

Abstract
In all eukaryotes, the mechanistic target of rapamycin (mTOR) signaling emerges as a master regulator of homeostasis, which integrates environmental inputs, including nutrients, energy, and growth factors, to regulate many fundamental cellular processes such as cell growth and metabolism. mTOR signaling functions through two structurally and functionally distinct complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), which correspond to two major branches of signal output. While mTORC1 is well characterized for its structure, regulation, and function in the last decade, information of mTORC2 signaling is only rapidly expanding in recent years, from structural biology, signaling network, to functional impact. Here we review the recent advances in many aspects of the mTORC2 signaling, with particular focus on its involvement in the control of cell metabolism and its physiological implications in metabolic diseases and aging.
1. Introduction
Mechanistic target of rapamycin (mTOR) is an evolutionarily conserved Ser/Thr kinase that belongs to the phosphatidylinositol-3-kinase-related kinase (PIKK) family. mTOR functions through two structurally and functionally distinct complexes, mTORC1 and mTORC2 [1]. Although the two mTOR complexes share three conserved subunits, mTOR, mLST8 (mammalian lethal with SEC13 protein 8, also known as GβL), and DEPTOR (DEP domain containing mTOR-interacting protein), they also contain complex-specific components. While mTORC1 comprises Raptor (regulatory-associated protein of mTOR) and PRAS40 (proline-rich Akt substrate of 40 kDa), mTORC2 distinctively contains Rictor (rapamycin-insensitive companion of mTOR) and mSin1 (mammalian stress-activated protein kinase-interacting protein 1) as well as Protors-1 and -2 (protein observed with Rictors-1 and -2, respectively) [2] (Figure 1(a)). In addition to this compositional difference, the two complexes also differ in their response to rapamycin with mTORC1 activity being acutely inhibited by rapamycin while mTORC2 only responding to long-term treatments [3].
Figure 1.

The protein composition of mTORC1 and mTORC2. (a) Schematic showing main molecular components and signals sensed by mTORC1 and mTORC2 (in the rectangles) and the processes they regulate to control cell growth and survival. With high sensitivity to rapamycin, mTORC1 senses oxygen, glucose, amino acids, energy, and growth factors to regulate cell growth by inhibiting autophagy and promoting several anabolic reactions, including synthesis of protein, lipids, and nucleotides. mTORC2 is insensitive to acute rapamycin treatment but responds to growth factors and insulin to regulate lipid and glucose metabolism, as well as survival and proliferation. (b) Schematic representation of mTORC2 core components. Domains of known function or structural motifs are indicated. Description of the abbreviations listed is contained within this review.
mTOR signaling has been considered as a master regulator of homeostasis, which controls many anabolic and catabolic processes in response to nutrient availability [1]. This is especially well established in mTORC1 signaling through its regulation of anabolic process. Notably, recent studies with the utilization of new pharmacological tools and genetic models demonstrate that mTORC2 also plays fundamental roles in regulating cell metabolism [4].
In this review, we will summarize the recent advances in mTORC2 signaling, including the structure and function of its subunits, its upstream regulators, and downstream effectors. Particularly, the emphasis will be given to the current understanding of mTORC2 in cellular metabolism, including glucose, lipids, amino acids, nucleotides, and reactive oxygen species (ROS), and their impact in metabolic disorders and longevity.
2. The mTORC2 Signaling
2.1. Composition of mTORC2
mTORC2 is composed of several protein subunits, of which four are considered as core, essential components: mTOR and mLST8 shared in both mTOR complexes and Rictor and mSin1 specific to mTORC2 (Figure 1(b)).
mTOR is the catalytic subunit of the mTOR complexes, composed of over 2500 amino acids and evolutionarily conserved in eukaryotes, from yeast to human [1]. Structurally, the N-terminal half of the mTOR protein contains more than 30 tandem prototypical HEAT (Huntingtin, EF3A, ATM, and TOR) repeats forming two α-solenoids, followed by a FAT (FRAP, ATM, and TRRAP) domain and a FRB (FKBP12/rapamycin binding) domain, which is linked to the kinase domain, ending with the FATC (FAT C-terminus) domain at the C-terminus (Figure 1(b)) [5]. The kinase domain of mTOR has a bilobal structure that is in an intrinsically active conformation, with a catalytic mechanism remarkably similar to canonical protein kinases [6]. However, its active site is usually recessed due to the FRB domain and an inhibitory helix protruding from the catalytic cleft, indicating that its accessibility is strictly regulated [6]. This regulation points to substrate recruitment as a major mechanism controlling the kinase activity.
mLST8 is another highly conserved subunit shared between the two mTOR complexes [7]. The entire protein of 326 amino acids is composed of seven WD40 repeats, which fold into a β-propeller that generally serves as a scaffold for protein interactions. Although mLST8 stably binds to the kinase domain of mTOR in both mTOR complexes, it appears to differentially regulate the kinase activity in the two complexes. Although deletion of mLST8 has no clear effect on mTORC1 activity and integrity, its absence completely abolishes mTORC2 activity, indicating that it is essential for mTORC2 function, not mTORC1 [8].
Rictor is a defining member of mTORC2, containing over 1700 amino acids [3, 9]. Despite of its large size, no clearly identifiable domains or motifs can be mapped to it except several sections that are conserved from yeast to human [3]. Nonetheless, Rictor contains several armadillo-like helical repeat clusters in its N-terminal portion, which extensively interact with mTOR and have a profound impact on mTORC2 assembly [10, 11]. The importance of Rictor in mTORC2 is determined by the observation that depletion of Rictor disrupts mTORC2 assembly and activity, suggesting that it plays a vital role in the integrity and stabilization of mTORC2 [12].
mSin1, encoded by the MAPKAP1 (mitogen-activated protein kinase-associated protein 1) gene, is another specific component of mTORC2, discovered by its interaction with Rictor [13, 14]. mSin1 is highly conserved in vertebrates and contains four possible domains (Figure 1(b)). The N-terminal TORC region is responsible for the interaction with mTOR and Rictor as its natural isoform without this region fails to constitute mTORC2 [14]. The C-terminal region contains a pleckstrin homology (PH) domain, which seems to be not necessarily required for mTORC2 assembly and activity but may function for its membrane localization [14]. The central region is the most conserved section and hence called conserved region in the middle (CRIM), which is linked to a RAS-binding (RB) domain at its C-terminal (Figure 1(b)). Structure analysis indicates that CRIM is an ubiquitin-like domain with a prominent acidic loop that is primarily responsible for the recruitment of mTORC2 substrates [15]. Therefore, mSin1 is considered to mainly function for substrate recruitment and selection, and the exact mechanisms, however, await for further investigation.
2.2. Structure of mTORC2
Human mTORC2 has an overall structural organization similar to that of mTORC1 and TORC2 of Saccharomyces cerevisiae [10, 11, 16]. It is a rhombohedron in shape formed by dimerization of two sets of the key subunits in the mTOR complexes, which are shaped in pseudo-2-fold symmetry and a prominent central cavity with the dimer of mTOR and mLST8 as a core [11]. In addition, the principal site for mTOR interacting with Rictor in this architecture is the same site occupied by Raptor in mTORC1, thereby sterically excluding Raptor incorporation in mTORC2. However, the current studies can only resolve the interaction between the N-terminal regions of mTOR and Rictor proteins in high resolution, whilst the interactions between the catalytic domain of mTOR and C-terminal part of Rictor and mSin1 have not been clearly determined due to their high flexibility and mobility [10, 11]. Although the structure of mTORC2 shares several features with that of mTORC1, the two complexes diverge remarkably in their overall size, the surface area of the protomer interface, the volume and shape of the central cavity, and, more importantly, the accessibility of the kinase domain. These differences are likely resulted from the distinct molecular compositions and have profound impact on the functions and regulations of the two complexes, rendering mTORC2 insensitive to rapamycin [11, 16]. The determination of the high-resolution mTORC2 architecture at its catalytic kinase domain is required to further decipher the structural organization, regulation, and activation of this important kinase.
2.3. Effectors of mTORC2
mTORC2 has a pleiotropic effect on cellular properties by means of activating/phosphorylating its downstream effectors, which are currently recognized to be mainly the members of the AGC kinase family, including PKB (protein kinase B, also called Akt), PKC (protein kinase C), and SGK1 (serum- and glucocorticoid-induced kinases 1) (Figure 2(a)) [17]. Upon activation by mTORC2, these AGC kinases also regulate multiple downstream targets, leading to cascading effects that impact on cell behavior and functions.
Figure 2.

The signaling network of mTORC2. (a) Schematic representation of AGC kinases downstream of mTORC2. The major positions for phosphorylation are indicated. (b) As ligands, growth factors bind to the membrane receptor, receptor tyrosine kinase (RTK), which activates PI3K to phosphorylate PIP2 to PIP3 at the plasma membrane. PTEN (phosphatase and tensin homolog) dephosphorylates PIP3 and is a key negative regulator of PI3K signaling. PIP3 or other unknown factors activate mTORC2 in distinct manners to promote the phosphorylation of conserved motifs in several AGC kinases (Akt, PKC, and SGK1). For maximal activation, Akt is phosphorylated at T308 and S473 by PDK1 and mTORC2, respectively, and subsequently promotes the activation of mTORC1, which is characterized by phosphorylation of several downstream effectors, including S6K, 4E-BP1, and ULK1. There are several other upstream regulators which can also regulate mTORC2 activity, including amino acids, ROS, ribosome, TSC complex, and GTPases, through distinct mechanisms. Description of the abbreviations listed is contained within this review.
2.3.1. Akt
Akt, a family of three members (Akt1, Akt2, and Akt3), is the most characterized effector of mTORC2 and a key factor downstream of PI3K signaling, which has important roles in cell survival, proliferation, and growth. Akt is located in cytosol in the absence of stimuli and is recruited to the plasma membrane via the interaction between its PH domain and phosphatidylinositol (3,4,5)-trisphosphate (PtdIns(3,4,5)P3) that is generated by PI3K activation. This leads to Akt phosphorylation at two residues, in the case of Akt1, T308 in the activation loop (A-loop) by PDK1 (phosphoinositide-dependent protein kinase 1) and S473 in hydrophobic motif (HM) by mTORC2 to ensure a full activation of Akt [18, 19]. Although Akt-S473 can also be phosphorylated by other kinases such as integrin-linked kinase (ILK) and DNA-dependent protein kinase (DNA-PK) in a cell/tissue-dependent manner [20, 21], it is predominantly phosphorylated by mTORC2 as deleting any of the mTORC2 key components results in dramatic abrogation of this phosphorylation [8, 12, 13]. Interestingly, although phosphorylation of both Akt-T308 and -S473 is required for a full activation of Akt, defective Akt-S473 phosphorylation affects only a subset of Akt targets in vivo, such as FoxO1/3a (forkhead box O1 and O3a), while other Akt targets, including TSC2 (tuberous sclerosis complex 2) and GSK3 (glycogen synthase kinase 3), and its downstream effector mTORC1 remain unaffected [13].
In addition to Akt-S473, mTORC2 has also been identified as the kinase for the phosphorylation of Akt-T450 in its turn motif (TM) to ensure proper folding and maturation of the Akt protein [22, 23]. Intriguingly, mTORC2 appears to phosphorylate Akt-T450 by a different mechanism from that of phosphorylating Akt-S473. Akt-S473 is phosphorylated by a canonical posttranslational mechanism and can be resembled by in vitro kinase assay with immunoprecipitated mTORC2, whereas Akt-T450 phosphorylation is thought to be performed by a cotranslational mechanism involving ribosomes and cannot simply be achieved by in vitro kinase assay [23, 24].
2.3.2. PKC
PKC is a protein family of 11 members, which are categorized into three classes: the conventional (c) PKC (α, β1, βII, and γ), the novel (n) PKC (δ, ε, θ, and η/Λ), and the atypical (a) PKC (ζ and ι/λ) [25]. Both cPKCs and nPKCs are identified as mTORC2 substrates, being phosphorylated by mTORC2 at their HM and TM motifs [22, 23, 26, 27]. Among them, PKCα is the first identified and the most characterized substrate of mTORC2 and plays an important role in regulating actin cytoskeleton function, thereby affecting cell shape and mobility [3, 9]. However, the precise mechanisms by which mTORC2 phosphorylates PKCα remain ambiguous as neither HM nor TM phosphorylation of PKCα could be achieved by in vivo kinase assay with immunoprecipitated mTORC2 [23]. Furthermore, the functions of these phosphorylations in PKCα are also not very clear, but they have a role in protein stabilization and solubility, thus affecting PKC activity [28].
2.3.3. SGK1
SGK1 is also phosphorylated and activated by mTORC2 at its S422 of HM, similar to that of Akt [29], and mSin1 has been shown to have a role in this phosphorylation, mediating the interaction between mTORC2 and SGK1 [30]. mTORC2-targeted SGK1 activation functions to regulate sodium transport as well as cell survival [31].
2.4. Regulation of mTORC2
The regulation of mTORC2 is much understudied and less understood than that of mTORC1 [1]. Nonetheless, increasing evidence in the last decade, particularly recent years, has emerged that mTORC2 activity could be affected by many factors via various mechanisms, which might directly or indirectly affect the localization, function, and stability of mTORC2 components as well as substrate availability.
2.4.1. Membrane Localization and mTORC2 Activation
The plasma membrane plays an important role in mTORC2 activation, and a proportion of mTORC2 is located at the plasma membrane of mammalian cells to activate Akt [32, 33]. However, how mTORC2 is activated at the plasma membrane has been a mystery. It has been long thought that growth factors are the major signals to activate mTORC2 by means of the PI3K pathway (Figure 2(b)). Upon PI3K activation by insulin or other growth factors, PtdIns(4,5)P2 is phosphorylated to PtdIns(3,4,5)P3 to enable its binding to mSin1-PH, which subsequently releases the mSin1-PH inhibition on the mTOR kinase domain to activate mTORC2 activity. This interaction also enables the recruitment of mTORC2 to the plasma membrane where it meets and phosphorylates Akt that is also recruited to the plasma membrane via the Akt-PH domain [34]. However, a recent study, using biochemical labelling and imaging approaches, showed that the plasma membrane-associated mTORC2 is constitutively active and is independent of growth factor/PI3K signaling and that only the recruitment of substrate Akt to the plasma membrane requires the activation of PI3K [35]. Nevertheless, the mTORC2 activity at endosomes is sensitive to PI3K, suggesting the existence of spatially distinct mTORC2 subpopulations in response to growth factors. These studies raise further questions on whether and how environmental cues, such as growth factors, regulate mTORC2 activity.
2.4.2. Component Modification and mTORC2 Activation
In response to upstream signals, mTORC2 components are susceptible to PTMs (posttranslational modifications), including phosphorylation, acetylation, and ubiquitination, which might serve to fine tune mTORC2 assembly, disassembly, activation, inactivation, or, perhaps, substrate recruitment.
The mTOR protein is phosphorylated at T2173 of the ATP-binding site in the kinase domain in an Akt-dependent manner, which impairs mTORC2 activity, thus functioning as a negative feedback to control mTORC2 activity [36]. The mTOR protein is also phosphorylated at S2481, and this phosphorylation is shown to depend on the intactness of mTORC2 activity but is unclear about its function, which is thus mainly used as a marker of mTORC2 [37]. The phosphorylation of mTOR at S2448 is thought to be associated with mTORC1 rather than mTORC2 [37].
The mTORC2 activity is also regulated by ubiquitination of mLST8 and DEPTOR. The K63-linked polyubiquitination of mLST8 by TRAF2 E3 ubiquitin ligase disrupts mTORC2 formation, thereby reducing mTORC2 activity. In contrast, deubiquitination of mLST8 by OTU domain-containing protein 7B (OTUD7B) deubiquitinase promotes its interaction with mSin1, facilitating the formation of mTORC2 and enhancing its activity [38]. DEPTOR, an inhibitory subunit of both mTOR complexes, is phosphorylated by mTOR and casein kinase 1 (CK1) in response to growth factor stimulation, which results in its degradation and mTOR activation [39–41].
Moreover, the mTORC2 scaffold protein Rictor also contains multiple modifiable sites, which might impact on mTORC2 activity. Rictor-T1135 has been found phosphorylated by S6K1, but whether this phosphorylation has any effect on mTORC2 activity remains controversial [42, 43]. In addition, Rictor has also been reported to be phosphorylated by GSK3 at S1235 or T1695. Although both appear to negatively affect mTORC2 activity, they function through distinct mechanisms [44, 45]. The phosphorylation of Rictor-S1235 interferes with the interaction between mTORC2 and the substrate Akt, whilst the phosphorylation of Rictor-T1695 promotes FBXW7-mediated degradation of Rictor [44, 45]. Furthermore, acetylation of Rictor at multiple sites between residues 1040–1140 has been reported to activate mTORC2 [46, 47].
Phosphorylation of mSin1 has also been shown to have an effect mTORC2 activity but with disputed results. In one report, mSin1-T86/T398 were shown to be phosphorylated by S6K or Akt, resulting in inhibited mTORC2 activity [48], whilst in other reports, Akt-mediated mSin1-T86 phosphorylation was shown to enhance mTORC2 [49, 50]. The reason for this discrepancy remains unclear. In addition, mSin1-S260 was shown to be phosphorylated by mTOR under energy-sufficient condition to stabilize mSin1, thus enhancing mTORC2 activity [51].
2.4.3. Other Factors
In addition to the above regulators, some other factors are also emerging to have a role in the regulation of mTORC2 activity, such as ribosomes, small GTPases, TSC complex, and amino acids (AAs). It is revealed that mTORC2 activity requires well-assembled intact ribosomes and that the insulin-stimulated PI3K pathway promotes their physical interaction [52]. The involvement of ribosomes in mTORC2 activation is supported by the finding of cotranslational phosphorylation of Akt-T450 [24]. Therefore, ribosomes might serve as a scaffolding platform that facilitates mTORC2 to phosphorylate some of its targets. However, the precise mechanism by which mTORC2 is activated by association with ribosomes remains to be elucidated. TSC complex and small GTPases (e.g., Rhy1, Rit, and Rac1) have been demonstrated to positively regulate mTORC2 activity by physically interacting with mTORC2 components [53–55]. Moreover, mTORC2 activity can be activated by glutamine depletion mediated by Sestrin2 with an unestablished underlying mechanism [54, 56].
3. mTORC2 in Cell Metabolism
3.1. Glucose Metabolism
Emerging evidence suggest that mTORC2 plays an important role in glucose metabolism and homeostasis (Figure 3(a)). mTORC2 signaling affects many aspects of glucose metabolism, including glucose uptake, glycolysis, gluconeogenesis, and oxidative phosphorylation [57–63].
Figure 3.

mTORC2 in cell metabolism. (a) mTORC2 promotes glucose metabolism via glycolysis and PPP (pentose phosphate pathway). In response to growth factors, mTORC2 activates glucose catabolism through two main factors, Akt and c-Myc. Akt activates glycolysis at both transcriptional and posttranslational levels. c-Myc enhances the expression of genes involved in glycolysis and PPP. Activated factors are shown in red letters; suppressed factors are shown in blue. Solid line indicates direct regulation; dash line indicates indirect regulation. (b) mTORC2 promotes lipogenesis via Akt-dependent and -independent mechanisms. mTORC2 activates, via Akt, SREBP and ChREBP, two transcription factors for the expression of lipogenic genes, such as ACLY, ACC, FAS, and SCD1. mTORC2 may also stimulate lipogenesis by activating mTORC1 in an Akt-independent manner. (c) mTORC2 regulates amino acids and nucleotide metabolism. mTORC2 regulates amino acid transport by modulating activity of amino acid transporters, SLC7A5 and SLC38A2, and antiporter, xCT. In addition, mTORC2 activates glutamine transporters, SLC1A5 and SLC38A5, via c-Myc to promote glutamine uptake. By activating glycolysis, mTORC2 increases the production of 3-phospholycerate and pyruvate, which can be used to synthesize serine and alanine. mTORC2 promotes glutaminolysis via c-Myc, which transforms glutamine to glutamate and aspartate. Purine synthesis can be stimulated by mTORC2 through Akt-mediated activation of transketolase. Description of the abbreviations listed is contained within this review.
3.1.1. Glucose Uptake
The regulation of glucose uptake is a key determinant of glucose metabolism [64]. Tissue-specific Rictor-knockout mice exhibit compromised glucose uptake in the liver, adipose tissue, and muscle while the underlying mechanisms seem to be divergent and tissue type dependent [58–61]. In the muscle, deletion of Rictor results in the failure of Akt-mediated AS160 phosphorylation in response to insulin stimulation, which subsequently decreases the plasma translocation of GLUT4, a glucose transporter [59]. In the liver, the decreased glucose uptake in Rictor-null cells is mainly resulted from a reduction in the expression and activity of glycolysis rate-limiting enzyme glucokinase (GK) [61]. In adipose tissue, mTORC2 contributes to glucose uptake mainly by regulating either hexokinase (HK, isoenzyme of GK) or GLUT4 [57, 58, 60]. In brown adipose tissue (BAT), mTORC2 activates cytosolic HK activity through an Akt-S473 phosphorylation-associated mechanism, which has no effect on GLUT1 and GLUT4 translocation [60], while, in white adipose tissue (WAT), mTORC2 stimulates the plasma membrane translocation and gene expression of a glucose transporter, GLUT1 or GLUT4, in an Akt-independent manner [58, 60].
3.1.2. Glycolysis
mTORC2 also regulates glucose metabolism by facilitating glycolysis. In Rictor-knockout liver, glycolysis is significantly reduced due to the downregulation of glycolytic genes, including GK, liver-type pyruvate kinase (L-PK), and carbohydrate response element-binding protein (ChREBP), and the decreased enzymatic activity of GK [61]. However, this Rictor deficiency seems to have no effect on overall glycolysis in the whole body [63]. In contrast, adipose-specific Rictor ablation causes decreased glycolysis in fat as well as in the whole body, possibly resulting from decreased HK activity [57, 58, 60]. mTORC2 is able to increase HK activity and glycolysis via Akt-dependent and -independent mechanisms [62]. In glioblastoma, mTORC2 upregulates the expression of glycolytic genes, including HK, by increasing the levels of transcription factor c-Myc. Mechanistically, mTORC2 phosphorylates and inactivates HDAC (histone deacetylase), leading to the increase of FoxO1 and FoxO3 acetylation. The acetylation in FoxOs attenuates their transcriptional activity and reduces the expression of miR-34-c that suppresses c-Myc expression [65]. Therefore, mTORC2 may also promote glycolysis through multiple mechanisms depending on tissue/cell types.
3.1.3. Gluconeogenesis
Gluconeogenesis is a glucose production process that utilizes pyruvate to produce glucose. Although sharing several enzymes with glycolysis implements reversible reactions, gluconeogenesis also has its specific enzymes that catalyze key irreversible steps (Figure 3(a)). For example, phosphoenolpyruvate carboxykinase (PEPCK) catalyzes the formation of phosphoenolpyruvate (PEP); fructose 1,6-bisphosphatase converts fructose 1,6-bisphosphatase to fructose 6-phosphate and glucose-6-phosphatase (G6Pase), finally turning glucose 6-phosphate to glucose. Gluconeogenesis is important for maintaining blood glucose levels during starvation and is reciprocally regulated with glycolysis [66]. In contrast to glycolysis, mTORC2 functions to suppress gluconeogenesis, and therefore, in the absence of Rictor, the mice exhibit hyperglycemia due to increased gluconeogenesis [58, 61, 63]. The effect of mTORC2 on gluconeogenesis mainly results from the upregulation of several key gluconeogenic enzymes, such as G6Pase and PEPCK, through Akt-mediated FoxO1-phosphorylation, which leads to nuclear exclusion of FoxO1 and reduction of its transcriptional activities [67, 68]. In addition, mTORC2-mediated Akt activation is also responsible for the phosphorylation and inhibition of PGC1α (peroxisome proliferator-activated receptor gamma coactivator 1-alpha), a transcription coactivator involved in activating gluconeogenic gene expression [69].
3.1.4. Oxidative Phosphorylation
It is noteworthy that both mTORC2 effectors of Akt and c-Myc have inhibitory effect on oxidative phosphorylation via activation of HK2 and pyruvate dehydrogenase kinase 1 (PDK1), respectively [62, 70, 71]. Indeed, Rictor-deficient cells and mice exhibit elevated mitochondrial respiration. Moreover, activated Akt signaling stimulates the oxidative to glycolytic metabolic shift in the muscle [72].
Taken together, mTORC2 regulates glucose metabolism by promoting glucose uptake and glycolysis and inhibiting gluconeogenesis and oxidative phosphorylation.
3.2. Lipid Metabolism
Increasing evidence demonstrates that mTORC2 also plays a pivotal role in lipid metabolism by increasing lipogenesis while suppressing lipolysis and fatty acid β-oxidation (Figure 3(b)) [73]. In liver-specific Rictor-knockout mice, both hepatic and serum triglyceride levels exhibited a reduction [61], while in adipose-specific Rictor-knockout mice, only serum triglycerides were decreased [57, 58]. Nonetheless, both knockout strains showed altered lipid composition and metabolism [57, 58, 61].
3.2.1. Lipogenesis
Rictor deficiency results in reduced lipogenesis, which is not only due to decreased glucose uptake (discussed above) but also due to decreased expression of genes encoding lipogenic enzymes, such as acetyl-CoA carboxylase (ACC), ATP-citrate lyase (ACL), fatty acid synthase (FAS), and stearoyl-CoA desaturase (SCD1) [58, 61, 63, 74]. Two transcription factors, ChREBP and SREBP1c (sterol regulatory element-binding protein 1c), regulate the expression of these key lipogenic enzymes [75, 76], and mTORC2 has been reported recently to be an upstream regulator of ChREBP and SREBP1c and therefore lipogenesis [58, 61].
In adipose-specific Rictor-knockout mice, ChREBP expression is decreased, and this reduction can be rescued by restoration of glucose uptake and glycolysis, suggesting that mTORC2 positively regulates ChREBP expression through modulating glucose flux and metabolism [77, 78]. ChREBP protein can undergo multiple different posttranslational modifications, which can impact on its nuclear localization and transcriptional activity [78]. It is thought that high glucose concentration leads to the dephosphorylation of ChREBP, enabling its nuclear entry, thus enhancing its transcriptional activity. Furthermore, ChREBP has two isoforms from alternative promoters, a canonical abundant α isoform and a novel β isoform. Glucose flux activates the α isoform, which induces the expression of the less abundant but more active β isoform to activate the gene expression of lipogenic enzymes [77]. Interestingly, Rictor-knockout adipose tissue exhibits reduced ChREBP-β expression without altering ChREBP-α protein levels [58], implying that mTORC2 affect ChREBP expression and activity probably through affecting ChREBP-α protein modification.
Unlike ChREBP, mTORC2-mediated SREBP1c activation is through a glucose uptake-independent mechanism [61, 74]. In both liver-specific and Myf5 lineage-specific Rictor-knockout mice, it showed reduced expression of SREBP1c, which may account for the changes detected in lipogenesis [61, 74]. In both studies, downregulation of SREBP1c expression by mTORC2 deficiency appeared to be associated with the defect in Akt-S473 phosphorylation as expression of Akt-S473D could rescue the expression in SREBP1c and the phenotype in lipogenesis [61, 74]. However, another study argued that mTORC2-mediated regulation of SREBP1c is through an Akt-independent mechanism [63]. Therefore, this discrepancy might require further studies to clarify.
The pentose phosphate pathway (PPP) is a metabolic pathway parallel to glycolysis and is considered as a major source of NADPH (nicotinamide adenine dinucleotide phosphate), which is required for reducing power during de novo lipid biosynthesis [79]. mTORC2 is involved in the regulation of PPP similar to that of glycolysis by promoting the expression of G6PD and PGD as discussed above. Therefore, it is likely that the connection between mTORC2 and PPP might contribute to lipogenesis and anabolic cell growth and proliferation.
3.2.2. Lipolysis and Fatty Acid β-Oxidation
In consistence with its positive effect on lipogenesis, mTORC2 suppresses lipolysis and fatty acid β-oxidation. Rictor-deficient mice exhibit increased serum glycerol and free fatty acids (FFA), which possibly resulted from the sustained activation of protein kinase A- (PKA-) dependent lipolysis, and the elevated expression of fatty acid oxidation-related genes, including acyl-CoA oxidase (ACO), carnitine palmitoyltransferase 1 (CPT1), and peroxisome proliferator-activated receptor alpha (PPARα) [57, 58, 61, 80]. Although elevated FFA is not necessarily associated with insulin resistance [81], increasing adipose lipolysis could increase hepatic glucose production and cause insulin resistance [82].
3.3. Other Metabolisms
3.3.1. Amino Acid Metabolism
Amino acids are essential nutrients as the building block of proteins and carbon/nitrogen sources of many metabolic pathways. Increasing evidence has emerged that mTORC2 is an important regulator participating in the amino acid metabolism (Figure 3(c)).
In response to insulin or EGF (epidermal growth factor) stimulation, mTORC2 regulates amino acid uptake by increasing FoxO-dependent c-Myc transcription and translation, which in turn upregulates the expression of glutamine transporters, SLC1A5 (SN1) and SLC38A5 (SN2) [65]. In addition, mTORC2 could also regulate amino acid metabolism by posttranslationally altering transporter/antiporter activity [83, 84]. For example, mTORC2 regulates cell surface abundance of specific transporter isoforms SLC7A5 and SLC38A2 without affecting their global protein expression [84], and mTORC2 phosphorylates of serine 26 and inhibits cystine-glutamate antiporter xCT-mediated cystine uptake and glutathione synthesis [83]. Moreover, mTORC2 can stimulate glutamine metabolism via c-Myc. c-Myc transcriptionally represses miR-23a and miR-23b, resulting in greater expression of mitochondrial glutaminase, thereby upregulating glutaminolysis [65, 71, 85]. Increased glutaminolysis can provide α-ketoglutarate and amidogen for the synthesis of nonessential amino acids [86]. Similarly, mTORC2 promotes the generation of pyruvate and 3-phosphoglycerate through glycolysis, which would provide carbon skeleton for nonessential amino acid synthesis [87]. Although speculative, mTORC2 inhibits autophagic and proteasomal protein turnover [88], the alteration of which may also participate in amino acid homeostasis [89].
3.3.2. Nucleotide Metabolism
mTORC2-activated glucose metabolism can provide nucleotide synthesis with essential materials [90]. First, mTORC2 stimulates glycolysis and thus supplies 3-phosphoglycerate and pyruvate, which serve as the precursors of serine, glycine, aspartate, glutamate, and glutamine synthesis. Along with CO2 and ATP, glycolysis provides the carbon/nitrogen units and energy for both purine and pyrimidine de novo synthesis. Second, mTORC2 directs glucose flux into the pentose phosphate pathway (both oxidative and nonoxidative phases). This guarantees the sufficient production of ribose-5-phosphate and thus phosphoribosyl pyrophosphate (PRPP), which forms the ribose unit of the nucleotide. In addition, the pentose phosphate pathway gives rise to an abundant pool of NADPH as the reducing power for desoxyribonucleic acid production. Consistently, a systematic translational analysis reveals that Rictor protein accumulates significantly during the S phase of the cell cycle rather than G1 and mitosis [91]. This suggests that mTORC2 may play an important role in providing dNTPs for DNA synthesis during the S phase.
mTORC2 positively regulates transketolase activity for purine synthesis via Akt-mediated phosphorylation [92]. Transketolase catalyzes the formation of ribose-5-phosphate from the nonoxidative pentose phosphate pathway, which is then transformed to PRPP for purine synthesis. It is of great importance that this discovery mechanistically links mTORC2-mediated glucose metabolism and nucleotide synthesis. This implies that mTORC2 contributes to nucleotide synthesis via providing essential substrates. Moreover, essential amino acids are indispensable for the activation of mTORC2 and purine synthesis. This also indicates that the existence of essential amino acids may manifest an energy-sufficient state so that cells can put lots of energy and materials into nucleotide production. Recent studies reveal the mechanism of how mTORC1 is involved in nucleotide synthesis [93]. The two mTOR complexes may coordinate to facilitate nucleotide synthesis.
3.3.3. ROS Metabolism
Aside from being activated by ROS, mTORC2 is, in turn, involved in ROS metabolism by regulating GSH (glutathione) and NADPH synthesis [71]. GSH pool is the important cellular redox buffer to protect the cell from oxidative damage, and NADPH can maintain the GSH in the reduction state. At the cost of glutamine, lowered mTORC2 activity facilitates cystine uptake via cystine-glutamate antiporter, which is then incorporated into GSH [83]. Although paradoxical, mTORC2 can regulate cellular content of glutamate, glycine, and cysteine, which form GSH. As for NADPH, mTORC2 facilitates its production through the pentose phosphate pathway. However, except for the antioxidant, it is not clear whether ROS scavenging enzymes are also under regulation of mTORC2. Even though the relationship between mTORC2 and ROS metabolism remains unclear, evidence is gathering, and the prospect is promising.
4. Impact of mTORC2-Mediated Metabolism on Diseases
mTORC2 signaling plays an important role in metabolic regulation, the dysregulation of which is closely related to human diseases, including T2DM (type 2 diabetes mellitus), cancer, and aging (Figure 4).
Figure 4.
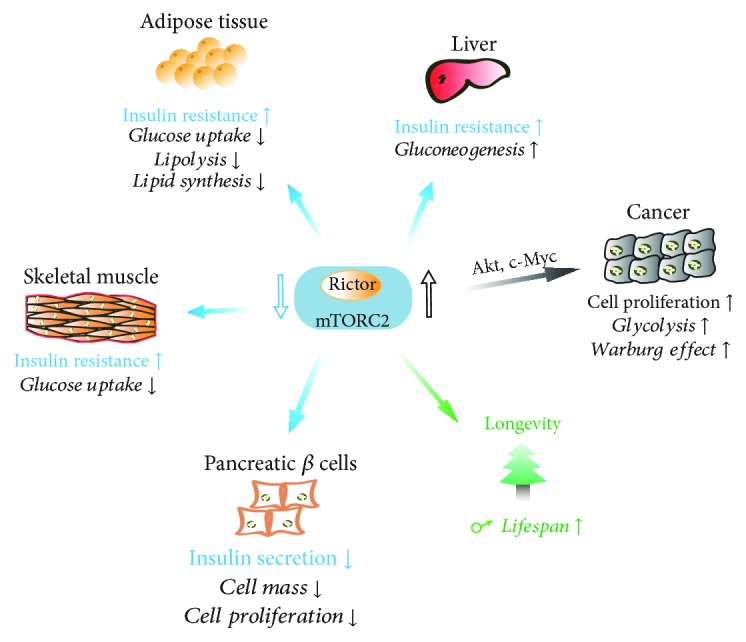
Impact of mTORC2-mediated metabolism on T2DM, cancer, and aging. In T2DM, the suppression of mTORC2 leads to gluconeogenesis in the liver and impaired glucose uptake in the muscle and adipose tissue, leading to insulin resistance. In pancreatic β cells, mTORC2 dysfunction also leads to reduced β-cell mass, proliferation, and impaired insulin secretion. In many types of cancers, mTORC2 activation promotes glucose uptake and glycolysis, which may contribute to the altered glucose metabolism and Warburg Effect, which fuels cell proliferation. In mammals, mTORC2 activity promotes longevity in males without established mechanism.
4.1. T2DM
T2DM is a metabolic disorder, characterized by dysregulation of carbohydrate, lipid, and protein metabolism as a result of impaired insulin secretion, insulin resistance, or both combined [94]. Insulin resistance, a fundamental mechanism causing T2DM, results in disturbed glucose homeostasis with increased glucose production in the liver and decreased glucose uptake in the muscle and adipose tissue. As a convergent node in insulin signaling cascade and a master regulator of homeostasis, deregulation of mTOR signaling, including mTORC2, impairs insulin signal transduction and its biological actions, leading to metabolic disorders, including T2DM. This has been clearly demonstrated in the studies of Rictor-knockout mice. Although germ-line deletion of Rictor in mice resulted in embryonic lethal at mid-gestation probably due to defects in placenta and fetal vascular development [8, 95], mice with tissue-specific Rictor-knockout in the liver, muscle, or adipose tissue all exhibited severe insulin resistance [57, 59, 61, 63]. This could be attributed to the fact that mTORC2 suppresses gluconeogenesis in the liver while promoting glucose uptake in the muscle and adipose tissue as discussed above. Therefore, disturbance in mTORC2 signaling may underlie the abnormality of glucose metabolism in T2DM, leading to hyperglycemia. Furthermore, mTORC2 could also contribute to T2DM through acting on pancreatic β cells as many mTORC2 downstream targets, including Akt, PKCα, FoxO1/3, and MST1 (mammalian sterile 20-like kinase 1), are essential for β-cell survival and insulin production [96]. Indeed, it has been reported that patients with T2DM have dramatically reduced mTORC2 activity in pancreatic β cells [97]. Therefore, mTORC2 is very important for maintaining overall glucose metabolism and homeostasis in response to nutrient fluctuations and metabolic demand.
In addition to glucose, lipid metabolism is also altered in T2DM. Independent studies suggest that adipose tissue-specific knockout of Rictor leads to decreased lipogenesis and increased lipolysis in adipose tissue, which appear to result in hepatic insulin resistance and hepatic steatosis in these mice [57, 58, 80]. The dysregulation of lipid metabolism in adipose tissue has also been associated with insulin resistance in humans [98]. Mechanistically, fat-specific ablation of Rictor may contribute to insulin resistance through multiple ways. First, Rictor deficiency in adipose tissue reduces the glucose uptake in this tissue, leading to a shortage of glucose supply, which reduces lipogenesis and increases the risk of hepatic insulin resistance [77, 99]. Second, it also increases lipolysis, leading to increased circulating FFAs, which then accumulate in tissues and impair insulin signaling [100]. Finally, Rictor ablation causes decreased lipid synthesis and altered lipid composition in adipose tissue, which may disrupt the synthesis of active lipid to improve insulin sensitivity [101].
4.2. Cancer
Deregulation of mTORC2, particularly hyperactivation, has been commonly observed in many types of human cancers. Mutations and aberrant amplifications of mTORC2 core components are two main factors contributing to its hyperactivation. For example, mutations in the mTOR-FAT domain decrease mTOR binding to the inhibitor DEPTOR, thereby conferring the hyperactivation of both mTOR complexes [102]. In addition, Rictor has also been identified to be highly mutated [102] and abnormally overexpressed through genetic and epigenetic regulations [103] in a variety of cancer types. The exact functions of these abnormalities in tumorigenesis and treatment await identification. Nonetheless, mTORC2 regulates AGC kinase family proteins, such as Akt and PKC, for their stabilization and activity, which have important roles in the cell proliferation, survival, and migration, thereby having a crucial role in cancer [2].
In terms of metabolism, mTORC2 activation promotes glucose uptake, facilitates glycolysis, and inhibits oxidative phosphorylation, which may contribute greatly to the alteration of glucose metabolism in cancer cells, known as the Warburg Effect, conferring to a high rate of cell proliferation [71, 79]. Furthermore, mTORC2-mediated lipogenesis is identified to promote hepatocellular carcinoma, particularly by stimulating sphingolipid and glycerophospholipid synthesis, which fuels cancer cell growth and energy production [104].
In addition, Rapamycin derivatives have been applied to cancer treatment in clinical trials with limited efficiency, which is thought to be due to the limitation to inhibit mTORC2 [2]. As such, the second-generation ATP-competitive inhibitors against mTOR kinase have entered clinical trials [105]. These mTOR kinase inhibitors show greater inhibitory effects on both complexes and are more effective in inhibiting cancer cell growth [106]. Moreover, dual mTOR/PI3K kinase inhibitors have been developed in order to fully suppress Akt and 4EBP1 activation [107]. Recently, selective inhibition of mTORC2 signaling by a nanoparticle-based RNAi therapy showed being able to effectively block breast cancer cell growth and survival [108]. All these suggest that mTORC2 might be a good therapeutic target for cancer treatment.
4.3. Aging
Aging is a natural process, commonly accompanied by a progressive loss of physiological functions and increased susceptibility to age-associated diseases, including cardiovascular diseases, neurodegenerative diseases, infectious diseases, and cancers, thus ultimately limiting the health and lifespan [109]. The velocity of aging is affected by a large number of environmental or genetic factors, such as nutrition sensing, DNA damage, stem cell maintenance, energy, and oxidative metabolism [110], and three sets of key proteins have been reported to modulate aging process: mTOR signaling, insulin/IGF (insulin-like growth factor) signaling (IIS), and sirtuin family proteins [111]. Using genetic or pharmacological intervention, mTOR was discovered as a regulator of lifespan in Caenorhabditis elegans [112], Drosophila melanogaster [113], and Saccharomyces cerevisiae [114] as well as mice [115]. Inhibition of mTOR by rapamycin showed 9–14% increase in the maximum lifespan of both male and female mice [115], indicating the importance of mTOR signaling in longevity.
Given the long administration of rapamycin in these mice, its effect could be the inhibition of both mTOR pathways, suggesting that mTORC2 might also have a role in aging. However, exact functions of mTORC2 remain unknown. In a separate study using mouse models in which Rictor has been reduced or deleted conditionally [116], it showed that the lifespan is significantly decreased in males with deficient Rictor but not in females and that the deleterious effect on lifespan was independent of the glucose intolerance resulting from Rictor deletion. Other studies of longevity on mTORC2 substrates yield conflicting results. In Caenorhabditis elegans, mTORC2-SGK1 activation affects the lifespan through two pathways and generates opposing outcomes depending on the cellular and environmental contexts [117]. In mice with heterozygous Akt1, it showed an increase in the lifespan [118].
Taken together, it suggests that mTORC2 signaling has important functions in regulating aging and aging-associated diseases, but the roles they play are complex and influenced by other factors.
5. Conclusion and Perspective
With the exciting progress accomplished over the last few years, it becomes clearer than before that mTORC2 signaling plays vital roles in tissue homeostasis and human health. These are implemented by its function as a crucial signaling hub, in response to both environmental and intracellular changes, to regulate several signaling networks and many essential cellular functions, such as cell proliferation, survival, migration, and metabolisms. However, our understanding of mTORC2 is far from complete. There are still several important questions awaiting to be addressed.
5.1. mTORC2 Localization and Structure
Unlike mTORC1, mTORC2 have been observed to localize at several distinct membranous compartments, e.g., plasma membrane, mitochondria, ER (endoplasmic reticulum), and MAM (mitochondria-associated ER membrane) [119]. However, why mTORC2 have so many subcellular distributions and whether mTORC2 at different compartments sense different inputs and regulate distinct substrates remain unclear. It is possible that mTORC2 utilizes various subcellular localizations to different membranous structures as a general principle to enable its spatiotemporal activation by diverse signals and mechanisms.
In addition, it remains incomplete in our understanding of the mTORC2 structure, particularly association between the mTOR kinase domain and other components of mTORC2, such as Rictor and mSin1 [11, 120]. A better elucidation of mTORC2 structure on this crucial part might aid us to determine the molecular mechanisms by which it is regulated and functions on its substrates.
5.2. Regulation of mTORC2
Currently, the regulatory mechanism governing mTORC2 activation is fairly controversial. Although growth factors have been recognized as the major activating signal, little is known about how exactly they control mTORC2 [34, 35]. Moreover, does mTORC2 respond to other environmental cues in addition to growth factors? For example, the activation of TORC2 in yeast is not responsive to growth factors, and instead, the lipid binding is a crucial requirement [121]. The ligand engagement of Toll-like receptor (TLR) is also able to activate mTORC2 [122, 123]. Furthermore, mTORC2 that localizes at the plasma membrane is constitutively active in mammalian cells [35]. In this context, could nutrients, cellular stress, or other metabolic signals generated from the plasma membrane be the primary regulator of mTORC2? The identification of upstream regulators of mTORC2 might provide novel insights into these uncertainties.
It should also be noted that several mTORC2 components undergo PTMs, including phosphorylation, ubiquitination, acetylation, and palmitoylation, to regulate mTORC2 stability and assembly. mTORC2 signaling network is so intricately intertwined with multiple signal and metabolic pathways. Crosstalk between these pathways may extensively modulate mTORC2 [124].
5.3. Targets of mTORC2 Signaling
Although it has long been regarded that mTORC2 exerts wide spectrum effects on cellular properties through members of the AGC kinase family, the exact mechanisms for mTORC2 governing their activation are incompletely understood. For example, why does mTORC2 phosphorylate Akt-HM and -TM through different mechanisms, and is ribosome required for both? Why is the immunoprecipitated mTORC2 only able to phosphorylate the Akt-HM but not the PKCa-HM in in vitro kinase assays? Moreover, many proteins, in addition to the three AGC kinases, have been found to be modified in a mTORC2-dependent manner, such as HDAC [65], ChREBP-α [58], and Smad2/3 [125]. However, it is unclear whether this is a direct or indirect modification as it is largely unknown the mechanism by which mTORC2 recruits its substrates, which might depend on the localization of mTORC2 and may also involve other factors.
Finding answers to these questions will greatly expand our knowledge on mTORC2 biology, aiding the design and development of mTORC2-specific agonists and inhibitors, which will also help the development of therapeutic avenues to treat human metabolic diseases. The future will undoubtedly continue to bring unexpected insight on this remarkable pathway.
Acknowledgments
We apologize to the many colleagues and contributors in the field whose work has added to our understanding of the mTORC2 signaling network and functions but could not be cited due to space limitations.
Contributor Information
Yongting Luo, Email: luo_yongting@163.com.
Wei Cui, Email: wei.cui@imperial.ac.uk.
Conflicts of Interest
The authors declare no conflict of interest.
Authors' Contributions
Yongting Luo, Wenyi Xu, and Guannan Li have contributed equally to this work.
References
- 1.Saxton R. A., Sabatini D. M. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169(2):361–371. doi: 10.1016/j.cell.2017.03.035. [DOI] [PubMed] [Google Scholar]
- 2.Zoncu R., Efeyan A., Sabatini D. M. mTOR: from growth signal integration to cancer, diabetes and ageing. Nature Reviews Molecular Cell Biology. 2011;12(1):21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sarbassov D. D., Ali S. M., Kim D.-H., et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Current Biology. 2004;14(14):1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 4.Gaubitz C., Prouteau M., Kusmider B., Loewith R. TORC2 structure and function. Trends in Biochemical Sciences. 2016;41(6):532–545. doi: 10.1016/j.tibs.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Knutson B. A. Insights into the domain and repeat architecture of target of rapamycin. Journal of Structural Biology. 2010;170(2):354–363. doi: 10.1016/j.jsb.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang H., Rudge D. G., Koos J. D., Vaidialingam B., Yang H. J., Pavletich N. P. mTOR kinase structure, mechanism and regulation. Nature. 2013;497(7448):217–223. doi: 10.1038/nature12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim D.-H., Sarbassov D. D., Ali S. M., et al. GβL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Molecular Cell. 2003;11(4):895–904. doi: 10.1016/S1097-2765(03)00114-X. [DOI] [PubMed] [Google Scholar]
- 8.Guertin D. A., Stevens D. M., Thoreen C. C., et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCα, but not S6K1. Developmental Cell. 2006;11(6):859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 9.Jacinto E., Loewith R., Schmidt A., et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nature Cell Biology. 2004;6(11):1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 10.Karuppasamy M., Kusmider B., Oliveira T. M., et al. Cryo-EM structure of Saccharomyces cerevisiae target of rapamycin complex 2. Nature Communications. 2017;8(1):p. 1729. doi: 10.1038/s41467-017-01862-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stuttfeld E., Aylett C. H., Imseng S., et al. Architecture of the human mTORC2 core complex. eLife. 2018;7 doi: 10.7554/eLife.33101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 13.Jacinto E., Facchinetti V., Liu D., et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127(1):125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 14.Frias M. A., Thoreen C. C., Jaffe J. D., et al. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Current Biology. 2006;16(18):1865–1870. doi: 10.1016/j.cub.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 15.Tatebe H., Murayama S., Yonekura T., et al. Substrate specificity of TOR complex 2 is determined by a ubiquitin-fold domain of the Sin1 subunit. eLife. 2017;6 doi: 10.7554/eLife.19594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aylett C. H. S., Sauer E., Imseng S., et al. Architecture of human mTOR complex 1. Science. 2016;351(6268):48–52. doi: 10.1126/science.aaa3870. [DOI] [PubMed] [Google Scholar]
- 17.Saxton R. A., Sabatini D. M. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alessi D. R., Andjelkovic M., Caudwell B., et al. Mechanism of activation of protein kinase B by insulin and IGF-1. The EMBO Journal. 1996;15(23):6541–6551. doi: 10.1002/j.1460-2075.1996.tb01045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang J., Cron P., Thompson V., et al. Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Molecular Cell. 2002;9(6):1227–1240. doi: 10.1016/S1097-2765(02)00550-6. [DOI] [PubMed] [Google Scholar]
- 20.Hannigan G., Troussard A. A., Dedhar S. Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nature Reviews Cancer. 2005;5(1):51–63. doi: 10.1038/nrc1524. [DOI] [PubMed] [Google Scholar]
- 21.Bozulic L., Surucu B., Hynx D., Hemmings B. A. PKBα/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Molecular Cell. 2008;30(2):203–213. doi: 10.1016/j.molcel.2008.02.024. [DOI] [PubMed] [Google Scholar]
- 22.Facchinetti V., Ouyang W., Wei H., et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. The EMBO Journal. 2008;27(14):1932–1943. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ikenoue T., Inoki K., Yang Q., Zhou X., Guan K. L. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. The EMBO Journal. 2008;27(14):1919–1931. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oh W. J., Wu C. -c., Kim S. J., et al. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. The EMBO Journal. 2010;29(23):3939–3951. doi: 10.1038/emboj.2010.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newton A. C. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochemical Journal. 2003;370(2):361–371. doi: 10.1042/bj20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gan X., Wang J., Wang C., et al. PRR5L degradation promotes mTORC2-mediated PKC-δ phosphorylation and cell migration downstream of Gα12. Nature Cell Biology. 2012;14(7):686–696. doi: 10.1038/ncb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomanetz V., Angliker N., Cloetta D., et al. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. Journal of Cell Biology. 2013;201(2):293–308. doi: 10.1083/jcb.201205030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parekh D. B., Ziegler W., Parker P. J. Multiple pathways control protein kinase C phosphorylation. The EMBO Journal. 2000;19(4):496–503. doi: 10.1093/emboj/19.4.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia-Martinez J. M., Alessi D. R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) Biochemical Journal. 2008;416(3):375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- 30.Lu M., Wang J., Ives H. E., Pearce D. mSIN1 protein mediates SGK1 protein interaction with mTORC2 protein complex and is required for selective activation of the epithelial sodium channel. Journal of Biological Chemistry. 2011;286(35):30647–30654. doi: 10.1074/jbc.M111.257592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lang F., Bohmer C., Palmada M., Seebohm G., Strutz-Seebohm N., Vallon V. (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiological Reviews. 2006;86(4):1151–1178. doi: 10.1152/physrev.00050.2005. [DOI] [PubMed] [Google Scholar]
- 32.Hill M. M., Feng J., Hemmings B. A. Identification of a plasma membrane Raft-associated PKB Ser473 kinase activity that is distinct from ILK and PDK1. Current Biology. 2002;12(14):1251–1255. doi: 10.1016/S0960-9822(02)00973-9. [DOI] [PubMed] [Google Scholar]
- 33.Gao X., Lowry P. R., Zhou X., et al. PI3K/Akt signaling requires spatial compartmentalization in plasma membrane microdomains. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(35):14509–14514. doi: 10.1073/pnas.1019386108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu P., Gan W., Chin Y. R., et al. PtdIns(3,4,5)P3-dependent activation of the mTORC2 kinase complex. Cancer Discovery. 2015;5(11):1194–1209. doi: 10.1158/2159-8290.CD-15-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ebner M., Sinkovics B., Szczygiel M., Ribeiro D. W., Yudushkin I. Localization of mTORC2 activity inside cells. Journal of Cell Biology. 2017;216(2):343–353. doi: 10.1083/jcb.201610060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halova L., Du W., Kirkham S., Smith D. L., Petersen J. Phosphorylation of the TOR ATP binding domain by AGC kinase constitutes a novel mode of TOR inhibition. Journal of Cell Biology. 2013;203(4):595–604. doi: 10.1083/jcb.201305103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Copp J., Manning G., Hunter T. TORC-specific phosphorylation of mammalian target of rapamycin (mTOR): phospho-Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Research. 2009;69(5):1821–1827. doi: 10.1158/0008-5472.CAN-08-3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang B., Jie Z., Joo D., et al. TRAF2 and OTUD7B govern a ubiquitin-dependent switch that regulates mTORC2 signalling. Nature. 2017;545(7654):365–369. doi: 10.1038/nature22344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duan S., Skaar J. R., Kuchay S., et al. mTOR generates an auto-amplification loop by triggering the βTrCP- and CK1α-dependent degradation of DEPTOR. Molecular Cell. 2011;44(2):317–324. doi: 10.1016/j.molcel.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao D., Inuzuka H., Tan M.-K. M., et al. mTOR drives its own activation via SCFβTrCP-dependent degradation of the mTOR inhibitor DEPTOR. Molecular Cell. 2011;44(2):290–303. doi: 10.1016/j.molcel.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao Y., Xiong X., Sun Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCFβTrCP E3 ubiquitin ligase and regulates survival and autophagy. Molecular Cell. 2011;44(2):304–316. doi: 10.1016/j.molcel.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boulbes D., Chen C. H., Shaikenov T., et al. Rictor phosphorylation on the Thr-1135 site does not require mammalian target of rapamycin complex 2. Molecular Cancer Research. 2010;8(6):896–906. doi: 10.1158/1541-7786.MCR-09-0409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dibble C. C., Asara J. M., Manning B. D. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Molecular and Cellular Biology. 2009;29(21):5657–5670. doi: 10.1128/MCB.00735-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen C.-H., Shaikenov T., Peterson T. R., et al. ER stress inhibits mTORC2 and Akt signaling through GSK-3β–mediated phosphorylation of Rictor. Science Signaling. 2011;4(161, article ra10) doi: 10.1126/scisignal.2001731. [DOI] [PubMed] [Google Scholar]
- 45.Koo J., Wu X., Mao Z., Khuri F. R., Sun S. Y. Rictor undergoes glycogen synthase kinase 3 (GSK3)-dependent, FBXW7-mediated ubiquitination and proteasomal degradation. Journal of Biological Chemistry. 2015;290(22):14120–14129. doi: 10.1074/jbc.M114.633057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Glidden E. J., Gray L. G., Vemuru S., Li D., Harris T. E., Mayo M. W. Multiple site acetylation of Rictor stimulates mammalian target of rapamycin complex 2 (mTORC2)-dependent phosphorylation of Akt protein. The Journal of Biological Chemistry. 2012;287(1):581–588. doi: 10.1074/jbc.M111.304337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Masui K., Tanaka K., Ikegami S., et al. Glucose-dependent acetylation of Rictor promotes targeted cancer therapy resistance. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(30):9406–9411. doi: 10.1073/pnas.1511759112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu P., Gan W., Inuzuka H., et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nature Cell Biology. 2013;15(11):1340–1350. doi: 10.1038/ncb2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Humphrey S. J., Yang G., Yang P., et al. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metabolism. 2013;17(6):1009–1020. doi: 10.1016/j.cmet.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang G., Murashige D. S., Humphrey S. J., James D. E. A positive feedback loop between Akt and mTORC2 via SIN1 phosphorylation. Cell Reports. 2015;12(6):937–943. doi: 10.1016/j.celrep.2015.07.016. [DOI] [PubMed] [Google Scholar]
- 51.Chen C.-H., Kiyan V., Zhylkibayev A. A., et al. Autoregulation of the mechanistic target of rapamycin (mTOR) complex 2 integrity is controlled by an ATP-dependent mechanism. Journal of Biological Chemistry. 2013;288(38):27019–27030. doi: 10.1074/jbc.M113.498055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zinzalla V., Stracka D., Oppliger W., Hall M. N. Activation of mTORC2 by association with the ribosome. Cell. 2011;144(5):757–768. doi: 10.1016/j.cell.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 53.Hatano T., Morigasaki S., Tatebe H., Ikeda K., Shiozaki K. Fission yeast Ryh1 GTPase activates TOR complex 2 in response to glucose. Cell Cycle. 2015;14(6):848–856. doi: 10.1080/15384101.2014.1000215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cai W., Andres D. A. mTORC2 is required for rit-mediated oxidative stress resistance. PLoS One. 2014;9(12, article e115602) doi: 10.1371/journal.pone.0115602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saci A., Cantley L. C., Carpenter C. L. Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Molecular Cell. 2011;42(1):50–61. doi: 10.1016/j.molcel.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Byun J. K., Choi Y. K., Kim J. H., et al. A positive feedback loop between Sestrin2 and mTORC2 is required for the survival of glutamine-depleted lung cancer cells. Cell Reports. 2017;20(3):586–599. doi: 10.1016/j.celrep.2017.06.066. [DOI] [PubMed] [Google Scholar]
- 57.Kumar A., Lawrence J. C., Jr., Jung D. Y., et al. Fat cell–specific ablation of rictor in mice impairs insulin-regulated fat cell and whole-body glucose and lipid metabolism. Diabetes. 2010;59(6):1397–1406. doi: 10.2337/db09-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tang Y., Wallace M., Sanchez-Gurmaches J., et al. Adipose tissue mTORC2 regulates ChREBP-driven de novo lipogenesis and hepatic glucose metabolism. Nature Communications. 2016;7, article 11365 doi: 10.1038/ncomms11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kumar A., Harris T. E., Keller S. R., Choi K. M., Magnuson M. A., Lawrence J. C., Jr. Muscle-specific deletion of rictor impairs insulin-stimulated glucose transport and enhances basal glycogen synthase activity. Molecular and Cellular Biology. 2007;28(1):61–70. doi: 10.1128/MCB.01405-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Albert V., Svensson K., Shimobayashi M., et al. mTORC2 sustains thermogenesis via Akt‐induced glucose uptake and glycolysis in brown adipose tissue. EMBO Molecular Medicine. 2016;8(3):232–246. doi: 10.15252/emmm.201505610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hagiwara A., Cornu M., Cybulski N., et al. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metabolism. 2012;15(5):725–738. doi: 10.1016/j.cmet.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 62.Betz C., Stracka D., Prescianotto-Baschong C., Frieden M., Demaurex N., Hall M. N. mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(31):12526–12534. doi: 10.1073/pnas.1302455110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yuan M., Pino E., Wu L., Kacergis M., Soukas A. A. Identification of Akt-independent regulation of hepatic lipogenesis by mammalian target of rapamycin (mTOR) complex 2. Journal of Biological Chemistry. 2012;287(35):29579–29588. doi: 10.1074/jbc.M112.386854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang S., Czech M. P. The GLUT4 glucose transporter. Cell Metabolism. 2007;5(4):237–252. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 65.Masui K., Tanaka K., Akhavan D., et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metabolism. 2013;18(5):726–739. doi: 10.1016/j.cmet.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xiong Y., Lei Q. Y., Zhao S., Guan K. L. Regulation of glycolysis and gluconeogenesis by acetylation of PKM and PEPCK. Cold Spring Harbor Symposia on Quantitative Biology. 2011;76:285–289. doi: 10.1101/sqb.2011.76.010942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Puigserver P., Rhee J., Donovan J., et al. Insulin-regulated hepatic gluconeogenesis through FOXO1–PGC-1α interaction. Nature. 2003;423(6939):550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 68.Matsumoto M., Pocai A., Rossetti L., DePinho R. A., Accili D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metabolism. 2007;6(3):208–216. doi: 10.1016/j.cmet.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 69.Li X., Monks B., Ge Q., Birnbaum M. J. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1α transcription coactivator. Nature. 2007;447(7147):1012–1016. doi: 10.1038/nature05861. [DOI] [PubMed] [Google Scholar]
- 70.Kim J. W., Tchernyshyov I., Semenza G. L., Dang C. V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metabolism. 2006;3(3):177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 71.Masui K., Cavenee W. K., Mischel P. S. mTORC2 in the center of cancer metabolic reprogramming. Trends in Endocrinology & Metabolism. 2014;25(7):364–373. doi: 10.1016/j.tem.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Meng Z.-X., Li S., Wang L., et al. Baf60c drives glycolytic metabolism in the muscle and improves systemic glucose homeostasis through Deptor-mediated Akt activation. Nature Medicine. 2013;19(5):640–645. doi: 10.1038/nm.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Caron A., Richard D., Laplante M. The roles of mTOR complexes in lipid metabolism. Annual Review of Nutrition. 2015;35(1):321–348. doi: 10.1146/annurev-nutr-071714-034355. [DOI] [PubMed] [Google Scholar]
- 74.Hung C.-M., Calejman C. M., Sanchez-Gurmaches J., et al. Rictor/mTORC2 loss in the Myf5 lineage reprograms brown fat metabolism and protects mice against obesity and metabolic disease. Cell Reports. 2014;8(1):256–271. doi: 10.1016/j.celrep.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shimano H. Sterol regulatory element-binding proteins (SREBPs): transcriptional regulators of lipid synthetic genes. Progress in Lipid Research. 2001;40(6):439–452. doi: 10.1016/S0163-7827(01)00010-8. [DOI] [PubMed] [Google Scholar]
- 76.Uyeda K., Repa J. J. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metabolism. 2006;4(2):107–110. doi: 10.1016/j.cmet.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 77.Herman M. A., Peroni O. D., Villoria J., et al. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature. 2012;484(7394):333–338. doi: 10.1038/nature10986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Filhoulaud G., Guilmeau S., Dentin R., Girard J., Postic C. Novel insights into ChREBP regulation and function. Trends in Endocrinology and Metabolism. 2013;24(5):257–268. doi: 10.1016/j.tem.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 79.Cairns R. A., Harris I. S., Mak T. W. Regulation of cancer cell metabolism. Nature Reviews Cancer. 2011;11(2):85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 80.Cybulski N., Polak P., Auwerx J., Rüegg M. A., Hall M. N. mTOR complex 2 in adipose tissue negatively controls whole-body growth. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(24):9902–9907. doi: 10.1073/pnas.0811321106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Karpe F., Dickmann J. R., Frayn K. N. Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes. 2011;60(10):2441–2449. doi: 10.2337/db11-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Perry R. J., Camporez J.-P. G., Kursawe R., et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015;160(4):745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gu Y., Albuquerque C. P., Braas D., et al. mTORC2 regulates amino acid metabolism in cancer by phosphorylation of the cystine-glutamate antiporter xCT. Molecular Cell. 2017;67(1):128–138.e7. doi: 10.1016/j.molcel.2017.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rosario F. J., Kanai Y., Powell T. L., Jansson T. Mammalian target of rapamycin signalling modulates amino acid uptake by regulating transporter cell surface abundance in primary human trophoblast cells. The Journal of Physiology. 2013;591(3):609–625. doi: 10.1113/jphysiol.2012.238014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gao P., Tchernyshyov I., Chang T.-C., et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458(7239):762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Altman B. J., Stine Z. E., Dang C. V. From Krebs to clinic: glutamine metabolism to cancer therapy. Nature Reviews Cancer. 2016;16(10):619–634. doi: 10.1038/nrc.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lunt S. Y., Vander Heiden M. G. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annual Review of Cell and Developmental Biology. 2011;27(1):441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 88.van der Vos K. E., Eliasson P., Proikas-Cezanne T., et al. Modulation of glutamine metabolism by the PI(3)K–PKB–FOXO network regulates autophagy. Nature Cell Biology. 2012;14(8):829–837. doi: 10.1038/ncb2536. [DOI] [PubMed] [Google Scholar]
- 89.Suraweera A., Munch C., Hanssum A., Bertolotti A. Failure of amino acid homeostasis causes cell death following proteasome inhibition. Molecular Cell. 2012;48(2):242–253. doi: 10.1016/j.molcel.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vander Heiden M. G., Cantley L. C., Thompson C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stumpf C. R., Moreno M. V., Olshen A. B., Taylor B. S., Ruggero D. The translational landscape of the mammalian cell cycle. Molecular Cell. 2013;52(4):574–582. doi: 10.1016/j.molcel.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Saha A., Connelly S., Jiang J., et al. Akt phosphorylation and regulation of transketolase is a nodal point for amino acid control of purine synthesis. Molecular Cell. 2014;55(2):264–276. doi: 10.1016/j.molcel.2014.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ben-Sahra I., Hoxhaj G., Ricoult S. J. H., Asara J. M., Manning B. D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 2016;351(6274):728–733. doi: 10.1126/science.aad0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.DeFronzo R. A., Ferrannini E., Groop L., et al. Type 2 diabetes mellitus. Nature Reviews Disease Primers. 2015;1, article 15019 doi: 10.1038/nrdp.2015.19. [DOI] [PubMed] [Google Scholar]
- 95.Shiota C., Woo J. T., Lindner J., Shelton K. D., Magnuson M. A. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Developmental Cell. 2006;11(4):583–589. doi: 10.1016/j.devcel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 96.Yuan T., Lupse B., Maedler K., Ardestani A. mTORC2 signaling: a path for pancreatic β cell’s growth and function. Journal of Molecular Biology. 2018;430(7):904–918. doi: 10.1016/j.jmb.2018.02.013. [DOI] [PubMed] [Google Scholar]
- 97.Yuan T., Rafizadeh S., Gorrepati K. D. D., et al. Reciprocal regulation of mTOR complexes in pancreatic islets from humans with type 2 diabetes. Diabetologia. 2017;60(4):668–678. doi: 10.1007/s00125-016-4188-9. [DOI] [PubMed] [Google Scholar]
- 98.Smith U., Kahn B. B. Adipose tissue regulates insulin sensitivity: role of adipogenesis, de novo lipogenesis and novel lipids. Journal of Internal Medicine. 2016;280(5):465–475. doi: 10.1111/joim.12540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Abel E. D., Peroni O., Kim J. K., et al. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 2001;409(6821):729–733. doi: 10.1038/35055575. [DOI] [PubMed] [Google Scholar]
- 100.Samuel V. T., Shulman G. I. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148(5):852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cao H., Gerhold K., Mayers J. R., Wiest M. M., Watkins S. M., Hotamisligil G. S. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell. 2008;134(6):933–944. doi: 10.1016/j.cell.2008.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Grabiner B. C., Nardi V., Birsoy K., et al. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discovery. 2014;4(5):554–563. doi: 10.1158/2159-8290.CD-13-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gkountakos A., Pilotto S., Mafficini A., et al. Unmasking the impact of Rictor in cancer: novel insights of mTORC2 complex. Carcinogenesis. 2018;39(8):971–980. doi: 10.1093/carcin/bgy086. [DOI] [PubMed] [Google Scholar]
- 104.Guri Y., Colombi M., Dazert E., et al. mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell. 2017;32(6):807–823.e12. doi: 10.1016/j.ccell.2017.11.011. [DOI] [PubMed] [Google Scholar]
- 105.Baselga J., Campone M., Piccart M., et al. Everolimus in postmenopausal hormone-receptor–positive advanced breast cancer. The New England Journal of Medicine. 2012;366(6):520–529. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Guichard S. M., Curwen J., Bihani T., et al. AZD2014, an inhibitor of mTORC1 and mTORC2, is highly effective in ER+ breast cancer when administered using intermittent or continuous schedules. Molecular Cancer Therapeutics. 2015;14(11):2508–2518. doi: 10.1158/1535-7163.MCT-15-0365. [DOI] [PubMed] [Google Scholar]
- 107.Alqurashi N., Hashimi S., Alowaidi F., Ivanovski S., Wei M. Dual mTOR/PI3K inhibitor NVP-BEZ235 arrests colorectal cancer cell growth and displays differential inhibition of 4E-BP1. Oncology Reports. 2018;40(2):1083–1092. doi: 10.3892/or.2018.6457. [DOI] [PubMed] [Google Scholar]
- 108.Werfel T. A., Wang S., Jackson M. A., et al. Selective mTORC2 inhibitor therapeutically blocks breast cancer cell growth and survival. Cancer Research. 2018;78(7):1845–1858. doi: 10.1158/0008-5472.CAN-17-2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lopez-Otin C., Blasco M. A., Partridge L., Serrano M., Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Riera C. E., Merkwirth C., De Magalhaes Filho C. D., Dillin A. Signaling networks determining life span. Annual Review of Biochemistry. 2016;85(1):35–64. doi: 10.1146/annurev-biochem-060815-014451. [DOI] [PubMed] [Google Scholar]
- 111.Pan H., Finkel T. Key proteins and pathways that regulate lifespan. Journal of Biological Chemistry. 2017;292(16):6452–6460. doi: 10.1074/jbc.R116.771915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jia K., Chen D., Riddle D. L. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004;131(16):3897–3906. doi: 10.1242/dev.01255. [DOI] [PubMed] [Google Scholar]
- 113.Kapahi P., Zid B. M., Harper T., Koslover D., Sapin V., Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Current Biology. 2004;14(10):885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kaeberlein M., Powers R. W., 3rd, Steffen K. K., et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310(5751):1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- 115.Harrison D. E., Strong R., Sharp Z. D., et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460(7253):392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lamming D. W., Mihaylova M. M., Katajisto P., et al. Depletion of Rictor, an essential protein component of mTORC2, decreases male lifespan. Aging Cell. 2014;13(5):911–917. doi: 10.1111/acel.12256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mizunuma M., Neumann-Haefelin E., Moroz N., Li Y., Blackwell T. K. mTORC2-SGK-1 acts in two environmentally responsive pathways with opposing effects on longevity. Aging Cell. 2014;13(5):869–878. doi: 10.1111/acel.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nojima A., Yamashita M., Yoshida Y., et al. Haploinsufficiency of Akt1 prolongs the lifespan of mice. PLoS One. 2013;8(7, article e69178) doi: 10.1371/journal.pone.0069178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Betz C., Hall M. N. Where is mTOR and what is it doing there? Journal of Cell Biology. 2013;203(4):563–574. doi: 10.1083/jcb.201306041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chen X., Liu M., Tian Y., et al. Cryo-EM structure of human mTOR complex 2. Cell Research. 2018;28(5):518–528. doi: 10.1038/s41422-018-0029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Berchtold D., Walther T. C. TORC2 plasma membrane localization is essential for cell viability and restricted to a distinct domain. Molecular Biology of the Cell. 2009;20(5):1565–1575. doi: 10.1091/mbc.e08-10-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Festuccia W. T., Pouliot P., Bakan I., Sabatini D. M., Laplante M. Myeloid-specific Rictor deletion induces M1 macrophage polarization and potentiates in vivo pro-inflammatory response to lipopolysaccharide. PLoS One. 2014;9(4, article e95432) doi: 10.1371/journal.pone.0095432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brown J., Wang H., Suttles J., Graves D. T., Martin M. Mammalian target of rapamycin complex 2 (mTORC2) negatively regulates toll-like receptor 4-mediated inflammatory response via FoxO1. Journal of Biological Chemistry. 2011;286(52):44295–44305. doi: 10.1074/jbc.M111.258053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yang Q., Vijayakumar A., Kahn B. B. Metabolites as regulators of insulin sensitivity and metabolism. Nature Reviews Molecular Cell Biology. 2018;19(10):654–672. doi: 10.1038/s41580-018-0044-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yu J. S. L., Ramasamy T. S., Murphy N., et al. PI3K/mTORC2 regulates TGF-β/activin signalling by modulating Smad2/3 activity via linker phosphorylation. Nature Communications. 2015;6(1):p. 7212. doi: 10.1038/ncomms8212. [DOI] [PMC free article] [PubMed] [Google Scholar]