

Abstract
α1-Antitrypsin (A1AT) purified from human plasma upregulates expression and release of angiopoietin-like protein 4 (Angptl4) in adherent human blood monocytes and in human lung microvascular endothelial cells, providing a mechanism for the broad immune-regulatory properties of A1AT independent of its anti-protease activity. Here we demonstrate that A1AT (Prolastin®), a potent inducer of Angptl4, contains significant quantities of the fatty acids (FA): linoleic (LA, C18:2) and oleic (OA, C18:1). However, only trace amounts of FAs were present in preparations that failed to increase Angplt4 expression, for example A1AT (Zemaira) or M-type A1AT purified by affinity chromatography. FA pull-down assays with western blot analysis revealed a FA-binding ability of A1AT. In human blood adherent monocytes A1AT‐FA conjugates up-regulated expression of Angptl4 (54.9-fold, p<0.001), fatty acid binding protein 4 (FABP4) (11.4-fold, p<0.001) and to a lesser degree, fatty acid translocase (CD36) (3.1-fold, p<0.001) relative to A1AT devoid of FA (A1AT‐0). These latter effects of A1AT-FA were blocked by inhibitors of PPARβ/δ (ST247) and PPARγ (GW9662). When compared to controls, cell pre-treatment with ST247 diminished the effect of A1AT-LA on Angptl4 mRNA (11.6- vs 4.1-fold, p<0.001) and FABP4 mRNA (5.4-vs 2.8-fold, p<0.001). Similarly, pre-incubation of cells with GW9662 inhibited inducing effect of A1AT-LA on Angptl4 mRNA (by 2-fold, p<0.001) and FABP4 mRNA (by 3-fold, p<0.001). Thus, A1AT binds to FA, and it is this form of A1AT that induces Angptl4 and FABP4 expression via a PPARs-dependent pathway. These findings provide a mechanism for the unexplored area of A1AT biology independent of its anti-protease properties.
Keywords: Alpha1-Antitrypsin, fatty acids, linoleic acid, oleic acid, pull-down assay, angiopoietin-like protein 4, albumin, PBMCs, fatty acid binding protein-4, CD36, peroxisome proliferator-activated receptors
Introduction
Angiopoietin-like protein 4 (Angptl4), also named PPARγ angiopoietin-related, fasting-induced adipose factor, was originally discovered as one of the target genes of PPARγ (1). The most well characterized function of Angptl4 is the regulation of lipid metabolism through the inhibition of lipoprotein lipase (LPL), an enzyme that hydrolyzes triglycerides from the apolipoprotein B-containing lipoproteins chylomicrons (2, 3). Studies using Angptl4-knockout mice suggest that Angptl4 plays a role in inflammation, atherosclerosis and wound healing (4-8), whereas data from Angptl4-overexpressing models imply that Angptl4 is involved in the development of cancer, nephrotic syndrome and cardiovascular diseases (9-15). Angptl4 also seems to play a role in type 2 diabetes mellitus and metabolic syndrome, both of which are associated with dyslipidemia (5, 16-18). Angptl4 is therefore a multifunctional protein and there is a considerable interest in identifying the regulators of Angptl4 expression and release.
The expression of Angplt4 is controlled by peroxisome proliferator-activated receptors (PPARs). Therefore, free fatty acids (FAs), which activate the lipid-sensing peroxisome proliferator-activated receptors (PPARs) α, β and γ can directly stimulate its expression (16). Regulation of Angptl4 by FA is also dependent on the type of FA. For instance, the unsaturated FAs, linoleic and oleic, are potent induces of Angptl4 (19), whereas saturated palmitic acid induces Angplt4 of markedly lower magnitude (4). Recent studies by Georgiadi and co-authors demonstrate that induction of Angptl4 and subsequent inhibition of LPL by dietary FA protects the heart against lipid overload, reduces lipotoxicity and inflammation (5, 14). Other authors suggest that induction of Angptl4 by FA promotes the use of plasma triglycerides, a fuel for the exercising muscles (16).
Normal sources of FA include diet, mobilization from adipose tissue and conversion of excess carbohydrates into fat by the liver. For transport and/or storage FAs can be coupled with glycerol to form triglycerides, and can be converted back into free FAs by lipases. In blood, FAs are mostly coupled non-covalently with albumin which can simultaneously bind up to ten FA molecules and serves as a vehicle to transport FA (18, 19). Besides albumin there are other so-called FA-binding proteins but none are known to bind FAs in such large amounts as albumin (17). Therefore, a mechanism for increased expression of Angptl4 by proteins carrying FAs remains uncharacterized.
Extensive studies show that α1-antitrypsin (A1AT) purified from human plasma possesses anti-inflammatory and immuno-regulatory properties across a broad spectrum of animal models for systemic or local inflammation (20, 21). Although A1AT is commonly used to treat patients with inherited Z (Glu342Lys) deficiency, A1AT has been administered to non-deficient patients with recent onset Type 1 diabetes (22) and ST-elevated myocardial infarction (23). Those studies revealed a distinct anti-inflammatory profile of reduced IL‐1β and C-reactive protein levels. In a small cohort of emphysema patients receiving therapy with A1AT (Prolastin®), we recently reported that plasma Angptl4 levels correlate with A1AT levels and were significantly higher relative to treatment naïve patients. More detailed in vitro investigation revealed that A1AT strongly up-regulates expression and release of Angplt4 in human blood adherent monocytic cells and in primary human lung microvascular endothelial cells (24). However, it came to our attention that the magnitude by which preparations of A1AT stimulated expression of Angptl4 varied significantly. These inconsistencies between pharmaceutical preparations of seemingly the same protein suggested variability in their constituents. Here, we provide unexpected evidence that A1AT binds FAs (linoleic and oleic) and only FA-bound forms of A1AT induce Angptl4.
Materials and Methods
A1AT preparation
Clinical grade A1AT preparations, Zemaira (CSL Behring) and Prolastin® (Grifols) were used in all experiments. Zemaira is also termed A1AT-0 to indicate that it is fatty acid free.
Purification of A1AT from human serum
Human serum was collected from volunteers with PiMM and PiZZ genotype of A1AT. Serum M-A1AT and Z-A1AT were isolated by affinity chromatography using A1AT specific Alpha-1 Antitrypsin Select matrix (GE Healthcare Life Sciences) according to the manufacturer's recommendations. In brief, pooled serum (PiMM or PiZZ) was diluted 1:3 with 20 mM Tris/HCl 150 mM NaCl pH 7.4 binding buffer and loaded onto Alpha-1 Antitrypsin Select columns. Serum A1AT binds to the ligand and unbound impurities were washed away with binding buffer. M- and Z-A1AT were eluted with 2 M MgCl2 in 20 mM Tris/HCl pH 7.6. A1AT concentration in elution fractions was determined from OD280 measured on Nanodrop 1000 Spectrophotometer (Thermo Fisher Scientific). Pools of M- or Z-A1AT containing fractions were concentrated and buffer was changed to PBS using 10 kDa cut-off centricons (Merck Millipore). M- and Z-A1AT purities were confirmed on Coomassie stained gels.
Lipid analysis of A1AT preparations
In collaboration with the Clinical Chemistry of Hannover Medical School, commercial A1AT preparation (Zemaira), and affinity isolated M- and Z-A1AT were probed for the content of cholesterol, triglycerides, LDL and HDL. For analysis, the cobas® 8000 modular analyzer series (Roche) was used.
Preparation of A1AT- and HSA-fatty acid complexes
A1AT-0 (Zemaira) and in some experiments fatty acid free HSA (Sigma-Aldrich), were spiked with linoleic acid or oleic acid (Sigma Aldrich). For spiking, proteins were directly mixed with linoleic acid or oleic acid (1:2.4 molar ratio) and incubated for 3 h at 37°C in a water bath. Post incubation, unbound fatty acids were removed using 10 kDa cut-off membranes (Millipore). Potential protein-fatty acid complexes were recovered in sterile PBS. Fatty acids alone prepared under the same conditions were used as controls. For all experiments, preparations were directly used and kept no longer than 48 h at 4°C.
Fatty acid analysis
Lipid extraction was performed as described previously (25). Briefly, 80 μg protein was subjected to an acidic Bligh&Dyer extraction in the presence of 200 pmol C17:0 fatty acid. Evaporated lipid extracts were resuspended in methanol. Samples subjected to MS analysis in negative ion mode were diluted 1:2 in 0.05% triethylamine in methanol. Mass spectrometric analysis of lipids was performed on a Q Exactive from Thermo Scientific. Samples were automatically injected via a TriVersa NanoMate device (Advion). Spray voltage was set to 1500 V with a capillary temperature of 200°C. Full MS scans (m/z 200-1000 Da) were obtained with automatic gain control target of 1x106 ions and maximal injection time of 200 ms.
Pull down assay with fatty acid beads
Coupling of fatty acid to agarose beads was performed according to Beck-Garcia et al. (26). Shortly, in a first step the carboxyl group of the fatty acid was activated by incubation of 46 μmol of fatty acid with 92 μmol of N,N-diisopropyl-ethylamine (DIEA) and 35 μmol O-(benzotriazol-1-yl)-tetramethylurnium tetrafluoroborate (TBTU) in 1 ml dimethylformamide (DMF) for 6 h at room temperature on a rotating mixer. Then 500 μl (bead volume) of ω -aminohexyl-agarose beads in DMF were added and incubated overnight at room temperature on a rotating mixer. Coupled beads were washed twice with DMF and three times with PBS and were diluted 1 to 10 with unbound ω-aminohexyl-agarose beads. For the pull down assay, 400 μl of protein solution in PBS was incubated with 15 μl diluted beads (bed volume) for 2 h at 4 °C on a rotating mixer. After washing beads five times with ice-cold PBS, bound protein was eluted from the beads by addition of 2 fold SDS loading buffer and heating at 95 °C for 5 min. Samples were separated on 7.5 % polyacrylamide SDS gels followed by Western Blot analysis.
Electrophoresis (SDS-PAGE) and Western blot analysis
For protein analysis, samples were run on 7.5 or 10 % native or sodium dodecyl sulfate–polyacrylamide gels (SDS-PAGE). For visualization of separated proteins, gel was stained with 0.1 % Coomassie Blue R250 in 10 % acetic acid, 50 % methanol and 40 % H2O and destained with 10 % acetic acid, 50 % methanol and 40 % H2O. From other gels proteins were transferred onto polyvinylidene fluoride membrane by semidry western blotting. For specific detection, the following primary antibodies were used: rabbit polyclonal anti A1AT (DAKO), mouse monoclonal anti A1AT (clone B9, Santa Cruz Biotechnology), mouse monoclonal anti polymeric A1AT (Clone 2C1, Hycult biotech), mouse monoclonal anti FABP4 (clone 1105CT1-1-1, Antibodies-Online GmbH), rabbit polyclonal anti ERK1/2 and mouse monoclonal anti p-ERK1/2 (both from Sigma-Aldrich), monoclonal anti β-actin (AC-15, Sigma-Aldrich). The immune-complexes were visualized with appropriate secondary horseradish peroxidase-conjugated antibodies (DAKO A/S) and ECL Western blotting substrate (Thermo Fisher Scientific). The density of the specific bands was quantified using ImageJ software (http://imagej.nih.gov/ij)
Isolation of Peripheral Blood Mononuclear Cells (PBMCs)
Human PBMCs were isolated from peripheral blood of healthy volunteers using Lymphosep discontinuous gradient centrifugation according to the manufacturer's instructions. PBMCs were resuspended in RPMI-1640 with 2 mM N-acetyl-L-alanyl-L-glutamine (Gibco, Life Technologies) supplemented with 1% nonessential amino acids, 2% sodium pyruvate and 20 mM HEPES and plated at a density of 4 - 10 x 106 cells/ml. Cells were incubated for 75 min at 37°C and 5% CO2 to allow monocytes to adhere to the cell culture plates. Afterwards, non-adherent cells were removed by washing with PBS containing Mg2+ and Ca2+ (Gibco, Life Technologies) and fresh medium without fetal calf serum was added. After 24 h, adherent monocytes were used for experiments.
Isolation of human peripheral blood monocytes
Primary monocytes were isolated by negative isolation from freshly prepared PBMCs using MACS magnetic separation system with Human Monocyte Isolation Kit II (Miltenyi Biotec) as described previously (27). Cells were >98% positive for CD14 as determined by flow cytometry.
Experimental conditions
Human blood adherent monocytes or purified monocytes were incubated directly or pre-treated for 30 min with specific MAPK/ERK kinase inhibitor UO126 (10 μM, Sigma Aldrich), with GW9662, a selective, irreversible PPARγ antagonist, or with ST247 (1 μM, Sigma Aldrich), a selective inverse agonist of PPARβ/δ. Afterwards, cells were cultured alone or together with the aforementioned A1AT preparations (0.5 or 1 mg/ml) or HSA (1 mg/ml) for various time points up to 24 h. At the end of incubation time, cell supernatants and lysates were collected for further analysis. In some experiments cells were subjected to fractionation using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific).
Detection of Cytotoxicity (LDH assay)
Treatment associated cytotoxicity was determined using the Cytotoxicity Detection Kit (LDH) from Roche according to the manufacturer's protocol. In brief, the assay quantifies LDH released from ruptured or dead cells into the culture supernatant by a colorimetric reaction. Cells were treated according to the experimental setting and cell supernatants were collected at the end of incubation time. Total cell lysate was used as high control. For low control and background control, supernatant from untreated cells and assay medium alone were used, respectively. Absorbance of colorimetric product of LDH reaction was measured at 490 nm using Infinite M200 microplate reader (Tecan). Measurements were carried out in triplicates.
Specific gene expression analysis by RT-PCR
Gene expression analysis was assessed as described earlier (24). Briefly, total RNA was isolated using the RNeasy® Mini Kit (Qiagen) according to the manufacturer's instructions. cDNA was synthesized by reverse transcription using the high-capacity cDNA reverse transcription kit (Applied Biosystems, Life Technologies). Expression levels of Angptl4, fatty acid binding protein 4 (FABP4), and fatty acid translocase (CD36) were analyzed by real time PCR (RT-PCR) using the TaqMan® Gene Expression Assay (Applied Biosystems, Life Technologies). The expression of the housekeeping gene, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used for normalization. All primers were purchased from Applied Biosystems. Relative gene expression was calculated according to the ΔΔ CT method.
Analysis of CD36 surface expression by flow cytometry
In all flow cytometry experiments 2x105 cells per condition were used. Surface expression of CD14, CD16, and CD36 was assessed in cells kept as control or pre-incubated with appropriate substance for 6 h. Cells were then labeled with fluorescein (FITC) conjugated monoclonal anti-human CD36 antibody, phycoerythrin (PE) conjugated monoclonal anti-human CD14 antibody, allophycocyanin (APC) conjugated monoclonal anti-human CD16 antibody, or the corresponding isotype controls (all mouse IgG1 mAb) (Immnotools, Friesoythe, Germany) for 35 min at 4°C. One set of cells was kept unstained for gating purpose. After labeling, cells were washed with FACS buffer (PBS, 1% bovine serum albumin, pH 7.4), re-suspended in PBS with 0.5% bovine serum albumin and examined using FACS Calibur (BD Biosciences). Data was analyzed using FACSdiva software (BD Biosciences).
In vitro measurement of elastase activity
The A1AT preparations were tested for their inhibitory activity towards pancreatic elastase (Sigma Aldrich). For quantification of elastase activity, degradation of elastase substrate N-succinyl-Ala-Ala-Ala-p-nitroanilide (Sigma Aldrich) was followed spectrophotometrically as described previously (28). In brief, A1AT preparations were pre-incubated with elastase at a molar ratio of 1:2.6 at 37°C in 0.1 M Tris buffer, pH 8. After 5 min, elastase substrate to a final concentration of 83 μM was added and absorbance was followed at 405 nm for 3 min on Infinite® M200 microplate reader (Tecan). Sample containing substrate and buffer alone was used for blank reduction. Elastase inhibition was calculated relative to samples containing only elastase and substrate.
Quantitative analysis of Angptl4
Cell culture supernatants collected from monocytes treated with different A1AT preparations for 24 h were analyzed for the concentration of Angptl4 protein using Duoset ELISA ki(R&D Systems) or ARP4 (Angiopoietin-like 4) Human ELISA kit (Abcam). Detection limit for Duoset ELISA was 1.25 ng/ml and for ARP4 ELISA < 20 pg/ml.
Statistical analysis
The differences in the means of experimental results were analysed for their statistical significance using one-way ANOVA combined with a multiple-comparison procedure (Scheffe multiple range test), with an overall significance level of p=0.05. An independent two-sample t-test was also used. Statistical Package (SPSS for Windows, release 21.0) was used for the statistical calculations.
Results
Analysis of FA content in different A1AT preparations
Fatty acid spectra in the A1AT solutions were determined by nano-electrospray ionization tandem mass spectrometry as previously described (25). The fatty acid anion fragments found at m/z 279 and 281 represent the anions of linoleic acid (LA) and oleic acid (OA), respectively. For quantitative determination of LA and OA an internal standard was added prior to lipid extraction. As fatty acid standard FA 17:0 was used that does not occur in a significant amount in the analyzed samples. As shown in Table 1, A1AT (Prolastin®) contained significant amounts of fatty acids C18:2 (linoleic acid, LA) and C18:1 (oleic acid, OA) whereas A1AT (Zemaira) contained only trace amounts of LA and OA (Table 1). Other studies (see Methods) confirmed that A1AT (Zemaira) contains no detectable amounts of other lipids, such as HDL, LDL, cholesterol or triglycerides. In light of the findings presented above, we prepared M- and Z-A1AT from pooled PiMM and PiZZ plasma by using one step alpha1-antitrypsin select affinity chromatography without employing organic solvents, detergents or heat inactivation steps (Supplementary Fig. 1A). Affinity purified M-A1AT contained only trace amounts of LA and OA whereas Z-A1AT contained 10 times or more LA and OA than M-A1AT (Table 1). Furthermore, FA free Zemaira (A1AT-0) was pre-incubated with LA (at 1: 2.4 molar ratio A1AT: LA) for 3 h at 37°C. Unbound LA was removed by repeated filtration using Centricon centrifugal filter with molecular weight cut-off of 10 kDa. Analysis of FA content revealed that A1AT-LA (Zemaira pre-incubated with LA) contained more LA than A1AT (Prolastin®) (Table 1).
Table. 1. FA content in different A1AT preparations.
| Sample | N | Linoleic acid (LA) | N | Oleic acid (OA) | ||
|---|---|---|---|---|---|---|
| pmol/μg protein | pmol/μg protein | |||||
| mean | SD | mean | SD | |||
| A1AT (Zemaira) | 6 | nd* | 6 | nd* | ||
| A1AT (Zemaira)-LA | 6 | 3.40 | 1.38 | 6 | 0.11 | 0.26 |
| A1AT (Prolastin®) | 6 | 1.00 | 0.55 | 6 | 2.65 | 1.51 |
| M-A1AT affinity purified | 6 | 0.03 | 0.02 | 6 | 0.02 | 0.04 |
| Z-A1AT affinity purified | 6 | 0.35 | 0.35 | 6 | 0.94 | 0.83 |
N-batches of pooled preparations of A1AT; SD, standard deviation; nd- not detected
in some samples trace amounts of FAs were detected producing values below of 0.02 pmol/μg protein with high SD.
Analysis of interactions between A1AT‐0 and FA using a fatty acid pull-down assay
Based on the above findings, we prepared linoleic (LA) and oleic acid (OA)-coupled agarose beads and using pull-down assays investigated a putative interaction between A1AT and FA. Human serum albumin (HSA), a known FA binding protein (19), was chosen to validate the FA pull-down assay. FA-bead-bound proteins were separated by SDS-PAGE and visualized on Western blots by using specific monoclonal antibodies against human A1AT or HSA. As shown in Fig. 1A both, LA and OA coupled beads enriched HSA-0 from 1mg/ml solution with a similar efficiency. Control beads produced in a coupling reaction without addition of FA, were used to evaluate non-specific binding to ω-aminohexyl-agarose. Non-specific binding of HSA to control beads was negligible low (Fig. 1A). Similar to HSA-0, A1AT-0 bound to LA and OA coupled beads from 1mg/ml A1AT solution (Fig. 1A). Non-specific binding of A1AT to control beads was low.
Figure 1.

Pull down of FA-free HSA-0 and A1AT-0 by FA-coupled agarose beads. (A) Fatty acid free HSA-0 or A1AT-0 (Zemaira) was mixed on a rotating mixer with ω-aminohexyl-agarose beads coupled to LA, OA or control (co.) beads exposed to coupling reaction in absence of FA. After incubation for 2h, the beads were washed thoroughly with PBS to remove unbound proteins, SDS sample buffer was added and bead-bound proteins were released by heating at 95°C for 5 min. (B) For competition experiments target protein was mixed with competing protein in the concentrations as indicated in the figure before addition of beads. One representative blot out of 3 independent experiments is shown. Densities were evaluated using ImageJ. Results are given in mean±SD.
Relative affinities of HSA and A1AT for LA binding
We next performed competition experiments of A1AT binding to LA-coupled beads in the presence of increasing concentrations of HSA and vice versa. Fig. 1B shows that HSA-0 dose-dependently competes with A1AT‐0 for binding to FA. Similarly, A1AT also competed for HSA binding to LA beads. When albumin was added into the mixture in a molar ratio of 1 to 0.8 (A1AT to HSA), the amount of bead-bound A1AT was reduced to 66±4% compared to 100% when A1AT-0 was added alone (Fig. 1B). Under similar experimental conditions, the addition of A1AT-0 in a molar ratio of 1 to 1.2 (HSA to A1AT) resulted in the loss of 41±9% bead-bound albumin. HSA-0 and A1AT-0 prevented their respective binding to LA beads by about 73% when added in a molar excess of 8-fold for HSA-0 and 12-fold for A1AT-0. Noticeably, with 33-fold molar excess of A1AT and 36-fold molar excess of HSA, both A1AT and HSA were capable of almost completely hindering binding of the other protein to LA beads (Fig. 1B).
Despite the high molar excess of HSA versus A1AT (37.9 mg/ml albumin versus 1.44 mg/ml A1AT, in PiMM serum), LA- and OA-bound beads captured and pulled down not only albumin, but also M-A1AT. Likewise, FA beads were capturing lower but clearly detectable amounts of Z-A1AT from PiZZ serum (Fig. 2).
Figure 2.

FA pull down of A1AT and HSA from PiMM and PiZZ serum. Pooled serum samples were pre-diluted with 3 volumes of PBS and incubated with LA-, OA- or control (co.) beads for 2 h on a rotating mixer. Beads were washed thoroughly with PBS to remove unbound proteins, SDS sample buffer was added and bead-bound proteins were released by heating at 95°C for 5 min. Sample load was 15 μl for A1AT blot and 5 μl for HSA blot. To facilitate comparability of the blots 200 ng target proteins were applied. Antibodies don't show any cross reactivity with competing proteins.
Qualitative analysis of A1AT preparations
It remained unclear whether the A1AT complex formation with FA alters the molecular form of A1AT protein. As illustrated in Fig. 3A, a polyclonal anti-A1AT antibody showed a similar pattern for A1AT-0 and A1AT-FA preparations, i.e. monomer and distinct higher molecular weight forms of A1AT. However, when the specific anti-A1AT (2C1) polymer antibody was used, 110 kDa and larger polymers were detected in A1AT-LA and A1AT-OA preparations (Fig. 3A). Qualitative characterization of M- and Z-type A1AT isolated from human serum by affinity chromatography revealed that the M-A1AT protein mainly appears as a monomer whereas Z-A1AT, which contains FAs, comprises a mixture of a monomeric and several polymers of different sizes (Fig. 3B). Importantly, mixing A1AT with LA did not change the property of A1AT to inhibit elastase activity (Supplementary Fig. 1B). In line with previously published data (29), the anti-elastase activity of affinity-purified M-A1AT was similar to that of commercial serum-derived A1AT-0 (Zemaira), whereas affinity-purified Z-A1AT showed lower anti-elastase activity (Supplementary Fig. 1B).
FIGURE 3.

Qualitative analysis of molecular forms of A1AT found in A1AT preparations by non-denaturing SDS-PAGE following Western blot analysis using rabbit polyclonal anti-A1AT and mouse monoclonal anti-A1AT polymer (2C1) antibodies. (A) A1AT-LA and –OA complexes were prepared by 3 h incubation at 37°C following separation from free FA with centricon-10 (MWCO 10 kDa). Each blot is representative out of n=3 independent experiments. (B) M- and Z-A1AT pools from Alph1-Antitrypsin Select affinity purification were analyzed by Western Blot. Each blot is representative out of n=3 independent experiments.
Time-dependent effects of A1AT-0 and A1AT-FA on Angptl4 and FABP4 expression
In the present study, adherent blood monocytes were used to compare the effect of A1AT-0 (Zemaira) to A1AT-LA (Zemaira LA complex) on Angptl4 expression. As demonstrated in Fig. 4A, exposure of cells to A1AT-LA for 2 h resulted in a 10-fold induction of Angptl4 expression, whereas at 6 h, Angptl4 expression was maximal relative to untreated or A1AT-0 treated cells. After 24 h, Angptl4 expression in A1AT-LA-treated cells still remained higher (4-fold; NS) than in untreated controls or A1AT-0 treated cells. As demonstrated in Fig. 4B, adherent blood monocytes incubated with A1AT‐LA increased FABP4 expression in a time-dependent manner. After only 4 h, there was a 3-fold induction of FABP4 expression (p=0.02) relative to untreated controls (Fig. 4B). However, unlike Angptl4, the maximal increase of FABP4 (9.7-fold, p<0.001) occurred after 24 h. A1AT-OA significantly induced transient expression of Angptl4 and FABP4 genes albeit with lower magnitude than A1AT-LA Supplementary figure 3). Both, Angptl4 and FABP4 mRNA levels did not change in cells treated with A1AT-0 (Fig. 4A, B and Supplementary figure 2). It is noteworthy, that exposure to A1AT‐FA did not affect cell viability as measured by lack of release of LDH (data not shown). Since A1AT‐LA maximally induces Angptl4 expression and significantly upregulated FABP4 at 6 h, further experiments used this time point.
Figure 4.

Time-dependent effect of A1AT-0 and A1AT-LA on Angptl4 and FABP4 expression of human adherent blood monocytes. Cells were treated with A1AT -0 or A1AT-LA (1 mg/ml) for different periods of time. Gene expression levels of Angptl4 (A) and FABP4 (B) were analyzed by RT-PCR and normalized to GAPDH. Each point represents mean ± SD of n=3 independent experiments, each with 3 repeats.
Effects of A1AT-0 and A1AT-FA on Angptl4, FABP4, CD36 and HIF-1α expression
At mRNA level, FABP4 was shown to correlate with CD36 in cells of monocyte/macrophage lineage (30). CD36 is a key protein involved in regulating the uptake of FA (31). In the following set of experiments, we compared effects of A1AT-LA and A1AT-OA on Angptl4, FABP4 and CD36 expression at 6h. To exclude the putative interference of free FA, adherent peripheral monocytes were treated with LA or OA solutions produced under the same conditions as A1AT-FA complexes (See Materials and Methods). As illustrated in Fig. 5, A1AT-LA preparation markedly increased mRNA levels of Angplt4 (54.9-fold, p<0.001), FABP4 (11.4-fold, p<0.001) and CD36 (3.1-fold, p<0.001) relative to cells treated with A1AT-0 or FA alone. Similarly, when compared to A1AT-0-treated cells, A1AT-OA induced expression of Angptl4 by 10.7-fold (p<0,001), FABP4 by 2.9-fold (p<0.001) and CD36 by 2-fold (p<0.001).
Figure 5.
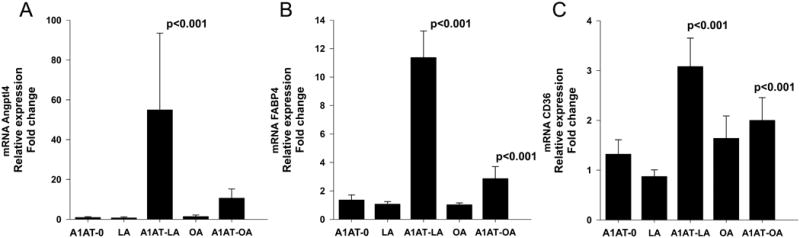
Effects of A1AT-LA and A1AT-OA complexes on Angptl4 and related gene expression in adherent blood monocytes compared to those of A1AT-0 and FAs alone. Cells were treated with A1AT-0, complexes of A1AT-LA and A1AT-OA (1 mg/ml) or with LA and OA preparations alone for 6 h. Gene expression levels of Angptl4 (A), FABP4 (B) and CD36 (C) were assessed by RT-PCR and normalized to GAPDH. Each bar represents mean ± SD of n=6 independent experiments in case of A1AT-0, A1AT-LA and LA and n=3 in case of A1AT-OA and OA. Measurements were performed in triplicates.
We previously found that A1AT (Prolastin®) induces HIF-1α expression in a time dependent manner, however blocking HIF-1α expression with CAY10585, a small molecule inhibitor of HIF-1α accumulation and gene transcriptional activity, had no effect on A1AT (Prolastin®)-induced Angplt4 expression (24). We then asked, if FA bound A1AT has any effect on HIF-1α expression. When compared to controls, treatment of adherent peripheral monocytes with A1AT-LA resulted in an upregulation of HIF-1α mRNA by 1.6 (0.34)-fold, p<0.01, n=3 independent experiments with 15 repeats. However, milder induction in HIF-1α expression was found in A1AT-OA treated cells [1.2 (0.38)-fold, n.s. relative to controls, n=3 independent experiments, 21 repeats].
Effects of A1AT-0 and A1AT-FA on Angptl4 and related gene expression in CD14-positive peripheral blood monocytes
In the next set of experiments, the effect of A1AT-0 and A1AT-FA complexes were studied in human peripheral blood monocytes isolated by negative selection (purity >98% CD14-positive cells). At 6 h, A1AT-LA and A1AT-OA, both increased Angptl4 gene expression by 28-fold and 23-fold (p<0.001) relative to controls of A1AT-0 treated cells (Supplementary Fig. 3A). Concomitantly, when compared to controls, cell treatment with A1AT-LA and A1AT-OA resulted in an increased FABP4 expression by 8.9 and 6.4-fold, respectively, p<0.001 (Supplementary Fig. 3B). As demonstrated in Supplementary Fig. 3C, both A1AT-FA preparations also increased CD36 mRNA (A1AT-LA: by 3-fold and A1AT-OA: by 1.7-fold, p<0.001, relative to controls). A1AT-0 didn't show any effects on Angptl4, FABP4 or CD36 mRNA levels (Supplementary Fig. 3). These results clearly indicate the contribution of CD14-positive monocytes to A1AT-FA-induced Angptl4, FABP4 and CD36 gene expression.
Effect of A1AT-FAs on Angptl4, FABP4 and CD36 protein levels
In accordance with our gene expression, we observed increased protein levels of Angptl4, FABP4 and CD36 in A1AT-FA treated adherent peripheral monocytes for 6 h as compared to untreated control cells and cells treated with A1AT-0 (Fig. 6). When compared Angptl4 levels in supernatant of control cells (13.9±11.6 pg/ml), treatment with A1AT-FAs remarkably increased the release of Angptl4 [A1AT-LA: 59.3±12.4 pg/ml, p=0.013; and A1AT-OA: 66.1±19.1 pg/ml, p=0.005] (Fig. 6A). Analysis of nuclear and cytoplasmic fractions of adherent peripheral monocytes revealed increased cytoplasmic FABP4 protein concentration in cells treated with A1AT-LA or A1AT-OA, but not in control cells or those treated with A1AT-0 (Fig. 6B). Notably, nuclear translocation of FABP4 was not found (Fig. 6B). Flow cytometry analysis showed increased CD36 surface levels on A1AT-LA (162±23 MFI in % of control, p<0.001) and A1AT-OA (153±12 MFI in % of control, p=0.012) treated cells compared to control cells. According to our results, no significant shift in monocyte populations occurred when cells were treated with A1AT-0 or A1AT-FA (Fig 6C).
Figure 6.

Effects of A1AT-LA and A1AT-OA complexes on Angptl4, FABP4 and CD36 protein levels. Adherent monocytes were kept untreated for the control or treated with A1AT-0, A1AT-LA or A1AT-OA complexes (1 mg/ml) for 6 h. (A) Supernatants were collected and analyzed for released Angptl4 by ARP4 ELISA. (B) Cells were fractionated into cytoplasmic and nuclear fractions using Cytoplasmic and Nuclear Fractionation kit. FABP4 protein levels in the fractions were determined by Western Blot analysis. β-Actin was stained for a loading control. Presented blot is representative of n=3 repeated experiments. Cell surface expression of CD36 was analyzed by flow cytometry (C). Cells were stained with anti CD36-FITC or respective FITC isotype. Mean fluorescence intensities of ungated cells were determined. Each bar represents mean ±SD of n=3 independent experiments, each performed in duplicates.
Effects of affinity-purified M-A1AT and Z-A1AT on Angptl4 and related gene expression
To demonstrate physiological relevance of A1AT-FA complexes and to confirm that /test if endogenous A1AT carrying different concentrations of LA and OA has diverse effects on expression of genes related with lipid homeostasis, we treated adherent blood monocytes with different naturally occurring A1ATs, namely affinity-purified M-A1AT binding only small amounts of FAs and Z-A1AT carrying considerable amounts of both LA and OA (Table 1). As demonstrated in Fig. 7A, C and D, M-A1AT did not affect Angptl4, FABP4 and CD36 gene expression whereas Z-A1AT significantly induced expression of Angptl4, FABP4 and CD36 by 4-, 3.7-and 2-fold, respectively, as compared to controls. In support of these latter, Z-A1AT also markedly induced the release of Angptl4 protein, whereas M-A1AT had only a negligible effect (Fig. 7B).
Figure 7.
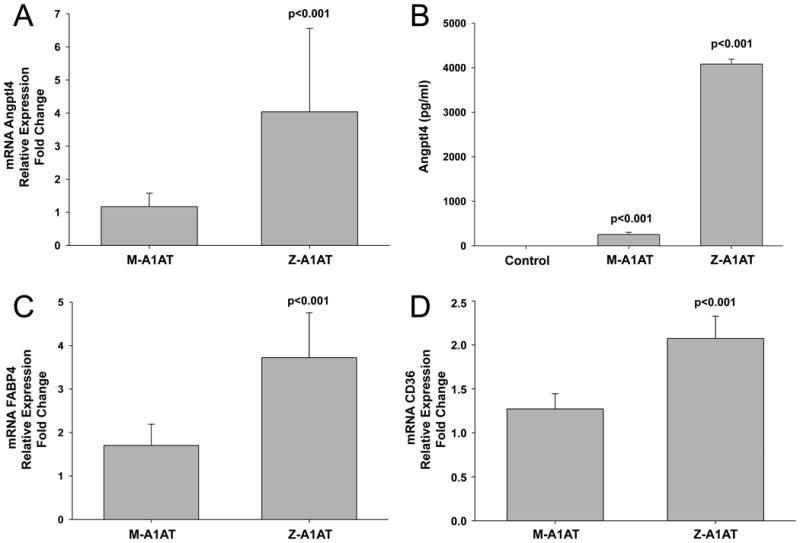
Effects of serum derived affinity purified M- and Z-A1AT on Angptl4 and related gene expression. Adherent blood monocytes were treated with 0.5 mg/ml of M- and Z-A1AT, isolated from pooled PiMM and PiZZ serum, for a total of 6 h. mRNA levels of Angptl4 (A), FABP4 (C) and CD36 (D) were analyzed by RT-PCR and normalized to GAPDH. Each point represents mean ±SD of n=4 independent experiments, each with 3 repeats. (B) Concentration of Angptl4 in cell culture supernatants was determined by Duoset ELISA. Each bar represents mean ± SD of n=4 independent experiments, each with 2 repeats.
Effects of GW9662 and ST247 on A1AT-LA-induced Angptl4 and FABP4 expression
We previously found that pre-incubation of adherent blood monocytes for 30 min with GW9662, a selective and irreversible PPARγ antagonist, dramatically lowers the ability of A1AT (Prolastin®) to induce Angplt4 expression (24). As shown in Fig. 8A, cells pre-treated with GW9662 (10 μM), significantly diminished the effect of A1AT-LA on Angplt4 mRNA compared to A1AT-LA effect without the inhibitor. Similarly, pre-incubation of cells with GW9662 also significantly diminished inducing effect of A1AT-LA on FABP4 mRNA (6.2- vs 2.5-fold, p<0.001) (Fig. 8B). Angptl4 expression is not exclusively regulated by PPARγ. Others report that Angplt4 is also regulated by the subtype PPARβ/δ (32), and that FAs induce Angptl4 expression via PPARβ/δ (33). Therefore, the effect of ST247, a selective and inverse agonist of PPARβ/δ, was studied. When compared to non-pre-treated cells, cell pre-treatment with ST247 (1 μM) clearly diminished the effect of A1AT-LA on Angptl4 mRNA (11.6- vs 4.1-fold, p<0.001) and FABP4 mRNA (5.4-vs 2.8-fold, p<0.001), respectively (Fig. 8C and D). Cell pre-treatment with ST247 also significantly lowered A1AT (Prolastin®)-induced Angptl4 and FABP4 expression relative to non-pre-treated cells (Angptl4 9.7- vs 2.6-fold, p=0.003; FABP4 4.6-vs 2.4-fold, p<0.001) (Fig. 8C and D). Hence, A1AT-FA induces Angptl4 through the PPARs pathway.
Figure 8.

A1AT-LA induced expression of Angptl4 and FABP4 is related to PPARγ and PPARβ/δ activity. Cells were pretreated for 30 min with 10 μM GW9662, an irreversible PPARγ antagonist (A, B), or with 1 μM ST247, a selective and inverse agonist of PPARβ/δ (C, D), prior to addition of 1 mg/ml A1AT-0, A1AT-LA, LA preparation or Prolastin® for another 6 h. Expression levels of Angptl4 (A, C) and FABP4 (B, D) were determined by RT-PCR and normalized to GAPDH. In (A, B) each point represents mean ±SD of n=3 independent experiments, each with 3 repeats. In (C, D) each point represents mean ±SD of n=2 independent experiments, each with 3 repeats.
Effects of A1AT-0 and A1AT-LA on transient ERK1/2 phosphorylation
PPARs activity is regulated by extracellular signal-regulated protein kinase (ERK), mitogen-activated protein (MAP) kinase, and PPAR ligands show varying effects on the activity of ERK (34). Our previous studies revealed that A1AT (Prolastin®) induces a rapid and transient activation of the MEK-ERK1/2 pathway (24). Elevated phosphorylation of ERK 1/2 was noted as early as 15-30 min following treatment with A1AT; however this increase was not detected after 1h. To address whether transient MEK-ERK1/2 activation by A1AT (Prolastin®) is due to complexes of A1AT with FAs (Table. 1), adherent blood monocytes were treated for 30 min with A1AT-0 or A1AT-LA. As illustrated in Fig. 9A, both A1AT-0 and A1AT-LA induced ERK1/2 phosphorylation suggesting that this effect of A1AT is independent of FA content. To observe an influence of ERK1/2 phosphorylation on A1AT-LA induced Angptl4 expression, we pre-incubated cells with MEK/ERK1/2 inhibitor, UO126. Consistent with our previous data, the A1AT-induced increase in ERK phosphorylation was absent by a 30 min pre-incubation with UO126. Concomitantly, we observed a significant reduction of the stimulating effect of A1AT-LA on Angplt4 mRNA (Fig. 9B)
Figure 9.

(A) Transient activation of MEK-ERK1/2 pathway is independent on A1AT complexation with LA. Western blot of cell lysates prepared from adherent blood monocytes illustrates phosphorylation of ERK1/2 at 30 min in response to treatment with 1 mg/ml A1AT-0 or A1AT-LA. Blots were stained for phosphorylated and total ERK1/2. β-Actin was used as a loading control. Pre-incubation of cells with MEK/ERK1/2 inhibitor UO126 (10 μM), completely abolished ERK1/2 activation in all samples. Presented blot is representative of n=4 repeated experiments. (B) Effect of ERK1/2 phosphorylation on A1AT-LA-induced Angptl4 expression. Adherent PBMCs were pre incubated for 30 min with UO126 (10 μM) prior to addition of 1 mg/ml A1AT-0 or A1AT-LA for another 6 h. Angptl4 mRNA levels were assessed by RT-PCR and normalized to GAPDH. Each point represents mean ±SD of n=3 independent experiments, each with 3 repeats.
Effects of GW9662, ST247 and UO126 on A1AT-OA-induced Angptl4 and FABP4 expression
Similarly to the A1AT-LA, targeting of PPARs with GW9662 and ST247 as well as blocking ERK phosphorylation using UO126, strongly decreased A1AT-OA-induced Angptl4 and FABP4 mRNA levels (Supplementary Figure 4) suggesting that A1AT-FA-induced Angptl4 expression is dependent on ERK-regulated PPARs activation.
Discussion
The clinical importance of A1AT is highlighted in individuals with inherited PiZZ (Glu342Lys) A1AT deficiency (plasma levels below 0.7 g/l, whereas normal values range between 1 and 2 g/l) due to the polymerization and a defective secretion of Z A1AT protein. These individuals have an increased risk of developing early onset emphysema, liver and pancreatic diseases at any age, and in rare cases panniculitis and vasculitis (35). Therapy with A1AT isolated from pooled human plasma is used to treat patients with inherited A1AT deficiency-related emphysema, and several preparations of A1AT are available. These A1AT preparations are administered intravenously every week, typically at a dose of 60mg/kg of body weight. They are well tolerated with no evidence of virus transmission, and show very similar effects in maintaining serum levels of A1AT. Differences in manufacturing process are known to result in analytical differences in the A1AT preparations. For example, Cowden et al, (36) demonstrated that A1AT (Zemaira) is 99% pure, contains the least contaminating proteins and has high activity as an elastase inhibitor. By contrast, A1AT (Prolastin®) has only 60% purity and contains significant amounts of inhibitory-inactive forms of A1AT. The significance to the patient, if any, of these differences in A1AT preparations is not known.
We previously reported that clinical-grade preparations of A1AT, such as Prolastin (Grifols) and Aralast (Baxter), as well as A1AT purchased from Sigma Aldrich, induce Angptl4 expression in human adherent monocytes and primary lung endothelial cells (24). However, under the same experimental conditions, A1AT (Zemaira) did not affect Angptl4 mRNA and protein release. This striking discrepancy between the plasma purified preparations of A1AT prompted us to investigate this further.
One of the biological functions of Angptl4 is the regulation of lipid metabolism (37-39). The expression of Angptl4 is also regulated through a synergistic induction of the lipid-sensing peroxisome proliferator-activated receptors (PPARs) α, β, and γ (40). Among the lipids, fatty acids (FA) are the best-recognized inducers of Angptl4 expression (16, 41-43). Although FA are an energy source, they are also recognized as regulators of inflammation, and it has been suggested that PPARs play an important role in FA-dependent gene regulation (44).
Depending on purification methods, therapeutic plasma protein products can contain variable amounts of FAs and lipids (45). Thus, we suspected that FA content might explain A1AT-induced Angptl4 expression. Indeed, lipidomic analysis revealed that A1AT (Prolastin®) contains significant amounts of FA, specifically linoleic acid (LA, C18:2) and oleic acid (OA, C18:1), two of the most abundant free FA in human plasma (46). In contrast, A1AT (Zemaira) was lipid and FA free-only trace amounts of LA and OA were detected. The presence of FA in A1AT (Prolastin®) preparations pointed to a putative property of A1AT to bind FA.
The solubility of FA in aqueous solutions, such as blood plasma, is far below 0.3 mM (47), therefore FA-binding and transporting proteins guarantee a sufficient transport of FA to the FA-consuming organs. The best characterized candidate for transport is serum albumin. Due to the high FA-binding capacity of albumin, it is assumed that even substantially reduced albumin levels during acute phase reaction can sufficiently perform this function. On the other hand, it is likely that a low albumin-FA binding capacity is compensated by other acute phase plasma proteins with FA binding properties. To test if A1AT, a positive acute phase protein, is a FA binding protein, we employed an FA bead-based pull-down assay (26). When LA or OA-coupled beads were pre-incubated with A1AT-0 or HSA-0 (as a positive control), FA-coupled beads pulled down A1AT and HSA with similar efficiency. Neither A1AT nor HSA demonstrated non-specific binding to uncoupled agarose beads. Both proteins in a concentration-dependent manner competed for the binding to the FA-coupled beads. We next took an unbiased approach by employing the LA bead pull-down assay for human plasma. Incubation of LA-coupled agarose beads with pooled plasma from MM (normal) or ZZ (deficient) A1AT subjects resulted in a specific binding of M and Z A1AT protein with LA. Plasma A1AT bound to LA despite the fact that normal serum contains about 26 times more albumin than A1AT and that albumin binding affinity to LA is high. Moreover, when we employed plasma from ZZ A1AT individuals, containing only 10% of A1AT relative to normal MM plasma, binding of A1AT to FA was still measurable. In fact, lipidomics analysis revealed that affinity purified plasma Z, but not M A1AT contains significant amount of fatty acids, LA and OA. The Z A1AT protein is characterized by an increased exposure of hydrophobic regions to the solvent and therefore, has enhanced tendency to hydrophobic interactions and oligomeric assemblies (48). As a consequence, Z A1AT may spontaneously interact with free FAs and form protein-FA polymers. Opposite, the amino acids involved in the polymerization are completely buried in native, M-type
A1AT (49). Likewise, M-A1AT probably binds FA only under interaction-favoring conditions, like during increased free FA concentrations and/or decrease in albumin concentration during acute phase reaction. These data constitute good evidence that A1AT exists in FA-free and FA-bound forms and therefore might express different biological activities.
Angptl4 is a potent anti-angiogenic and anti-inflammatory factor, which is under the transcriptional control of PPARγ (50). FABP4 and CD36 are also PPARγ target genes involved in lipid metabolism, and PPARγ activation was found to induce FABP4 mRNA in human monocytes (51). As expected, GW9662, a selective and irreversible PPARγ antagonist (52), significantly inhibited A1AT-FA-induced Angptl4, FABP4 and CD36 expression confirming involvement of PPARγ pathway. However, Angptl4 expression can also be regulated by other PPAR isotypes. For example, Kersten et al reported that in rat hepatoma cells FA induce Angplt4 mRNA via PPARα, in mouse intestinal cells via PPARβ/δ whereas in human myocytes both, PPARα and PPARβ/δ, likely play a role (53). Krey et al showed that the affinity of PPARδ for linoleic acid is higher than for oleic acid, and that linoleic acids are the most potent natural ligands for PPARδ (54). These findings imply that Angptl4 is one of the PPARβ/δ target genes. ST247 is an inverse agonist of PPARβ/δ, which has the ability to enter cells and to inhibit the transcriptional activity of PPARβ/δ but it does not affect activated PPARγ (55). Likewise, pre-incubation of adherent blood monocytes with ST247 resulted in a significant inhibition of A1AT-LA-induced Angptl4 and FABP4 expression. Hence, both GW9662 and ST247 inhibit effect of A1AT-FA on Angptl4 expression supporting a notion that Angptl4 transcription is regulated by several members of the PPAR family.
Angptl4 transcription, at least partially, can be regulated by ERK-mediated PPARs activation (56). Knowing that A1AT can be in FA-free or FA-bound forms, we wanted to re-investigate effects of A1AT on ERK1/2 activation. We previously published that A1AT (Prolastin®)-induces transient ERK1/2 phosphorylation and that blocking the ERK1/2 pathway with MEK inhibitor (UO126) markedly diminishes effect of A1AT on Angptl4 expression (24). Here in adherent blood monocyte cultures we show that both, A1AT-0 and A1AT-FA, induce a rapid and transient ERK1/2 activation. Thus, the FA bound form of A1AT does not seem to be a prerequisite for ERK1/2 activation, although only A1AT-FA up-regulates Angptl4 expression. Because inhibition of ERK1/2 by UO126 also decreases PPARs activity (57) and, as discussed above, Angptl4 is the downstream target gene of PPARs, it is logical to assume that the regulation of Angptl4 expression by A1AT-FA is dependent on PPARs rather than the MEK/ERK1/2 pathway. This conclusion is in line with our previous findings that GW9662, which does not alter the property of A1AT-FA to induce ERK1/2 phosphorylation, strongly inhibits induction of Angptl4 expression (24).
To our knowledge, in vitro FA binding of A1AT has not been previously reported, although interestingly, A1AT does possess lipid binding capacity. For example, A1AT occurs in lipid rafts (58, 59), forms complexes with low-density-lipoprotein (LDL) and high-density lipoprotein (HDL) (60, 61). Moreno and et al. found that intravenous therapy with HDL-A1AT affords a better protection against elastase-induced pulmonary emphysema in mice than A1AT alone (62). The increased expression of Angptl4 in response to fetal HDL was also reported (63). The finding that A1AT binds FA, specifically LA, and upregulates expression of Angptl4 through PPARs pathway, suggests its role in lipid homeostasis and immune regulation (44). To date various studies show that FAs directly or indirectly regulate many cellular processes including membrane receptors, ion channels, and gene expression (64). However, there is a paucity of data on FA binding proteins/translocases in FA uptake, intracellular trafficking, and signaling. Relevant to the putative efficacy of A1AT replacement therapy to reduce lung inflammation via inhibition of neutrophil elastase, the interaction between FA and A1AT has no effect on the anti-elastase activity of A1AT protein. This suggests that FA binding does not interfere with inhibitory conformation of the reactive loop of A1AT. However, aside from its elastase inhibitory function A1AT expresses other anti-inflammatory activities (65).
The interest of healthcare providers in A1AT preparations recently has increased because of the beneficial effects of A1AT therapy in single cases and in small cohorts with clinical conditions other than lung emphysema (66). Novel data provide evidence that therapy with A1AT modulates or prevents tissue injury in experimental animal models of human diseases, including graft-versus-host-disease, rheumatoid arthritis, autoimmune diabetes, and renal ischemia–reperfusion injury, among others (20). Nevertheless, despite these many effects ascribed to the A1AT protein, the mechanisms of its effects remain incompletely understood. Our data illustrate that A1AT might occur in FA-free and FA-bound forms, which express diverse effects on the regulation of Angptl4 expression, an inhibitor of plasma triglyceride clearance and anti-inflammatory protein. Notably, we observed high donor-dependent variability in Angptl4 mRNA levels, which is in accordance with findings in vivo (24). This latter might be mediated by plasma free FA levels and/or FA-protein complexes, and warrants further investigations. In addition it is of interest to consider the role of A1AT-FA complexes as exogenous PPARs ligands.
According to the current theory, Z A1AT forms polymers in the endoplasmic reticulum of hepatocytes leading to liver disease, whereas the lack of active protein leaves lung parenchyma unprotected against neutrophil elastase leading to early-onset emphysema (67). However, clinical phenotypes in Z A1AT deficiency can be expressed in different ways, including early onset (at age of 30 years) pulmonary emphysema, childhood or adult liver cirrhosis, lung and liver diseases simultaneously during adulthood, or no clinical symptoms of any disease. This shows that clinical phenotypes are driven by additional factor(s). Our finding that Z A1AT contains FAs and upregulates expression of Angplt4 and FABP4 suggests a novel role for Z A1AT-FA polymers in lipid metabolism and inflammation. For example, FABP4 is expressed in adipocytes, monocytes/macrophages and in human bronchial epithelial cells (68, 69) and plays a role in cholesterol ester accumulation, uptake of fatty acids and cholesterol. Several studies have shown that abolishing expression of FABP4 protects against atherosclerosis (70) and macrophage activation (71). Opposite, the induction of FABP4 in human bronchial epithelial cells was related to inflammation and the development of asthma (72). Based on these examples above, it is possible to hypothesize that, in particular circumstances, Z A1AT-FA polymers may constantly activate Angptl4 and FABP4, and thus express pro-inflammatory effects. Taken together, our data provide new insights about Z- and M-A1AT properties, which might add to the knowledge about the biological mechanisms behind Z-A1AT-related pathologies.
Our finding that A1AT binds FA and regulates Angptl4 expression opens a new field for investigations of A1AT role in health and disease, and provides a new opportunity for evaluating effects of A1AT therapy.
Supplementary Material
Acknowledgments
We thank laboratory technician Helena Lickei for excellent technical assistance in performing various assays. We also thank colleagues at the Institute for Transfusion Medicine for providing laboratory facilities.
These studies are supported by the NIH Grant AI 15614 (to CAD).
References
- 1.Yoon JC, Chickering TW, Rosen ED, Dussault B, Qin Y, Soukas A, Friedman JM, Holmes WE, Spiegelman BM. Peroxisome proliferator-activated receptor gamma target gene encoding a novel angiopoietin-related protein associated with adipose differentiation. Mol Cell Biol. 2000;20:5343–5349. doi: 10.1128/mcb.20.14.5343-5349.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yoshida K, Shimizugawa T, Ono M, Furukawa H. Angiopoietin-like protein 4 is a potent hyperlipidemia-inducing factor in mice and inhibitor of lipoprotein lipase. J Lipid Res. 2002;43:1770–1772. doi: 10.1194/jlr.c200010-jlr200. [DOI] [PubMed] [Google Scholar]
- 3.Sukonina V, Lookene A, Olivecrona T, Olivecrona G. Angiopoietin-like protein 4 converts lipoprotein lipase to inactive monomers and modulates lipase activity in adipose tissue. Proc Natl Acad Sci U S A. 2006;103:17450–17455. doi: 10.1073/pnas.0604026103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lichtenstein L, Mattijssen F, de Wit NJ, Georgiadi A, Hooiveld GJ, van der Meer R, He Y, Qi L, Koster A, Tamsma JT, Tan NS, Muller M, Kersten S. Angptl4 protects against severe proinflammatory effects of saturated fat by inhibiting fatty acid uptake into mesenteric lymph node macrophages. Cell Metab. 2010;12:580–592. doi: 10.1016/j.cmet.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Georgiadi A, Wang Y, Stienstra R, Tjeerdema N, Janssen A, Stalenhoef A, van der Vliet JA, de Roos A, Tamsma JT, Smit JW, Tan NS, Muller M, Rensen PC, Kersten S. Overexpression of angiopoietin-like protein 4 protects against atherosclerosis development. Arterioscler Thromb Vasc Biol. 2013;33:1529–1537. doi: 10.1161/ATVBAHA.113.301698. [DOI] [PubMed] [Google Scholar]
- 6.Adachi H, Fujiwara Y, Kondo T, Nishikawa T, Ogawa R, Matsumura T, Ishii N, Nagai R, Miyata K, Tabata M, Motoshima H, Furukawa N, Tsuruzoe K, Kawashima J, Takeya M, Yamashita S, Koh GY, Nagy A, Suda T, Oike Y, Araki E. Angptl 4 deficiency improves lipid metabolism, suppresses foam cell formation and protects against atherosclerosis. Biochem Biophys Res Commun. 2009;379:806–811. doi: 10.1016/j.bbrc.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 7.Goh YY, Pal M, Chong HC, Zhu P, Tan MJ, Punugu L, Tan CK, Huang RL, Sze SK, Tang MB, Ding JL, Kersten S, Tan NS. Angiopoietin-like 4 interacts with matrix proteins to modulate wound healing. J Biol Chem. 2010;285:32999–33009. doi: 10.1074/jbc.M110.108175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galaup A, Cazes A, Le Jan S, Philippe J, Connault E, Le Coz E, Mekid H, Mir LM, Opolon P, Corvol P, Monnot C, Germain S. Angiopoietin-like 4 prevents metastasis through inhibition of vascular permeability and tumor cell motility and invasiveness. Proc Natl Acad Sci U S A. 2006;103:18721–18726. doi: 10.1073/pnas.0609025103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clement LC, Avila-Casado C, Mace C, Soria E, Bakker WW, Kersten S, Chugh SS. Podocyte-secreted angiopoietin-like-4 mediates proteinuria in glucocorticoid-sensitive nephrotic syndrome. Nat Med. 2011;17:117–122. doi: 10.1038/nm.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galaup A, Gomez E, Souktani R, Durand M, Cazes A, Monnot C, Teillon J, Le Jan S, Bouleti C, Briois G, Philippe J, Pons S, Martin V, Assaly R, Bonnin P, Ratajczak P, Janin A, Thurston G, Valenzuela DM, Murphy AJ, Yancopoulos GD, Tissier R, Berdeaux A, Ghaleh B, Germain S. Protection against myocardial infarction and no-reflow through preservation of vascular integrity by angiopoietin-like 4. Circulation. 2012;125:140–149. doi: 10.1161/CIRCULATIONAHA.111.049072. [DOI] [PubMed] [Google Scholar]
- 11.Xu A, Lam MC, Chan KW, Wang Y, Zhang J, Hoo RL, Xu JY, Chen B, Chow WS, Tso AW, Lam KS. Angiopoietin-like protein 4 decreases blood glucose and improves glucose tolerance but induces hyperlipidemia and hepatic steatosis in mice. Proc Natl Acad Sci U S A. 2005;102:6086–6091. doi: 10.1073/pnas.0408452102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mehta N, Qamar A, Qu L, Qasim AN, Mehta NN, Reilly MP, Rader DJ. Differential association of plasma angiopoietin-like proteins 3 and 4 with lipid and metabolic traits. Arterioscler Thromb Vasc Biol. 2014;34:1057–1063. doi: 10.1161/ATVBAHA.113.302802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruge T, Sukonina V, Kroupa O, Makoveichuk E, Lundgren M, Svensson MK, Olivecrona G, Eriksson JW. Effects of hyperinsulinemia on lipoprotein lipase, angiopoietin-like protein 4, and glycosylphosphatidylinositol-anchored high-density lipoprotein binding protein 1 in subjects with and without type 2 diabetes mellitus. Metabolism. 2012;61:652–660. doi: 10.1016/j.metabol.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 14.Georgiadi A, Lichtenstein L, Degenhardt T, Boekschoten MV, van Bilsen M, Desvergne B, Muller M, Kersten S. Induction of cardiac Angptl4 by dietary fatty acids is mediated by peroxisome proliferator-activated receptor beta/delta and protects against fatty acid-induced oxidative stress. Circ Res. 2010;106:1712–1721. doi: 10.1161/CIRCRESAHA.110.217380. [DOI] [PubMed] [Google Scholar]
- 15.Kersten S, Lichtenstein L, Steenbergen E, Mudde K, Hendriks HF, Hesselink MK, Schrauwen P, Muller M. Caloric restriction and exercise increase plasma ANGPTL4 levels in humans via elevated free fatty acids. Arterioscler Thromb Vasc Biol. 2009;29:969–974. doi: 10.1161/ATVBAHA.108.182147. [DOI] [PubMed] [Google Scholar]
- 16.Catoire M, Alex S, Paraskevopulos N, Mattijssen F, Evers-van Gogh I, Schaart G, Jeppesen J, Kneppers A, Mensink M, Voshol PJ, Olivecrona G, Tan NS, Hesselink MK, Berbee JF, Rensen PC, Kalkhoven E, Schrauwen P, Kersten S. Fatty acid-inducible ANGPTL4 governs lipid metabolic response to exercise. Proc Natl Acad Sci U S A. 2014;111:E1043–52. doi: 10.1073/pnas.1400889111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spector AA. Fatty acid binding to plasma albumin. J Lipid Res. 1975;16:165–179. [PubMed] [Google Scholar]
- 18.Fujiwara S, Amisaki T. Molecular dynamics study of conformational changes in human serum albumin by binding of fatty acids. Proteins. 2006;64:730–739. doi: 10.1002/prot.21053. [DOI] [PubMed] [Google Scholar]
- 19.van der Vusse GJ. Albumin as fatty acid transporter. Drug Metab Pharmacokinet. 2009;24:300–307. doi: 10.2133/dmpk.24.300. [DOI] [PubMed] [Google Scholar]
- 20.Sabina J, Tobias W. Augmentation therapy with alpha1-antitrypsin: novel perspectives. Cardiovasc Hematol Disord Drug Targets. 2013;13:90–98. doi: 10.2174/1871529x11313020002. [DOI] [PubMed] [Google Scholar]
- 21.Stockley RA. Alpha1-antitrypsin review. Clin Chest Med. 2014;35:39–50. doi: 10.1016/j.ccm.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 22.Gottlieb PA, Alkanani AK, Michels AW, Lewis EC, Shapiro L, Dinarello CA, Zipris D. alpha1-Antitrypsin therapy downregulates toll-like receptor-induced IL-1beta responses in monocytes and myeloid dendritic cells and may improve islet function in recently diagnosed patients with type 1 diabetes. J Clin Endocrinol Metab. 2014;99:E1418–26. doi: 10.1210/jc.2013-3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abbate A, Van Tassell BW, Christopher S, Abouzaki NA, Sonnino C, Oddi C, Carbone S, Melchior RD, Gambill ML, Roberts CS, Kontos MC, Peberdy MA, Toldo S, Vetrovec GW, Biondi-Zoccai G, Dinarello CA. Effects of Prolastin C (Plasma-Derived Alpha-1 Antitrypsin) on the acute inflammatory response in patients with ST-segment elevation myocardial infarction (from the VCU-alpha 1-RT pilot study) Am J Cardiol. 2015;115:8–12. doi: 10.1016/j.amjcard.2014.09.043. [DOI] [PubMed] [Google Scholar]
- 24.Frenzel E, Wrenger S, Immenschuh S, Koczulla R, Mahadeva R, Deeg HJ, Dinarello CA, Welte T, Marcondes AM, Janciauskiene S. Acute-phase protein alpha1-antitrypsin--a novel regulator of angiopoietin-like protein 4 transcription and secretion. J Immunol. 2014;192:5354–5362. doi: 10.4049/jimmunol.1400378. [DOI] [PubMed] [Google Scholar]
- 25.Ozbalci C, Sachsenheimer T, Brugger B. Quantitative analysis of cellular lipids by nano-electrospray ionization mass spectrometry. Methods Mol Biol. 2013;1033:3–20. doi: 10.1007/978-1-62703-487-6_1. [DOI] [PubMed] [Google Scholar]
- 26.Beck-Garcia E, Beck-Garcia K, Schlosser A, Schamel WW. Analysis of interactions between proteins and fatty acids or cholesterol using a fatty acid/cholesterol pull-down assay. Anal Biochem. 2013;436:75–77. doi: 10.1016/j.ab.2013.01.026. [DOI] [PubMed] [Google Scholar]
- 27.Dorresteijn MJ, Paine A, Zilian E, Fenten MG, Frenzel E, Janciauskiene S, Figueiredo C, Eiz-Vesper B, Blasczyk R, Dekker D, Pennings B, Scharstuhl A, Smits P, Larmann J, Theilmeier G, van der Hoeven JG, Wagener FA, Pickkers P, Immenschuh S. Cell-type-specific downregulation of heme oxygenase-1 by lipopolysaccharide via Bach1 in primary human mononuclear cells. Free Radic Biol Med. 2015;78:224–232. doi: 10.1016/j.freeradbiomed.2014.10.579. [DOI] [PubMed] [Google Scholar]
- 28.Janciauskiene SM, Nita IM, Stevens T. Alpha1-antitrypsin, old dog, new tricks. Alpha1-antitrypsin exerts in vitro anti-inflammatory activity in human monocytes by elevating cAMP. J Biol Chem. 2007;282:8573–8582. doi: 10.1074/jbc.M607976200. [DOI] [PubMed] [Google Scholar]
- 29.Nukiwa T, Brantly M, Ogushi F, Fells G, Satoh K, Stier L, Courtney M, Crystal RG. Characterization of the M1(Ala213) type of alpha 1-antitrypsin, a newly recognized, common “normal” alpha 1-antitrypsin haplotype. Biochemistry. 1987;26:5259–5267. doi: 10.1021/bi00391a008. [DOI] [PubMed] [Google Scholar]
- 30.Agardh HE, Folkersen L, Ekstrand J, Marcus D, Swedenborg J, Hedin U, Gabrielsen A, Paulsson-Berne G. Expression of fatty acid-binding protein 4/aP2 is correlated with plaque instability in carotid atherosclerosis. J Intern Med. 2011;269:200–210. doi: 10.1111/j.1365-2796.2010.02304.x. [DOI] [PubMed] [Google Scholar]
- 31.Bonen A, Campbell SE, Benton CR, Chabowski A, Coort SL, Han XX, Koonen DP, Glatz JF, Luiken JJ. Regulation of fatty acid transport by fatty acid translocase/CD36. Proc Nutr Soc. 2004;63:245–249. doi: 10.1079/PNS2004331. [DOI] [PubMed] [Google Scholar]
- 32.Kaddatz K, Adhikary T, Finkernagel F, Meissner W, Muller-Brusselbach S, Muller R. Transcriptional profiling identifies functional interactions of TGF beta and PPAR beta/delta signaling: synergistic induction of ANGPTL4 transcription. J Biol Chem. 2010;285:29469–29479. doi: 10.1074/jbc.M110.142018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lichtenstein L, Kersten S. Modulation of plasma TG lipolysis by Angiopoietin-like proteins and GPIHBP1. Biochim Biophys Acta. 2010;1801:415–420. doi: 10.1016/j.bbalip.2009.12.015. [DOI] [PubMed] [Google Scholar]
- 34.Chana RS, Lewington AJ, Brunskill NJ. Differential effects of peroxisome proliferator activated receptor-gamma (PPAR gamma) ligands in proximal tubular cells: thiazolidinediones are partial PPAR gamma agonists. Kidney Int. 2004;65:2081–2090. doi: 10.1111/j.1523-1755.2004.00624.x. [DOI] [PubMed] [Google Scholar]
- 35.Parr DG, Stewart DG, Hero I, Stockley RA. Panniculitis secondary to extravasation of clarithromycin in a patient with alpha 1-antitrypsin deficiency (phenotype PiZ) Br J Dermatol. 2003;149:410–413. doi: 10.1046/j.1365-2133.2003.05530.x. [DOI] [PubMed] [Google Scholar]
- 36.Cowden DI, Fisher GE, Weeks RL. A pilot study comparing the purity, functionality and isoform composition of alpha-1-proteinase inhibitor (human) products. Curr Med Res Opin. 2005;21:877–883. doi: 10.1185/030079905X46395. [DOI] [PubMed] [Google Scholar]
- 37.Sanderson LM, Degenhardt T, Koppen A, Kalkhoven E, Desvergne B, Muller M, Kersten S. Peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) but not PPARalpha serves as a plasma free fatty acid sensor in liver. Mol Cell Biol. 2009;29:6257–6267. doi: 10.1128/MCB.00370-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mandard S, Zandbergen F, van Straten E, Wahli W, Kuipers F, Muller M, Kersten S. The fasting-induced adipose factor/angiopoietin-like protein 4 is physically associated with lipoproteins and governs plasma lipid levels and adiposity. J Biol Chem. 2006;281:934–944. doi: 10.1074/jbc.M506519200. [DOI] [PubMed] [Google Scholar]
- 39.Robciuc MR, Tahvanainen E, Jauhiainen M, Ehnholm C. Quantitation of serum angiopoietin-like proteins 3 and 4 in a Finnish population sample. J Lipid Res. 2010;51:824–831. doi: 10.1194/jlr.M002618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jonker JT, Smit JW, Hammer S, Snel M, van der Meer RW, Lamb HJ, Mattijssen F, Mudde K, Jazet IM, Dekkers OM, de Roos A, Romijn JA, Kersten S, Rensen PC. Dietary modulation of plasma angiopoietin-like protein 4 concentrations in healthy volunteers and in patients with type 2 diabetes. Am J Clin Nutr. 2013;97:255–260. doi: 10.3945/ajcn.112.043687. [DOI] [PubMed] [Google Scholar]
- 41.Wang J, Zhu X, Wang Z, Yao J, Zhao B, Liu G. Non-esterified fatty acids promote expression and secretion of angiopoietin-like protein 4 in calf hepatocytes cultured in vitro. Mol Cell Biochem. 2015;401:141–146. doi: 10.1007/s11010-014-2301-2. [DOI] [PubMed] [Google Scholar]
- 42.Staiger H, Haas C, Machann J, Werner R, Weisser M, Schick F, Machicao F, Stefan N, Fritsche A, Haring HU. Muscle-derived angiopoietin-like protein 4 is induced by fatty acids via peroxisome proliferator-activated receptor (PPAR)-delta and is of metabolic relevance in humans. Diabetes. 2009;58:579–589. doi: 10.2337/db07-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alex S, Lange K, Amolo T, Grinstead JS, Haakonsson AK, Szalowska E, Koppen A, Mudde K, Haenen D, Al-Lahham S, Roelofsen H, Houtman R, van der Burg B, Mandrup S, Bonvin AM, Kalkhoven E, Muller M, Hooiveld GJ, Kersten S. Short-chain fatty acids stimulate angiopoietin-like 4 synthesis in human colon adenocarcinoma cells by activating peroxisome proliferator-activated receptor gamma. Mol Cell Biol. 2013;33:1303–1316. doi: 10.1128/MCB.00858-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Varga T, Czimmerer Z, Nagy L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim Biophys Acta. 2011;1812:1007–1022. doi: 10.1016/j.bbadis.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gardner CD, Fortmann SP, Krauss RM. Association of small low-density lipoprotein particles with the incidence of coronary artery disease in men and women. JAMA. 1996;276:875–881. [PubMed] [Google Scholar]
- 46.Richieri GV, Kleinfeld AM. Unbound free fatty acid levels in human serum. J Lipid Res. 1995;36:229–240. [PubMed] [Google Scholar]
- 47.Brunaldi K, Huang N, Hamilton JA. Fatty acids are rapidly delivered to and extracted from membranes by methyl-beta-cyclodextrin. J Lipid Res. 2010;51:120–131. doi: 10.1194/jlr.M900200-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kass I, Knaupp AS, Bottomley SP, Buckle AM. Conformational properties of the disease-causing Z variant of alpha1-antitrypsin revealed by theory and experiment. Biophys J. 2012;102:2856–2865. doi: 10.1016/j.bpj.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cabrita LD, Bottomley SP. How do proteins avoid becoming too stable? Biophysical studies into metastable proteins. Eur Biophys J. 2004;33:83–88. doi: 10.1007/s00249-003-0356-1. [DOI] [PubMed] [Google Scholar]
- 50.Inoue T, Kohro T, Tanaka T, Kanki Y, Li G, Poh HM, Mimura I, Kobayashi M, Taguchi A, Maejima T, Suehiro J, Sugiyama A, Kaneki K, Aruga H, Dong S, Stevens JF, Yamamoto S, Tsutsumi S, Fujita T, Ruan X, Aburatani H, Nangaku M, Ruan Y, Kodama T, Wada Y. Cross-enhancement of ANGPTL4 transcription by HIF1 alpha and PPAR beta/delta is the result of the conformational proximity of two response elements. Genome Biol. 2014;15:R63. doi: 10.1186/gb-2014-15-4-r63. 2014-15-4-r63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Savage DB, Sewter CP, Klenk ES, Segal DG, Vidal-Puig A, Considine RV, O'Rahilly S. Resistin / Fizz3 expression in relation to obesity and peroxisome proliferator-activated receptor-gamma action in humans. Diabetes. 2001;50:2199–2202. doi: 10.2337/diabetes.50.10.2199. [DOI] [PubMed] [Google Scholar]
- 52.Seargent JM, Yates EA, Gill JH. GW9662, a potent antagonist of PPARgamma, inhibits growth of breast tumour cells and promotes the anticancer effects of the PPARgamma agonist rosiglitazone, independently of PPARgamma activation. Br J Pharmacol. 2004;143:933–937. doi: 10.1038/sj.bjp.0705973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koliwad SK, Kuo T, Shipp LE, Gray NE, Backhed F, SO AY, Farese RV, Jr, Wang JC. Angiopoietin-like 4 (ANGPTL4, fasting-induced adipose factor) is a direct glucocorticoid receptor target and participates in glucocorticoid-regulated triglyceride metabolism. J Biol Chem. 2009;284:25593–25601. doi: 10.1074/jbc.M109.025452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krey G, Braissant O, L'Horset F, Kalkhoven E, Perroud M, Parker MG, Wahli W. Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivator-dependent receptor ligand assay. Mol Endocrinol. 1997;11:779–791. doi: 10.1210/mend.11.6.0007. [DOI] [PubMed] [Google Scholar]
- 55.Naruhn S, Toth PM, Adhikary T, Kaddatz K, Pape V, Dorr S, Klebe G, Muller-Brusselbach S, Diederich WE, Muller R. High-affinity peroxisome proliferator-activated receptor beta/delta-specific ligands with pure antagonistic or inverse agonistic properties. Mol Pharmacol. 2011;80:828–838. doi: 10.1124/mol.111.074039. [DOI] [PubMed] [Google Scholar]
- 56.Stapleton CM, Joo JH, Kim YS, Liao G, Panettieri RA, Jetten AM. Induction of ANGPTL4 expression in human airway smooth muscle cells by PMA through activation of PKC and MAPK pathways. Exp Cell Res. 2010;316:507–516. doi: 10.1016/j.yexcr.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Prusty D, Park BH, Davis KE, Farmer SR. Activation of MEK/ERK signaling promotes adipogenesis by enhancing peroxisome proliferator-activated receptor gamma (PPARgamma) and C/EBPalpha gene expression during the differentiation of 3T3-L1 preadipocytes. J Biol Chem. 2002;277:46226–46232. doi: 10.1074/jbc.M207776200. [DOI] [PubMed] [Google Scholar]
- 58.Subramaniyam D, Zhou H, Liang M, Welte T, Mahadeva R, Janciauskiene S. Cholesterol rich lipid raft microdomains are gateway for acute phase protein, SERPINA1. Int J Biochem Cell Biol. 2010;42:1562–1570. doi: 10.1016/j.biocel.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 59.Bergin DA, Reeves EP, Meleady P, Henry M, McElvaney OJ, Carroll TP, Condron C, Chotirmall SH, Clynes M, O'Neill SJ, McElvaney NG. alpha-1 Antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8. J Clin Invest. 2010;120:4236–4250. doi: 10.1172/JCI41196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vaisar T, Pennathur S, Green PS, Gharib SA, Hoofnagle AN, Cheung MC, Byun J, Vuletic S, Kassim S, Singh P, Chea H, Knopp RH, Brunzell J, Geary R, Chait A, Zhao XQ, Elkon K, Marcovina S, Ridker P, Oram JF, Heinecke JW. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J Clin Invest. 2007;117:746–756. doi: 10.1172/JCI26206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Talmud PJ, Martin S, Steiner G, Flavell DM, Whitehouse DB, Nagl S, Jackson R, Taskinen MR, Frick MH, Nieminen MS, Kesaniemi YA, Pasternack A, Humphries SE, Syvanne M, Diabetes Atherosclerosis Intervention Study Investigators Progression of atherosclerosis is associated with variation in the alpha1-antitrypsin gene. Arterioscler Thromb Vasc Biol. 2003;23:644–649. doi: 10.1161/01.ATV.0000065196.61663.8D. [DOI] [PubMed] [Google Scholar]
- 62.Moreno JA, Ortega-Gomez A, Rubio-Navarro A, Louedec L, Ho-Tin-Noe B, Caligiuri G, Nicoletti A, Levoye A, Plantier L, Meilhac O. High-density lipoproteins potentiate alpha1-antitrypsin therapy in elastase-induced pulmonary emphysema. Am J Respir Cell Mol Biol. 2014;51:536–549. doi: 10.1165/rcmb.2013-0103OC. [DOI] [PubMed] [Google Scholar]
- 63.Augsten M, Hackl H, Ebner B, Chemelli A, Glatter O, Marsche G, Lang U, Desoye G, Wadsack C. Fetal HDL/apoE: a novel regulator of gene expression in human placental endothelial cells. Physiol Genomics. 2011;43:1255–1262. doi: 10.1152/physiolgenomics.00109.2011. [DOI] [PubMed] [Google Scholar]
- 64.Stremmel W, Pohl L, Ring A, Herrmann T. A new concept of cellular uptake and intracellular trafficking of long-chain fatty acids. Lipids. 2001;36:981–989. doi: 10.1007/s11745-001-0809-2. [DOI] [PubMed] [Google Scholar]
- 65.Jonigk D, Al-Omari M, Maegel L, Muller M, Izykowski N, Hong J, Hong K, Kim SH, Dorsch M, Mahadeva R, Laenger F, Kreipe H, Braun A, Shahaf G, Lewis EC, Welte T, Dinarello CA, Janciauskiene S. Anti-inflammatory and immunomodulatory properties of alpha1-antitrypsin without inhibition of elastase. Proc Natl Acad Sci U S A. 2013;110:15007–15012. doi: 10.1073/pnas.1309648110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lewis EC. Expanding the clinical indications for alpha(1)-antitrypsin therapy. Mol Med. 2012;18:957–970. doi: 10.2119/molmed.2011.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lomas DA, Carrell RW. Serpinopathies and the conformational dementias. Nat Rev Genet. 2002;3:759–768. doi: 10.1038/nrg907. [DOI] [PubMed] [Google Scholar]
- 68.Furuhashi M, Hotamisligil GS. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat Rev Drug Discov. 2008;7:489–503. doi: 10.1038/nrd2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Makowski L, Hotamisligil GS. The role of fatty acid binding proteins in metabolic syndrome and atherosclerosis. Curr Opin Lipidol. 2005;16:543–548. doi: 10.1097/01.mol.0000180166.08196.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Makowski L, Boord JB, Maeda K, Babaev VR, Uysal KT, Morgan MA, Parker RA, Suttles J, Fazio S, Hotamisligil GS, Linton MF. Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nat Med. 2001;7:699–705. doi: 10.1038/89076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Makowski L, Brittingham KC, Reynolds JM, Suttles J, Hotamisligil GS. The fatty acid-binding protein, aP2, coordinates macrophage cholesterol trafficking and inflammatory activity. Macrophage expression of aP2 impacts peroxisome proliferator-activated receptor gamma and IkappaB kinase activities. J Biol Chem. 2005;280:12888–12895. doi: 10.1074/jbc.M413788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shum BO, Mackay CR, Gorgun CZ, Frost MJ, Kumar RK, Hotamisligil GS, Rolph MS. The adipocyte fatty acid-binding protein aP2 is required in allergic airway inflammation. J Clin Invest. 2006;116:2183–2192. doi: 10.1172/JCI24767. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.