

Graphical abstract

Method name: Quantitative imaging of cell micropolarity
Keywords: Spectral phasors, Nile Red, Membranes micropolarity, Metabolic imaging, Lipids, Confocal microscopy, Fatty acids, Triglycerides, Lipid droplets
Abstract
Intracellular micropolarity is essential in several metabolic processes, as it controls membrane permeability, regulating the fluxes of molecules and energy. Here we describe a method for the determination of the micropolarity in living cells using spectral confocal microscopy. The method is based on a phasor analysis of spectrally resolved images of live cells, labelled with the solvatochromic probe Nile Red. An application is provided to extract a polarity profile from the acquired Spectral datasets, which represent the contribution of hyperpolar, polar and non-polar lipids, and to generate a micropolarity map at submicrometric spatial resolution. A metabolic parameter, representing a quantitative index of the fatty acid-triacylglycerol turnover, is also furnished. This method allows a functional profiling of cells and tissues and the detection of metabolic imbalances between lipid storage and usage.
-
•
Use of spectral resolved confocal microscopy of Nile Red labelled cells for pixel resolved determination of the membranes micropolarity.
-
•
Spectral acquisition increases the specificity and sensitivity of the detection to provide a polarity profile and a metabolic index for fatty acid-TG turnover.
-
•
Use of spectral resolved confocal microscopy of Nile Red labelled cells for pixel resolved determination of the membranes micropolarity.
Specification Table
| Subject area: | Biochemistry, Genetics and Molecular Biology |
| More specific subject area: | Biophysics of lipids |
| Method name: | Quantitative imaging of cell micropolarity |
| Name and reference of original method: | G. Maulucci, F. Di Giacinto, C. De Angelis, O. Cohen, B. Daniel, C. Ferreri, M. De Spirito, S. Sasson, Real time quantitative analysis of lipid storage and lipolysis pathways by confocal spectral imaging of intracellular micropolarity, Biochim. Biophys. Acta - Mol. Cell Biol. Lipids. 1863 (2018) 783–793. doi:10.1016/J.BBALIP.2018.04.004. |
| Resource availability: | The PhasorM program and the supplementary data and materials are available in Mendeley Data at the following link: https://data.mendeley.com/datasets/z3g572hvjs/draft?a=01d35be1-2cf2-4223-ba09-e4f15375de5b |
| Application file for the phasor analysis (Windows) | PhasorM Windows.zip. The archive contains: PhasorM_installer.exe Application file for the phasor analysis (Mac OS): PhasorM MacOS.zip The archive contains: MyAppIInstaller_mcr |
| Sample data and mask | supplementary materials.zip The archive contains: A folder “data” which contains: 1. Reference lipids TO.tif: 32 channel spectral image of nile red labelled Triolein 2. Spectral image cells.tiff: 32 channel spectral image of Nile red labelled cells A folder “masks” which contains 3. The files of the masks which can be directly used for the analysis (NP_Mask_95_noOverlap.mat,P_Mask_95_noOverlap.mat and HP_Mask_95_noOverlap.mat) if these requirements are met: 32 channels spectral detector in the range 580-660 nm and an excitation laser of 488 nm wavelength. |
Method details
Membrane micropolarity is one of the main regulators of the fluxes of molecules and energy in several metabolic processes. Lipid metabolism can determine changes in intracellular micropolarity by altering the composition of intracellular membranes and disorders in the lipid balance has been frequently associated to many different kinds of metabolic, cardiovascular and neurodegenerative diseases. Indeed, Free Fatty Acids (FFA) and acyl-CoA, that can be used to produce energy, have highly polar head groups, whereas lipids as cholesterol esters and triglycerides, which are mainly used as energy storage, have neutral head groups. Phospholipids and other biosynthetic products, with structural and signaling roles, are generally characterised by an intermediate polarity. In this article, we report the detailed protocol for the determination of the intracellular micropolarity by a confocal spectral imaging approach combined with the phasor-based analysis. The method, presented in [1], is designed to provide a fast and accurate technique for the classification of the intracellular micropolarity relying on the spectrally resolved confocal microscopy. The physical phenomenon underlying this method is the highly solvatochromic behaviour of the lipophilic fluorophore Nile Red, whose emission spectra shifts from yellow to red with the increasing degree of polarity of the environment [2]. This property is employed for mapping and characterizing intracellular micropolarity as well as its regulation by lipid metabolic processes [1]. The emission spectrum can be measured pixel by pixel with a confocal microscope equipped with a spectral detector, and quantified with a phasor based algorithm [3]. The phasor method [4,5] assigns to each pixel spectrum two coordinates (G and S), obtained from the normalized Fourier Discrete transform of the emission spectrum:
| (1) |
| (2) |
In the Eqs. (1) and (2) Ii is the intensity of the signal recorded at the i-th channel, Δλ is the width of the spectral detector channels ((λmax−λmin)/nch). The G and S coordinates of each pixel are used to create a 2-D histogram, which is called the phasor plot [1]. Reference spectra of pure lipids allows to identify and select three regions in this plot, classified according to their micropolarity (Non-Polar (NP), Polar (P) and Hyper-Polar (HP)). The phasor analysis enables a polarity driven segmentation for hyperpolar, polar and non-polar micro-environments in the membranes visualization, and for a polarity profile of the sample. Moreover, since the overall state of intracellular micropolarity reflects the activation state of the lipid storage and catabolism network, a quantitative index is provided, which summarise the metabolic state of the network regulating fatty acid-TG turnover. This index consists in the average angle θp formed by the points laying in the phasor plot and the G axis [1]. The whole protocol is schematized in the graphical abstract. This simple classification in three groups and the θp index can give a direct and immediate tool to visualize and quantify changes and differences in the membranes composition and state with a pixel resolution but it cannot provide an exact characterization of the whole lipidic species which can be present in the environment or their fraction within a single pixel. In this manuscript we will show data and measurements obtained on cells cultures, but the method is directly applicable also to tissues which are easily labeled by the cell permeant probe Nile Red [6].
Required equipment and software
Materials
-
•
Nile red probe (Thermo Fisher Scientific, USA, CN N1142).
-
•
Sample (cultured cells, tissues or living organisms).
-
•
Slides, plates or dishes for confocal microscopy (VWR, USA, CN 470210-568).
-
•
Standard 250 mm-square mesh grids for transmission electron microscopy (Sigma–Aldrich, USA, CN G1403).
-
•
Reference lipids (see Table 1).
Table 1.
Reference lipids.
| Molecule | Linear Formula | Structure Formula | Polar Head | Charge(pH = 7) | Class |
|---|---|---|---|---|---|
| Cholesteryl Stearate | C45H80O2 | 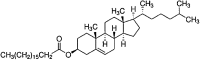 |
aliphatic | Neutral | NP |
| Oleic acid triglyceride (Trioleyn) | C57H104O6 | 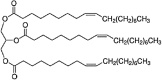 |
aliphatic | Neutral | NP |
| Cholesterol | C27H45OH | 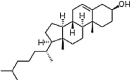 |
hydroxyl | Neutral | P |
| Dipalmitoleoyl-Glycero3-Phosphocholine (DPPC) Glycerophospholipid |
C40H80NO8P | 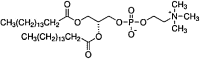 |
Phosphate + Cholin | Neutral (zwitterionic) | P |
| Dipalmitoyl-Phosphatidyl-l-serine (DPPS) Glycerophospholipid |
C38H73NO10P | 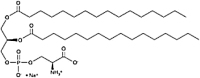 |
Phosphate + ammonium + carboxyl | Negative | P |
| Dipalmitoyl-Phosphatidyl-glycerol (DPPG) Glycerophospholipid |
C38H75O10P | 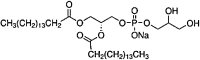 |
Phosphate+2 hydroxil | Neutral | P |
| 1-2-dioleoyl-rac-glycerol (dioleyn) | C39H72O5 | 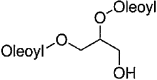 |
hydroxyl | Neutral | P |
| Dipalmitoyl-Phosphatidyl-ethanolammine (DPPE) Glycerophospholipid |
C37H74NO8P | Phosphate + ammonium | Neutral (zwitterionic) | P | |
| 1-Oleoyl-lisophosphatidic acid (LPHA) Glycerophospholipid |
C21H40O7P | 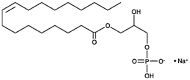 |
Phosphate | Negative | HP |
| Dioleoyl phosphatidic acid Glycerophospholipid |
C39H73O8P | 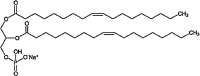 |
Phosphate | Negative | HP |
| Glycerol monoleate (monoleyn) | C21H40O4 | 2 hydroxil | Neutral | HP | |
| Oleic acid | C18H34O2 | Carboxyl | Negative | HP | |
| Palmitic Acid | C16H32O2 | Carboxyl | Negative | HP |
Equipment
A Confocal microscope equipped with
-
•
An excitation laser line in the range 450–500 nm, to achieve the best sensitivity.
-
•
A spectral detector with a detection range from 580 nm to 660 nm, or larger.
For this study we have used the A1-MP confocal head (NIKON, Japan), an inverted microscope (Eclipse Ti-e, NIKON, Japan) equipped with a 60x objective (1.4 NA), a 32 channels spectral detector (A1 DUS spectral detector Unit, NIKON, Japan), and a laser excitation module tuned to 488 nm (LU-N4 laser unit, LD laser, Output power at the fiber end of 15 mW, Nikon, Japan). Since the aim of the method is to characterize the micro-polarity of the lipidic environment with pixel resolution is important to use an objective with a high magnification (40x or higher). Nile Red emission spectrum range from 500 nm to about 750 nm, which are the lower and upper limits of the detectors that is possible to use. The method was tested with a variable number of channels (from 16 to 32) with different channel width (from 2.5 nm to 10 nm). The system is also equipped with an on-stage incubator (OKOLAB, Italy) set at T = 37 ± 0.3 °C, %5 CO2. The on-stage incubator is optional, but room-temperature has to be kept fixed to avoid temperature induced spectral-shifts in Nile red (tolerance 1%).
Software
-
•
Standard Acquisition Software for confocal microscope. For this study we employed NIS-Element (Nikon, Japan).
-
•
ImageJ or another similar program for the image pre-processing.
-
•
Provided software PhasorM.
Samples preparation
Probe Preparation, cells plating and labelling
Prepare 1 mM stocks of Nile Red in Dimethyl sulfoxide (DMSO) (Merck Millipore, USA, CN 102,952).
-
1
Prepare cultured cells, tissue or living organisms with the appropriate culture medium and plate them on the confocal dishes with the desired density. In this article we used cultured HT-29 cells, prepared as in [1].
-
2
Label cells with Nile red at 1μM final concentration (dilution 1:1000 in culture medium of the 1 mM stock) and incubate them in the dark at 37 °C for 30 min. If tissues are labelled, keep the same concentration if dimensions don’t exceed 2 mm × 2 mm × 2mm. Otherwise raise the concentration up to 5 μM until an enough signal to noise ratio is reached.
Reference Lipids
The measurements on reference lipids are strictly necessary only if there are some differences from our settings in the excitation wavelength (488 nm) or in the number and the range of the spectral detectors (32 channels, from 580 to 660 nm). Otherwise, it is possible to directly use the masks we provide with this article, avoiding buying the references lipids and repeat the measurements.
We used the reference lipids listed in the Table 1. Briefly, lipids were dissolved in ethanol (1 mg/ml). Then, 10 ml of lipid/ethanol solutions were dropped onto standard 250mm-square mesh grids for transmission electron microscopy. Ethanol evaporation (30 min at room temperature followed by 30 min at 37 °C) resulted in the progressive concentration and accumulation of lipids along the grid borders (Graphical abstract, A). Grids were subsequently covered with 50 ml of 300 nM Nile Red in PBS for 15 min and then observed with the same confocal spectral imaging settings used for observing cells.
Image acquisition
The procedure here reported is based on the use of the software NIS-Elements (Nikon, Japan) and the experimental setup described in the previous paragraph, but it can be easily adapted to similar equipment. These procedures are thought for helping any researcher to make the image acquisition, especially the ones who are not expert in the confocal microscopy field. The procedure is almost the same for both the acquisitions on the references and the samples, and minor differences will be underlined in the following procedure.
-
1
Turn on the microscope and launch the NIS-Elements AR 4.60.00 (or newer) acquisition software (Nikon, Japan).
-
2
To achieve optimal results, choose the objective with the highest magnification. We have utilised a 60x oil immersion objective (1.4 NA); in this case, it is necessary to put a drop of oil in the interface between the dish and the objective lens before starting the measurements.
-
3
Place the dish, or other kinds of support, on the microscope sample holder. (Note: As outlined above, it is important to avoid strong variations in temperature during measurements, to avoid temperature induced Nile red shifts and a wrong interpretation of results).
-
4
Set the excitation and acquisition parameters with the NIS software. Open the ribbon "View" > "Acquisition Control" > "A1plus Compact GUI" from the main window (Fig. 1). Push the SD button to switch on the spectral acquisition mode, and then click on the wheel icon in the same line of the panel (indicated with a red square in Fig. 1A). The spectral settings window opens (Fig. 1B), allowing to choose the excitation line and the detector range. Select the desired laser line (in the range 460–550 nm) from the first pop-up menu in the upper-right panel (blue square 1). In our settings, we used the cyan laser (wavelength 488 nm). Make sure to check the box at the left of the selected wavelength and to uncheck the box of the other excitation lines. From the same panel, it is also possible to set the number of the detector channels (32) and the wavelength resolution (2.5 nm) (upper left blue square 2), as well as the Start and the End of the range (respectively 580 nm, and 660 nm, lower right blue square 3 and 4). Push OK to save the settings and return to the Main Window. It is fundamental to use these same settings (excitation wavelength, range, number of channels and resolution of the detector) for all the measurements, both on the reference lipids and on the samples. Even a slight difference in the settings could lead to significant shifts in the phasor plot.
-
5
Before searching the focus and the field for the acquisition, set the pinhole radius dimension to achieve an optical sectioning of the sample. A common practical compromise to have both good z-resolution and a strong signal is to set the pinhole radius value close to 1 Airy units (AU). If the signal is weak it is possible to slightly increase the pinhole radius, but it will also enhance the number of photons from outside the focus plane which reach the detector, therefore the noise. The images showed in this manuscript were acquired using a pinhole radius of 1.2 AU (A1plus Compact GUI window on the NIS software, Fig. 1A). Set the Si HV (the detection system gain, bottom of Fig. 1A) at high values but not at the extreme ones (less than 90% of the whole range) and the laser power (bottom of Fig. 1A, below the Si HV slide bar) at low values, i.e. we use to keep it below 10.
-
6
If the sample is thin and transparent enough use the transmitted light to roughly find using the oculars the focus (eye light path). After that, turn off the lamp, switch to the laser path, press on the Remove Interlock button on the NIS A1plus Compact GUI window (Fig. 1A) and push on Scan to start lasering. Move slowly in the vertical axes to adjust the focus and then search in the horizontal directions a good field for an acquisition. For the images acquisition on the reference's lipids is necessary to focus on the regions where lipids accumulate, usually close to the grid borders. At this stage, set the number of pixels per column/line (Size) to 512 and the line average value to Normal (line below). These settings are required to speed up the image update and facilitate the search. If even at focus the signal is too low, turn up the Si HV and/or the laser power (bottom of Fig. 1A). In regulating these values, the user should consider that the overall aim is to get the highest signal to noise ratio (SNR) without damage the sample and saturate pixels, especially during the image acquisition (point 7). An higher operating voltage of the PMT (si HV) increase image brightness, but at the same time augment electric noise, and it is also possible to enter in the non-linear region of the detector. We usually keep the value below the 80%–90% of the range, which means less than 225 in the NIS settings. Otherwise, increasing the laser power improves the SNR, but can damage the sample and induce photo-bleaching effects. We usually use low values of the laser power, if possible below 10% of the maximum, especially while regulating the focus or searching for the field of acquisition.
-
7
Once the field is found, stop the Scan and change the parameters for the acquisition. Adjust the resolution to have a pixel size of about 200 nm (we fix Size to 1024, Fig. 1A). To reduce the signal to noise ratio, perform line averaging (line average to ∅2x or even more,see the line below of the same panel) and set the optimal dwell time (we set Frame/sec to ½, if the signal is low increase this value). Then, open the window A1 plus Scan Area ("View" > "Acquisition Control" > "A1plus Scan Area", Fig. 1C) to get a 2X optical zoom (by setting Zoom = 2). Select the acquisition region by moving the red square on the desired part of the field, and then right click on the square to activate the selected area (the colour of the square change to green). Push again on the Scan button and, if needed, optimise the Si HV and the laser power values. Variations in the excitation power as well as in the detector gain are allowed since the analysis relies on normalised emission spectrum values. It is crucial regulating both these values to limit as much as possible the presence of saturated pixels in every channel because they can significantly alter the shape of the emission spectrum; the NIS software, like all the other principal software used for the image acquisition, allows to directly visualize them on the images both during live visualization of the sample and on the acquired images.
-
8
Push on Capture button (Fig. 1A) to acquire the image.
Fig. 1.

The three panels of the NIS software from which it is possible to configure the most important settings of the acquisition. The A1plus Compact GUI window (A) of the SD mode acquisition allows to select the resolution (1024), the number of the line averages (∅2x), the Pinhole dimension (1.2 AU) and both the Si HV gain and the laser power through the laser bars. Clicking on the settings icon (red square in A) opens the window (B) for the settings of the excitation line and the spectral detector range. We selected the 488 nm laser line (blue rectangle 1) and a 32 channels spectral detector (blue rectangle 2) with a resolution of 2.5 nm (blue rectangle 2), from 580 nm (blue rectangle 3) to 660 nm (blue rectangle 4). (C) The A1 plus Scan Area window allows to select the acquisition region and the zoom level.
The success in segmentation of the lipids which will be described in the next sections is based on the quality of the images acquired, both of references and of samples. If the noise component is relevant, the distribution of the point in the phasor plot is wider and shifted closer to the origin of the G and S axis. In this case the segmentation can lose specificity. The “despeckle” filtering of the images that will be described in the next sections can just reduce the impact of the stochastic noise on the calculation of the phasor coordinates, so it is important to follow the suggestion given in the previous points and all the precautions to make good acquisitions and reduce background and noise.
Installation of the analysis software
Image processing and the whole phasor-based analysis can be performed with the stand-alone application PhasorM that we furnished with this article. The software was originally written using Matlab language (Mathworks, USA), and then converted to an executable file runnable even on computers which do not have Matlab through the automatic installation of the MATLAB Runtime library.
-
1
Double click on the PhasorM_installer file and wait until it installs the PhasorM.exe.
-
2
Open the PhasorM application by clicking on PhasorM.exe icon.
Analysis of the reference ROIs phasor plot for the masks creation
Image pre-processing
The first step consists in the determination, on the phasor plot, of regions of interests (ROIs) correspondent to the spectral emissions of reference lipids (see Table 1). These lipids are grouped according to the polarity headgroup in three classes: polar (P), non-polar (NP) and hyperpolar (HP). If the same excitation wavelength (488 nm) and spectral detectors (32 channels, from 580 to 660 nm) are used, there is no need to perform this step, since it is possible to relay on already determined regions. In this case skip ahead to Image Analysis section, in the next paragraph and use the files NP_Mask_95_noOverlap.mat, P_Mask_95_noOverlap.mat and HP_Mask_95_noOverlap.mat for recalling the masks.
To get the phasor fingerprint of the reference's lipids, we need to isolate them in the spectral images by cropping the sections where they accumulate. This operation has to be made before the analysis, by using any image processing software. Here we have used the open-source program ImageJ [7] (NIH).
-
1
Open the image in Image-J
-
2
Perform a “despeckle” of the images (menu “Process” > “Noise” > “Despeckle”) to ease the noise contribution in the spectral references data. This led to a more accurate determination of the region in the phasor plot associated to a specific micropolarity, by reducing the dispersion and the shift of the G and S values induced by the effect of noise. This operation can also be done with other similar software or directly with PhasorM.
-
3
As shown in the Graphical abstract (A), select a region within the grid where the lipid density is high and in which there are no artifacts (impurities or scratches). Lipids accumulations are clearly visible along the grid borders or at the centre part of the grid.
-
4
Crop the selected area (menu “Image” > “Crop”)
Masks creation
This step consists in processing reference images to build the masks on the phasor plot that will be used for identifying three different classes of lipids in the spectral images of cells.
Important: the application can only read tiff files, so if the images stacks are saved with other extension, convert them in a tiff format with other image analysis program or directly from the software used for the acquisition (i.e. NIS).
You can follow these instructions and create your own masks:
-
1
Open PhasorM application.
-
2
Select the ribbon “File” > “Open stack of images” and choose a stack of images of the reference lipids. This operation generates the phasor plot of the selected stack which is shown in the main window (at the left) with the overlap of the images (at the right).
-
3
Save the phasor data in a. mat file (for example test1.mat) that will be used in the next phase of the protocol to create the masks (“File” > “Save Data”).
-
4
Repeat the operations described in the points 2 and 3 for all the stacks of the references lipids.
For creating the masks that we used for the micropolarity classification we divided the reference lipids into three different classes. Here we show how to generate the Non-Polar mask from the phasor data, but the process is identical for the Polar and Hyper-Polar lipids. We identify and draw the position of the NP ROI considering data from two reference lipids, the Cholesteryl Stearate (CS), the Oleic acid triglyceride (TO) and a mixture of these two lipids (TO + CS).
-
5
Select the ribbon “Apps” > “Create Mask From Data”. This command opens the window Generate Masks that allows generating the masks from the reference lipid data.
-
6
Press the “Load Data” button and select the file you want to import (i.e. test1.mat). A window opens allowing to associate these data to a reference lipid. In this particular case TO is associated to I Data and CS to II Data. Confirm the operation by pressing OK. In the case of multiple acquisitions of the same reference, it is possible to store up to four acquisitions in the same group (Fig. 2C and D). After the CS and TO reference data are imported, the phasor plot that is characteristic for a Non-Polar lipid environment is displayed in the window (Fig. 2A). The pseudo-coloured scale of the 2-D histogram is proportional to the number of pixel having that particular position in the phasor plot. To delete the outliers and the less significant zones from the plot by excluding the points with lower statistical weight in the histogram, it is possible to cancel out the values below the 5th percentile. This operation can be done by setting the value 0.95 in the "Probability" box.
-
7
Save the data pushing the “Save Data” button (i.e. NP.mat).
-
8
Push on “Clear all data” and repeat the points 6-7-8 for the other two groups (P and HP).
Fig. 2.

The window for the masks creation from the measurements of the reference lipids. In the upper blue panel (A and B) is shown the generation of the phasor plot through the collection and the processing of the data from the NP reference lipids. Load Data button opens the window that allow to group the data if multiple measurements was made on the same kind of lipid or to create a new group (as shown for the CS between A and B). In C and D the final merge of the data for generating the P and NP masks respectively are reported.
It is very likely that some regions of the three areas in the phasor plot obtained through the previous operations may belong to two or more groups. This is essentially due to the noise, which widens the distribution of the points in the phasor plot and, in few cases, causes a fraction of point to fall closer to regions associated to another reference lipid. This must be avoided because the method is intrinsically based on the decision to divide the lipids in a discrete number of groups according to the micropolarity value which, on the contrary, varies continuously.
So, as the objective is to assign each point of the phasor plot to at most one group (NP, P, HP), it is essential to avoid these overlaps. For this purpose:
-
9
Select the ribbon “Options” > “Modify existing masks to avoid their overlap” in the same window that we used before ("Generate Mask") and open the three data files pushing the "Add" buttons in the "Manage the masks overlap" window (Fig. 3). The overlap between the different groups is automatically quantified and reported by the software (blue rectangle 1 in Fig. 3)
-
10
To avoid the overlap, select the masks in the pop-up menu at the bottom of the window and push the last "Ok" button ("Avoid the overlap following the % criteria"). The algorithm assigns the different pixels of the phasor plot to a specific class (HP, P or NP) based on the weight that the pixel has in them. For example, if both the 0.5% of the NP region points and the 0.05% of the P region points fall in a certain point of the phasor plot, that point will be assigned to the NP ROI.
-
11
Save the three masks using the button “Save Mask” (i.e. NPmask.mat, Pmask.mat and HPmask.mat).
Fig. 3.

The window allows to manage the masks to avoid the masks overlap. After loading the masks data of the reference lipids NP, P and HP, the software automatically calculates and reports the percentage of area intersections of the different groups (blue rectangle 1). Select the number of the masks from the pop-up menus and eliminate the overlap by pushing on the lower Ok button in the figure (Avoid the overlap following the % criteria). This avoid the intersections between the three masks.
Phasor-based analysis of cells: micropolarity map creation, polarity profile, metabolic index
Image analysis
After the creation of the masks, we can start the analysis of the cells stacks of images for the division and the quantification of their lipids into the three different classes.
-
1
Open one of the stacks of images that you want to analyse, as described in the point 2 of the paragraph above (remember: PhasorM can only opens tiff files). To reduce the impact of the stochastic noise in the calculation of the phasor coordinates make a “despeckle” by selecting the ribbon “Image Processing” > “Median Filter” (by default it is suggested to fix the value 3 for both n and m parameters). This process thickens the points distribution in the phasor plot. After this it is necessary to set a threshold (“Image Processing” > “Threshold”) which will depend on the image characteristics. This operation removes the presence of the background pixels in the image and hence in the phasor plot, without altering the contribution of the pixels carrying information about lipids micropolarity.
-
2
Segment the image by recalling the masks that you have created before and drawing them on the phasor plot: select the box "From Mask" on the top and left of the image and push the "Fix Roi" button, this opens a window for choosing the file of the saved mask (i.e. NPmask.mat).
-
3
Repeat this operation for each of the three classes: we have first recalled the Non-Polar lipids data, then the Polar and the Hyper Polar.
Data extraction
The image segmentation is now complete, as shown in the right panel of the main window (Fig. 4B). The software assigns three different colours (blue, green and red) to the pixels which fall respectively in the first, the second or the third area limited by the mask's boundaries. The image reveals the distribution of different classes of lipids within the cells, aiding to distinguish intracellular membranes, vesicles and lipid droplets. Brightness and contrast can be regulated through the panel in the upper right of the main window (Fig. 4C), and just to the left of it, there is the "Save Image" button (red rectangle on the right of Fig. 4A) which can be used for saving the image in the desired format. Similarly, to save the phasor plot, push on "Save Phasor Plot" button (red rectangle on the left of Fig. 4A) that will open a window, from which is it possible to make many different operations on the image. The image processing and the operations described above also produce some relevant quantitative information, which indicates the metabolic state of the cells and the fraction of the lipids classes within the sample. As described in [1], the most significant index which summarises the condition of the lipidic metabolism is the φav value. It is defined as the arctangent of the ratio between the average value of the sine and cosine, calculated for the angles between the points in the phasor plot and the G axis (graphic explanation and equation in Fig. 4E). Other relevant values reported in the left of the main window (Fig. 4D) are Rav, the average of the second polar coordinates, Gav and Sav, the average of the two coordinates in the Cartesian system. Just below the panels that report the previous four values, other three boxes display the fractions of lipids belong to the different groups identified by the masks (#1 ROI, #2 ROI, #3 ROI in Fig. 4D). To find, for example, the percentage of the blue pixels in the image compared with all those enlightened it is necessary to take the value in the corresponding box, namely the #1 ROI, and multiply by 100.
Fig. 4.

An overview of the software main window (A) and the principal quantitative results which derive from the application of the workflow (D and E). The phasor analysis algorithm implemented to a spectral stack of cells or tissues generate the phasor plot (A). By recalling the masks created before, it is possible to segment the image and locate the position of the different kind of lipids in the original image (NP blue, P green and HP red, panel B). The appearance of the image can be controlled through the commands in the upper right of the main window (C). The fractions of the NP, P and HP lipids are reported at the left (panel D, #1 ROI, #2 ROI and #3 ROI respectively), as well as other important quantities calculated by the phasor analysis: Gav, Sav, φav and Rav. In particular, the φav value (graphically and mathematically described in the E panel) emerges as the most important index which can better summarize the metabolic state of the sample.
All of these quantities can be exported to a new or an existent Excel file (.xlxs) by selecting the ribbon “File” > “Export Data to Excel” and then choosing from the displayed menu what data would save. It is also possible to collect all the data in a Matlab file, including the phasor plot and the original images, by choosing the option "Save Data" in the "File" ribbon from the main window. In this case, all the data will be collected in a structure within the Matlab file (.m) which is called Phasor.
Conflict of Interest
The authors declare no conflicts of interest related to this work.
Acknowledgements
This work was supported by grants from Universita Cattolica del Sacro Cuore contributed to the funding of this research project and its publication(Fondi di Ateneo, Linea D1 2017 to GM and Linea D.3.1 2018 to GM. The confocal analysis has been performed at Labcemi, UCSC, Rome. The authors thank Mr. Mario Amici for the excellent technical support, and the reviewers for providing valuable input in this submission.
Footnotes
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.mex.2018.10.010.
Appendix A. Supplementary data
The following are Supplementary data to this article:
References
- 1.Maulucci G., Di Giacinto F., De Angelis C., Cohen O., Daniel B., Ferreri C., De Spirito M., Sasson S. Real time quantitative analysis of lipid storage and lipolysis pathways by confocal spectral imaging of intracellular micropolarity. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids. 2018;1863:783–793. doi: 10.1016/j.bbalip.2018.04.004. [DOI] [PubMed] [Google Scholar]
- 2.Greenspan P., Mayer E.P., Fowler S.D. Nile red: a selective fluorescent stain for intracellular lipid droplets. J. Cell Biol. 1985;100:965–973. doi: 10.1083/jcb.100.3.965. http://www.ncbi.nlm.nih.gov/pubmed/3972906 (Accessed 29 September, 2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fereidouni F., Bader A.N., Colonna A., Gerritsen H.C. Phasor analysis of multiphoton spectral images distinguishes autofluorescence components of in vivo human skin. J. Biophotonics. 2014;7(8):589–596. doi: 10.1002/jbio.201200244. [DOI] [PubMed] [Google Scholar]
- 4.Jameson D.M., Gratton E., Hall R.D. The measurement and analysis of heterogeneous emissions by multifrequency phase and modulation fluorometry. Appl. Spectrosc. Rev. 1984;20:55–106. doi: 10.1146/annurev.bb.13.060184.000541. [DOI] [PubMed] [Google Scholar]
- 5.Stringari C., Nourse J.L., Flanagan L.A., Gratton E. Phasor fluorescence lifetime microscopy of free and protein-bound NADH reveals neural stem cell differentiation potential. PLoS One. 2012;7 doi: 10.1371/journal.pone.0048014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crociati M., Di Giacinto F., Manuali E., Stradaioli G., Sylla L., Monaci M., Maulucci G., De Spirito M. Systemic profiling of ectopic fat deposits in the reproductive tract of dairy cows. Theriogenology. 2018;114:46–53. doi: 10.1016/j.theriogenology.2018.03.026. [DOI] [PubMed] [Google Scholar]
- 7.Rueden C.T., Schindelin J., Hiner M.C., DeZonia B.E., Walter A.E., Arena E.T., Eliceiri K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics. 2017;18:529. doi: 10.1186/s12859-017-1934-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.