the 3′ end of histone mRNA when the mRNA is stable, as well as the degradation intermediates, it is likely recruited by the SLBP/3′hExo complex bound to the stemloop RNA. The specific sequences in the TUT7 and the SLBP/3′hExo that participate in this selective recruitment are not known.
How is the exosome specifically recruited to the degradation intermediate that contains the uridylated stemloop, and which exosome subunit actively degrades histone mRNA?
How is the same degradation pathway activated at the end of S-phase and when DNA replication is inhibited in mid-S-phase? ATR, which is activated when DNA synthesis is inhibited in mid-S-phase, is not activated at the end of S-phase, suggesting that there must be other pathways that result in the same changes to the histone mRNP that activate histone mRNA degradation at the end of S-phase.
(e). Role of uridylation in metabolism of other RNAs
Uridylation also plays important roles in the metabolism of other RNAs. It has been most thoroughly studied in miRNAs and pre-miRNAs. Many of these contain non-templated uridines, and in the regulation of the let7 pre-miRNA, addition of a single uridine to the pre-miRNA promotes maturation while the addition of multiple uridines results in the degradation of the pre-miRNA [43,44]. The switch in uridylation from monouridylation to oligouridylations is caused by binding of Lin28 to either TUT4 or TUT7 [45]. Uridylation clearly plays a critical role in degradation of structural RNAs, such as snoRNAs, in mammalian cells, probably as a quality control mechanism to remove non-functional RNAs. Uridylation triggers 3′–5′ degradation followed by the processive 3′–5′ exonuclease Dis3L2 [46–48]. A similar pathway degrades mRNAs during apoptosis in mammalian cells [49]. Dis3L2 also degrades the oligouridylated let7 pre-miRNA [50].
Polyadenylated mRNAs are also uridylated, and these were first detected in fission yeast [51]. Most of the uridylation is on ‘deadenylated’ mRNAs. The first step in mRNA degradation is the shortening of the polyA tail by deadenylation. This intermediate can be uridylated, usually by the addition of a small number of uridines [52,53]. Uridylation has been implicated in the degradation of polyadenylated mRNAs in mammals [52], although its precise biochemical role in degradation is unclear. In plants, uridylation of deadenylated mRNAs can clearly function to stabilize the deadenylated RNA, restoring it to a size able to bind PABP [54]. In plants, the 5′ product of cleavage of mRNAs by miRNAs is uridylated and in mammals the 5′ cleavage product of the Hoxb8 mRNA by miR196 is uridylated [55]. A potential common theme of the variety of uridylations is a difference in function between short U-tails and long U-tails, as in pre-let7 RNA and histone mRNAs. There seems to be a general role for uridylation in degradation of RNAs that have been previously damaged, which may include endonucleolytic cleavage product of mRNAs.
5. Conclusion
Oligouridylation of histone mRNA is important in maintaining the levels of histone mRNA during S-phase by maintaining a 3′ nt tail after the stemloop on histone mRNAs. TUT7 and 3′hExo activities work together, resulting in the removal of the original templated nts at the 3′ end and replacing them with uridines [32]. Oligouridylation by TUT7 also plays a role in the rapid degradation of histone mRNA, where extensive oligouridylation occurs on 3′–5′ degradation intermediates, both when degradation is paused in the stemloop, and again when it is stalled by the terminating ribosome. Note that removal of the stemloop will result in inhibition of further translation initiation on histone mRNA, and histone protein production may initially be stopped by inhibition of translation termination on histone mRNA, resulting in activation of 3′–5′ degradation.
Regulation of histone mRNA half-life can function to rapidly alter histone mRNA levels. Treating S-phase cells with low levels of DNA synthesis inhibitors to partially inhibit DNA replication results in a reduction in histone mRNA, while treatment with low levels of protein synthesis inhibitors to partially inhibit histone protein synthesis results in a rapid increase in histone mRNA levels [56], as a result of changing the half-life of histone mRNAs. In each case, the cell is adjusting the rates of DNA replication and histone protein synthesis to optimize the packaging of newly replicated DNA into chromatin. This mechanism likely functions throughout the cell cycle to maintain the balance between histone protein production and the rate of DNA replication. One potential way the cell could determine whether the synthesis of histones is balanced appropriately with DNA replication would be to monitor the amount of free histone protein (not bound to either chromatin or histone chaperones). Ultimately, there must be a signal generated (likely in the nucleus) that then leads to the initiation of degradation of histone mRNA in the cytoplasm.
Acknowledgements
We thank Dr Josh Welch (Univ. Michigan) for developing the App-End analysis platform for analysis of degradation intermediates; Dr Nancy Fisher, Director of the UNC FLOW Cytometry Facility for assistance; Dr Chelsea K. Raulerson for assistance with the analysis of the sequencing data; and Dr Esther Griesbach for assistance with the EdU labelling.
Data accessibility
DNA sequencing data has been deposited in GEO, Accession no. GSE121461.
Authors' contributions
S.A.M. designed and performed all the experiments in figures 2 and 3, participated in writing the paper and prepared the final figures. C.E.H. performed the experiments in figure 4 and participated in writing the paper. W.F.M. directed the project and designed the overall strategy in conjunction with S.A.M. and C.E.H. W.F.M. also took the lead in writing the paper.
Competing interests
The authors declare that they have no competing interests.
Funding
This work was supported by NIH grant GM29832 to W.F.M. S.A.M. was supported by postdoctoral fellowship F32GM80007.
References
- 1.Whitfield ML, Zheng L-X, Baldwin A, Ohta T, Hurt MM, Marzluff WF. 2000. Stem-loop binding protein, the protein that binds the 3′ end of histone mRNA, is cell cycle regulated by both translational and posttranslational mechanisms. Mol. Cell. Biol. 20, 4188–4198. ( 10.1128/MCB.20.12.4188-4198.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris ME, Böhni R, Schneiderman MH, Ramamurthy L, Schümperli D, Marzluff WF. 1991. Regulation of histone mRNA in the unperturbed cell cycle: evidence suggesting control at two posttranscriptional steps. Mol. Cell. Biol. 11, 2416–2424. ( 10.1128/MCB.11.5.2416) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scharl EC, Steitz JA. 1994. The site of 3′ end formation of histone messenger RNA is a fixed distance from the downstream element recognized by the U7 snRNP. EMBO J. 13, 2432–2440. ( 10.1002/j.1460-2075.1994.tb06528.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skrajna A, Yang XC, Dadlez M, Marzluff WF, Dominski Z. 2018. Protein composition of catalytically active U7-dependent processing complexes assembled on histone pre-mRNA containing biotin and a photo-cleavable linker. Nucleic Acids Res. 46, 4752–4770. ( 10.1093/nar/gky133) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tan D, Marzluff WF, Dominski Z, Tong L. 2013. Structure of histone mRNA stem-loop, human stem-loop binding protein, and 3'hExo ternary complex. Science 339, 318–321. ( 10.1126/science.1228705) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang XC, Purdy M, Marzluff WF, Dominski Z. 2006. Characterization of 3'hExo, a 3′ exonuclease specifically interacting with the 3′ end of histone mRNA. J. Biol. Chem. 281, 30447–30454. ( 10.1074/jbc.M602947200) [DOI] [PubMed] [Google Scholar]
- 7.Welch JD, Slevin MK, Tatomer DC, Duronio RJ, Prins JF, Marzluff WF. 2015. EnD-Seq and AppEnD: Sequencing 3′ ends to identify nontemplated tails and degradation intermediates. RNA 21, 1375–1389. ( 10.1261/rna.048785.114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sittman DB, Graves RA, Marzluff WF. 1983. Histone mRNA concentrations are regulated at the level of transcription and mRNA degradation. Proc. Natl Acad. Sci. USA 80, 1849–1853. ( 10.1073/pnas.80.7.1849) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whitfield ML, Kaygun H, Erkmann JA, Townley-Tilson WHD, Dominski Z, Marzluff WF. 2004. SLBP is associated with histone mRNA on polyribosomes as a component of histone mRNP. Nucleic Acids Res. 32, 4833–4842. ( 10.1093/nar/gkh798) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marzluff WF, Gongidi P, Woods KR, Jin JP, Maltais L. 2002. The human and mouse replication-dependent histone genes. Genomics 80, 487–498. ( 10.1006/geno.2002.6850) [DOI] [PubMed] [Google Scholar]
- 11.Kaygun H, Marzluff WF. 2005. Translation termination is involved in histone mRNA degradation when DNA replication is inhibited. Mol. Cell. Biol. 25, 6879–6888. ( 10.1128/MCB.25.16.6879-6888.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Graves RA, Pandey NB, Chodchoy N, Marzluff WF. 1987. Translation is required for regulation of histone mRNA degradation. Cell 48, 615–626. ( 10.1016/0092-8674(87)90240-6) [DOI] [PubMed] [Google Scholar]
- 13.Kaygun H, Marzluff WF. 2005. Regulated degradation of replication-dependent histone mRNAs requires both ATR and Upf1. Nat. Struct. Mol. Biol. 12, 794–800. ( 10.1038/nsmb972) [DOI] [PubMed] [Google Scholar]
- 14.Maquat LE. 2002. Nonsense-mediated mRNA decay. Curr. Biol. 12, R196–R197. ( 10.1016/S0960-9822(02)00747-9) [DOI] [PubMed] [Google Scholar]
- 15.Kim YK, Furic L, LesGroseillers L, Maquat LE. 2005. Mammalian Staufen1 recruits Upf1 to specific mRNA 3'UTRs so as to elicit mRNA decay. Cell 120, 195–208. ( 10.1016/j.cell.2004.11.050) [DOI] [PubMed] [Google Scholar]
- 16.Ross J, Kobs G. 1986. H4 histone messenger RNA decay in cell-free extracts initiates at or near the 3′ terminus and proceeds 3′ to 5′. J. Mol. Biol. 188, 579–593. ( 10.1016/S0022-2836(86)80008-0) [DOI] [PubMed] [Google Scholar]
- 17.Ross J, Peltz SW, Kobs G, Brewer G. 1986. Histone mRNA degradation in vivo: the first detectable step occurs at or near the 3′ terminus. Mol. Cell. Biol. 6, 4362–4371. ( 10.1128/MCB.6.12.4362) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ross J, Kobs G, Brewer G, Peltz SW. 1987. Properties of the exonuclease activity that degrades H4 histone mRNA. J. Biol. Chem. 262, 9374–9381. [PubMed] [Google Scholar]
- 19.Caruccio N, Ross J. 1994. Purification of a human polyribosome-associated 3′ to 5′ exonuclease. J. Biol. Chem. 269, 31 814–31 821. [PubMed] [Google Scholar]
- 20.Dominski Z, Yang X, Kaygun H, Marzluff WF. 2003. A 3′ exonuclease that specifically interacts with the 3′ end of histone mRNA. Mol. Cell 12, 295–305. ( 10.1016/S1097-2765(03)00278-8) [DOI] [PubMed] [Google Scholar]
- 21.Mullen TE, Marzluff WF. 2008. Degradation of histone mRNA requires oligouridylation followed by decapping and simultaneous degradation of the mRNA both 5′ to 3′ and 3′ to 5′. Genes Dev. 22, 50–65. ( 10.1101/gad.1622708) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoefig KP, Rath N, Heinz GA, Wolf C, Dameris J, Schepers A, Kremmer E, Ansel KM, Heissmeyer V. 2013. Eri1 degrades the stem-loop of oligouridylated histone mRNAs to induce replication-dependent decay. Nat. Struct. Mol. Biol. 20, 73–81. ( 10.1038/nsmb.2450) [DOI] [PubMed] [Google Scholar]
- 23.Slevin MK, Meaux S, Welch JD, Bigler R, de Marval PLM, Su W, Rhoads RE, Prins JF, Marzluff WF. 2014. Deep sequencing shows multiple oligouridylations are required for 3′ to 5′ degradation of histone mRNAs on polyribosomes. Mol. Cell 53, 1020–1030. ( 10.1016/j.molcel.2014.02.027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Camp ND, et al. 2012. Wilms tumor gene on X chromosome (WTX) inhibits degradation of NRF2 protein through competitive binding to KEAP1 protein. J. Biol. Chem. 287, 6539–6550. ( 10.1074/jbc.M111.316471) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zund D, Gruber AR, Zavolan M, Muhlemann O. 2013. Translation-dependent displacement of UPF1 from coding sequences causes its enrichment in 3′ UTRs. Nat. Struct. Mol. Biol. 20, 936–943. ( 10.1038/nsmb.2635) [DOI] [PubMed] [Google Scholar]
- 26.Brooks L III, Lyons SM, Mahoney JM, Welch JD, Liu Z, Marzluff WF, Whitfield ML. 2015. A multiprotein occupancy map of the mRNP on the 3′ end of histone mRNAs. RNA 21, 1943–1965. ( 10.1261/rna.053389.115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nicholson P, Yepiskoposyan H, Metze S, Zamudio OR, Kleinschmidt N, Muhlemann O. 2010. Nonsense-mediated mRNA decay in human cells: mechanistic insights, functions beyond quality control and the double-life of NMD factors. Cell. Mol. Life Sci. 67, 677–700. ( 10.1007/s00018-009-0177-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Azzalin CM, Lingner J. 2006. The double life of UPF1 in RNA and DNA stability pathways. Cell Cycle 5, 1496–1498. ( 10.4161/cc.5.14.3093) [DOI] [PubMed] [Google Scholar]
- 29.Azzalin CM, Lingner J. 2006. The human RNA surveillance factor UPF1 is required for S phase progression and genome stability. Curr. Biol. 16, 433–439. ( 10.1016/j.cub.2006.01.018) [DOI] [PubMed] [Google Scholar]
- 30.Yamashita A, Ohnishi T, Kashima I, Taya Y, Ohno S. 2001. Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes Dev. 15, 2215–2228. ( 10.1101/gad.913001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z, Johnson MR, Ke Z, Chen L, Welte MA. 2014. Drosophila lipid droplets buffer the H2Av supply to protect early embryonic development. Curr. Biol. 24, 1485–1491. ( 10.1016/j.cub.2014.05.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lackey PE, Welch JD, Marzluff WF. 2016. TUT7 catalyzes the uridylation of the 3′ end for rapid degradation of histone mRNA. RNA 22, 1663–1672. ( 10.1261/rna.058107.116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Graves RA, Marzluff WF. 1984. Rapid, reversible alterations in histone gene transcription and histone mRNA levels in mouse myeloma cells. Mol. Cell. Biol. 4, 351–357. ( 10.1128/MCB.4.2.351) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heintz N, Sive HL, Roeder RG. 1983. Regulation of human histone gene expression: kinetics of accumulation and changes in the rate of synthesis and in the half-lives of individual histone mRNAs during the HeLa cell cycle. Mol. Cell. Biol. 3, 539–550. ( 10.1128/MCB.3.4.539) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin L, Meier M, Lyons SM, Sit RV, Marzluff WF, Quake SR, Chang HY. 2012. Systematic reconstruction of RNA functional motifs with high-throughput microfluidics. Nat. Methods 9, 1192–1194. ( 10.1038/nmeth.2225) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lyons SM, Ricciardi A, Guo AY, Kambach C, Marzluff WF. 2014. The C-terminal tail of Lsm4 interacts directly with the 3′ end of the histone mRNP and is required for efficient histone mRNA degradation. RNA 20, 88–102. ( 10.1261/rna.042531.113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song MG, Kiledjian M. 2007. 3′ Terminal oligo U-tract-mediated stimulation of decapping. RNA 13, 2356–2365. ( 10.1261/rna.765807) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tharun S, Muhlrad D, Chowdhury A, Parker R. 2005. Mutations in the Saccharomyces cerevisiae LSM1 gene that affect mRNA decapping and 3′ end protection. Genetics 170, 33–46. ( 10.1534/genetics.104.034322) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tharun S. 2009. Lsm1-7-Pat1 complex: a link between 3′ and 5'-ends in mRNA decay? RNA Biol. 6, 228–232. ( 10.4161/rna.6.3.8282) [DOI] [PubMed] [Google Scholar]
- 40.Choe J, Kim KM, Park S, Lee YK, Song OK, Kim MK, Lee B-G, Song HK, Kim YK. 2013. Rapid degradation of replication-dependent histone mRNAs largely occurs on mRNAs bound by nuclear cap-binding proteins 80 and 20. Nucleic Acids Res. 41, 1307–1318. ( 10.1093/nar/gks1196) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hwang J, Sato H, Tang Y, Matsuda D, Maquat LE. 2010. UPF1 association with the cap-binding protein, CBP80, promotes nonsense-mediated mRNA decay at two distinct steps. Mol. Cell 39, 396–409. ( 10.1016/j.molcel.2010.07.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uchida N, Hoshino SI, Imataka H, Sonenberg N, Katada T. 2002. A novel role of the mammalian GSPT/eRF3 associating with poly(A)-binding protein in Cap/Poly(A)-dependent translation. J. Biol. Chem. 277, 50 286–50 292. ( 10.1074/jbc.M203029200) [DOI] [PubMed] [Google Scholar]
- 43.Heo I, Ha M, Lim J, Yoon MJ, Park JE, Kwon SC, Chang H, Kim VN. 2012. Mono-uridylation of pre-microRNA as a key step in the biogenesis of group II let-7 microRNAs. Cell 151, 521–532. ( 10.1016/j.cell.2012.09.022) [DOI] [PubMed] [Google Scholar]
- 44.Kim B, et al. 2015. TUT7 controls the fate of precursor microRNAs by using three different uridylation mechanisms. EMBO J. 34, 1801–1815. ( 10.15252/embj.201590931) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Faehnle CR, Walleshauser J, Joshua-Tor L. 2017. Multi-domain utilization by TUT4 and TUT7 in control of let-7 biogenesis. Nat. Struct. Mol. Biol. 24, 658–665. ( 10.1038/nsmb.3428) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pirouz M, Du P, Munafo M, Gregory RI. 2016. Dis3l2-mediated decay is a quality control pathway for noncoding RNAs. Cell Rep. 16, 1861–1873. ( 10.1016/j.celrep.2016.07.025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ustianenko D, Pasulka J, Feketova Z, Bednarik L, Zigackova D, Fortova A, Zavolan M, Vanacova S. 2016. TUT-DIS3L2 is a mammalian surveillance pathway for aberrant structured non-coding RNAs. EMBO J. 35, 2179–2191. ( 10.15252/embj.201694857) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Labno A, Warkocki Z, Kulinski T, Krawczyk PS, Bijata K, Tomecki R, Dziembowski A. 2016. Perlman syndrome nuclease DIS3L2 controls cytoplasmic non-coding RNAs and provides surveillance pathway for maturing snRNAs. Nucleic Acids Res. 44, 10 437–10 453. ( 10.1093/nar/gkw649) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomas MP, Liu X, Whangbo J, McCrossan G, Sanborn KB, Basar E, Zavolan M, Vanacova S. 2015. Apoptosis triggers specific, rapid, and global mRNA decay with 3′ uridylated intermediates degraded by DIS3L2. Cell Rep. 11, 1079–1089. ( 10.1016/j.celrep.2015.04.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang HM, Triboulet R, Thornton JE, Gregory RI. 2013. A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28-let-7 pathway. Nature 497, 244–248. ( 10.1038/nature12119) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rissland OS, Norbury CJ. 2009. 3′ uridylation precedes decapping in a novel pathway of bulk mRNA turnover. Nat. Struct. Mol. Biol. 16, 616–623. ( 10.1038/nsmb.1601) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lim J, Ha M, Chang H, Kwon SC, Simanshu DK, Patel DJ, Kim VN. 2014. Uridylation by TUT4 and TUT7 marks mRNA for degradation. Cell 159, 1365–1376. ( 10.1016/j.cell.2014.10.055) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sement FM, Ferrier E, Zuber H, Merret R, Alioua M, Deragon JM, Bousquet-Antonelli C, Lange H, Gagliardi D. 2013. Uridylation prevents 3′ trimming of oligoadenylated mRNAs. Nucleic Acids Res. 41, 7115–7127. ( 10.1093/nar/gkt465) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zuber H, Scheer H, Ferrier E, Sement FM, Mercier P, Stupfler B, Gagliardi D. 2016. Uridylation and PABP cooperate to repair mRNA deadenylated ends in Arabidopsis. Cell Rep. 14, 2707–2717. ( 10.1016/j.celrep.2016.02.060) [DOI] [PubMed] [Google Scholar]
- 55.Shen B, Goodman HM. 2004. Uridine addition after microRNA-directed cleavage. Science 306, 997 ( 10.1126/science.1103521) [DOI] [PubMed] [Google Scholar]
- 56.Sariban E, Wu RS, Erickson LC, Bonner WM. 1985. Interrelationships of protein and DNA syntheses during replication of mammalian cells. Mol. Cell. Biol. 5, 1279–1286. ( 10.1128/MCB.5.6.1279) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
DNA sequencing data has been deposited in GEO, Accession no. GSE121461.