

Abstract
Sepsis is a leading cause of death worldwide. After initial trials modulating the hyperinflammatory phase of sepsis failed, generations of researchers have focused on evaluating hypo-inflammatory immune phenotypes. The main goal has been to develop prognostic biomarkers and therapies to reduce organ dysfunction, nosocomial infection, and death. The depressed host defense in sepsis has been characterized by broad cellular reprogramming including lymphocyte exhaustion, apoptosis, and depressed cytokine responses. Despite major advances in this field, our understanding of the dynamics of the septic host response and the balance of inflammatory and anti-inflammatory cellular programs remains limited. This review aims to summarize the epidemiology of nosocomial infections and characteristic immune responses associated with sepsis, as well as immunostimulatory therapies currently under clinical investigation.
Keywords: sepsis, compensatory anti-inflammatory response, priming, nosocomial infection, immunosuppression, SIRS, immunostimulation
Introduction
In 1996, following several failed trials aimed at treating the systemic inflammatory response syndrome (SIRS) (1–3), Roger C. Bone first urged a paradigm shift toward understanding the “compensatory anti-inflammatory response (CARS)” in sepsis (4). Decades later, sepsis remains a leading cause of morbidity and mortality affecting over 30 million people worldwide each year (5), yet not a single immune modulating therapy is actively being used in the clinical setting today. Though mortality rates have declined over time with advances in supportive care, improvements in sepsis therapy are needed to combat persistently high mortality (6, 7). The most recent focus has been on delivery of precision medicine through immunomodulation of the altered host response in sepsis.
Early observations of immune dysfunction in the critically ill come from the trauma and surgical literature (8–10). Decreased cytokine responses to stimulation were later identified and associated with decreased survival in patients with septic shock (11). Though a correlation between sepsis and nosocomial infection has been clinically recognized for decades, it is mostly within the last 10–15 years that this clinical entity has been strongly associated with depressed host immune responses. Large observational studies examining the impact of secondary infection on morbidity and mortality in patients with sepsis are limited and at times data are conflicting. Here, we review the evidence for increased susceptibility to secondary infection in sepsis, mechanisms of depressed host immunity, and promising therapies to modulate the host response.
Epidemiology of nosocomial infection after sepsis
Nosocomial infections after sepsis are common. However, there are wide variations in the reported incidence and associated morbidity. Sepsis-related immunosuppression in the form of depressed cytokine responses and lymphocyte apoptosis has been hypothesized as the main factor contributing to this complication, though evidence supporting causation is found mainly in experimental models of sepsis. A number of small retrospective observational studies have demonstrated increased risk for nosocomial bacterial and fungal infections in patients with sepsis (12, 13). Likewise, reactivation of dormant viral infections has also been well-recognized and occurs concomitantly with other nosocomial infections (14). For the purposes of this review, we will focus on two large observational studies that have examined this topic in detail.
One large retrospective study estimated that 1 in 3 patients with sepsis will develop a nosocomial infection and half of these infections will occur in the lung (15). A larger prospective study found that 1 in 8 patients will develop nosocomial infection and one-quarter of these will be pulmonary infections (16). In both studies, nosocomial infection developed in the late phase of sepsis at a median of 9 days from admission. In the study by Zhao and colleagues, the most common site of secondary infection was pulmonary (52.5%) and there was no association between primary site of infection (e.g., pulmonary, abdominal, skin/soft tissue, urinary) and the development of secondary infection. In the study of van Vught and colleagues, the most common site of secondary infection was cardiovascular (35.3%). The distribution of primary and secondary infection sites in both studies were distinct, suggesting that secondary infection resulted from a new infectious insult rather than inadequately managed primary infection. Patient risk factors for development of nosocomial infection were similar and included older age, higher illness severity score, longer intensive care unit (ICU) length of stay (LOS), and respiratory insufficiency. ICU-specific exposures such as central venous catheterization and endotracheal intubation also increased risk. The most common causative pathogens were bacterial in both studies. As list of typical sites of nosocomial infection and pathogens are shown Table 1.
Table 1.
Primary and secondary sites of infection and etiology of secondary nosocomial infection in patients presenting with sepsis.
| Infection site | Primary | Secondary |
|---|---|---|
| Pulmonary | 48% | 25.4% |
| Cardiovascular* | 7.3% | 35.3% |
| Abdominal | 19% | 15.9% |
| Neurological | 2.2% | 12.7% |
| Skin/Soft tissue | 2.2% | 3.9% |
| Urinary | 4.3% | 1.2% |
| Other¥ | 16.8% | 19% |
| TYPICAL NOSOCOMIAL PATHOGENS | ||
| Gram positive (45.2%) | ||
| • Staphylococcus epidermidis (14.7%) | ||
| • Enterococcus faecalis (12.0%) | ||
| • Enterococcus faecium (6.3%) | ||
| • Staphylococcus aureus (6.0%) | ||
| • Others (6.2%) | ||
| Gram negative (26.6%) | ||
| • Pseudomonas aeruginosa (9.0%) | ||
| • Escherichia coli (3.9%) | ||
| • Klebsiella pneumonia (2.7%) | ||
| • Stenotrophomonas maltophilia (2.7%) | ||
| • Others (8.3%) | ||
| Fungi (9.6%) | ||
| • Candida albicans (2.7%) | ||
| • Candida glabrata (1.2%) | ||
| • Others (5.7%) | ||
| Viruses (9.9%) | ||
| • Herpes simplex (3.9%) | ||
| • Cytomegalovirus (2.1%) | ||
| • Others (3.9%) | ||
Data from Van vught et al. (16) Table 2 and Supplementary Table 3. *Cardiovascular site of infection included bacteremia and catheter-related bloodstream infections ¥Other sites of infection included lung abscess, sinusitis, pharyngitis, tracheobronchitis, endocarditis, mediastinitis, myocarditis, postoperative wound infection, bone and joint infection, oral infection, eye infection, reproductive tract infection.
Nosocomial infection was associated with increased hospital LOS in both studies, but the effect on mortality varied between 15 and 21%. The adjusted absolute increase in mortality specifically attributable to nosocomial infection (population attributable mortality fraction) was only 2% (16). These data suggest that a significant portion of the difference in mortality after sepsis is actually due to competing factors such as higher admission illness severity rather than nosocomial infection. Other studies have made similar observations linking illness severity to outcome, rather than nosocomial infection (17). Furthermore, critically ill patients without sepsis had similarly high rates of nosocomial infection suggesting that ICU exposure, rather than sepsis itself, contributes largely to the development of nosocomial infections. However, infections in patients with sepsis were more commonly due to opportunistic pathogens (enterococci, Pseudomonas aeruginosa and viruses) implying there still may be a link to sepsis-related immunosuppression.
Both exposures and host susceptibility play a role in development of nosocomial infection. As such, differences among studies in nosocomial infection and mortality rates are likely due to differences in patient selection, ICU type, primary type of sepsis, infectious diagnostics/definitions, infection prevention practices, and geographical location. Regardless of their impact on mortality, nosocomial infections are common and remain a significant factor in morbidity during recovery from sepsis. In addition, they are a burden on the health care system and account for an additional $20,000–40,000 dollars per episode (18). Whether immunostimulatory therapies will reduce rates of secondary infection in patients with sepsis will be determined in ongoing clinical trials.
Does the biphasic model explain the heterogeneity of response in sepsis?
The biphasic model
The biphasic model of sepsis has been hypothesized for nearly two decades (19). This model depicts an initial hyperinflammatory response followed by prolonged immune paralysis resulting in morbidity and mortality. However, it is well recognized that the septic immune response does not fit a linear timeline of enhanced inflammation with progression to impaired immunity. Evidence to support this comes from multiple studies, including systematic reviews of gene expression microarray data from blood leukocytes over the course of sepsis (20). In this study, no clear immunosuppressive phase was identified. In fact, at any given timepoint pro- and anti-inflammatory genes were expressed simultaneously in the same patient. Similarly, others have demonstrated circulating anti-inflammatory mediators, such as IL-10, in conjunction with prototypical inflammatory cytokines (TNFα, IL-6, IL-8) at the onset of septic shock (21, 22). Sepsis is therefore a heterogeneous continuum of pro and anti-inflammatory immune programs occurring concurrently. There is also evidence supporting primed immune programs, discussed later in this section, that may occur during the course of sepsis or recovery. A revised model of the inflammation is therefore necessary to illustrate the simultaneous nature of these processes (Figure 1).
Figure 1.
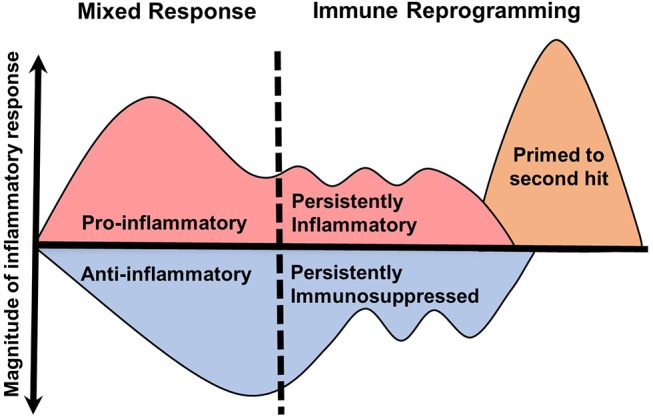
Revised model of inflammation in sepsis. The traditional biphasic model of sepsis (19) plots the immune system on a timeline with an initial hyperinflammatory cytokine storm followed by hypoinflammatory immune paralysis. However, clinical evidence does not support well-demarcated immune phases. In this revised model, the initial immune response to sepsis is a continuous mix of pro- and anti-inflammatory processes that lead to specific immune reprogramming. These programs include persistently pro- or anti-inflammatory and primed responses. The duration and magnitude of each inflammatory program is likely result of many determining factors.
To complicate matters further, cytokines often do not behave in a dichotomized manner (23). A single cytokine may contribute to survival or death depending on the context in which it is examined (23). For example, IL-10 neutralization 24 h after cecal ligation and puncture (CLP) is protective against secondary Pseudomonas aeruginosa pneumonia (24). However, IL-10 neutralization performed prior to endotoxemia or CLP (25, 26) is lethal. A cytokine may also act pleiotropically depending on the environmental context. For instance, IL-10 has suppressive and priming effects depending on the adherent state of a monocyte (27). Non-adherent monocytes are actually primed by this “anti-inflammatory” cytokine to produce more TNFα, IL-6, and IL-1ra upon endotoxin challenge ex vivo. The net effect of an inflammatory mediator is therefore highly contextual.
Moreover, the inflammatory response is tissue-specific (28). Differential expression of inflammatory mediators has been noted in mouse tissues in response to systemic endotoxin. IL-1α production is maximally increased in lung, spleen and liver, while IL-6 is increased in the heart, muscle, brain and kidney (29). In a rat model of CLP examining the molecular response to sepsis, though a subset of gene expression was shared, each organ had a distinct molecular fingerprint (30). Tissue-level responses to secondary stimulation may also be discrepant in the septic host. For example, hemorrhage prior to CLP in mice caused primed responses in alveolar macrophages and Kupffer cells, whereas splenic macrophages and peripheral blood mononuclear cells demonstrated decreased cytokine production consistent with endotoxin tolerance (31). The magnitude of response to an inflammatory stimulus is also variable, as in-vivo imaging of lipopolysaccharide (LPS)-induced NF-κB activation has shown heterogenous intracellular activation more prominently in the skin, lungs, spleen, and small intestine as compared to other organs (32). These observations illustrate two important conceptual points: (1) the (tissue) compartmental response to sepsis and other inflammatory stimuli is highly variable both in quality and magnitude and (2) primed and suppressed responses may be present simultaneously within the same organism following a single exposure. These experimental studies raise important questions that require further investigation in humans.
Priming
Primed immune responses in sepsis may contribute to the heterogenous inflammatory response and are not accounted for in the biphasic model. Priming requires an initial exposure to the host that results in an enhanced inflammatory response to secondary stimulation. For example, Kupffer cells are primed to produce more TNFα in response to endotoxin when femur fracture precedes CLP (33). Similarly, hemorrhage prior to CLP enhances production of plasma IL-6 and TNFα (31). Priming during sepsis may be a protective mechanism, as seen in a model of enteral Enterococcus faecalis infection (34). In this study, mice were pre-exposed to mild or severe sterile systemic inflammation using varying degrees of pancreatitis or thermal injury. Mice experiencing mild inflammation were protected from E. faecalis related mortality, this was associated with primed IL-12 production and enhanced phagocytic function in peritoneal macrophage.
Priming may also represent a mechanism of late organ injury in survivors of sepsis. It is well recognized that survivors of sepsis are at increased risk for long-term cognitive impairment (35), new cardiac events (myocardial infarction, cerebrovascular accident, sudden cardiac death) (36), and new renal failure (37). The cause of these complications remains unclear, though persistent inflammation and primed immune responses have been hypothesized to contribute. Murine CLP models have demonstrated enhanced TNFα production in splenic inflammatory monocytes and brain microglia for at least 2 weeks after sepsis (38, 39) suggesting a possible link between primed cells and long-term organ dysfunction. Meanwhile, persistent inflammation may influence patient outcome, as observations of persistent elevation of IL-6 in patients with pneumonia have been associated with increased risk for long-term mortality due to cardiovascular disease or renal failure (40). Similarly, models of sepsis survival have demonstrated progressive atherosclerosis in the setting of low-grade circulating inflammation (41) and neurocognitive dysfunction associated with persistent neuroinflammation (38, 42) weeks to months after polymicrobial sepsis. In a mouse model of pneumococcal pneumonia, recruitment of inflammatory monocytes to the brain was associated with microglial activation and long-term cognitive impairment (43). When monocyte recruitment was abolished, neuroinflammation was reduced and cognitive impairment was improved. While these initial findings are exciting, the relationship between persistent inflammation, immune priming and long-term organ injury needs to be understood in more detail.
There is strong experimental evidence supporting the conclusion that the inflammatory response of sepsis is heterogeneous at a molecular, cellular, tissue compartment, and individual level. Though further studies are needed in human sepsis, the possibility of “within patient” compartment specific immune heterogeneity warrants consideration. A new conceptual model of the patient experiencing sepsis is required (Figure 2), as one may have reduced immune response on peripheral blood assays, yet in other compartments the net immune response may be mixed or primed. With the introduction of immune stimulating therapies, one must consider that disproportionately primed organs may be harmed by this therapy. Decisions on how to modulate the immune response may be informed by ex vivo stimulation assays, but consideration should be taken to survey for multiple cellular programs in multiple tissue compartments. In addition, these programs are not limited to the acute phase of sepsis, as immune reprogramming influences the entire clinical course including recovery (Figure 3). Pre-sepsis immune status is likely to be an important determinant of which predominant cellular program manifests during acute illness. Post-sepsis immune status may also be impacted by the preceding phases of illness. Likewise, chronic comorbidities may influence the magnitude and evolution of both responses. Despite the evidence supporting sepsis-related immunosuppression in this review, there are large pieces of the immune response to sepsis that remain a mystery and our understanding of sepsis heterogeneity is only in its infancy.
Figure 2.

Conceptual model of the compartmentalization and heterogeneity of sepsis. This conceptual model is derived from studies in experimental sepsis that have demonstrated tissue-specific inflammatory responses. In this model, acute sepsis in one compartment (abdomen) leads to specific and dynamic changes in proximal (blood) and distal (lungs) compartments. Assessment of the immune response by ex vivo stimulation assays (second hit) may then reveal the predominant cellular program. In this case, each compartment responds differently to secondary stimulation based on the severity and composition of the preceding inflammatory insult.
Figure 3.

Clinical phases of sepsis and factors influencing outcome. The clinical course of sepsis is characterized by accelerated progression in severity of illness leading to the development of clinical sepsis. The outcome of each clinical phase of sepsis is influenced by multiple factors. The pre-sepsis phase is influenced primarily by the baseline functional state of the patient. Pre-sepsis functionality directly affects the course of acute sepsis including onset, magnitude and duration. Furthermore, properties inherent to the type of sepsis and exposures occurring during management of acute sepsis continue to affect outcome. Recovery follows and is largely dependent on the severity of prior phases, though continued exposure to the healthcare system places patients at risk for nosocomial complications. Throughout each phase the specific immune program is heterogenous and influences outcome.
The immunosuppressive cellular program of sepsis
Critically ill patients with sepsis, trauma, and burns experience similar immunosuppressive phenotypes. Broadly, these include enhanced cellular apoptosis, suppressed cytokine production, decreased major histocompatibility complex (MHC) class II surface markers, reduced antigen presentation, anergy, and diminished cytotoxic effector cell function. Collectively, these septic leukocyte responses are known as the immunoparalysis or immune exhaustion. While these phenotypes may be associated with increased risk of nosocomial infections and death, they are not inclusive of broader immune changes in sepsis such as potentially primed cells. Moreover, though an exhausted cell may down-regulate cytokine responses, other cellular functions may be simultaneously upregulated (44, 45). As such, the term “cellular reprogramming” is more appropriate to describe general immunophenotypic changes occurring during sepsis. Several examples of immunosuppressive cellular program are described below and are summarized in Figure 4.
Figure 4.

Cellular and molecular mechanisms of immune reprogramming in sepsis. TLR, toll-like receptor; miRNA, microRNA; PAMP, pathogen associated molecular pattern; DAMP, damage associated molecular pattern; PRR, pathogen recognition receptor; MDSC, myeloid derived suppressor cell; T-reg, regulator T-cell; mHLA-DR, monocyte Human Leukocyte Antigen-DR.
Cellular apoptosis
Apoptosis of T and B lymphocytes has been demonstrated in models of sepsis (46), post-mortem analysis of septic patients (47–49), and in the circulation of patients with septic shock (50). Resultant lymphopenia is associated with increased mortality and risk of nosocomial infection (50, 51) and occurs commonly in patients with persistent critical illness (52). Enhanced apoptosis also occurs in myeloid and epithelial cells including blood monocytes (53, 54), dendritic cells (55), intestinal (48) and pulmonary epithelial cells (56), but not neutrophils (57). Apoptosis of monocytes may be a protective mechanism, as acute apoptosis of these cells has been associated with improved mortality (54). Broad reversal of lymphocyte apoptosis through caspase inhibition or over-expression of the antiapoptotic B-cell lymphoma 2 (Bcl-2) protein in experimental sepsis has subsequently been shown to improve mortality (58, 59). As such, improving sepsis-induced lymphocyte apoptosis and lymphopenia has been a primary target for immunomodulation.
Suppressed cytokine release
Immunodepression, immune tolerance, and immunoparalysis are all terms used to describe the decreased production of various cytokines, including TNFα, IL-1β, IL-6, and IFNγ, after ex vivo stimulation of leukocytes with endotoxin or other pattern recognition receptor (PRR) agonists. This phenotype in sepsis is similar to the in vitro phenomenon of endotoxin tolerance, whereby stimulating with high concentration endotoxin results in decreased responses upon secondary stimulation. In sepsis, decreased cytokine production in response to endotoxin stimulation has been demonstrated in whole blood (60), peripheral blood mononuclear cells (61), adherent monocytes (11), neutrophils (62), and NK cells (63). This phenotype occurs early in the course of sepsis and resolves in survivors. However, failure to resolve immune tolerance is associated with increased mortality (11), and expression of endotoxin-tolerant gene signature has been associated with sepsis induced organ failure (64). Though immune tolerance is associated with increased risk of nosocomial infection in trauma patients (65), the association in sepsis is less clear (66, 67).
Alterations in HLA-DR expression and other surface markers
Changes in cellular surface markers occurs during sepsis, with reduced expression of MHC class II molecules on monocytes being the most well studied. Low monocyte human leukocyte antigen (mHLA)-DR correlates with endotoxin tolerance and suppression of antigen-specific T cell responses (68). Early studies found increased rates of sepsis in trauma patients with low mHLA-DR expression (69). Subsequently, low mHLA-DR on admission for sepsis or septic shock has been associated with increased mortality (70, 71) and nosocomial infections (72). Failure to restore mHLA-DR expression over the course of illness is also associated with worse outcome in patients with severe sepsis (66). While highly predictive of mortality and nosocomial infection, mHLA-DR expression dynamics are contextual and dependent on the infectious agent (73) with Gram-positive infections showing lower monocytic mHLA-DR expression than Gram-negative bloodstream infections. The reliability of mHLA-DR to predict outcome and its dynamics of expression are currently under investigation as part of two large observational cohort studies (74, 75). Neutrophil surface markers are also altered, including CD88 expression which is associated with reduced phagocytic function and increased risk for nosocomial infection (76). Importantly, the presence of multiple surface marker abnormalities (low mHLA-DR, low neutrophil CD88 and increased T regulatory cell markers) was most associated with nosocomial infection than any single marker alone (77).
Gene expression endotypes
Functional assays and cell surface marker assessments remain the standard method to assess immunosuppression in sepsis. However, whole-genome expression endotypes correlating with survival of sepsis have been discovered using advanced statistical techniques (78, 79). Davenport et al. found increased 14-day mortality and illness severity associated with a gene expression endotype that was characterized by functional changes in T cell activation, apoptosis, endotoxin tolerance, and down-regulation of HLA class II molecules. Variation in genomic DNA sequence was also associated with specific gene expression patterns, supporting a genetic mechanism for individual heterogeneity. In a follow-up study of patients with community-acquired pneumonia (CAP) and fecal peritonitis (FP), the genomic response to sepsis was largely similar between types of infection with only a modest number of genes differentially expressed between CAP and FP (80). Serial sampling over the course of sepsis demonstrated that patients may switch between the defined endotypes during the course of disease. These studies confirm what is known about clinical sepsis, that phenotype is both heterogenous and dynamic. Classification of patient immune responses quickly and dynamically remains a priority for precision medicine in sepsis. In addition, while mixed leukocytes in whole blood have been reliably used for gene expression analysis, cell-type specific gene expression in sepsis may reveal additional endotypes and help us understand therapies that may reliably be used to modify them.
Lung-specific changes in immunity
The lung is uniquely and continuously exposed to the external environment and acts as a first line of defense against environmental pathogens, especially in critically ill patients with respiratory failure requiring mechanical ventilation. Consequently, the lung is a primary site for nosocomial infection in patients with sepsis. The alveolar macrophage (AM) represents the predominant immune effector cell in the alveolus. Similar to the dysfunction of blood monocytes, decreased TNFα production in AM of mice and humans with sepsis has been recognized for quite some time (81). In polymicrobial sepsis models, AM display both depressed cytokine responses to endotoxin challenge and decreased phagocytic capacity (82). Neutrophil recruitment to the alveolus is decreased (83) and recruited neutrophil ROS production is depressed (84). In an Escherichia coli pneumonia model, lung parenchymal dendritic cells demonstrated decreased antigen presenting capacity and reduced immunostimulatory responses during recovery from sepsis (85). Moreover, these depressed responses were specific to the pulmonary compartment and were mediated by local inflammatory factors released upon organ injury. Disruption of the epithelial barrier leads to alveolus permeability, leak, and decreased mucocilliary clearance all of which may predispose to development of nosocomial infection (86). General loss of pulmonary epithelial barrier function is noted with pulmonary epithelial cell apoptosis in polymicrobial sepsis models (87) and in patients with acute lung injury due to sepsis (56).
Anergy and cytotoxicity
Anergy due to a failure of T cell proliferation or elaboration of cytokines in response to specific antigens has also been described. An increased risk of post-operative sepsis and death was initially described in patients with anergy to delayed-type hypersensitivity skin testing (88). Similarly, patients with lethal post-operative peritonitis had reduced T cell proliferation and secretion of both IL-2 and TNFα in response to CD3/CD28 cross-linking (89). In sepsis models, development of anergy is mediated via a population of TNF-related apoptosis-inducing ligand (TRAIL) expressing CD8+ T cells (90). In humans, CD4+, CD25+, CD127lo regulatory T cells (Treg) have been associated with reduced mitogen responses and development of anergy (91). Depressed cytotoxic responses have been reported in various cell types. Impaired NK cell function with reduced IFNγ secretion and cytotoxicity has been reported in patients with sepsis (92), while others have found normal NK cytotoxic function in severe sepsis (93).
Mechanisms of altered immunity
There are many potential mechanisms for altered immunity, both suppressed and primed, in patients with sepsis. Here we highlight several important mechanisms with a primary focus on mechanisms of immunosuppression (Figure 4).
Anti-inflammatory cytokines and soluble receptors
Three decades of research examining the cytokine response in sepsis is too broad a topic to review here and extensive reviews have already been published on the subject (21). Several cytokines and anti-inflammatory mediators are associated with worse outcomes in septic patients. IL-10 suppresses the pro-inflammatory immune response through deactivating innate immune cells (94, 95). IL-10 is elevated early in the course of sepsis (96) and persistent elevations increase risk of death (97). As discussed previously, this cytokine has pleiotropic roles in experimental models of sepsis, though it may have a particular importance in sepsis-induced impairment of lung immunity (24). IL-10 is actively secreted by multiple cell types that are expanded in septic patients including Treg and myeloid derived suppressor cells (MDSC), which are discussed in more detail below. Though IL-10 has direct immunosuppressive effects, its association with development of nosocomial infection is less straight forward (96). Enhanced IL-10 signaling has been associated with the development of nosocomial infection in at least one study (98).
Soluble receptors for cytokines are additional anti-inflammatory mediators that have been long recognized in sepsis. These molecules are shed cell-surface receptors that neutralize the activity of pro-inflammatory cytokines and are largely viewed as a protective mechanism. TNF soluble receptors I and II (sTNFR-I, sTNFR-II) levels are increased in septic patients and are associated with mortality (97, 99, 100). Though there is minimal data linking sTNFR to nosocomial infections in sepsis, they are chronically elevated in the elderly and therefore elevated levels may represent a predisposition to infection (101).
IL-1 receptor antagonist (IL-1ra) is a naturally occurring antagonist to IL-1. IL-1ra levels are significantly elevated in patients with septic shock (97, 102) and are associated with increased mortality (103). Recent retrospective analysis of the IL-1 pathway in a previously completed trial of anti-IL1 therapy in sepsis showed a mortality benefit of anti-IL1 antibody administration in patients with the highest levels of circulating IL-1ra (104). These data suggest that the levels of soluble receptors and receptor antagonists may be markers of mortality by indicating the magnitude of the pro-inflammatory response. In addition, recent data have suggested a link between an initial dysregulated hyperinflammation and subsequent development of nosocomial infection (98). Gene expression analysis of leukocytes from patients developing nosocomial infection has demonstrated overactivation of IL-1 signaling (16) supporting a potential relationship between elevation of IL-1ra and nosocomial infection in sepsis.
Pathogen recognition receptor signaling inhibitors
The pro-inflammatory host response to microbial mediators occurs through PRRs including the Toll-like receptor (TLR) family. Negative regulators of TLR signaling are induced during sepsis. These regulators selectively inhibit the downstream inflammatory response via interactions with one or multiple TLR pathways. Single immunoglobulin IL-1R-related protein (SIGIRR) interferes with binding of IL-1 and LPS extracellularly and interferes with complexing of IRAK-1 and TRAF-6 intracellularly, resulting in profound effects on NF-κB and MAPK-dependent signaling (105). MyD88 short (MyD88s) splice variant is upregulated in response to LPS and is defective in its ability to phosphorylate IRAK resulting in reduced NF-κB activation (106). Both SIGIRR and MyD88s expression were found to be elevated in septic monocytes and associated with depressed cytokine responses (107). Interleukin-1 receptor associated kinase-M (IRAK-M, also known as IRAK-3) negatively regulates TLR signaling through inhibiting the dissociation of IRAK-1 from the Toll-IL-1 signaling domain. In experimental sepsis, IRAK-M is upregulated in alveolar macrophages and mediates supressed cytokine responses and impaired clearance of P. aeruginosa (108). IRAK-M is also elevated in monocytes from septic patients (109). MicroRNAs (miRNA) are small non-coding RNA and have been found to exert negative regulatory effects on TLR signaling. Multiple miRNAs are dysregulated in sepsis. In particular, elevated circulating levels of miRNA 155 have been associated with poor outcome and expansion of regulatory T cells in patients with sepsis (110) indicating a possible link to sepsis immunosuppression and nosocomial infection.
Pathogen associated molecular patterns
Pathogen associated molecular patterns (PAMPs) are exogenous microbial factors derived from infectious organisms that activate PRRs. In sepsis, PAMPs, such as cell wall and intracellular microbial components, are the primary factors initiating the inflammatory response. PAMPs are therefore critical to the reprogramming of immune cells in sepsis, this reprogramming is likely dependent on the specific antigen (PAMP) and receptor (PRR) combinations that are engaged on a particular cell. For example, in vitro stimulation of human monocytes with various PAMPs has demonstrated that the fungal cell wall component β-glucan induces primed (trained) responses through nucleotide-binding oligomerization domain-like receptors (NLRs) (111). In contrast, engagement of PAMP-TLR combinations, such as LPS with its receptor Toll-like receptor 4 (TLR4), induced predominantly tolerant programs with depressed cytokine production. Interestingly, the TLR ligands administered at low dose caused primed responses while inducing tolerance at higher doses, suggesting the presence of an inflammatory rheostat guiding secondary responses. In the context of sepsis, pathogen specific ligands such as LPS (Gram-negative organism) or lipotechoic acid (Gram-positive organism) and endogenous PRR ligands (discussed in the next section) form a complex network of PRR signaling that is likely to contribute to the inflammatory cellular program. In addition to exposure to infectious pathogens, sepsis and critical illness are associated with collapse of the host microbial community, a term known as dysbiosis. This occurs through a combination of ecological factors that are drastically altered in the critically ill (112). While there is no direct evidence linking the dysbiosis that occurs in septic patients to subsequent nosocomial infection, population level studies have demonstrated an increased risk of severe sepsis within 90 days following hospitalizations known to result in dysbiosis (113). A second study demonstrated an increased risk of severe sepsis and septic shock in the 90 days following a hospital-related antibiotic exposure (114). These studies suggest PAMP-PRR interactions via primary infection or continued dysbiosis may promote changes in the immune program that predispose critically ill patients to secondary infection and sepsis, although further investigation is required to establish causal relationships.
Damage-associated molecular patterns
Damage-associated molecular patterns (DAMPs) are endogenous pattern recognition receptor agonists that initiate inflammatory responses but have distinct biological roles in non-inflammatory states. These proteins are released upon host injury, either passively from necrotic cells or actively secreted into the extracellular space (115). DAMPs are released during injured states, including sepsis, trauma, and burns. As such, DAMPs are appealing candidates as mediators of altered immune programs observed in these patients. Several DAMPs have been shown to correlate with sepsis morbidity and mortality including S100A8/A9, high-mobility group box-1 (HMGB1), mitochondrial DNA, nuclear DNA, histones and heat shock proteins (HSP) (115). HMGB1 and S100A8/A9 are acutely elevated in patients presenting with sepsis and are associated with worse outcome (116, 117). Their functions are pleiotropic, including induction of immunosuppressive MDSC expansion and priming of immune cells in sepsis models (38, 39, 118). HMGB1 also stimulates the expansion of Treg in chronic inflammatory diseases (119). HSPs are intracellular molecular chaperone proteins that can have anti-inflammatory properties. Several HSPs (HSP27, 60, 70, 90) are increased in patients with sepsis and are associated with enhanced neutrophil oxidative activity and reduced apoptosis (120). HSP70 levels increase with the degree of hyperinflammatory response and are associated with increased risk of mortality in patients with sepsis (121, 122). HSP70 also promotes adaptive immune dysfunction through enhanced Treg suppressor activities and secretion of anti-inflammatory cytokines (123). In many of the studies referenced elevated levels of certain DAMPs persist in the circulation of survivors for many days, indicating a potential for ongoing modulation of the immune system after acute sepsis has resolved.
Expansion of regulatory T cells
Cellular subsets that have roles in homeostasis are expanded during sepsis and may contribute to nosocomial infection susceptibility. Treg are a T cell population that are able to negatively regulate the adaptive and innate immune response (124). There are several subsets of Treg which can be identified by cell surface markers. The CD4+, CD25+, CD127lo subset is one of several studied in sepsis. These cells are expanded in patients with sepsis (125) and contribute to lymphocyte anergy in septic shock patients (91). Higher Treg numbers are also associated with the development of nosocomial infections in critically ill patients with and without sepsis (77). In sepsis models, Treg expansion results in systemic immunosuppression potentiating tumor growth (126). Data is conflicting though as models of sepsis have demonstrated that antibody mediated depletion of Treg does not improve mortality (127) and adoptive transfer of CD4+ CD25+ Treg early in the course of sepsis actually improves bacterial clearance and mortality (128). Similarly, in one human study, increased Treg in patients with severe sepsis was associated with improved survival, though this finding may have been driven by higher total T cell counts in these patients (129). Further investigation such as depletion or inhibiting expansion of Treg in humans is required to establish a detrimental role for Treg in patients with sepsis.
Myeloid-derived suppressor cells
Myeloid-derived suppressor cells (MDSC) are a heterogenous group of immature innate immune cells that exert predominantly suppressive effects. Their expansion is a component of the emergency myelopoietic response to injury, infection and malignancy (130). This population consists of a mix of immature granulocytes, monocytes, and dendritic cells with the ability to suppress T cell function through enhanced arginase, nitric oxide synthase, and reactive oxygen species activity (131). MDSCs can produce a number of pro- and anti-inflammatory cytokines upon secondary stimulation, including IL-10 (132). As such, they represent important effector cells in sepsis recovery. Their release is driven mainly by STAT-3 signaling through inflammatory mediators including IL-6 and colony stimulating factors. Activated STAT-3 also induces release of S100A8/9 which both prevents maturation of MDSCs and promotes additional expansion in a feed forward loop (118, 133). In mouse models of sepsis, MDSCs exert pleiotropic effects, with protective or detrimental properties depending on phase of sepsis in which they are examined (134, 135). MDSCs are expanded acutely in patients with sepsis (136, 137) and are associated with the development of nosocomial infection (138). Their persistence in severe sepsis and septic shock is also associated with increased risk for persistent critical illness and mortality (139).
Co-inhibitory molecules
Co-inhibitory molecules of the B7-CD28 family function to maintain homeostasis in the host by negatively regulating the immune response (140). In sepsis, these molecules have been postulated to be responsible for immune exhaustion. Post-mortem studies of patients dying of sepsis has demonstrated elevations in co-inhibitory receptors PD-1 and CTLA-4 in splenic T cells, while the ligand PD-L1 was elevated in antigen presenting cells and tissue macrophages (47). T cells and dendritic cells isolated from the lung also expressed increased PD-1 and PD-L1, respectively. Circulating T cells in patients with severe sepsis demonstrated non-statistically significant elevations in PD-1, with a decrease in CTLA-4 (61). Others demonstrated marked elevation in T cell PD-1 and monocyte PD-L1 in septic shock. In vitro treatment with anti-PD1 antibody resulted in restoration of monocyte proinflammatory responses and decreased apoptosis of T cells (141). Furthermore, higher monocyte PD-L1 has been associated with mortality, while higher PD-1 and PD-L2 are associated with increased risk of nosocomial infection (142). Others have shown that monocyte PD-L1, and not T cell PD1, expression on day 3–4 of septic shock is an independent predictor of death (143). In mice, neutralization of PD-1 or PD-L1 24 h after sepsis reduced apoptosis of lymphocytes and improved survival (144, 145). In leukocytes isolated from septic patients, in vitro blockade of PD1 and PDL1 reverses T cell exhaustion and restores neutrophil and monocyte phagocytic function (146, 147).
Epigenetic reprogramming
Epigenetic mechanisms of immune dysfunction have also been proposed. Histone modification at inflammatory loci alters the accessibility of DNA to transcription factors and therefore gene transcription. Alterations in chromatin structure are determined by gene activating and repressing histone marks, which are in turn regulated by chromatin modifying enzymes (CME) (148). Specific changes in chromatin are associated with exposure to inflammatory stimuli (149). Not surprisingly, chromatin remodeling occurs in the monocytes of septic patients (150). In models of sepsis survival, dendritic cell cytokine responses were suppressed for up to 6 weeks and were associated with alterations in histone H3 lysine-4 (H3K4) and histone H3 lysine-27 (H3K27) methylation at IL-12 promoter regions. These changes were also mediated by interactions with histone methyltransferases (151). We have demonstrated an IRAK-M mediated reduction in histone H4 acetylation and H3K4 methylation (H3K4me) in immune tolerant AM 24 h after induction of sepsis (152). These studies provide compelling evidence for epigenetic changes in sepsis leading to immunosuppressive phenotypes in both the acute and recovery phases of illness. As such dynamic changes in histones and their CMEs have high potential for use as prognostic markers and therapeutic targets. Epigenetic reprogramming of inflammatory cells may also result in primed phenotypes. This concept is also known as “trained immunity” whereby exposure to specific PAMPs (β-glucan, BCG) leads to specific modifications in chromatin structure and enhanced inflammatory responses to secondary stimulation (153, 154). These modifications include genome wide changes in H3K4me3, H3K4me1, and H3K27 acetylation. Our understanding of trained immunity and its potential implications in the pathophysiology of sepsis-related immune priming remains limited.
Immunostimulation
Though the immune response to sepsis is dynamic and contextual, there is a large body of evidence supporting an association between immunosuppressive cellular programs and poor outcomes in patients with sepsis. We know that supportive care is inadequate in addressing the complex immunology of sepsis. As such, immunostimulatory therapies have been evaluated to improve sepsis outcomes. Precision approaches in sepsis through immunomodulation require the development of immune monitoring strategies and suitable immunomodulating agents that can be deployed quickly and dynamically at the bedside. The first step is accurately predicting which patients are at risk for secondary infection and mortality related to immune exhaustion. Large observational studies aiming to predict poor outcomes through cellular phenotyping and cell surface marker expression are already underway (74, 75). Given the heterogenous and rapidly evolving immune programs occurring during sepsis, safety and tolerability of immune therapies with a focus on monitoring for hyperinflammatory consequences is essential. Moreover, assessment of long-term outcomes will be important. In addition, recent in vitro studies of PD-1/PD-L1 blockade in mononuclear cells from septic patients has demonstrated significant variability in response to immunostimulation (146, 147). Non-response of mononuculear cells to in vitro immunostimulation was associated with increased mortality (155). As such, response rates and mechanisms of response will also need to be examined in further detail, similar to the current practices of immune modulation in oncology (156). There are a number of ongoing trials of immunostimulation in sepsis (Table 2).
Table 2.
Current clinical evidence for immunostimulation in patients with sepsis.
| Therapy | Goal of therapy | Human Evidence | References |
|---|---|---|---|
| G-CSF/ GM-CSF |
|
|
|
| IFNγ |
|
|
|
| IL-7 |
|
|
|
| Anti-PD-1/ PD-L1 |
|
|
|
| Tα1 |
|
|
|
| MSC |
|
|
|
Tα1, Thymosin alpha 1; G-CSF, granulocyte colony stimulating factor; GM-CSF, granulocyte-macrophage stimulating factor; IFNγ, interferon gamma; MSC, mesynchymal stem cell; NCT, clinicaltrials.gov identifier.
Immunostimulatory cytokines
Early clinical trials attempting to simulation the immune system used granulocyte colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) to enhance phagocyte production, function and improve bacterial clearance. A meta-analysis of trials performed between 1998 and 2011 failed to find a mortality benefit of these therapies, though there was some improvement in clinical endpoints such as ICU LOS (157). IFNγ has similarly been used to stimulate the innate immune response in patients with bacteremia and chronic fungal infections (159, 160). Both are being used in patients with sepsis as part of ongoing phase III clinical trials.
IL-7 is potent inducer of the antiapoptotic protein Bcl-2 that has the ability to both stimulate lymphocyte production and reduce apoptosis. A recent multicenter, randomized and controlled phase IIb trial examined IL-7 administration in patients with septic shock and lymphopenia (161). Patients received low or high dosing regimens of IL-7 for a total of 4 weeks or until discharge. These investigators found an increase in CD4+ and CD8+ T cells, enhanced T cell activation and potentially T cell trafficking as compared to placebo. In this small trial, there was no difference in mortality or rate of nosocomial infection. The drug was well tolerated with minimal adverse events. Cytokine profiles were measured serially over the course of IL-7 administration and there were no signs of patients developing cytokine storm. Also, the effect of enhanced lymphocyte production persisted for weeks following treatment. These promising results will require confirmation in larger studies to determine if therapy is efficacious and without long-term consequences.
Checkpoint inhibition
Checkpoint inhibitors such as anti-PD1 and anti-PD-L1, improve monocyte and lymphocyte function and reduce apoptosis through disruption of negative cell-cell interactions. These inhibitors have revolutionized management of malignancy and are now first line therapy for many types of cancer (165). However, immune related adverse events (iRAE) with checkpoint inhibition are not infrequent, with rates of grade ≥3 toxicity approaching 7% (166). Early human studies have demonstrated feasibility and reversal of sepsis induced immunosuppression with in vitro blockade (146, 147). A phase I clinical trial of the novel PD-L1 inhibitor, BMS-936559, has been completed (162). A phase I clinical trial of PD-1 inhibitor, Nivolumab, is currently ongoing (NCT02960854).
Endogenous immunostimulatory proteins
Thymosin alpha 1 (Tα1) is an endogenous thymic peptide that regulates the innate and adaptive immune system. Initial studies in sepsis have shown improvement in mHLA-DR (163). Continued study of this agent is ongoing in a phase III clinical trial (NCT02883595).
Cellular therapies
Mesenchymal stem cells (MSCs) reduce mortality and organ dysfunction in models of sepsis through modulation of the inflammatory cascade, pathogen clearance and promotion of tissue repair (167). Following completion of a phase I clinical trial in septic shock, administration of MSCs appears to be safe (164). Further research is ongoing in a phase II clinical trial (NCT02883803).
Conclusions
The clinical phases of sepsis are associated with specific and dynamic changes in the immune programming of multiple cell types. Suppressed inflammatory responses to stimulation have been demonstrated extensively in patients with sepsis, however the dynamics, pathogen, and compartmental specificity of these findings requires additional investigation. Primed immune responses have also been demonstrated in animal models of sepsis survival. This cellular program has not been examined extensively in humans and its contributions to sepsis related morbidity and mortality remains unknown. Molecular mechanisms of immune reprogramming in sepsis still require further investigation. In particular the complexities of PAMP/DAMP-PRR interactions, the role of MDSC and T-reg, and alterations in the epigenome are prime targets for evaluation.
Patients at higher risk for nosocomial infection and mortality frequently experience an immunosuppressed state characterized by defects in immune tolerance, exhaustion, and apoptosis. While reversal of these immunosuppressive phenotypes has improved outcomes in animal models, a direct causal relationship between sepsis-related immunosuppression and nosocomial infection/death has not yet been established in humans. Furthermore, rates of nosocomial infection and its attributable mortality in sepsis may not be as high as previously estimated, suggesting that reasons why septic patients die despite best supportive care still need to be explored. Regardless, nosocomial infections are common and carry such significant morbidity that sepsis patients may benefit from immunostimulatory therapies. Early stage trials of immune therapy have shown reversal of leukocyte dysfunction and good safety profiles, both promising for the potential future of these therapies. However, as sepsis has long-term consequences that are still not well understood and immune therapies may have lasting effects, long-term outcomes of patients receiving immunostimulatory therapy require attention.
Author contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Michigan Interdisciplinary Critical Care Research Workgroup for their continued support. SMART Servier medical art (smart.servier.com) was used with permission through the creative commons license in Figures 2, 4, significant editing was performed on images used in Figure 4.
Footnotes
Funding. This work was supported by National Institutes of Health grants T32HL00774921 (to SD), K08NS101054 (to BS), R01HL123515 (to TS).
References
- 1.Abraham E, Wunderink R, Silverman H, Perl TM, Nasraway S, Levy H, et al. Efficacy and safety of monoclonal antibody to human tumor necrosis factor α in patients with sepsis syndrome. A randomized, controlled, double-blind, multicenter clinical trial JAMA. J Am Med Assoc. (1995) 273:934–41. 10.1001/jama.273.12.934 [DOI] [PubMed] [Google Scholar]
- 2.Dhainaut JF, Tenaillon A, Le Tulzo Y, Schlemmer B, Solet JP, Wolff M, et al. Platelet-activating factor receptor antagonist BN 52021 in the treatment of severe sepsis: a randomized, double-blind, placebo-controlled, multicenter clinical trial. BN 52021 Sepsis Study Group. Crit. Care Med. (1994) 22:1720–8. [PubMed] [Google Scholar]
- 3.Fisher CJ, Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA (1994) 271:1836–43. 10.1001/jama.1994.03510470040032 [DOI] [PubMed] [Google Scholar]
- 4.Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. (1996) 24:1125–6. 10.1097/00003246-199607000-00010 [DOI] [PubMed] [Google Scholar]
- 5.Fleischmann C, Scherag A, Adhikari NKJ, Hartog CS, Tsaganos T, Schlattmann P, et al. Assessment of global incidence and mortality of hospital-treated sepsis current estimates and limitations. Am J Respir Crit Care Med. (2016) 193:259–72. 10.1164/rccm.201504-0781OC [DOI] [PubMed] [Google Scholar]
- 6.Rhee C, Dantes R, Epstein L, Murphy DJ, Seymour CW, Iwashyna TJ, et al. Incidence and trends of sepsis in US hospitals using clinical vs claims data, 2009-2014. JAMA - J Am Med Assoc. (2017) 318:1241–9. 10.1001/jama.2017.13836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stevenson EK, Rubenstein AR, Radin GT, Wiener RS, Walkey AJ. Two decades of mortality trends among patients with severe sepsis: a comparative meta-analysis*. Crit Care Med. (2014) 42:625–31. 10.1097/CCM.0000000000000026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faist E, Kupper TS, Baker CC, Chaudry IH, Dwyer J, Baue AE. Depression of cellular immunity after major injury. Its association with posttraumatic complications and its reversal with immunomodulation. Arch. Surg. (1986) 121:1000–5. [DOI] [PubMed] [Google Scholar]
- 9.Richardson JD, DeCamp MM, Garrison RN, Fry DE. Pulmonary infection complicating intra-abdominal sepsis: clinical and experimental observations. Ann. Surg. (1982) 195:732–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stephan RN, Kupper TS, Geha AS, Baue AE, Chaudry IH. Hemorrhage without tissue trauma produces immunosuppression and enhances susceptibility to sepsis. Arch. Surg. (1987) 122:62–8. [DOI] [PubMed] [Google Scholar]
- 11.Munoz C, Carlet J, Fitting C, Misset B, Blériot JP, Cavaillon JM. Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest. (1991) 88:1747–54. 10.1172/JCI115493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Landelle C, Lepape A, Français A, Tognet E, Thizy H, Voirin N, et al. Nosocomial infection after septic shock among intensive care unit patients. Infect Control Hosp Epidemiol. (2008) 29:1054–65. 10.1086/591859 [DOI] [PubMed] [Google Scholar]
- 13.Otto GP, Sossdorf M, Claus RA, Rödel J, Menge K, Reinhart K, et al. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit Care (2011) 15:R183. 10.1186/cc10332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walton AH, Muenzer JT, Rasche D, Boomer JS, Sato B, Brownstein BH, et al. Reactivation of multiple viruses in patients with sepsis. PLoS ONE (2014) 9:e98819. 10.1371/journal.pone.0098819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao G, Li D, Zhao Q, Song J, Chen X, Hong G, et al. Incidence, risk factors and impact on outcomes of secondary infection in patients with septic shock: an 8-year retrospective study. Sci Rep. (2016) 6:38361. 10.1038/srep38361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Vught LA, Klein Klouwenberg PMC, Spitoni C, Scicluna BP, Wiewel MA, Horn J, et al. Incidence, risk factors, and attributable mortality of secondary infections in the intensive care unit after admission for sepsis. Jama (2016) 315:1469. 10.1001/jama.2016.2691 [DOI] [PubMed] [Google Scholar]
- 17.Bonten MJ, Froon AH, Gaillard CA, Greve JW, de Leeuw PW, Drent M, et al. The systemic inflammatory response in the development of ventilator-associated pneumonia. Am J Respir Crit Care Med. (1997) 156:1105–13. 10.1164/ajrccm.156.4.9610002 [DOI] [PubMed] [Google Scholar]
- 18.Schmier JK, Hulme-Lowe CK, Semenova S, Klenk JA, DeLeo PC, Sedlak R, et al. Estimated hospital costs associated with preventable health care-associated infections if health care antiseptic products were unavailable. Clinicoecon Outcomes Res. (2016) 8:197–205. 10.2147/CEOR.S102505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. (2003) 348:138–50. 10.1056/NEJMra021333 [DOI] [PubMed] [Google Scholar]
- 20.Tang BM, Huang SJ, McLean AS. Genome-wide transcription profiling of human sepsis: a systematic review. Crit Care (2010) 14:R237. 10.1186/cc9392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cavaillon JM, Adib-Conquy M, Fitting C, Adrie C, Payen D. Cytokine cascade in sepsis. Scand J Infect Dis. (2003) 35:535–44. 10.1080/00365540310015935 [DOI] [PubMed] [Google Scholar]
- 22.Tamayo E, Fernández A, Almansa R, Carrasco E, Heredia M, Lajo C, et al. Pro- and anti-inflammatory responses are regulated simultaneously from the first moments of septic shock. Eur Cytokine Netw. (2011) 22:82–7. 10.1684/ecn.2011.0281 [DOI] [PubMed] [Google Scholar]
- 23.Cavaillon JM. Pro- versus anti-inflammatory cytokines: myth or reality. Cell. Mol. Biol. (2001) 47:695–702. [PubMed] [Google Scholar]
- 24.Steinhauser ML, Hogaboam CM, Kunkel SL, Lukacs NW, Strieter RM, Standiford TJ. IL-10 is a major mediator of sepsis-induced impairment in lung antibacterial host defense. J. Immunol. (1999) 162:392–9 [PubMed] [Google Scholar]
- 25.Standiford TJ, Strieter RM, Lukacs NW, Kunkel SL. Neutralization of IL-10 increases lethality in endotoxemia. Cooperative effects of macrophage inflammatory protein-2 and tumor necrosis factor. J. Immunol. (1995) 155:2222–9. [PubMed] [Google Scholar]
- 26.Walley KR, Lukacs NW, Standiford TJ, Strieter RM, Kunkel SL. Balance of inflammatory cytokines related to severity and mortality of murine sepsis. Infect. Immun. (1996) 64:4733–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adib-Conquy M, Petit AF, Marie C, Fitting C, Cavaillon JM. Paradoxical priming effects of IL-10 on cytokine production. Int Immunol. (1999) 11:689–98. 10.1093/intimm/11.5.689 [DOI] [PubMed] [Google Scholar]
- 28.Cavaillon J-M, Annane D. Compartmentalization of the inflammatory response in sepsis and SIRS. J Endotoxin Res. (2006) 12:151–70. 10.1179/096805106X102246 [DOI] [PubMed] [Google Scholar]
- 29.Hacham M, Cristal N, White RM, Segal S, Apte RN. Complementary organ expression of IL-1 vs. IL-6 and CSF-1 activities in normal and LPS-injected mice. Cytokine (1996) 8:21–31. 10.1006/cyto.1995.0004 [DOI] [PubMed] [Google Scholar]
- 30.Chinnaiyan AM, Huber-Lang M, Kumar-Sinha C, Barrette TR, Shankar-Sinha S, Sarma VJ, et al. Molecular signatures of sepsis: multiorgan gene expression profiles of systemic inflammation. Am J Pathol. (2001) 159:1199–209. 10.1016/S0002-9440(10)62505-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suzuki T, Shimizu T, Szalay L, Choudhry MA, Rue LW, Bland KI, et al. Androstenediol ameliorates alterations in immune cells cytokine production capacity in a two-hit model of trauma-hemorrhage and sepsis. Cytokine (2006) 34:76–84. 10.1016/j.cyto.2006.04.007 [DOI] [PubMed] [Google Scholar]
- 32.Carlsen H, Moskaug JO, Fromm SH, Blomhoff R. In vivo imaging of NF- B activity. J Immunol. (2002) 168:1441–6. 10.4049/jimmunol.168.3.1441 [DOI] [PubMed] [Google Scholar]
- 33.Huynh T, Lemasters JJ, Bracey LW, Baker CC. Proinflammatory kupffer cell alterations after femur fracture trauma and sepsis in rats. Shock (2000) 14:555–60. 10.1097/00024382-200014050-00010 [DOI] [PubMed] [Google Scholar]
- 34.Takahashi H, Tsuda Y, Takeuchi D, Kobayashi M, Herndon DN, Suzuki F. Influence of systemic inflammatory response syndrome on host resistance against bacterial Infections. Crit Care Med. (2004) 32:1879–85. 10.1097/01.CCM.0000139606.34631.61 [DOI] [PubMed] [Google Scholar]
- 35.Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA (2010) 304:1787–94. 10.1001/jama.2010.1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Corrales-Medina VF, Alvarez KN, Weissfeld LA, Angus DC, Chirinos JA, Chang C-CH, et al. Association Between Hospitalization for Pneumonia and Subsequent Risk of Cardiovascular Disease. JAMA (2015) 313:264. 10.1001/jama.2014.18229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prescott HC, Langa KM, Iwashyna TJ. Readmission diagnoses after hospitalization for severe sepsis and other acute medical conditions. JAMA (2015) 313:1055–7. 10.1001/jama.2015.1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Denstaedt SJ, Spencer-Segal JL, Newstead MW, Laborc K, Zhao AP, Hjelmaas A, et al. S100A8/A9 Drives neuroinflammatory priming and protects against anxiety-like behavior after sepsis. J. Immunol., (2018) 200:3188–200. 10.4049/jimmunol.1700834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Valdés-Ferrer SI, Rosas-Ballina M, Olofsson PS, Lu B, Dancho ME, Ochani M, et al. HMGB1 mediates splenomegaly and expansion of splenic CD11b+ Ly-6C(high) inflammatory monocytes in murine sepsis survivors. J Intern Med. (2013) 274:381–90. 10.1111/joim.12104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yende S, Angelo GD, Kellum JA, Weissfeld L, Fine J, Welch RD, et al. Inflammatory markers at hospital discharge predict subsequent mortality after pneumonia and sepsis. (2008) 177:1242–7. 10.1164/rccm.200712-1777OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaynar AM, Yende S, Zhu L, Frederick DR, Chambers R, Burton CL, et al. Effects of intra-abdominal sepsis on atherosclerosis in mice. Crit Care (2014) 18:469. 10.1186/s13054-014-0469-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singer BH, Newstead MW, Zeng X, Cooke CL, Thompson RC, Singer K, et al. Cecal ligation and puncture results in long- term central nervous system myeloid inflammation. PLoS ONE (2016) 11:e0149136. 10.1371/journal.pone.0149136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andonegui G, Zelinski EL, Schubert CL, Knight D, Craig LA, Winston BW, et al. Targeting inflammatory monocytes in sepsis-associated encephalopathy and long-term cognitive impairment. JCI insight (2018) 3:99364. 10.1172/jci.insight.99364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature (2007) 447:972–8. 10.1038/nature05836 [DOI] [PubMed] [Google Scholar]
- 45.Shalova IN, Lim JY, Chittezhath M, Zinkernagel AS, Beasley F, Hernández-Jiménez E, et al. Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1α. Immunity (2015) 42:484–98. 10.1016/j.immuni.2015.02.001 [DOI] [PubMed] [Google Scholar]
- 46.Hotchkiss RS, Swanson PE, Cobb JP, Jacobson A, Buchman TG, Karl IE. Apoptosis in lymphoid and parenchymal cells during sepsis: Findings in normal and T- and B-celldeficient mice. Crit Care Med. (1997) 25:1298–307. 10.1097/00003246-199708000-00015 [DOI] [PubMed] [Google Scholar]
- 47.Boomer JS, To K, Chang KC, Ii SDJ, Kreisel D. Immunsuppression in patients who die of sepsis and multiple organ failure. JAMA (2011) 306:2594–605. 10.1001/jama.2011.1829.Immunosuppression [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit. Care Med. (1999) 27:1230–51. [DOI] [PubMed] [Google Scholar]
- 49.Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE, Hui JJ, Chang KC, et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol. (2001) 166:6952–63. 10.4049/jimmunol.166.11.6952 [DOI] [PubMed] [Google Scholar]
- 50.Le Tulzo Y, Pangault C, Gacouin A, Guilloux V, Tribut O, Amiot L, et al. Early circulating lymphocyte apoptosis in human septic shock is associated with poor outcome. Shock (2002) 18:487–94. 10.1097/01.shk.0000043614.39685.6f [DOI] [PubMed] [Google Scholar]
- 51.Drewry AM, Samra N, Skrupky LP, Fuller BM, Compton SM, Hotchkiss RS. Persistent lymphopenia after diagnosis of sepsis predicts mortality. Shock (2014) 42:383–91. 10.1097/SHK.0000000000000234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stortz JA, Murphy TJ, Raymond SL, Mira JC, Ungaro R, Dirain ML, et al. Evidence for persistent immune suppression in patients who develop chronic critical illness after sepsis. Shock (2018) 49:249–58. 10.1097/SHK.0000000000000981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adrie C, Bachelet M, Vayssier-Taussat M, Russo-Marie F, Bouchaert I, Adib-Conquy M, et al. Mitochondrial membrane potential and apoptosis peripheral blood monocytes in severe human sepsis. Am J Respir Crit Care Med. (2001) 164:389–95. 10.1164/ajrccm.164.3.2009088 [DOI] [PubMed] [Google Scholar]
- 54.Giamarellos-Bourboulis EJ, Routsi C, Plachouras D, Markaki V, Raftogiannis M, Zervakis D, et al. Early apoptosis of blood monocytes in the septic host: Is it a mechanism of protection in the event of septic shock? Crit Care (2006) 10:1–8. 10.1186/cc4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hotchkiss RS, Tinsley KW, Swanson PE, Grayson MH, Osborne DF, Wagner TH, et al. Depletion of dendritic cells, but not macrophages, in patients with sepsis. J. Immunol. (2002) 168:2493–500. 10.4049/jimmunol.168.5.2493 [DOI] [PubMed] [Google Scholar]
- 56.Albertine KH, Soulier MF, Wang Z, Ishizaka A, Hashimoto S, Zimmerman GA, et al. Fas and fas ligand are up-regulated in pulmonary edema fluid and lung tissue of patients with acute lung injury and the acute respiratory distress syndrome. Am J Pathol. (2002) 161:1783–96. 10.1016/S0002-9440(10)64455-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wesche DE, Lomas-Neira JL, Perl M, Chung C-S, Ayala A. Leukocyte apoptosis and its significance in sepsis and shock. J Leukoc Biol. (2005) 78:325–37. 10.1189/jlb.0105017 [DOI] [PubMed] [Google Scholar]
- 58.Hotchkiss RS, Swanson PE, Knudson CM, Chang KC, Cobb JP, Osborne DF, et al. Overexpression of Bcl-2 in transgenic mice decreases apoptosis and improves survival in sepsis. J Immunol. (1999) 162:4148–56. [PubMed] [Google Scholar]
- 59.Hotchkiss RS, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, et al. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci USA. (1999) 96:14541–6. 10.1073/pnas.96.25.14541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ertel W, Kremer JP, Kenney J, Steckholzer U, Jarrar D, Trentz O, et al. Downregulation of proinflammatory cytokine release in whole blood from septic patients. Blood (1995) 85:1341–7. [PubMed] [Google Scholar]
- 61.Boomer JS, Shuherk-Shaffer J, Hotchkiss RS, Green JM. A prospective analysis of lymphocyte phenotype and function over the course of acute sepsis. Crit Care (2012) 16:R112. 10.1186/cc11404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McCall CE, Grosso-Wilmoth LM, LaRue K, Guzman RN, Cousart SL. Toerance to endotoxin-induced expression of the interleukin- beta gene in blood neutrophils of humans with sepsis syndrome. Joural Clin. Investig. (1993) 91:853–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Souza-Fonseca-Guimaraes F, Parlato M, Philippart F, Misset B, Cavaillon J-M, Adib-Conquy M, et al. Toll-like receptors expression and interferon-gamma production by NK cells in human sepsis. Crit Care (2012) 16:R206. 10.1186/cc11838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pena OM, Hancock DG, Lyle NH, Linder A, Russell JA, Xia J, et al. An endotoxin tolerance signature predicts sepsis and organ dysfunction at initial clinical presentation. EBioMedicine (2014) 1:64–71. 10.1016/j.ebiom.2014.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Muszynski JA, Nofziger R, Greathouse K, Nateri J, Hanson-Huber L, Steele L, et al. Innate immune function predicts the development of nosocomial infection in critically injured children. Shock (2014) 42:313–21. 10.1097/SHK.0000000000000217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Drewry AM, Ablordeppey EA, Murray ET, Beiter ER, Walton AH, Hall MW, et al. Comparison of monocyte human leukocyte antigen-DR expression and stimulated tumor necrosis factor alpha production as outcome predictors in severe sepsis: a prospective observational study. Crit Care (2016) 20:334. 10.1186/s13054-016-1505-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Van Vught LA, Wiewel MA, Hoogendijk AJ, Scicluna BP, Belkasim-Bohoudi H, Horn J, et al. Reduced responsiveness of blood leukocytes to lipopolysaccharide does not predict nosocomial infections in critically ill patients. Shock (2015) 44:110–4. 10.1097/SHK.0000000000000391 [DOI] [PubMed] [Google Scholar]
- 68.Wolk K, Döcke WD, von Baehr V, Volk HD, Sabat R. Impaired antigen presentation by human monocytes during endotoxin tolerance. Blood (2000) 96:218–23. 10.1182/blood.v98.13.3800 [DOI] [PubMed] [Google Scholar]
- 69.Hershman MJ, Cheadle WG, Wellhausen SR, Davidson PF, Polk HC. Monocyte HLA-DR antigen expression characterizes clinical outcome in the trauma patient. Br J Surg. (1990) 77:204–7. 10.1002/bjs.1800770225 [DOI] [PubMed] [Google Scholar]
- 70.Monneret G, Lepape A, Voirin N, Bohé J, Venet F, Debard AL, et al. Persisting low monocyte human leukocyte antigen-DR expression predicts mortality in septic shock. Intensive Care Med. (2006) 32:1175–83. 10.1007/s00134-006-0204-8 [DOI] [PubMed] [Google Scholar]
- 71.Wu JF, Ma J, Chen J, Ou-Yang B, Chen MY, Li LF, et al. Changes of monocyte human leukocyte antigen-DR expression as a reliable predictor of mortality in severe sepsis. Crit Care (2011) 15:R220. 10.1186/cc10457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Landelle C, Lepape A, Voirin N, Tognet E, Venet F, Bohé J, et al. Low monocyte human leukocyte antigen-DR is independently associated with nosocomial infections after septic shock. Intensive Care Med. (2010) 36:1859–66. 10.1007/s00134-010-1962-x [DOI] [PubMed] [Google Scholar]
- 73.Cajander S, Rasmussen G, Tina E, Magnuson A, Söderquist B, Källman J, et al. Dynamics of monocytic HLA-DR expression differs between bacterial etiologies during the course of bloodstream infection. PLoS ONE (2018) 13:e192883. 10.1371/journal.pone.0192883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Conway Morris A, Datta D, Shankar-Hari M, Weir CJ, Rennie J, Antonelli J, et al. Predictive value of cell-surface markers in infections in critically ill patients: Protocol for an observational study (ImmuNe FailurE in Critical Therapy (INFECT) Study). BMJ Open (2016) 6:e011326. 10.1136/bmjopen-2016-011326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rol ML, Venet F, Rimmele T, Moucadel V, Cortez P, Quemeneur L, et al. The REAnimation Low Immune Status Markers (REALISM) project: A protocol for broad characterisation and follow-up of injury-induced immunosuppression in intensive care unit (ICU) critically ill patients. BMJ Open (2017) 7:e015734. 10.1136/bmjopen-2016-015734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Conway Morris A, Brittan M, Wilkinson TS, McAuley DF, Antonelli J, McCulloch C, et al. C5a-mediated neutrophil phagocytic dysfunction is RhoA-dependent and predicts nosocomial infection in critically ill patients. Blood (2011) 117:5178–89. 10.1182/blood-2010-08-304667 [DOI] [PubMed] [Google Scholar]
- 77.Conway Morris A, Anderson N, Brittan M, Wilkinson TS, Mcauley DF, Antonelli J, et al. Combined dysfunctions of immune cells predict nosocomial infection in critically ill patients. Br J Anaesth. (2013) 111:778–87. 10.1093/bja/aet205 [DOI] [PubMed] [Google Scholar]
- 78.Davenport EE, Burnham KL, Radhakrishnan J, Humburg P, Hutton P, Mills TC, et al. Genomic landscape of the individual host response and outcomes in sepsis: A prospective cohort study. Lancet Respir Med. (2016) 4:259–71. 10.1016/S2213-2600(16)00046-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Scicluna BP, van Vught LA, Zwinderman AH, Wiewel MA, Davenport EE, Burnham KL, et al. Classification of patients with sepsis according to blood genomic endotype: a prospective cohort study. Lancet Respir Med. (2017) 5:816–26. 10.1016/S2213-2600(17)30294-1 [DOI] [PubMed] [Google Scholar]
- 80.Burnham KL, Davenport EE, Radhakrishnan J, Humburg P, Gordon AC, Hutton P, et al. Shared and distinct aspects of the sepsis transcriptomic response to fecal peritonitis and pneumonia. Am J Respir Crit Care Med. (2017) 196:328–39. 10.1164/rccm.201608-1685OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Simpson SQ, Modi HN, Balk RA, Bone RC, Casey LC. Reduced alveolar macrophage production of tumor necrosis factor during sepsis in mice and men. Crit. Care Med. (1991) 19:1060–6. [DOI] [PubMed] [Google Scholar]
- 82.Reddy RC, Chen GH, Newstead MW, Zeng X, Tateda K, Theodore J, et al. Alveolar macrophage deactivation in murine septic peritonitis : role of interleukin 10 alveolar macrophage deactivation in murine septic peritonitis : role of interleukin 10. Infect Immun. (2001) 69:1394–401. 10.1128/IAI.69.3.1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hiruma T, Tsuyuzaki H, Uchida K, Trapnell BC, Yamamura Y, Kusakabe Y, et al. Interferon-β improves sepsis-related alveolar macrophage dysfunction and post-septic ARDS-related mortality. Am J Respir Cell Mol Biol. (2018) 59:45–55. 10.1165/rcmb.2017-0261OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Delano MJ, Thayer T, Gabrilovich S, Kelly-Scumpia KM, Winfield RD, Scumpia PO, et al. Sepsis Induces Early Alterations in Innate Immunity That Impact Mortality to Secondary Infection. J Immunol. (2011) 186:195–202. 10.4049/jimmunol.1002104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roquilly A, McWilliam HEG, Jacqueline C, Tian Z, Cinotti R, Rimbert M, et al. Local modulation of antigen-presenting cell development after resolution of pneumonia induces long-term susceptibility to secondary infections. Immunity (2017) 47:135–147.e5. 10.1016/j.immuni.2017.06.021 [DOI] [PubMed] [Google Scholar]
- 86.Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol. (2015) 16:27–35. 10.1038/ni.3045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hiramatsu M, Hotchkiss RS, Karl IE, Buchman TG. Cecal ligation and puncture (CLP) induces apoptosis in thymus, spleen, lung, and GUT by an endotoxin and TNF- independent pathway. Shock (1997) 7:247–53. 10.1097/00024382-199704000-00002 [DOI] [PubMed] [Google Scholar]
- 88.Meakins JL, Pietsch JB, Bubenick O, Kelly R, Rode H, Gordon J, et al. Delayed hypersensitivity: indicator of acquired failure of host defenses in sepsis and trauma. Ann Surg. (1977) 186:241–50. 10.1097/00000658-197709000-00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Heidecke CD, Hensler T, Weighardt H, Zantl N, Wagner H, Siewert JR, et al. Selective defects of T lymphocyte function in patients with lethal intraabdominal infection. Am J Surg. (1999) 178:288–92. 10.1016/S0002-9610(99)00183-X [DOI] [PubMed] [Google Scholar]
- 90.Unsinger J, Kazama H, McDonough JS, Griffith TS, Hotchkiss RS, Ferguson TA. Sepsis-induced apoptosis leads to active suppression of delayed-type hypersensitivity by CD8+ regulatory T cells through a TRAIL-dependent mechanism. J Immunol. (2010) 184:6766–72. 10.4049/jimmunol.0904054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Venet F, Chung CS, Kherouf H, Geeraert A, Malcus C, Poitevin F, et al. Increased circulating regulatory T cells (CD4+CD25+CD127-) contribute to lymphocyte anergy in septic shock patients. Intensive Care Med. (2009) 35:678–86. 10.1007/s00134-008-1337-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chiche L, Forel J-M, Thomas G, Farnarier C, Cognet C, Guervilly C, et al. Interferon-γ production by natural killer cells and cytomegalovirus in critically ill patients*. Crit Care Med. (2012) 40:3162–9. 10.1097/CCM.0b013e318260c90e [DOI] [PubMed] [Google Scholar]
- 93.Forel JM, Chiche L, Thomas G, Mancini J, Farnarier C, Cognet C, et al. Phenotype and functions of natural killer cells in critically-ill septic patients. PLoS ONE (2012) 7:e50446. 10.1371/journal.pone.0050446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J. Exp. Med. (1989) 170:2081–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O'Garra A. IL-10 inhibits cytokine production by activated macrophages. J. Immunol. (1991) 147:3815–22. [PubMed] [Google Scholar]
- 96.Gomez HG, Gonzalez SM, Londoño JM, Hoyos NA, Niño CD, Leon AL, et al. Immunological characterization of compensatory anti-inflammatory response syndrome in patients with severe sepsis: a longitudinal study. Crit Care Med. (2014) 42:771–80. 10.1097/CCM.0000000000000100 [DOI] [PubMed] [Google Scholar]
- 97.Gogos CA, Drosou E, Bassaris HP, Skoutelis A. Pro- versus anti-inflammatory cytokine profile in patients with severe sepsis: a marker for prognosis and future therapeutic options. J Infect Dis. (2000) 181:176–80. 10.1086/315214 [DOI] [PubMed] [Google Scholar]
- 98.van Vught LA, Wiewel MA, Hoogendijk AJ, Frencken JF, Scicluna BP, Klein Klouwenberg PMC, et al. The host response in sepsis patients developing intensive care unit-acquired secondary infections. Am J Respir Crit Care Med. (2017) 196:1–55. 10.1164/rccm.201606-1225OC [DOI] [PubMed] [Google Scholar]
- 99.Froon AH, Bemelmans MH, Greve JW, van der Linden CJ, Buurman WA. Increased plasma concentrations of soluble tumor necrosis factor receptors in sepsis syndrome: correlation with plasma creatinine values. Crit. Care Med. (1994) 22:803–9. [DOI] [PubMed] [Google Scholar]
- 100.Schröder J, Kremer B, Stüber F, Gallati H, Schade FU. Pattern of soluble TNF receptors I and II in sepsis. Infection (1995) 23:143–8. 10.1007/BF01793854 [DOI] [PubMed] [Google Scholar]
- 101.Hasegawa Y, Sawada M, Ozaki N, Inagaki T, Suzumura A. Increased soluble tumor necrosis factor receptor levels in the serum of elderly people. Gerontology (2000) 46:185–8. 10.1159/000022157 [DOI] [PubMed] [Google Scholar]
- 102.Goldie AS, Fearon KC, Ross JA, Barclay GR, Jackson RE, Grant IS, et al. Natural cytokine antagonists and endogenous antiendotoxin core antibodies in sepsis syndrome. The Sepsis Intervention Group. JAMA (1995) 274:172–7. 10.1001/jama.274.2.172 [DOI] [PubMed] [Google Scholar]
- 103.De Pablo R, Monserrat J, Reyes E, Diaz-Martin D, Rodriguez Zapata M, Carballo F, et al. Mortality in patients with septic shock correlates with anti-inflammatory but not proinflammatory immunomodulatory molecules. J Intensive Care Med. (2011) 26:125–32. 10.1177/0885066610384465 [DOI] [PubMed] [Google Scholar]
- 104.Meyer NJ, Reilly JP, Anderson BJ, Palakshappa JA, Jones TK, Dunn TG, et al. Mortality benefit of recombinant human interleukin-1 receptor antagonist for sepsis varies by initial interleukin-1 receptor antagonist plasma concentration. Crit Care Med. (2018) 46:21–8. 10.1097/CCM.0000000000002749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ballinger MN, Standiford TJ. Innate immune responses in ventilator-associated pneumonia. In: Prince A, editor. Mucosal Immunology of Acute Bacterial Pneumonia. New York NY: Springer; (2013). p. 185–212. [Google Scholar]
- 106.Janssens S, Burns K, Tschopp J, Beyaert R. Regulation of interleukin-1- and lipopolysaccharide-induced NF-κB activation by alternative splicing of MyD88. Curr Biol. (2002) 12:467–71. 10.1016/S0960-9822(02)00712-1 [DOI] [PubMed] [Google Scholar]
- 107.Adib-Conquy M, Adrie C, Fitting C, Gattolliat O, Beyaert R, Cavaillon JM. Up-regulation of MyD88s and SIGIRR, molecules inhibiting Toll-like receptor signaling, in monocytes from septic patients. Crit Care Med. (2006) 34:2377–85. 10.1097/01.CCM.0000233875.93866.88 [DOI] [PubMed] [Google Scholar]
- 108.Deng JC, Cheng G, Newstead MW, Zeng X, Kobayashi K, Flavell RA, et al. Sepsis-induced suppression of lung innate immunity is mediated by IRAK-M. J Clin Invest. (2006) 116:2532–42. 10.1172/JCI28054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Escoll P, Del Fresno C, García L, Vallés G, Lendínez MJ, Arnalich F, et al. Rapid up-regulation of IRAK-M expression following a second endotoxin challenge in human monocytes and in monocytes isolated from septic patients. Biochem Biophys Res Commun. (2003) 311:465–72. 10.1016/j.bbrc.2003.10.019 [DOI] [PubMed] [Google Scholar]
- 110.Liu J, Shi K, Chen M, Xu L, Hong J, Hu B, et al. Elevated miR-155 expression induces immunosuppression via CD39+regulatory T-cells in sepsis patient. Int J Infect Dis. (2015) 40:e135–41. 10.1016/j.ijid.2015.09.016 [DOI] [PubMed] [Google Scholar]
- 111.Ifrim DC, Quintin J, Joosten LAB, Jacobs C, Jansen T, Jacobs L, et al. Trained immunity or tolerance: opposing functional programs induced in human monocytes after engagement of various pattern recognition receptors. Clin Vaccine Immunol. (2014) 21:534–45. 10.1128/CVI.00688-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dickson RP. The microbiome and critical illness. Lancet Respir Med. (2016) 4:59–72. 10.1016/S2213-2600(15)00427-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Prescott HC, Dickson RP, Rogers MAM, Langa KM, Iwashyna TJ. Hospitalization type and subsequent severe sepsis. Am J Respir Crit Care Med. (2015) 192:581–8. 10.1164/rccm.201503-0483OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Baggs J, Jernigan JA, Halpin AL, Epstein L, Hatfield KM, McDonald LC. Risk of subsequent sepsis within 90 days after a hospital stay by type of antibiotic exposure. Clin Infect Dis. (2018) 66:1004–12. 10.1093/cid/cix947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ma KC, Schenck EJ, Pabon MA, Choi AMK. The role of danger signals in the pathogenesis and perpetuation of critical illness. Am J Respir Crit Care Med. (2017) 197:300–9. 10.1164/rccm.201612-2460PP [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gao S, Yang Y, Fu Y, Guo W, Liu G. Diagnostic and prognostic value of myeloid-related protein complex 8/14 for sepsis. Am J Emerg Med. (2015) 33:1278–82. 10.1016/j.ajem.2015.06.025 [DOI] [PubMed] [Google Scholar]
- 117.Gibot S, Massin F, Cravoisy A, Barraud D, Nace L, Levy B, et al. High-mobility group box 1 protein plasma concentrations during septic shock. Intensive Care Med. (2007) 33:1347–53. 10.1007/s00134-007-0691-2 [DOI] [PubMed] [Google Scholar]
- 118.Dai J, Kumbhare A, Youssef D, McCall CE, El Gazzar M. Intracellular S100A9 promotes myeloid-derived suppressor cells during late sepsis. Front Immunol. (2017) 8:1565. 10.3389/fimmu.2017.01565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li J, Wang F, She W, Yang C, Li L, Tu C, et al. Enhanced high-mobility group box 1 (HMGB1) modulates regulatory T cells (Treg)/ T helper 17 (Th17) balance via toll-like receptor (TLR)−4-interleukin (IL)−6 pathway in patients with chronic hepatitis B. J Viral Hepat. (2014) 1:129–40. 10.1111/jvh.12152 [DOI] [PubMed] [Google Scholar]
- 120.Hashiguchi N, Ogura H, Tanaka H, Koh T, Nakamori Y, Noborio M, et al. Enhanced expression of heat shock proteins in activated polymorphonuclear leukocytes in patients with sepsis. J Trauma (2001) 51:1104–9. 10.1097/00005373-200112000-00015 [DOI] [PubMed] [Google Scholar]
- 121.Delogu G, Lo Bosco L, Marandola M, Famularo G, Lenti L, Ippoliti F, et al. Heat shock protein (HSP70) expression in septic patients. J Crit Care (1997) 12:188–92. 10.1016/S0883-9441%2897%2990031-9 [DOI] [PubMed] [Google Scholar]
- 122.Gelain DP, De Bittencourt Pasquali MAM, Comim C, Grunwald MS, Ritter C, Tomasi CD, et al. Serum heat shock protein 70 levels, oxidant status, and mortality in sepsis. Shock (2011) 35:466–70. 10.1097/SHK.0b013e31820fe704 [DOI] [PubMed] [Google Scholar]
- 123.Wachstein J, Tischer S, Figueiredo C, Limbourg A, Falk C, Immenschuh S, et al. HSP70 enhances immunosuppressive function of CD4+CD25+FoxP3+ T regulatory cells and cytotoxicity in CD4+CD25- T Cells. PLoS ONE (2012) 7:e51747. 10.1371/journal.pone.0051747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Venet F, Chung C-S, Monneret G, Huang X, Horner B, Garber M, et al. Regulatory T cell populations in sepsis and trauma. J Leukoc Biol. (2007) 83:523–35. 10.1189/jlb.0607371 [DOI] [PubMed] [Google Scholar]
- 125.Monneret G, Debard AL, Venet F, Bohe J, Hequet O, Bienvenu J, et al. Marked elevation of human circulating CD4+CD25+ regulatory T cells in sepsis-induced immunoparalysis. Crit Care Med. (2003) 31:2068–71. 10.1097/01.CCM.0000069345.78884.0F [DOI] [PubMed] [Google Scholar]
- 126.Cavassani KA, Carson WF, Moreira AP, Wen H, Schaller MA, Ishii M, et al. The post sepsis-induced expansion and enhanced function of regulatory T cells create an environment to potentiate tumor growth The post sepsis-induced expansion and enhanced function of regulatory T cells create an environment to potentiate tumor growth. Blood (2010) 115:4403–11. 10.1182/blood-2009-09-241083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Scumpia PO, Delano MJ, Kelly KM, O'Malley KA, Efron PA, McAuliffe PF, et al. Increased natural CD4+CD25+ regulatory T cells and their suppressor activity do not contribute to mortality in murine polymicrobial sepsis. J Immunol (2006) 177:7943–9. 10.4049/jimmunol.177.11.7943 [DOI] [PubMed] [Google Scholar]
- 128.Heuer JG, Zhang T, Zhao J, Ding C, Cramer M, Justen KL, et al. Adoptive Transfer of in vitro-stimulated CD4+CD25+ regulatory T cells increases bacterial clearance and improves survival in polymicrobial sepsis. J Immunol. (2005) 174:7141–6. 10.4049/jimmunol.174.11.7141 [DOI] [PubMed] [Google Scholar]
- 129.Wu H-P, Chung K, Lin C-Y, Jiang B-Y, Chuang D-Y, Liu Y-C. Associations of T helper 1:2, 17 and regulatory T lymphocytes with mortality in severe sepsis. Inflamm Res. (2013) 62:751–63. 10.1007/s00011-013-0630-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:1–10. 10.1038/ncomms12150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ost M, Singh A, Peschel A, Mehling R, Rieber N, Hartl D. Myeloid-Derived Suppressor Cells in Bacterial Infections. Front Cell Infect Microbiol. (2016) 6:37. 10.3389/fcimb.2016.00037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, et al. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med. (2007) 204:1463–74. 10.1084/jem.20062602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, et al. Inhibition of dendritic cell diff erentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. (2008) 205:2235–49. 10.1084/jem.20080132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Brudecki L, Ferguson DA, McCall CE, El Gazzar M. Myeloid-derived suppressor cells evolve during sepsis and can enhance or attenuate the systemic inflammatory response. Infect Immun. (2012) 80:2026–34. 10.1128/IAI.00239-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Derive M, Bouazza Y, Alauzet C, Gibot S. Myeloid-derived suppressor cells control microbial sepsis. Intensive Care Med. (2012) 38:1040–9. 10.1007/s00134-012-2574-4 [DOI] [PubMed] [Google Scholar]
- 136.Darcy CJ, Minigo G, Piera KA, Davis JS, McNeil YR, Chen Y, et al. Neutrophils with myeloid derived suppressor function deplete arginine and constrain T cell function in septic shock patients. Crit Care (2014) 18:1–12. 10.1186/cc14003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Janols H, Bergenfelz C, Allaoui R, Larsson AM, Rydén L, Björnsson S, et al. High frequency of myeloid-derived suppressor cells in sepsis patients, with the granulocytic subtype dominating in Gram-positive cases. Crit Care (2014) 96:1–53. 10.1186/cc14006 [DOI] [PubMed] [Google Scholar]
- 138.Uhel F, Azzaoui I, Grégoire M, Pangault C, Dulong J, Tadié JM, et al. Early expansion of circulating granulocytic myeloid-derived suppressor cells predicts development of nosocomial infections in patients with sepsis. Am. J. Respir. Crit. Care Med. (2017) 196:315–27. 10.1164/rccm.201606-1143OC [DOI] [PubMed] [Google Scholar]
- 139.Mathias B, Delmas AL, Ozrazgat-Baslanti T, Vanzant EL, Szpila BE, Mohr AM, et al. Human myeloid-derived suppressor cells are associated with chronic immune suppression after severe sepsis/septic shock. Ann Surg. (2017) 265:827–34. 10.1097/SLA.0000000000001783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. (2004) 4:336–47. 10.1038/nri1349 [DOI] [PubMed] [Google Scholar]
- 141.Zhang Y, Li J, Lou J, Zhou Y, Bo L, Zhu J, et al. Upregulation of programmed death-1 on T cells and programmed death ligand-1 on monocytes in septic shock patients. Crit Care (2011) 15:1–9. 10.1186/cc10059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Guignant C, Lepape A, Huang X, Kherouf H, Denis L, Poitevin F, et al. Programmed death-1 levels correlate with increased mortality, nosocomial infection and immune dysfunctions in septic shock patients. Crit Care (2011) 15:R99. 10.1186/cc10112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shao R, Fang Y, Yu H, Zhao L, Jiang Z, Li CS. Monocyte programmed death ligand-1 expression after 3-4 days of sepsis is associated with risk stratification and mortality in septic patients: a prospective cohort study. Crit Care (2016) 20:1–10. 10.1186/s13054-016-1301-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Brahmamdam P, Inoue S, Unsinger J, Chang KC, McDunn JE, Hotchkiss RS. Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J Leukoc Biol. (2010) 88:233–40. 10.1189/jlb.0110037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhang Y, Zhou Y, Lou J, Li J, Bo L, Zhu K, et al. PD-L1 blockade improves survival in experimental sepsis by inhibiting lymphocyte apoptosis and reversing monocyte dysfunction. Crit Care (2010) 14:1–9. 10.1186/cc9354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chang K, Svabek C, Vazquez-guillamet C, Sato B, Rasche D, Wilson S, et al. Targeting the programmed cell death 1 : programmed cell death ligand 1 pathway reverses T cell exhaustion in patients with sepsis. Crit Care (2014) 18:R3. 10.1186/cc13176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Patera AC, Drewry AM, Chang K, Beiter ER, Osborne D, Hotchkiss RS. Frontline science: defects in immune function in patients with sepsis are associated with PD-1 or PD-L1 expression and can be restored by antibodies targeting PD-1 or PD-L1. J Leukoc Biol. (2016) 100:1239–54. 10.1189/jlb.4HI0616-255R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Carson WF, Cavassani KA, Dou Y, Kunkel SL. Epigenetic regulation of immune cell functions during post-septic immunosuppression. Epigenetics (2011) 6:273–83. 10.4161/epi.6.3.14017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hamon MA, Cossart P. Histone Modifications and Chromatin Remodeling during Bacterial Infections. Cell Host Microbe (2008) 4:100–9. 10.1016/j.chom.2008.07.009 [DOI] [PubMed] [Google Scholar]
- 150.Weiterer S, Uhle F, Lichtenstern C, Siegler BH, Bhuju S, Jarek M, et al. Sepsis induces specific changes in histone modification patterns in human monocytes. PLoS ONE (2015) 10:e121748. 10.1371/journal.pone.0121748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wen H, Dou Y, Hogaboam CM, Kunkel SL. Epigenetic regulation of dendritic cell – derived interleukin-12 facilitates immunosuppression after a severe innate immune response. Blood (2008) 111:1797–804. 10.1182/blood-2007-08-106443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lyn-Kew K, Rich E, Zeng X, Wen H, Kunkel SL, Newstead MW, et al. IRAK-M regulates chromatin remodeling in lung macrophages during experimental sepsis. PLoS ONE (2010) 5:e11145. 10.1371/journal.pone.0011145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kleinnijenhuis J, Quintin J, Preijers F, Joosten LAB, Ifrim DC, Saeed S, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci USA. (2012) 109:17537–42. 10.1073/pnas.1202870109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe (2012) 12:223–32. 10.1016/j.chom.2012.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Thampy LK, Remy KE, Walton AH, Hong Z, Liu K, Liu R, et al. Restoration of T Cell function in multi-drug resistant bacterial sepsis after interleukin-7, anti-PD-L1, and OX-40 administration. PLoS ONE (2018) 13:e0199497. 10.1371/journal.pone.0199497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer (2018) 118:9–16. 10.1038/bjc.2017.434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bo L, Wang F, Zhu J, Li J, Deng X. Granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) for sepsis: A meta-analysis. Crit Care (2011) 15:1–12. 10.1186/cc10031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Scott JP, Ji Y, Kannan M, Wylam ME. Inhaled granulocyte-macrophage colony-stimulating factor for Mycobacterium abscessus in cystic fibrosis. Eur Respir J. (2018) 51:1702127. 10.1183/13993003.02127-2017 [DOI] [PubMed] [Google Scholar]
- 159.Nalos M, Santner-Nanan B, Parnell G, Tang B, McLean AS, Nanan R. Immune effects of interferon gamma in persistent staphylococcal sepsis. Am J Respir Crit Care Med. (2012) 185:110–2. 10.1164/ajrccm.185.1.110 [DOI] [PubMed] [Google Scholar]
- 160.Delsing CE, Gresnigt MS, Leentjens J, Preijers F, Frager FA, Kox M, et al. (2014). Interferon-gamma as adjunctive immunotherapy for invasive fungal infections : a case series. BMC Infect Dis. 14:166. 10.1186/1471-2334-14-166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Francois B, Jeannet R, Daix T, Walton AH, Shotwell MS, Unsinger J, et al. Interleukin-7 restores lymphocytes in septic shock: the IRIS-7 randomized clinical trial. JCI insight (2018) 3:98960. 10.1172/jci.insight.98960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hotchkiss R, Colston E, Yende S, Angus D, Moldawer L, Crouser E, et al. 1504: Immune checkpoint inhibitors in sepsis A phase 1B Trial Of Anti-PD-L1 (BMS-936559). Crit. Care Med. (2018) 46:736 10.1097/01.ccm.0000529506.43290.de [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wu J, Zhou L, Liu J, Ma G, Kou Q, He Z, et al. The efficacy of thymosin alpha 1 for severe sepsis (ETASS): a multicenter, single-blind, randomized and controlled trial. Crit Care. (2013) 17:R8. 10.1186/cc11932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.McIntyre LA, Stewart DJ, Mei SHJ, Courtman D, Watpool I, Granton J, et al. Cellular immunotherapy for septic shock. A phase I clinical trial. Am J Respir Crit Care Med. (2018) 197:337–47. 10.1164/rccm.201705-1006OC [DOI] [PubMed] [Google Scholar]
- 165.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (2018) 359:1350–5. 10.1126/science.aar4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Osta B, El HF, Sadek R, Chintalapally R, Tang S. Not all immune-checkpoint inhibitors are created equal : Meta-analysis and systematic review of immune-related adverse events in cancer trials. Crit Rev Oncol Hematol. (2017) 119:1–12. 10.1016/j.critrevonc.2017.09.002 [DOI] [PubMed] [Google Scholar]
- 167.Mei SHJ, Haitsma JJ, Dos Santos CC, Deng Y, Lai PFH, Slutsky AS, et al. Mesenchymal stem cells reduce inflammation while enhancing bacterial clearance and improving survival in sepsis. Am J Respir Crit Care Med. (2010) 182:1047–57. 10.1164/rccm.201001-0010OC [DOI] [PubMed] [Google Scholar]