

Abstract
Introduction:
We investigated the clinical differences between familial and sporadic frontotemporal dementia (FTD), screening for mutations in known FTD genes.
Methods:
We diagnosed 22 affected individuals belonging to 8 families and 43 sporadic cases with FTD in Apulia, Southern Italy in two years. Mutations in common causative FTD genes (GRN, MAPT, VCP and TARDBP) and C9ORF72 expansions were screened.
Results:
Behavioural variant of FTD was the most common clinical subtype (50% and 69% in familial and sporadic cases, respectively). Social conduct impairment/disinhibition, loss of insight, and inflexibility were the most frequent clinical features observed at onset. One new mutation was identified in GRN in family A.
Conclusion:
Disease onset in sporadic FTD was more frequently characterized by a clustering of behavioral symptoms with apathy and loss of personal hygiene. Mutations in common causative FTD genes are not a major cause of familial and sporadic FTD in the Southern Italian population.
Keywords: frontotemporal dementia, behavioural variant of FTD, semantic dementia, primary progressive aphasia, familial, sporadic
1. INTRODUCTION
Frontotemporal dementia (FTD) is a genetically and pathologically complex disorder [1, 2] with a heterogeneous clinical presentation [3]. FTD is characterized by progressive changes in behavior, personality, and/or language functions associated with degeneration of the frontal and temporal lobes[3–5]. FTD occurs both in familial and sporadic forms, with 30–50% of cases being familial [6]. Mutations in numerous genes have been associated with FTD [7–10]. Mutations in the genes that encode tau (MAPT), progranulin (GRN), and C9ORF72 are the most common causes of FTD. Rare mutations have been identified in other genes such as valosin-containing protein (VCP) and transactive response DNA binding protein 43 (TARDBP). Nonetheless, a large proportion of familial and sporadic forms still have an unknown genetic origin.
An earlier age at onset and a more rapid progression of disease have been reported in familial Alzheimer’s disease (AD) [11]. For FTD, several studies have compared mutation carriers with non-carriers with the aim to find a genotype-phenotype correlation [12–15]. Nonetheless, the genetic contribution to the clinical phenotype in FTD, including age at onset, symptoms, and progression of disease is not clear. Familial FTD can present with various extrapyramidal signs in addition to behavioural changes, thus suggesting that familial FTD may differ clinically from sporadic FTD [16]. Conversely, a recent study of 22 FTD families, of which half presented tau gene mutations, reported identical clinical presentations in both tau mutation carriers and non carriers [17]. Piguet and colleagues reported that familial and non familial FTD cases are similar for age at onset, disease onset-diagnosis interval, and infrequent presence of extrapyramidal signs [12]. Nevertheless, in the same study, psychiatric disorders such as depression, paranoid psychosis, delusions, hallucinations were more frequently found at onset in sporadic patients and apathy was reported as less frequent in tau positive familial cases compared to tau negative familial and sporadic cases [12]. High prevalence of behavioural variant of FTD (bvFTD) with psychiatric features were also reported in C9ORF72 expansion carriers [13]. In contrast, GRN mutation carriers more often present primary progressive aphasia (PPA) compared to C9ORF72 mutation carriers (8%) and patients without mutation (14% of familial FTD; 14% of sporadic FTD). Furthermore, GRN mutations carriers (3% with familial FTD and 6% with sporadic FTD) developed limb apraxia more frequently than C9ORF72 mutation carriers [13]. In the present study, we aimed to assess the clinical presentation and the genetics of familial and sporadic FTD cases from the population-based Apulia FTD Registry.
2. METHODS
2.1. Participants and data collection
Patients were identified through the Apulia FTD Registry, a network of neurologists and geriatricians expert in the care of cognitive disorders including rare dementias, established in Apulia, a region in Southern Italy, with about 4 million inhabitants. The Apulia FTD registry included thirteen clinical hospital sites and about thirty clinicians working in outpatient services. A detailed clinical history was obtained from the caregivers, generally the spouse or a first-degree relatives (frequently children or brothers and sisters) using a structured checklist of behavioral and cognitive changes. Family history was defined as the presence of one or more subject(s) with FTD in the same family [18]. Pedigrees were designed based on interviews with family relatives. Patients presented with clinical features matching the diagnostic criteria proposed by Neary and colleagues [19], including initial behavioural changes and/or language problems and relatively preserved memory and orientation. We assessed the following clinical variables: disease duration (onset-death or onset-end of the study with censoring date, April 2014), age at onset, a complete list of specific features observed by a close relative. We defined the “time of shift” as the interval from behavior symptoms to the first language symptom or vice versa. All patients diagnosed with FTD were evaluated with neuropsychological tests (for bedridden patients or patients in advanced stage of dementia, information about the main investigated domains as memory and attentive-executive functions were obtained from previous neuropsychological examinations or personal history), and electromyography if neurological examination showed evidence of second motor neuron signs. Imaging was obtained through Magnetic Resonance (MR) 1.5 or 3 Tesla and/or Single Photon Emission Computerized Tomography (SPECT).
At the time of enrollment of the probands, many affected family members were deceased. Information about progressive behavior, language and/or cognitive dysfunctions for these subjects were obtained from both medical records and family reports. Parkinsonism was defined if at least one of the cardinal signs (rigidity, bradykinesia, tremor, postural instability) was present. The presence of pyramidal signs (increased deep tendon reflexes, spastic hypertone, Babinski sign, ankle clonus) was also actively explored. The study was approved by the Ethics Committee on Human Research of all hospitals in Apulia involved in the FTD genetic epidemiologic project. Written consent was obtained from each subject enrolled in the study.
2.2. Genetic analysis
Blood samples were collected from 22 patients and 34 first-degree healthy subjects belonging to 8 families and 43 sporadic cases. DNA was extracted from blood following standard procedures. Mutation screening in MAPT, GRN, VCP and TARDBP was performed via Sanger sequencing. C9ORF72 repeat expansions were assessed with a repeat-primed PCR.
2.2.1. Sanger sequencing
All exons and exon-intron boundaries of MAPT, GRN, VCP and TARDBP were amplified by PCR. PCR products were purified using the AMPure purification kit (Agencourt Bioscience Corporation, MA, USA) and directly sequenced with ABI BigDye Terminator Cycle Sequencing Kit on an ABI 3730 sequencer. Sequence traces were analyzed using Sequencher (version 4.2; Gene Codes Corporation, Ann Arbor, MI, USA).
2.2.2. C9ORF72 repeat expansion study
A repeat-primed PCR was performed to screen for the presence of the GGGGCC hexanucleotide repeat expansion in C9ORF72 as previously described [17]. Positive and negative controls were added to the polymerase chain reaction plate to assure accurate repeat analysis. Fragment length analysis was performed on an ABI 3730xl genetic analyzer (Applied Biosystems, Foster City, CA, USA) and data were analyzed using GeneScan software (version 4, ABI). The assay allows samples to be categorized into those that carry a pathogenic repeat expansion (greater than 30 repeats) and those that carry only wild-type alleles (less than 20 repeats).
2.3. Statistical Analysis
Normal distribution assumption was checked using the Kolomogorov-Smirnov test. Group comparisons in terms of demographic and clinical features were performed using the Pearson Chi-squared Test (or Fisher’s exact test when appropriate) or the Mann-Whitney U-test test, for categorical and continuous variables, respectively. A p-value less than 0.05 was considered as statistically significant. All analyses were performed using SAS 9.3 software.
3. RESULTS
3.1. Clinical findings
Twenty two affected individuals belonging to 8 families and 43 sporadic FTD patients were studied. Family history was positive in 22/65 (34%) of FTD patients. All families showed an autosomal dominant pattern of inheritance (Figure 1). Clinical and demographic information for the familial and sporadic groups are given in Table 1. There were no significant differences between sporadic and familial FTD in terms of age at onset, onset-diagnosis interval and time of shift. Mean age at onset was 64.5 and 62.5 years in familial and sporadic cases, respectively, with a wide range of variability in both groups. Disease duration, at first inclusion in the study, was 9.6 years in familial cases (range: 4 to 18 years) and 6.0 years in sporadic cases (range: 2 to 11 years). There were more females in the familial group (68%) and more males in the sporadic group (59%) (p=0.04) (Table 1).
Figure 1.
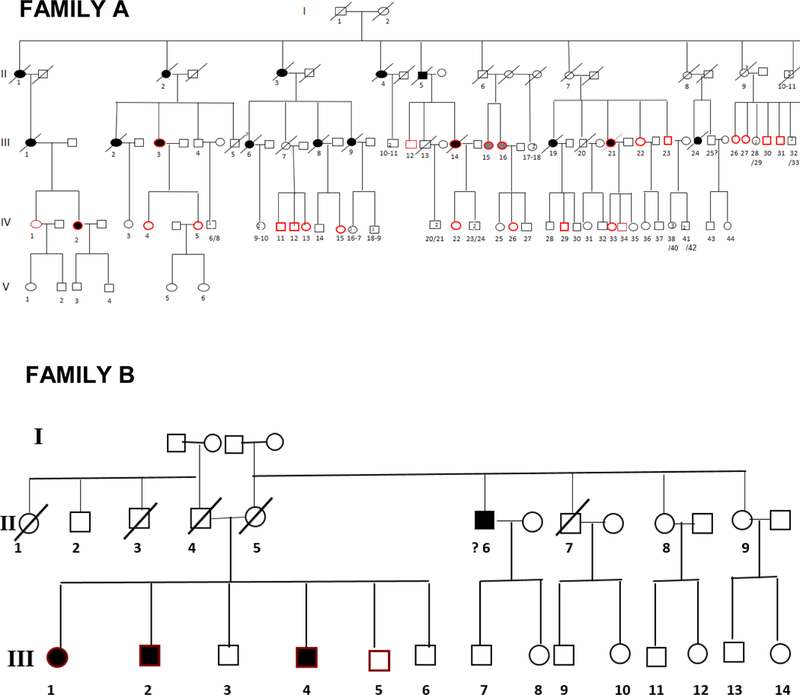
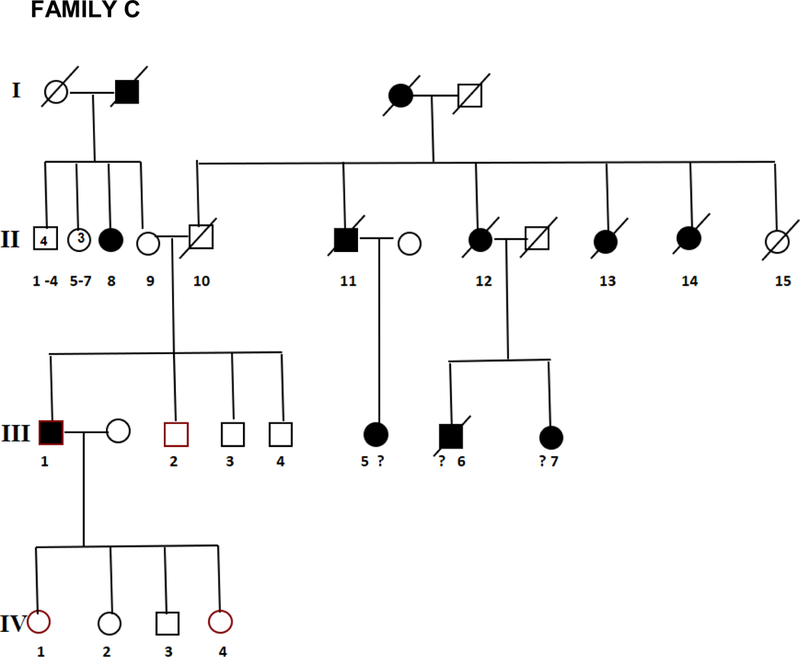

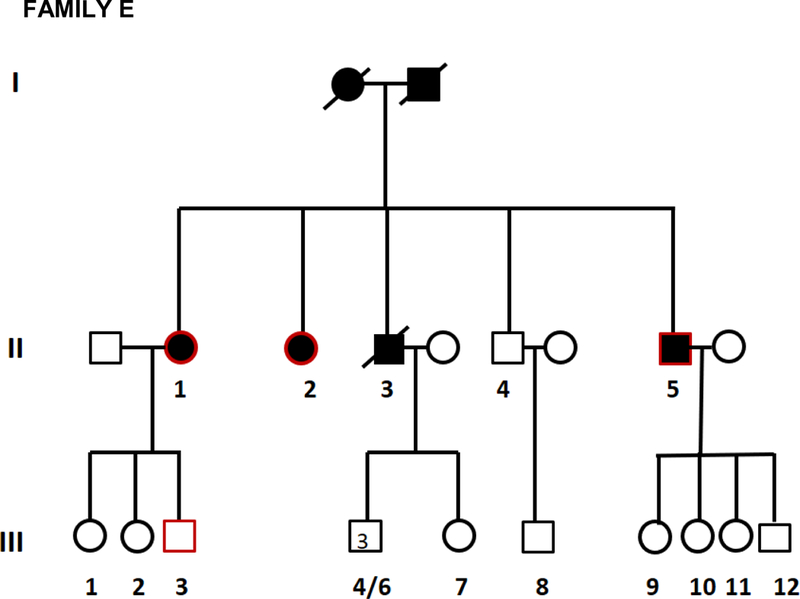
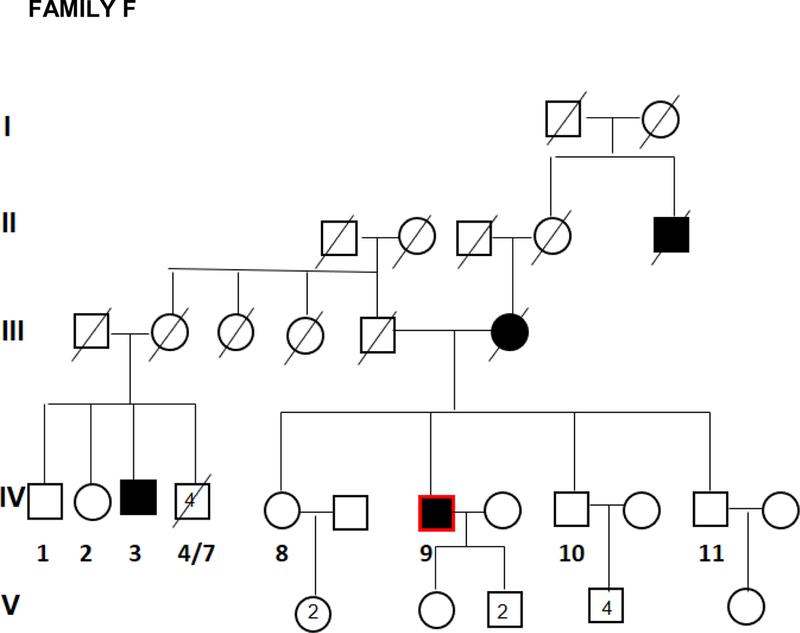
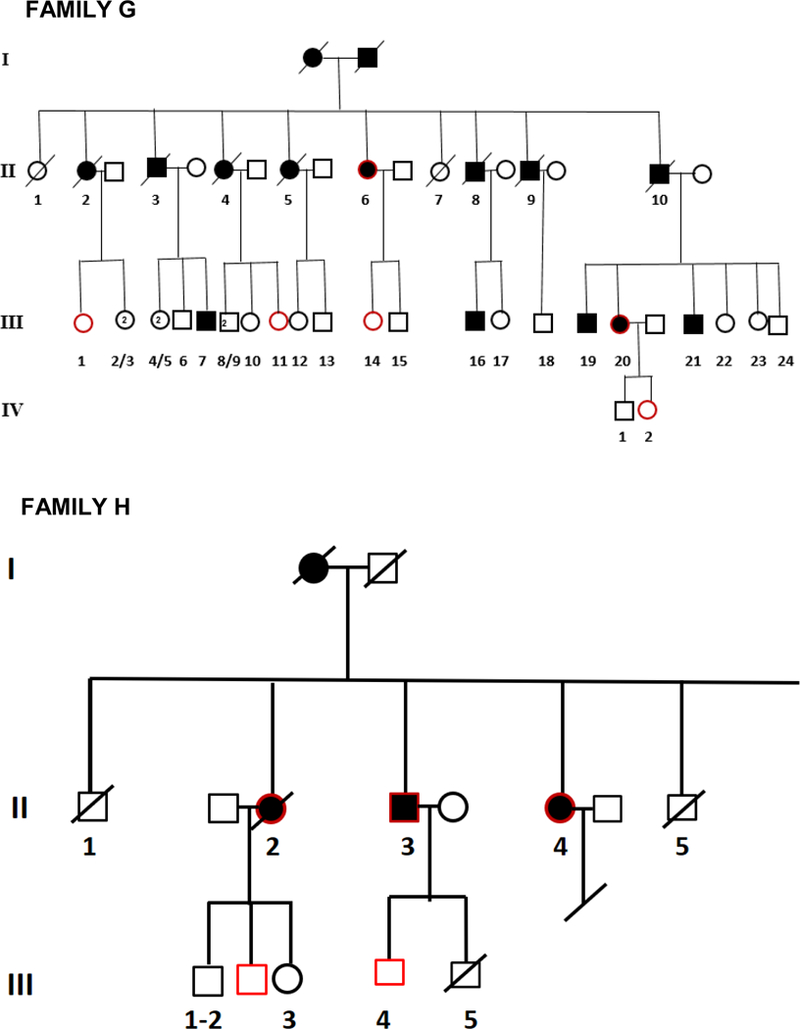

Pedigrees of frontotemporal dementia (FTD) families A,B,C,D,E,F,G, and H.
Table 1.
Clinical and demographic features of sporadic and familial patients with frontotemporal dementia (FTD).
| Variable | Category | Mean±SD, (min-max) or Freq. (Pere. %) | p-value* | |
|---|---|---|---|---|
| GROUPS | ||||
| Familial | Sporadic | |||
| n | 22 (33.85) | 43 (66.15) | ||
| Sex | F | 15(68.18) | 16(44.00) | 0.0416 |
| M | 7(31.82) | 24 (60.00) | ||
| Type of | bvFTD | 11 (50.00) | 30 (69.77) | <0.0001** |
| diagnosis | PPA | 4(18.18) | 13 (30.23) | |
| FTD-memory onset | 7(31.82) | 0 (0.00) | ||
| Type of | LANGUAGE | 2 (9.09) | 11 (25.58) | <0.0001** |
| onset | BEHAVIOUR | 13 (59.09) | 32 (74.42) | |
| MEMORY | 7(31.82) | 0 (0.00) | ||
| Onset age | 64.55±7.70, (49.0–75.0) | 62.57–10.06,(43.0–82.0) | 0.4244 | |
| Time | 4.29±2.97, (0.62–12.0) | 3.84±2.02, (0.50–9.0) | 0.4709 | |
| onset - diagnosis | ||||
| Disease duration | 9.62±3.85, (4.29–18.15) | 6.07±2.13, (2.29–11.29) | <0.0001 | |
| Time of shift | 1.78±1.07, (0.42–4.0) | 2.50±1.72,( 0.41–7.0) | 0.182 | |
Two sided p-values for continuous variables refer to unpaired t-tests or Two sided p-values for categorical variables refer to Pearson Chi-squared
Fisher’s Exact Test.
bvFTD = behavioral variant of frontotemporal dementia; PPA = primary progressive aphasia
BvFTD was the most common clinical subtype in both familial and sporadic groups. PPA was diagnosed in 18% of familial and 31% of sporadic cases. None of the cases met the Neary criteria for semantic dementia (SD).The distribution of clinical subtypes diagnosed in our FTD cohort is shown in Figure 2. Memory problems at onset were present in seven out of 22 familial cases. All these cases were diagnosed as AD in a previous neurological evaluation. A complete neuropsychological assessment was not possible since these patients had severe cognitive decline when included in the study. All these patients with memory problems, presented also at onset disorientation and predominant attentive-executive dysfunctions. Later in the disease, these patients developed progressive severe behavioral changes (apathy, reduced personal hygiene, aggressiveness) and language disorders (anomies, reduced speech), broadly falling in the clinical spectrum of FTD. On the other hand, none of the sporadic cases diagnosed as FTD presented with memory impairment at onset.
Figure 2.
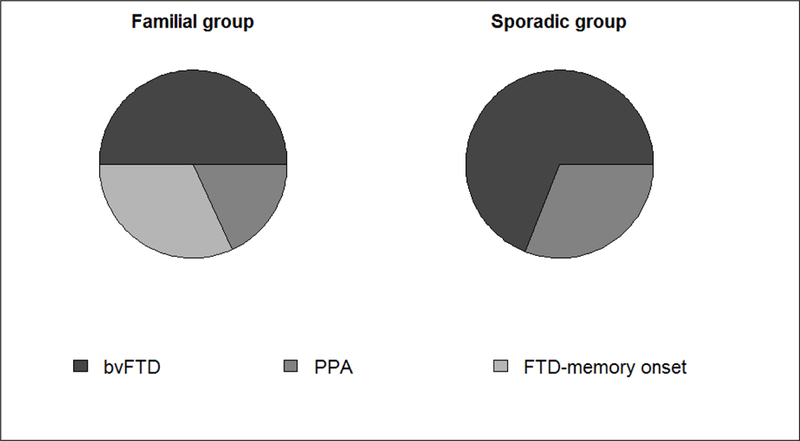
Clinical frontotemporal dementia (FTD) subtypes [familial: behavioral variant of frontotemporal dementia (bvFTD) 50%, primary progressive aphasia (PPA) 18%, semantic dementia (SD) 0%, FTD-memory onset 32%; sporadic: bvFTD 69%, PPA 31%, SD 0%].
The frequency of behavioral and language features observed at onset in the whole cohort are shown in Figure 3. The most common behavioral changes were social conduct impairment/ disinhibition (familial 36%, sporadic 66%; p=0.020), loss of insight (familial 24 %, sporadic 64%; p=0.002) and inflexibility (familial 14 %, sporadic 43 %; p=0.027). Sporadic cases showed changes in dietary preferences as well. When symptoms classified in language domains were analyzed, no significant differences were found in the two study groups. Noteworthy, language stereotypies, stuttering, reading and writing difficulties were more often observed in sporadic FTD patients (Figure 3). Furthermore, we analyzed what was the specific symptom of onset within the two main clinical phenotypes (behavioral and language impairment) both in familial and sporadic cases (Figure 4). The most prevalent behavioral symptom was apathy (81% and 93% in familial and sporadic cases, respectively) while the most frequent language symptom was difficulty in naming (anomies) present in all patients with both familial and sporadic PPA (Figure 4). A polysymptomatic onset was more frequent in sporadic bvFTD cases compared to familial bvFTD, (p=0.06) (Figure 5). Likewise, PPA with familial history showed a paucisymptomatic onset compared to sporadic PPA (p=0.61) (Figure 5).
Figure 3.
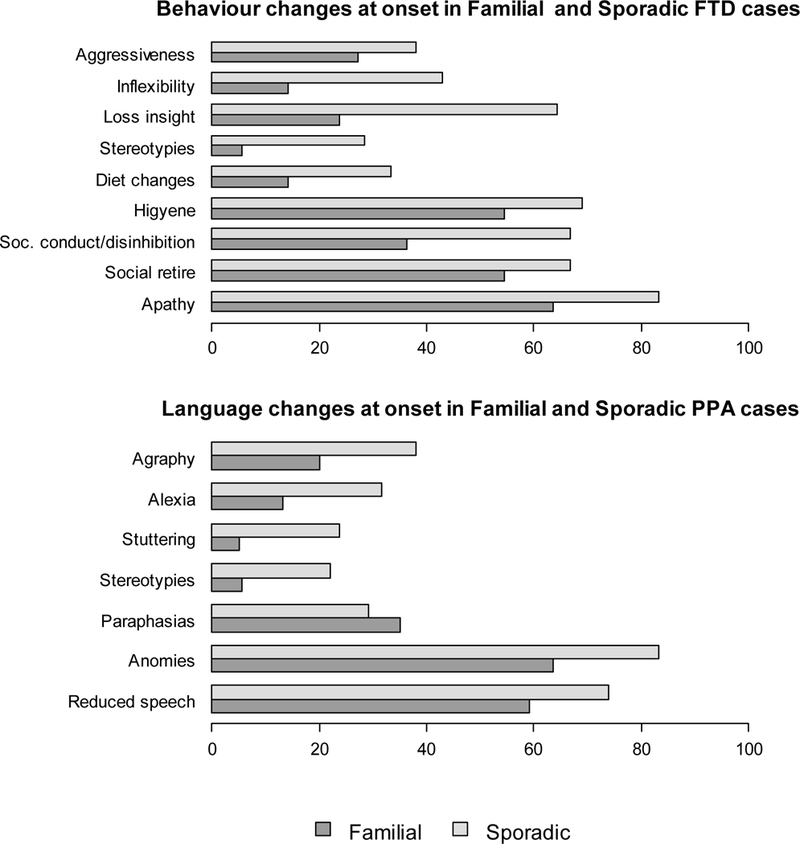
Behaviour and language changes observed at onset in the whole cohort of familial and sporadic frontotemporal dementia cases without considering diagnostic subtypes.
Figure 4.
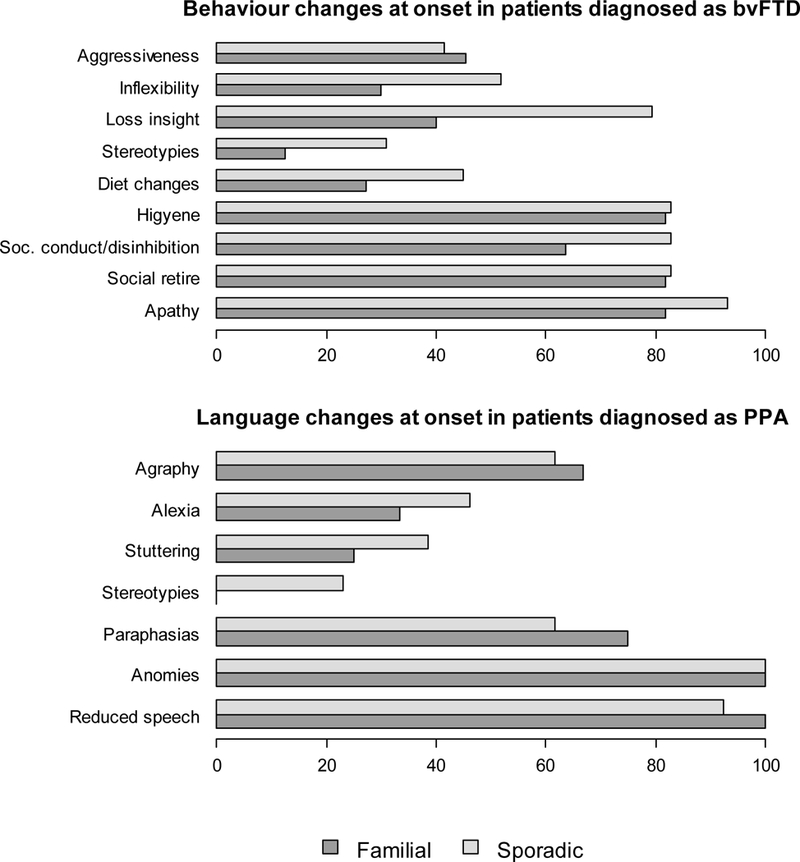
Frequencies of behaviour and language changes at onset respectively in behavioral variant of frontotemporal dementia (bvFTD) and primary progressive aphasia (PPA) both in familial and sporadic forms.
Figure 5.

Number of symptoms (1–3; 4–6; 7–9) referred at onset in familial and sporadic frontotemporal dementia patients.
The time of shift from behaviour to the first language symptom in bvFTD was 1.78 ±1.07 years while from language to the first behavioural symptom in PPA was 2.5 ±1.72 years. At neurological examination, we found parkinsonism in 1 out of 22 familial subjects (4.5%) and in 10 out of 43 sporadic subjects (23.8%, p=0.080). Pyramidal signs were present in 9 out of 22 familial subjects (40.9%) and 2 out of 43 (4.7%) sporadic subjects (p=<0.0001). None of the study subjects showed signs of second motor neuron involvement at neurological examination. We also performed neuroimaging scan in all the patients (structural and/or functional ). All patients showed predominant frontotemporal atrophy (Supplementary Figure 6 Panels a-d) and/or anterior hypoperfusion on SPECT.
3.2. Genetic findings
A novel GRN splice-acceptor site mutation (c.709–2 A>T) was identified in family A. For all the remaining cases, no variants were identified in GRN, MAPT, VCP and TARDBP. Repeats in C9ORF72 were below 20 for all familial and sporadic cases. It can be concluded that none of the patients carried a pathogenic repeat expansion.
4. DISCUSSION
In the present study, bvFTD was the most common clinical subtype in both familial and sporadic cases. No SD case was diagnosed in this series. A clustering of behavioral symptoms (social conduct impairment/disinhibition, loss of insight, and inflexibility) was the most frequent clinical feature observed at disease onset, more frequently present in sporadic FTD cases. There was no sporadic case with memory onset, suggesting that probably sporadic cases with memory-onset were not diagnosed as FTD. Mutations in common causative FTD genes are not a major cause of familial and sporadic FTD in this Southern Italian population.
To the best of our knowledge, most of previous studies on FTD were based on cases recruited in tertiary referral centers. This study is based on the collection of cases through a network in a defined geographic area, in a population-based setting. The referral was both from neurological clinical centers, from specialist working in clinical outpatient care (neurologists, geriatricians, psychiatrists) and sometimes also from general practitioners aware of the presence of the Apulia FTD registry. In our series, 30% of cases were familiar, similarly to previous reports [6, 20]. The presence of a family history in FTD may, however, be underestimated especially in second-degree relatives. In the present study, when we asked the first question of our interview: “Are there any other people affected by dementia in your family?, a large number of relatives interviewed on family history answered “We don’t know”.
There were no significant differences in clinical presentation between sporadic and familial FTD cases although the age at onset dispersion was wider in sporadic cases (43 to 82 years) compared to familial cases. Although FTD is considered as a leading cause of early onset dementia21, we reported an age at onset > 65 years in 43% of FTD patients (28/65). A previous study exploring demographic features of a large cohort of FTD patients reported an age at onset ranging from 35 to 80 years [22], thus suggesting that more attention should be paid to a possible diagnosis of FTD at older age. Moreover, in a population-based study of 85 year olds, Gliason and colleagues reported a bvFTD prevalence of 3% among subjects 85 years olds in population based setting, significantly higher than previously expected in that age group [23]. Finally, a recent population-based study found that the prevalence of FTD among those older than 65 years was more than double that of those aged 40 to 64 years, with the bvFTD having the youngest peak age at diagnosis (median age at diagnosis 63 years) [24].
In our cohort study, disease duration was longer in familial than in sporadic FTD subjects. Symptom duration at diagnosis was similar between familial and sporadic cases enrolled in this study. Hodges and colleagues analyzed survival in a large series of pathologically proven patients with FTD observing that the median disease duration from symptoms onset was 6 years [22], while Van Langenhove and colleagues did not report any differences in disease duration between familial and sporadic FTD [13]. Conversely, in familial AD forms, an earlier age at onset and a more rapid progression have been reported [11]. For FTD, the causal mechanism may be oligogenic, with several risk factor alleles on different genes mutually interacting and producing variation in clinical presentation or convergence with similar effects [12]. An inadequate collection of familial history during clinical examination may likely contribute to the lack of significant differences in early symptoms duration at first diagnostic evaluation between familial and sporadic cases. A detailed study of pedigrees may guide specialists in clinical diagnosis and help to promptly identify other affected family members, especially with mild symptoms. The onset of cases is monosymptomatic or paucisymptomatic in the same phenotypic domain. The patients often experienced the addition of one symptom of another domain (language or behavior) and this may indicate the spread of disease to other areas of the brain.
In the present study, we analyzed the time of shift from behaviour to the first language symptom in bvFTD (1.78 ±1.07 years) and from language to the first behavioural symptom in PPA (2.5 ±1.72 years). The time of shift may likely be considered as an indicator of disease progression and the pathological spread of neurodegeneration. A similar attempt to identify time of generalization has been recently proposed in amyotrophic lateral sclerosis [25]. Actually, there is no consistent method to determine disease stage or severity in FTD. Mioshi and colleagues, by developing a 30-item questionnaire, the Frontotemporal Dementia Rating Scale (FRS), identified six clinical severity stages, from very mild to profound, in FTD [25]. The bvFTD patients were the most severely impaired and showed the most rapid progression through the stages [26]. Attempt to identify progression has been also searched in pathology and imaging studies. A recent study reported preliminary evidences for a sequential regional dissemination of TAR DNA-binding protein with molecular weight 43 kDa (TDP-43) pathology as the mechanism determining the progression in FTD [27]. AD has been shown to present over time greater cortical atrophy in the inferior parietal and posterior cingulate cortex, while bvFTD patients showed greater atrophy in the striatum than AD over time. The atrophy in the posterior cingulate and the striatum may be considered a potential diagnostic marker for tracking rates of progression respectively of AD and FTD. The identification of clinical and imaging milestones may also represent a useful tool in the future to monitor drug efficacy and their ability to modulate disease progression [28].
In the present study cohort, bvFTD was the most common clinical diagnosis in both familial and sporadic groups followed by PPA in sporadic group and FTD with memory onset in familial group. In our clinical series, we did not report SD diagnosis neither in familial nor in sporadic groups, although in some cohort studies SD was, in term of frequency, the second diagnosis after bvFTD [29]. Similar clinical and demographic features, compared to our study cohort, have been described in a large cohort of SD patients [29]. The fact that most patients have been examined in later stages of disease when specific key diagnostic features of SD (loss of word meaning, semantic paraphasias, preserved word repetition) were not more valuable, may likely contribute to explain missing SD diagnosis in this study. Moreover, since the primary complaint of most SD patients was ‘memory loss’ and they perform poorly on several memory tests including verbal episodic memory, the mainstay of dementia screening test, it is easy to imagine that patients could be misdiagnosed with AD [29].
In the present cohort, none of sporadic FTD cases was diagnosed as FTD with memory onset. Memory impairment was traditionally reported to be preserved in FTD. Therefore, patients complaining of “memory loss” at onset and apparently without a familial history are likely misdiagnosed as having AD. Memory impairment was the most common initial symptom in patients with FTD and in patients with AD, although more prevalent in AD [30]. In FTD cases, harboring C9orf72 expansion, memory dysfunction may vary from mild to marked and often present early in the course of the disease [31]. Patients with a mixed amnesic/behavior/language picture involve diagnostic challenges due to the clinical-pathological overlap with AD. A recent brain SPECT perfusion study showed paradoxically a more severe temporal lobe hypoperfusion in FTD than in AD patients [32]. FTD features may occur in pathologically confirmed AD. In fact, approximately 30% of patients with PPA showed the neuropathological features of AD [33]. Furthermore, severe amnesia at presentation have been described in FTD pathologically confirmed patients [34].
Overall, familial and sporadic FTD patients showed no significant differences in clinical presentation, although the sporadic subjects presented at onset some behavioral features such as conduct impairment, disinhibition, behaviour stereotypies, diet changes and inflexibility and some language changes such as stuttering, language stereotypies, reading and writing abnormalities more frequently. The present series suggested that there were no clinical features that can reliably distinguish familial and sporadic FTD. The small sample may limit our conclusions. This study may be further limited by the lack of neuropathological information and limited cerebrospinal fluid (CSF) biomarkers evaluation (CSF was examined in 36% and 23% of familial and sporadic, respectively). We had insufficient biomarker information to attempt a classification of pathological subtypes in sporadic and familial FTD with different protein inclusions such as frontotemporal lobar degeneration-tau, frontotemporal lobar degeneration-TDP-43, and frontotemporal lobar degeneration-fused-in-sarcoma (FUS ). This may be relevant especially in the differential diagnosis between bvFTD and the frontal variant of AD [35]. Furthermore, we did not have follow-up imaging with MR or SPECT to analyze the disease progression and eventual imaging-related abnormalities that could discriminate between familial and sporadic FTD. Nonetheless, the present population-based study has been based on active surveillance, enrolling patients after clinical evaluation, with secondary clinical advice from the tertiary centre of motor neuron disease thus providing a valid support to diagnostic accuracy and minimizing the possibility of misdiagnosis after clinical evaluation.
In the present study, we investigated whether any potentially pathogenic DNA variants were present in MAPT, GRN, VCP and TARDBP in the geographical area of Southern Italy. Only a novel GRN splice-acceptor site mutation (c.709–2 A>T), segregating with bvFTD was identified in family A [36]. No known or novel mutations were found in the remaining cases. In a recent Turkish study with a clinical-based sample of 95 dementia cases from 92 families collected in a tertiary referral center (54 AD cases, 28 FTD cases and other 13 cases of other forms of dementia) revealed a mutation in both the MAPT and GRN genes in 3.6% (1/28) of the FTD cohort with an overall sample size similar to that of the present study [37]. However, other clinical-based samples collected in different populations worldwide showed an occurrence of MAPT mutations between 0 [38] and 2.9% [39] and GRN mutations between 1.3 and 11.7% [40]. Furthermore, C9ORF72 hexanucleotide repeat expansion is by far the most common mutation in apparently sporadic ALS collected in a tertiary referral center in Italy [41]. Because of the clinical and genetic overlap between FTD and ALS, C9ORF72 expansions were expected to be found in our cohort, but none was pathologic in our population-based cohort. Therefore mutations in common causative FTD genes are not a major cause of familial and sporadic FTD in the Apulia population, in Southern Italy. The screening of larger cohorts might lead to the identification of cases carrying the repeat expansion or pathogenic variants in common FTD genes. The screening for mutations in other uncommon FTD genes such as the charged multivesicular body protein 2B (CHMP2B) [42], FUS [43, 44], dynactin (DCTN1) [45], sequestosome 1 (SQSTM1) [46], colony-stimulating factor 1 receptor (CFS1R) [47], triggering receptor expressed on myeloid cells (TREM2) [48], ubiquilin-2 (UBQLN2) [49] and heterogeneous nuclear ribonucleoprotein A2B1 (hnRNPA2B1) [50] might be the next step to perform. Ultimately, next generation sequencing methods seem like the efficient strategy to unravel the genetic origin of FTD in our cohort. The present findings warrant further examination of larger FTD cohorts. Given the FTD low prevalence and the diagnostic challenges, the population-based approach in a defined geographic area, with a multicentre case ascertainment and collaboration based on uniform methods of disease phenotyping and genetic analysis, might represent a strategy to better identify cases at an earlier stage and possibly determine both the whole spectrum of clinical phenotypes and the natural history of this condition.
Supplementary Material
Footnotes
Supplementary Figure 6 (Panels 6 a, b) Severe atrophy of frontotemporal cortices with expansion of sylvian valles and temporal hornes in a 62 year-old man with sporadic behavioral variant of frontotemporal dementia (bvFTD/21/52); (Panels 6 c, d) severe asymmetric atrophy of bilateral frontotemporo-insular cortex in a 75 years-old woman with primary progressive aphasia (subject H II4).
REFERENCES
- 1.Neary D, Snowden J, Mann D. Frontotemporal dementia. Lancet Neurol 2005; 4:771–80. [DOI] [PubMed] [Google Scholar]
- 2.Grossman M Frontotemporal dementia: a review. J Int Neuropsychol Soc 2002;8:566–83. [DOI] [PubMed] [Google Scholar]
- 3.Laforce R Jr. Behavioral and language variants of frontotemporal dementia: a review of key symptoms. Clin Neurol Neurosurg 2013;115:2405–10. [DOI] [PubMed] [Google Scholar]
- 4.Shinagawa S Phenotypic variety in the presentation of frontotemporal lobar degeneration. Int Rev Psychiatry 2013;25:138–4. [DOI] [PubMed] [Google Scholar]
- 5.Hodges JR. Frontotemporal dementia (Pick’s disease): clinical features and assessment. Neurology 2001;56:S6–10. [DOI] [PubMed] [Google Scholar]
- 6.Rohrer JD, Guerreiro R, Vandrovcova J, Uphill J, Reiman D, Beck J, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology 2009;73:1451–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006;442:916–9. [DOI] [PubMed] [Google Scholar]
- 8.Holm IE, Isaacs AM, Mackenzie IR. Absence of FUS-immunoreactive pathology in frontotemporal dementia linked to chromosome 3 (FTD-3) caused by mutation in the CHMP2B gene. Acta Neuropathol 2009;118:719–20. [DOI] [PubMed] [Google Scholar]
- 9.See TM, LaMarre AK, Lee SE, Miller BL. Genetic causes of frontotemporal degeneration. J Geriatr Psychiatry Neurol 2010;23:260–8. [DOI] [PubMed] [Google Scholar]
- 10.Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wisniewski T, Dowjat WK, Buxbaum JD, Khorkova O, Efthimiopoulos S, Kulczycki J, et al. A novel Polish presenilin-1 mutation (P117L) is associated with familial Alzheimer’s disease and leads to death as early as the age of 28 years. Neuroreport 1998;9:217–21. [DOI] [PubMed] [Google Scholar]
- 12.Piguet O, Brooks WS, Halliday GM, Schofield PR, Stanford PM, Kwok JB, et al. Similar early clinical presentations in familial and non familial frontotemporal dementia. J Neurol Neurosurg Psychiatry 2004;75:1743–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Langenhove T, van der Zee J, Gijselinck I, Engelborghs S, Vandenberghe R, Vandenbulcke M, et al. Distinct clinical characteristics of C9orf72 expansion carriers compared with GRN, MAPT, and nonmutation carriers in a Flanders-Belgian FTLD cohort. JAMA Neurol 2013;70:365–73. [DOI] [PubMed] [Google Scholar]
- 14.Rohrer JD, Warren JD. Phenotypic signatures of genetic frontotemporal dementia. Curr Opin Neurol 2011;24:542–9. [DOI] [PubMed] [Google Scholar]
- 15.Pickering-Brown SM, Rollinson S, Du Plessis D, Morrison KE, Varma A, Richardson AM, et al. Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain 2008;131:721–31. [DOI] [PubMed] [Google Scholar]
- 16.Janssen JC, Warrington EK, Morris HR, Lantos P, Brown J, Revesz T, et al. Clinical features of frontotemporal dementia due to the intronic tau 10(+16) mutation. Neurology 2002;58:1161–8. [DOI] [PubMed] [Google Scholar]
- 17.Morris HR, Khan MN, Janssen JC, Brown JM, Perez-Tur J, Baker M, et al. The genetic and pathological classification of familial frontotemporal dementia. Arch Neurol 2001;58:1813–16. [DOI] [PubMed] [Google Scholar]
- 18.Goldman JS, Farmer JM, Wood EM, Johnson JK, Boxer A, Neuhaus J, et al. Comparison of family histories in FTLD subtypes and related tauopathies. Neurology 2005;65:1817–9. [DOI] [PubMed] [Google Scholar]
- 19.Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998;51:1546–54. [DOI] [PubMed] [Google Scholar]
- 20.Garre-Olmo J, Genís Batlle D, del Mar Fernández M, Marquez Daniel F, de Eugenio Huélamo R, et al. Incidence and subtypes of early-onset dementia in a geographically defined general population. Neurology 2010; 75:1249–55. [DOI] [PubMed] [Google Scholar]
- 21.Johnson JK, Diehl J, Mendez MF, Neuhaus J, Shapira JS, Forman M, et al. Frontotemporal Lobar Degeneration Demographic Characteristics of 353 Patients. Arch Neurol 2005;62:925–30. [DOI] [PubMed] [Google Scholar]
- 22.Hodges JR, Davies R, Xuereb J, Kril J, Halliday G. Survival in frontotemporal dementia. Neurology 2003;61:349–54. [DOI] [PubMed] [Google Scholar]
- 23.Gislason TB, Sjögren M, Larsson L, Skoog I. The prevalence of frontal variant frontotemporal dementia and the frontal lobe syndrome in a population based sample of 85 year olds. J Neurol Neurosurg Psychiatry 2003;74:867–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coyle-Gilchrist IT, Dick KM, Patterson K, et al. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology 2016;86:1736–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tortelli R, Copetti M, Panza F, Fontana A, Cortese R, Capozzo R, et al. Time to generalization and prediction of survival in patients with amyotrophic lateral sclerosis: a retrospective observational study. Eur J Neurol 2016;23:1117–25. [DOI] [PubMed] [Google Scholar]
- 26.Mioshi E, Hsieh S, Savage S, Hornberger M, Hodges JR. Clinical staging and disease progression in frontotemporal dementia. Neurology 2010;74:1591–7. [DOI] [PubMed] [Google Scholar]
- 27.Brettschneider J, Del Tredici K, Irwin DJ, Grossman M, Robinson JL, Toledo JB, et al. Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol 2014;127:423–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Landin-Romero R, Kumfor F, Leyton CE, Irish M, Hodges JR, Piguet O. Disease-specific patterns of cortical and subcortical degeneration in a longitudinal study of Alzheimer’s disease and behavioural-variant frontotemporal dementia. Neuroimage 2016; pii: S1053–8119(16)00237–8 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 29.Hodges JR, Mitchell J, Dawson K, Spillantini MG, Xuereb JH, McMonagle P, et al. Semantic dementia: demography, familial factors and survival in a consecutive series of 100 cases. Brain 2010;133:300–6. [DOI] [PubMed] [Google Scholar]
- 30.Binetti G, Locascio JJ, Corkin S, Vonsattel JP, Growdon JH. Differences between Pick disease and Alzheimer disease in clinical appearance and rate of cognitive decline. Arch Neurol 2000;57 225–32. [DOI] [PubMed] [Google Scholar]
- 31.Boeve BF, Boylan KB, Graff-Radford NR, DeJesus-Hernandez M, Knopman DS, Pedraza O, et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 2012;135:765–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Basely M, Ceccaldi M, Boyer L, Mundler O, Guedj E. Distinct patterns of medial temporal impairment in degenerative dementia: a brain SPECT perfusion study in Alzheimer’s disease and frontotemporal dementia. Eur J Nucl Med Mol Imaging 2013;40:932–42. [DOI] [PubMed] [Google Scholar]
- 33.Ahmed S, de Jager CA, Haigh AM, Garrard P. Logopenic aphasia in Alzheimer’s disease: clinical variant or clinical feature? J Neurol Neurosurg Psychiatry 2012;83:1056–62. [DOI] [PubMed] [Google Scholar]
- 34.Graham A, Davies R, Xuereb J, Halliday G, Kril J, Creasey H, et al. Pathologically proven frontotemporal dementia presenting with severe amnesia. Brain 2005;128:597–605. [DOI] [PubMed] [Google Scholar]
- 35.Johnson JK, Head E, Kim R, Starr A, Cotman CW. Clinical and pathological evidence for a frontal variant of Alzheimer disease. Arch Neurol 1999;56:1233–9. [DOI] [PubMed] [Google Scholar]
- 36.Sassi C, Capozzo R, Crews C, Zecca C, Arcuti S, Copetti M, et al. A novel splice-acceptor site mutation in GRN (c.709–2 a>t) in an italian family with FTD. J Alzheimers Dis 2016;53:475–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guven G, Lohmann E, Bras J, Gibbs JR, Gurvit H, Bilgic B, et al. Mutation Frequency of the Major Frontotemporal Dementia Genes, MAPT, GRN and C9ORF72 in a Turkish Cohort of Dementia Patients. PLoS One 2016;11:e0162592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fabre SF, Forsell C, Viitanen M, Sjögren M,Wallin A, Blennow K, et al. Clinic-based cases with frontotemporal dementia show increased cerebrospinal fluid tau and high apolipoprotein E epsilon4 frequency, but no tau gene mutations. Exp Neurol 2001;168:413–8. [DOI] [PubMed] [Google Scholar]
- 39.Le Ber I, van der Zee J, Hannequin D, Gijselinck I, Campion D, Puel M, et al. Progranulin null mutations in both sporadic and familial frontotemporal dementia. Hum Mutat 2007;28:846–55. [DOI] [PubMed] [Google Scholar]
- 40.Gijselinck I, Van Broeckhoven C, Cruts M. Granulin mutations associated with frontotemporal lobar degeneration and related disorders: an update. Hum Mutat 2008; 29:1373–86. [DOI] [PubMed] [Google Scholar]
- 41.Sabatelli M, Conforti FL, Zollino M, Mora G, Monsurrò MR, Volanti P, et al. C9ORF72 hexanucleotide repeat expansions in the Italian sporadic ALS population. Neurobiol Aging 2012;33:1848.e15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clayton EL, Mizielinska S, Edgar JR, Nielsen TT, Marshall S, Norona FE, et al. Frontotemporal dementia caused by CHMP2B mutation is characterised by neuronal lysosomal storage pathology. Acta Neuropathol 2015;130:511–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Broustal O, Camuzat A, Guillot-Noël L, Guy N, Millecamps S, Deffond D, et al. FUS mutations in frontotemporal lobar degeneration with amyotrophic lateral sclerosis. J. Alzheimers Dis. 2010;22:765–9. [PubMed] [Google Scholar]
- 44.Van Langenhove T, van der Zee J, Sleegers K, Engelborghs S, Vandenberghe R, Gijselinck I, et al. Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology 2010;74:366–71. [DOI] [PubMed] [Google Scholar]
- 45.Münch C, Rosenbohm A, Sperfeld A-D, Uttner I, Reske S, Krause BJ, et al. Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann Neurol 2005;58:777–80. [DOI] [PubMed] [Google Scholar]
- 46.Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, et al. SQSTM1mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 2011;68:1440–6. [DOI] [PubMed] [Google Scholar]
- 47.Guerreiro R, Kara E, Le Ber I, Bras J, Rohrer JD, Taipa R, et al. Genetic analysis of inherited leukodystrophies: genotype-phenotype correlations in the CSF1R gene. JAMA Neurol 2013;70:875–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guerreiro R, Bilgic B, Guven G, Bras J, Rohrer J, Lohmann E, et al. Novel compound heterozygous mutation in TREM2 found in a Turkish frontotemporal dementia-like family. Neurobiol Aging 2013;34:e1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gellera C Tiloca C, Del Bo R, Corrado L, Pensato V, Agostini J, et al. Ubiquilin 2 mutations in Italian patients with amyotrophic lateral sclerosis and frontotemporal dementia. J Neurol Neurosurg Psychiatry 2013;84:183–7. [DOI] [PubMed] [Google Scholar]
- 50.Kim HJ, Kim NC, Wang Y-D, Scarborough EA, Moore J, Diaz Z, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013;495:467–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.