

Abstract
Protein phosphatase 2A (PP2A) is a heterotrimer composed of single catalytic and scaffolding subunits and one of several possible regulatory subunits. We identified PPTR-2, a regulatory subunit of PP2A, as a binding partner for the giant muscle protein UNC-89 (obscurin) in Caenorhabditis elegans. PPTR-2 is required for sarcomere organization when its paralogue, PPTR-1, is deficient. PPTR-2 localizes to the sarcomere at dense bodies and M-lines, colocalizing with UNC-89 at M-lines. PP2A components in C. elegans include one catalytic subunit LET-92, one scaffolding subunit (PAA-1), and five regulatory subunits (SUR-6, PPTR-1, PPTR-2, RSA-1, and CASH-1). In adult muscle, loss of function in any of these subunits results in sarcomere disorganization. rsa-1 mutants show an interesting phenotype: one of the two myosin heavy chains, MHC A, localizes as closely spaced double lines rather than single lines. This “double line” phenotype is found in rare missense mutants of the head domain of MHC B myosin, such as unc-54(s74). Analysis of phosphoproteins in the unc-54(s74) mutant revealed two additional phosphoserines in the nonhelical tailpiece of MHC A. Antibodies localize PPTR-1, PAA-1, and SUR-6 to I-bands and RSA-1 to M-lines and I-bands. Therefore, PP2A localizes to sarcomeres and functions in the assembly or maintenance of sarcomeres.
INTRODUCTION
More than half of human proteins are subjected to reversible phosphorylation (Hornbeck et al., 2012). Most of this phosphorylation occurs on serine or threonine, but some occurs on tyrosines. The human genome encodes at least 500 different protein kinases with variably specific substrates (Kostich et al., 2002). In contrast, relatively few genes encode the catalytic subunits of serine/threonine phosphatases (Virshup and Shenolikar, 2009). Protein kinases and phosphatases both are classified based on their specificity for different amino acids, for example, serine–threonine, tyrosine, or both. Whereas the myriad of protein kinases achieve a fair amount of specificity for their targets via recognition of consensus sequences, some of the limited number of phosphatases achieve specificity through the combinatorial formation of multisubunit holoenzymes containing one or two regulatory subunits in addition to a catalytic subunit. A major serine–threonine phosphatase with conservation from yeast to humans is protein phosphatase 2A (PP2A; Shi, 2009). PP2A functions in a wide variety of cellular functions and primarily exists as a heterotrimer consisting of a catalytic subunit (C), a scaffolding subunit (A), and one of many regulatory/targeting (B) subunits. Humans have two alternative C subunits (Cα, Cβ), two alternative A subunits (Aα, Aβ), and four families of B subunits: B (B55 or PPP2R2), B’ (B56 or PPP2R5), B” (B72 or PPP2R3), and B’’’ (striatin family).
The fundamental functional unit of striated muscle is the sarcomere, which consists of a highly ordered assembly of hundreds of different proteins. At least in mammalian cardiac muscle, the activities of several sarcomeric proteins are regulated by phosphorylation. These include troponin I of the thin filament (Solaro and Kobayashi, 2011), myosin binding protein C (MyBP-C) of A-bands (Barefield and Sadayappan, 2010), and “regulatory” myosin light chains (Aoki et al. 2000; Du et al., 2003). In addition, several proteins key to calcium regulation of cardiac muscle activity are also regulated by phosphorylation, including the calcium release channel of the sarcoplasmic reticulum (RyR2; Terentyev and Hamilton, 2016). For these proteins, the major protein kinases and protein phosphatases have been identified. For example, in the case of the cardiac isoform of MyBP-C, Ca2+-dependent calmodulin kinase II, protein kinase A and protein kinase C (Barefield and Sadayappan, 2010), and PP1 and PP2A (Weber et al., 2015) are involved.
The nematode Caenorhabditis elegans has been and continues to be an excellent platform for discovery of new aspects of the assembly, maintenance and function of striated muscle (Gieseler et al., 2017). In general, the phosphorylation status of nematode muscle proteins has not been studied. However, it is known that a small N-terminal nonhelical segment of paramyosin, an invertebrate-specific thick filament protein, is phosphorylated on serine by an unidentified thick filament-associated protein kinase (Schriefer and Waterston, 1989; Dey et al., 1992). The N-terminus of paramyosin contains several copies of the motif S-S-A, which is also found in multiple copies at the C-terminal nonhelical tailpieces of the myosin heavy chains MHC A and MHC B (Schriefer and Waterston, 1989), leading to the possibility that the nematode myosin heavy chains are also phosphorylated. A candidate for the protein kinase involved is UNC-82, a 1600-residue polypeptide containing a protein kinase domain homologous to human ARK5 and SNARK protein kinases (Hoppe et al., 2010a).
As we were completing a yeast two-hybrid screen for binding partners for the giant sarcomeric polypeptide UNC-89 in C. elegans (homologous to obscurin in mammals; reviewed in Manring et al., 2017), we discovered an unexpected linkage to PP2A. We found that a C-terminal portion of UNC-89 interacts with PPTR-2, a B’ regulatory subunit of PP2A, and that PPTR-2 is a component of the sarcomere, partly localizing with UNC-89 at the M-line. This prompted us to investigate the function of the other PP2A subunits in C. elegans muscle. Loss of function of any one component of the PP2A holoenzyme results in a muscle phenotype, and most of these PP2A components localize to various regions of the muscle sarcomere.
RESULTS
A yeast two-hybrid screen reveals that PPTR-1 and PPTR-2, B’ subunits of PP2A, are binding partners for UNC-89 (obscurin)
To understand how UNC-89 (obscurin) assembles into the sarcomere and performs its functions, we have been identifying its binding partners by systematically using portions of the largest isoform of UNC-89, UNC-89B, to screen a yeast two-hybrid library (Qadota et al., 2008a,b, 2016; Xiong et al., 2009; Wilson et al., 2012a; Warner et al., 2013). We have now completed the process using the remaining 13 segments of UNC-89B and potentially identified 15 new interacting proteins (Supplemental Figure S1; Supplemental Tables S1 and S2). [Although 16 proteins are listed, one of them, LIM-9 was reported as an interactor previously (Xiong et al., 2009)]. Because the library consists of cDNAs representing mRNAs from whole nematodes, we sought evidence for body-wall muscle expression for these genes by querying SAGE data (Meissner et al., 2009) and WormBase for data on the expression pattern of a gene’s putative promoter region. (SAGE data are found in “Dataset S1” at the following site: https://doi.org/10.1371/journal.pgen.1000537.s002.) For 14 out of 15 genes, data were available, and of these 13, nine are expressed in body-wall muscle (Supplemental Table S2). Two of these muscle-expressed genes encode homologous proteins, that is, PPTR-1 and PPTR-2. As shown in Supplemental Figure S2, these two proteins, PPTR-1 (543 residues) and PPTR-2 (561 residues), are 55% identical in amino acid sequence. Moreover, each is homologous to the B’ subunits (B56) of human protein phosphatase 2A. There are five isoforms of human B56 (alpha through epsilon). Nematode PPTR-1 shows greatest identity to human B56alpha (78%), and nematode PPTR-2 shows greatest identity to human B56gamma (74%; Supplemental Figure S2). Four prey clones were isolated for PPTR-1, and two prey clones were isolated for PPTR-2. For PPTR-1, all the clones span residues 75-543; for PPTR-2, one prey clone spans residues 10-558, and one is full-length (Supplemental Figure S3).
PPTR-1 and PPTR-2 were isolated with a bait including domains just N-terminal of the kinase 2 domain of UNC-89, specifically, the C-terminal third of the interkinase region-Ig53-Fn2 (1/3 IK-Ig53-Fn2). To determine how specific this region of UNC-89 is for binding to PPTR-1 and PPTR-2, we assayed the binding of the additional 16 segments of UNC-89 to PPTR-1 and PPTR-2. As indicated in Figure 1A, 1/3 IK-Ig53-Fn2 is the only segment that interacts with either PPTR-1 or PPTR-2. By using deletion derivatives of 1/3 IK-Ig53-Fn2 in two-hybrid assays, we found that the minimal interacting fragment for PPTR-2 is Ig53-Fn2, but we were not able to narrow the interacting fragment for PPTR-1 further (Figure 1B).
FIGURE 1:

Mapping of interaction sites of PPTR-1 and PPTR-2 for UNC-89, and of UNC-89 for PPTR-1 and PPTR-2. (A) When tested by yeast two-hybrid assays with segments that cover all of UNC-89B, PPTR-1 and PPTR-2 only interact with UNC-89 1/3 IK-Ig53-Fn2. Pink rectangles, Ig domains; green rectangles, Fn3 domains; other domains are as indicated. (B) Domain mapping using yeast two-hybrid assays indicates that the minimal region of UNC-89 that interacts with PPTR-1 is 1/3 IK-Ig53-Fn2, but that the minimal region of UNC-89 that interacts with PPTR-2 is Ig53-Fn2.
PPTR-1 and PPTR-2 interact with a portion of UNC-89 as recombinant proteins in vitro
To obtain additional evidence for the interactions discovered by the yeast two-hybrid screen, we assayed for direct interactions using purified recombinant proteins (Supplemental Figure S4), using two methods. First, we employed a far-Western assay (Figure 2A). Neither His-tagged PPTR-1 nor His-tagged PPTR-2 on the membrane interact with maltose binding protein (MBP). However, both His-PPTR-1 and His-PPTR-2 interact with MBP-UNC-89 1/3 IK-Ig53-Fn2. In addition, His-PPTR-1 and His-PPTR-2 also interact with MBP-UNC-89-Ig53-Fn2, although His-PPTR1, does so with less apparent affinity. However, in a far-Western assay, since much of the protein on a membrane is denatured, and this could lead to nonphysiological interactions, we performed an in-solution binding experiment (Figure 2B). His-PPTR-1 or His-PPTR-2 was mixed in solution with either MBP or MBP-UNC-89-Ig53-Fn2, immunoprecipitated with anti-His beads, washed, separated on a gel, blotted, and incubated with anti-MBP. As shown in Figure 2B, this pull-down experiment indicates that UNC-89-Ig53-Fn2 can interact in-solution with PPTR-1 and PPTR-2, but with more apparent affinity with PPTR-2.
FIGURE 2:
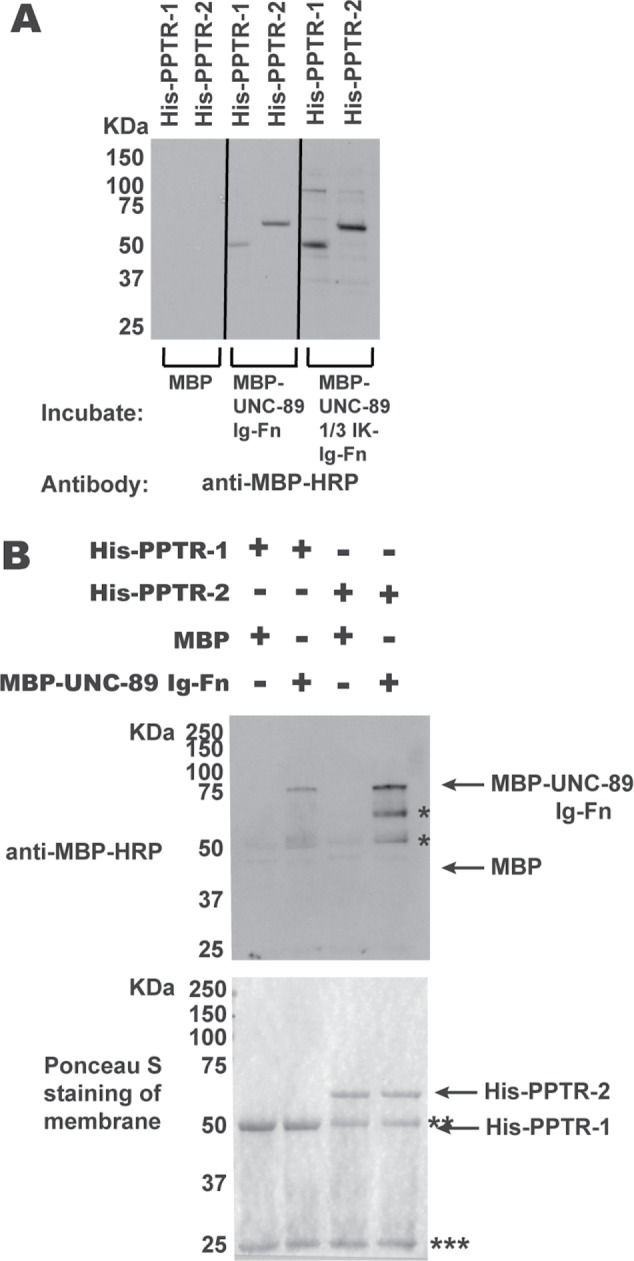
Segments of UNC-89 interact with PPTR-1 and PPTR-2 in vitro. (A) Far-Western assay. His-tagged PPTR-1 and PPTR-2 were separated by SDS–PAGE, transferred to membrane, incubated with MBP, MBP-UNC-89 Ig53-Fn2, or MBP-UNC-89 1/3 IK-Ig53-Fn2, washed, incubated with anti–MBP-HRP, washed, and detected by ECL. Both MBP fusions of these portions of UNC-89, but not MBP itself, bind to either His-PPTR-1 or His-PPTR-2. (B) In-solution pull-down assay. The indicated proteins were incubated together, and then the His-tagged proteins were pelleted using anti-6His beads. Proteins were eluted from the beads and separated by SDS–PAGE and transferred to a membrane, and then a Western blot was performed using anti–MBP-HRP to detect any copelleting MBP or MBP-UNC-89-Ig-Fn. At the bottom is shown the blot after Ponceau S staining, and above it is the Western blot. Both His-PPTR-1 and His-PPTR-2 pull down MBP-UNC-89-Ig-Fn, but not MBP. Positions of proteins are indicated by arrows; * likely degradation products from MBP-UNC-89-Ig-Fn; ** Ig heavy chain; *** Ig light chain. The predicted molecular weights of the bacterially expressed purified proteins are as follows: His-PPTR-1, 51.5 kDa; His-PPTR-2, 60 kDa; MBP, 42.5 kDa; MBP-UNC-89 Ig53-Fn2, 73.5 kDa; and MBP-UNC-89 1/3 IK-Ig53-Fn2, 97.3 kDa.
PPTR-1 and PPTR-2 are required for proper sarcomere organization
For each gene, pptr-1 and pptr-2, mRNAs are expressed in muscle (Meissner et al. 2009), and putative promoter sequences drive GFP expression in muscle (WormBase). Therefore, we wondered whether loss of function of either gene would result in a defect in sarcomere organization. To examine this question, we determined the organization of thick filaments by immunostaining with antibodies to one of the myosin heavy chain isoforms, MHC A (Miller et al. 1983). As indicated in Figure 3A, body-wall muscle from wild-type animals displays straight and parallel A-bands in each spindle-shaped cell. However, the pptr-1(tm3103) mutant, which has a 209–base pair deletion that removes 153 base pairs upstream of the initiator ATG and 56 base pairs of the first intron, displays disorganized A-bands (two representative examples are shown). We also examined this mutant by immunofluorescence microscopy using markers for actin thin filaments (phalloidin), dense bodies (antibodies to ATN-1 [α-actinin]), additional components of thick filaments (MHC B and UNC-15 [paramyosin]), M-lines (UNC-89), and the bases of M-lines and dense bodies (UNC-95). With all of these markers, perhaps except for MHC B, these components of the sarcomere showed disorganized localization in pptr-1(tm3103), in contrast to wild type (Supplemental Figure S5). However, pptr-1(abc19), which has an amino acid substitution, C95Y (residing in the B56 domain of PPTR-1), shows normal organization of MHC A (Figure 3B, first row). As shown in Supplemental Table S2, RNA interference (RNAi) of pptr-1 shows primarily embryonic lethality, but adult escapers show MHC A disorganization.
FIGURE 3:
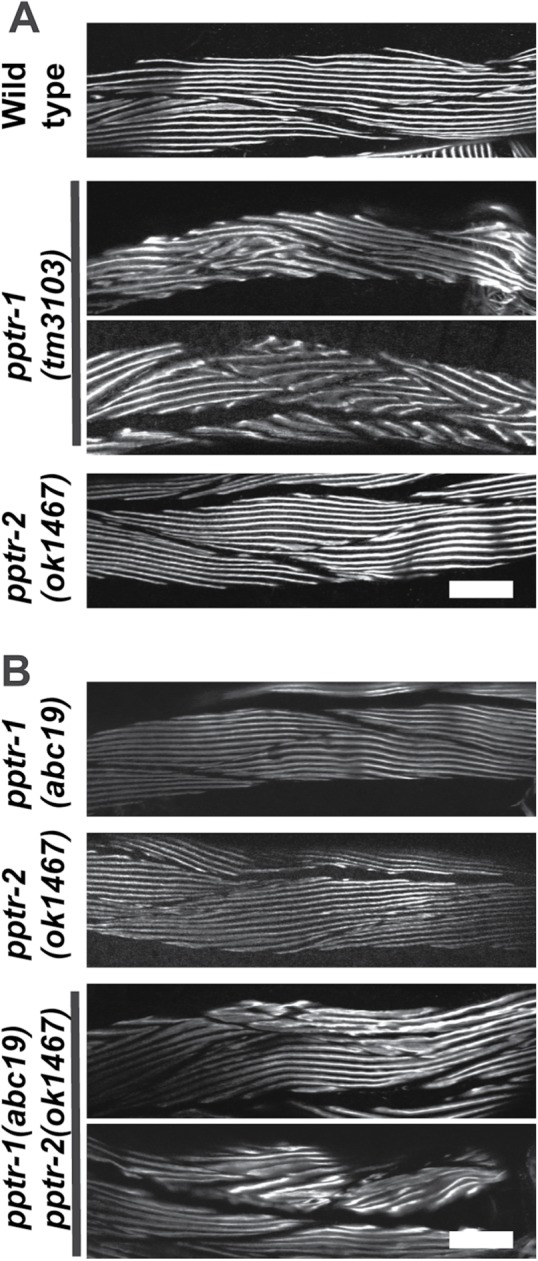
Effects of loss of function mutations in pptr-1 and pptr-2 on thick filament organization in body-wall muscle. (A) Immunostaining using a monoclonal to the thick filament component MHC A of wild type, pptr-1(tm3103), or pptr-2(ok1467) grown at 20°C. In each panel, portions of several spindle-shaped body-wall muscle cells are shown. (Two representative examples of images from pptr-1(tm3103) are shown.) In wild-type muscle, the A-bands are straight and parallel. Although the deletion allele pptr-2(ok1467) shows normal thick-filament organization, the deletion allele pptr-1(tm3103) shows disorganization of thick filaments (two representative examples are shown). (B) Immunostaining using anti-MHC A of missense allele pptr-1(abc19), deletion allele pptr-2(ok1467), and the double mutant, pptr-1(abc19) pptr-2(ok1467), grown at 15°C. Two representative examples of images from pptr-1(abc19) pptr-2(ok1467) animals are shown. Note that both single mutants have normal A-band organization, but the double mutant shows some A-band disorganization (two examples shown). The double mutant pptr-1(abc19) pptr-2(ok1467) is 90% sterile at 20°C, but only 75% sterile at 15°C. Therefore, it was easier for us to grow the double for immunostaining at 15°C and for comparison to the single mutants, which were also grown at 15°C. Scale bars, 20 μm.
We also examined sarcomeric organization in the loss-of-function mutant pptr-2(ok1467), which is a 593–base pair deletion within the 1062–base pair fourth exon of pptr-2b, resulting in a premature stop codon. As shown in Figure 3, A and B, the organization of MHC A is normal in pptr-2(ok1467). pptr-1(tm3103) pptr-2(RNAi) animals are embryonic lethal and preclude examining muscle structure in adults. However, when mutant alleles for each gene that have no muscle phenotype on their own are combined in the same animal, pptr-1(abc19) pptr-2(ok1467), many animals reach adulthood. Examination of these adults reveals a mild and variable disorganization of A-bands (Figure 3B; bottom two rows show two examples). This demonstrates that reducing the function of pptr-1 uncovers a function for pptr-2 in muscle.
PPTR-1 localizes to I-bands, and PPTR-2 localizes to M-lines and dense bodies
Given that PPTR-1 and PPTR-2 are required for proper sarcomere organization, we wondered if these proteins are localized in the sarcomere. Although PPTR-1 and PPTR-2 are highly homologous, their C-terminal tails are unique. Thus, we raised rabbit antibodies to GST fusions of the 80 C-terminal residues of PPTR-1 and the 116 C-terminal residues of PPTR-2b (Supplemental Figure S2; Figure 4A). After affinity purification, these antibodies were tested by Western blot. As shown in Figure 4B, each antibody reacts with a polypeptide of expected size, and such a protein is absent from the respective deletion mutant. These antibodies were then used with immunofluorescence microscopy to determine the location of these proteins in muscle. As shown in Figure 4C, anti–PPTR-1 localizes to I-bands, and anti–PPTR-2 localizes to M-lines and dense bodies. Because UNC-89 (Benian et al., 1996; Small et al., 2004) and PPTR-2 are localized at M-lines (Figure 4C), it is most likely that PPTR-2 interacts with UNC-89 in vivo. This result also suggests that during the two-hybrid screen using an UNC-89 bait, PPTR-1 was recovered because of its high sequence homology to PPTR-2.
FIGURE 4:

Immunolocalization of PPTR-1 and PPTR-2 to sarcomeres of body-wall muscle. (A) Schematic representation of PPTR-1 and PPTR-2 polypeptides, each having a single domain, the B56 domain (shown in red). Blue bars indicate the unique C-termini used as immunogens to generate rabbit antibodies. (B) Western blots demonstrate that antibodies to PPTR-1 or PPTR-2 detect expected-size proteins from the wild type but not from the respective deletion alleles. The numbers on the left side of each blot show the position of molecular weight markers in kDa. (C) By immunofluorescence microscopy, anti–PPTR-1 localizes to sarcomeric I-bands, and anti–PPTR-2 localizes to sarcomeric dense bodies and M-lines. Portions of a single body-wall muscle cell are shown in each row, costained with antibodies to UNC-89, which localizes to M-lines. Arrowheads mark the positions of dense bodies, and arrows mark the positions of M-lines. In the two-color image at bottom right, white indicates colocalization of PPTR-2 with UNC-89 at M-lines. Scale bar, 10 μm.
UNC-89 is a major determinant for localization of PPTR-2 at M-lines
If PPTR-2 and UNC-89 interact at the M-line, and if UNC-89 acts as a scaffold for PPTR-2 assembly, we expect that in a unc-89 mutant lacking the PPTR-2-interacting region (i.e., Ig53-Fn2), PPTR-2 would be missing or show reduced levels. unc-89(tm752) is a 531–base pair deletion expected to virtually eliminate expression of all protein kinase domain-containing isoforms. Indeed, by Western blot using an antibody to the interkinase region, the kinase-containing UNC-89 isoforms are not detectable in tm752 (Ferrara et al., 2005). A quantitative Western blot using whole worm extracts showed no difference in PPTR-2 protein levels in wild type versus unc-89(tm752) (unpublished data). This is probably explained by the fact that the pptr-2 promoter is widely expressed in neurons (ventral cord, pharyngeal, nerve ring), anal depressor muscle, and unidentified cells in head and tail, in addition to body-wall muscle (WormBase). However, immunostaining with anti–PPTR-2 of tm752 reveals reduced levels of PPTR-2 at the M-line as compared with the wild type, but approximately normal levels of PPTR-2 at dense bodies (Figure 5A). This observation is supported by an unbiased intensity profile of the fluorescence of the images (Figure 5B). This result is consistent with an interaction of UNC-89 with PPTR-2 at the M-lines in body-wall muscle and indicates that UNC-89 is a major determinant for the localization of PPTR-2 at M-lines.
FIGURE 5:
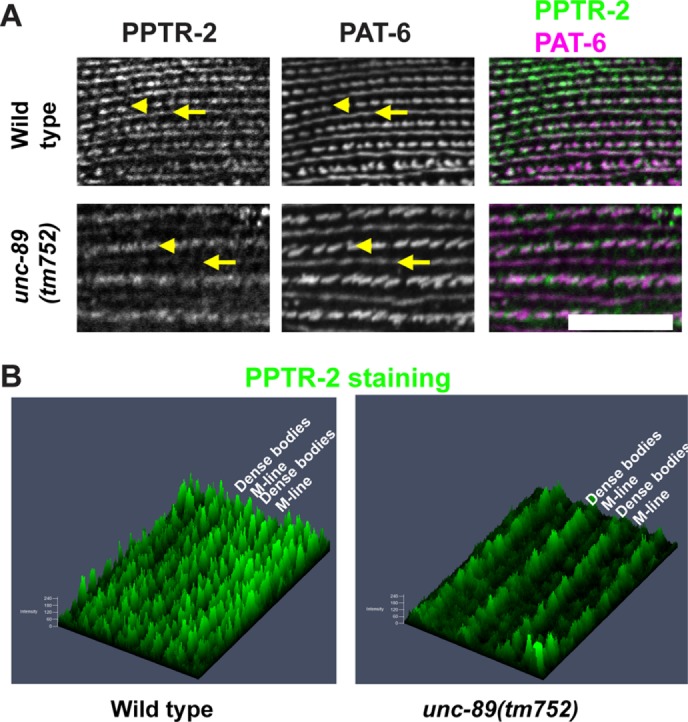
In an unc-89 mutant expressing UNC-89 protein that lacks the PPTR-2 binding region, there are reduced levels of PPTR-2 at M-lines. (A) Each row shows a portion of a single body-wall muscle cell immunostained with anti–PPTR-2 and anti–PAT-6 (α-parvin) of wild type and unc-89(tm752). PAT-6 is a marker for dense bodies (arrowheads) and M-lines (arrows). (B) Intensity profiles, created by Zeiss ZEN software, of the PPTR-2 images shown in A. The images were rotated to improve viewability. Note that in unc-89(tm752) as compared with wild type, the level of PPTR-2 at M-lines is reduced, but the level of PPTR-2 at dense bodies is unchanged. Scale bar, 10 μm.
Six additional PP2A subunits are each required for sarcomere organization
Based on current literature, C. elegans expresses PP2A catalytic (LET-92) and scaffolding (PAA-1) subunits, as well as at least one member of each of the mammalian regulatory subunits (B, SUR-6; B’, PPTR-1 and PPTR-2; B”, RSA-1; B’’’, CASH-1; Figure 6; Sieburth et al., 1999; Kao et al., 2004; Schlaitz et al., 2007; Padmanabhan et al., 2009; Ogura et al., 2010; Wang et al., 2014; Pal et al., 2017). So far, these genes/proteins have been reported to be important for various aspects of C. elegans biology (vulval development, axon guidance, germline development, early embryonic cell division, and insulin signaling), but not for muscle. As indicated in Table 1, there are SAGE and promoter data supporting body-wall muscle expression of all seven of these PP2A subunits. We next sought genetic evidence for the muscle function of these proteins. As shown in Table 1, we could find a muscle structural defect in loss of function mutants or by RNAi for each of these genes. We have already noted that mutants in the B’ subunits, PPTR-1 and PPTR-2, have sarcomere defects. For the B subunit protein SUR-6, the loss-of-function temperature-sensitive mutant sur-6(or550) is embryonic lethal at the restrictive temperature, but when grown at the permissive temperature until the L1 larval stage and then shifted to the restrictive temperature, it reaches adulthood and its sarcomeres are highly disorganized (Figure 7, second row). For the B” subunit protein RSA-1, there are maternal-effect lethal alleles (rsa-1(dd10) and rsa-1(dd13)), but when adult muscle is examined, it shows an unusual muscle phenotype that we call “split A-bands” (Figure 7, third and fourth rows, Figure 8, and explained below). For the B’’’ subunit CASH-1, no mutant alleles are available, but it has been reported that RNAi results in sterility (Pal et al., 2017). We performed our own RNAi experiments, and as shown in Supplemental Table S3, we confirmed sterility, but also found that in addition to defects in the vulva, we observed adults that are slow-moving (“uncoordinated” or Unc). Immunostaining of cash-1(RNAi) animals revealed that they have disorganized sarcomeres (Figure 7, lowest row).
FIGURE 6:
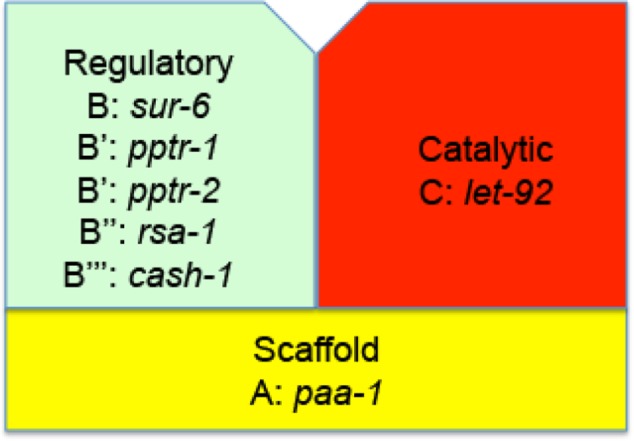
PP2A holoenzyme in C. elegans. There is a single catalytic subunit, C, encoded by the gene let-92, a single scaffolding subunit, A, encoded by paa-1, and at least the following regulatory subunits and their encoded genes: B by sur-6, B’ by pptr-1 or B’ by pptr-2, B” by rsa-1, and B’’’ by cash-1.
TABLE 1:
PP2A components in muscle
| Muscle expression | |||||
|---|---|---|---|---|---|
| Gene | Encoding | Promoter | SAGE | Muscle localization | Muscle phenotype |
| let-92 | Catalytic subunit (C) | Yes | Yes | ND | Detach muscle, jack-knife (L1 feeding RNAi) |
| paa-1 | Scaffolding subunit (A) | Yes | Yes | I-band | Detach muscle, jack-knife (L1 feeding RNAi) |
| sur-6 | Regulatory subunit (B) | No | Yes | I-band | Disorganized myofilaments (ts mutant) |
| pptr-1 | Regulatory subunit (B’)/B56 | Yes | No | I-band | Disorganized myofilaments (deletion mutant) |
| pptr-2 | Regulatory subunit (B’)/B56 | Yes | Yes | M-lines and dense bodies | Normal muscle (deletion mutant) |
| rsa-1 | Regulatory subunit (B’’) | ND | Yes | M-lines and I-bands | Split MHCA (maternal effect lethal mutants) |
| cash-1 | Regulatory subunit (B’’’)/striatin | No | Yes | ND | Disorganized myofilaments (RNAi) |
FIGURE 7:
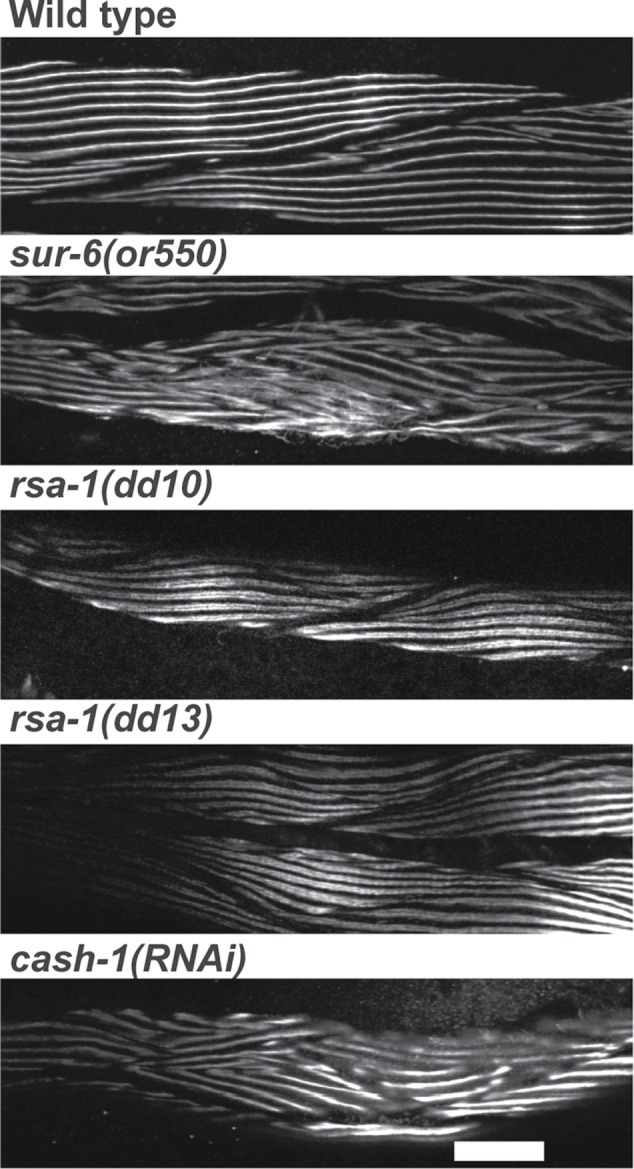
Disorganization of sarcomeres in loss-of-function mutants for PP2A B (SUR-6), B” (RSA-1), and B”’(CASH-1) subunits. Immunofluorescence images of portions of several body-wall muscle cells reacted with antibodies to MHC A from wild type, the temperature-sensitive mutant sur-6(or550), the maternal-effect lethal mutants rsa-1(dd10) and rsa-1(dd13), and cash-1(RNAi). There is clear disorganization of A-bands in sur-6(or550) and cash-1(RNAi). In the case of the rsa-1 mutants, although there are normal straight parallel A-bands, these A-bands appear as doublets, seen at higher resolution in Figures 9 and 10. Scale bar, 20 μm.
FIGURE 8:
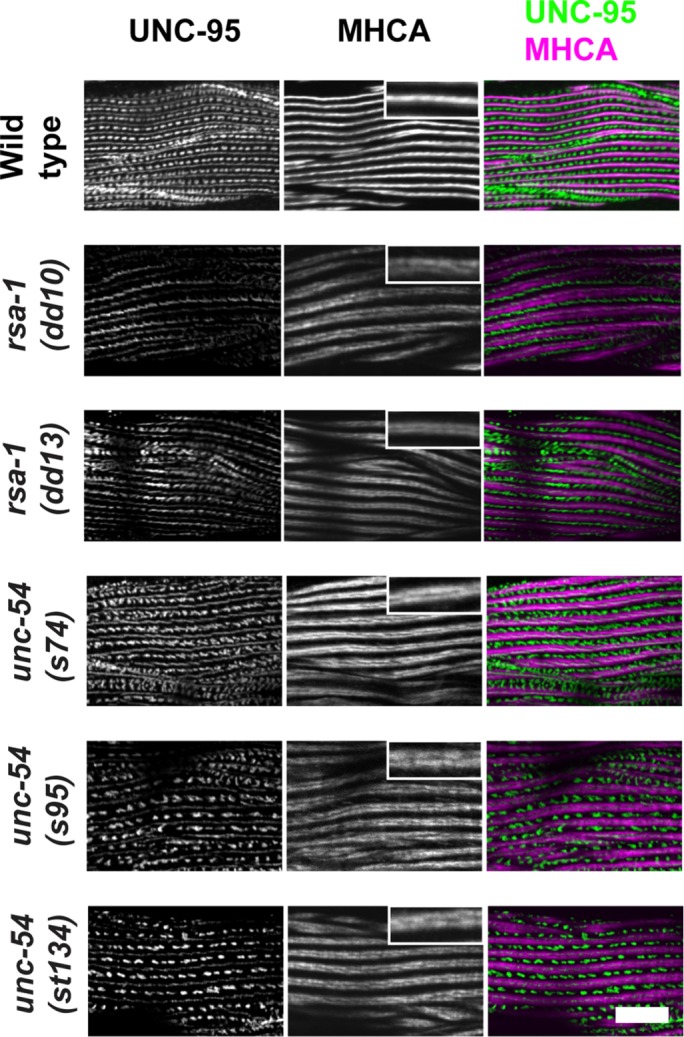
Split or doublet MHC A localization is also found in MHC B myosin head mutants. Immunolocalization of UNC-95 (marker for M-lines and dense bodies) and MHC A in muscle of wild type, rsa-1, and special unc-54 mutants. Insets show higher-magnification views. Whereas wild type shows single tight MHC A localization, both rsa-1 mutants and unc-54 mutants show closely spaced doublet MHC A localization. unc-54(s74), unc-54(s95), and unc-54(st134) are rare missense mutations in the head domain of MHC B. Scale bar, 10 μm.
Rare missense mutations in the MHC B head have the same characteristic effect on MHC A distribution as mutation of RSA-1, a B’ PP2A regulatory subunit
The phenotype of the rsa-1 mutants is particularly interesting. Although the localization of the M-line and dense body protein UNC-95 is normal or near normal, and overall there is normal myosin thick filament organization (parallel A-bands), MHC A localizes as a closely spaced double line rather than a single line (Figure 8, top three rows). Nematode body-wall muscle expresses two myosin heavy chain genes, unc-54 (encoding MHC B) and myo-3 (encoding MHC A); immunostaining has shown that MHC A is restricted to the central region of the A-band, whereas MHC B is present only on the outer portions of the A-band (Miller et al., 1983). We have observed this “double-line” MHC A staining in one other set of mutants: rare missense mutants (unc-54(s74), unc-54(s95), unc-54(st134)) of the head domain of MHC B myosin (Figure 8, bottom three rows). These mutants show normal or nearly normal sarcomere organization by polarized light or EM, but are slow-moving and stiff (Moerman et al. 1982; Moerman and Fire, 1997). In fact, these mutations all reside near the ATP-binding site of the myosin head in residues conserved between nematode and vertebrate myosin (s74, R273C; s95, G118R; st134, S117F; Moerman and Fire, 1997). We also examined this MHC A double-line phenotype at higher resolution. To do this, we used structured illumination microscopy (SIM), a type of superresolution microscopy, which affords ∼120 nm resolution in the xy plane. As shown in Figure 9, both rsa-1(dd10) and unc-54(s95) images reveal very clear double-line localization of MHC A, which extends throughout the depth of the A-band. The double-line localization of MHC A in rsa-1 and unc-54 mutants appears somewhat like the localization of MHC B in the wild type. However, when either rsa-1(dd10) or unc-54(s74) was stained with anti-MHC B, the patterns were similar to the wild type, and different from MHC A staining. In these mutants, the double-line staining of MHC A is more narrow than the broader distribution of MHC B (Figure 10). As RSA-1 is a regulatory subunit of PP2A, our results suggest that PP2A containing RSA-1 is normally involved in dephosphorylating myosin heavy chains or myosin light chains.
FIGURE 9:
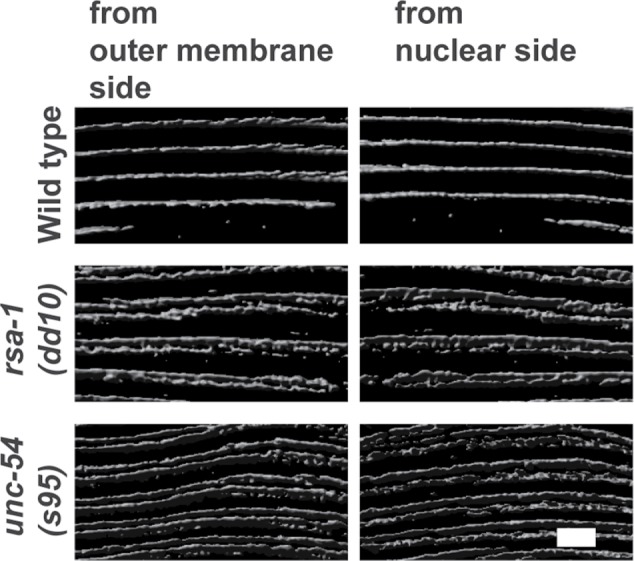
Superresolution images of MHC A staining in wild type, rsa-1, and unc-54 mutants. Nematodes with the indicated genotypes were immunostained with anti-MHC A and imaged with N-SIM. A z-series, planes 0.2 μm apart, was taken, and then 3D rendering was used to create the views of the middle of the A-bands where MHC A resides. For each strain, views from opposite sides of a body-wall muscle cell are shown. Note the single line in wild type and the closely spaced double line in rsa-1 and unc-54. Scale bar, 3 μm.
FIGURE 10:
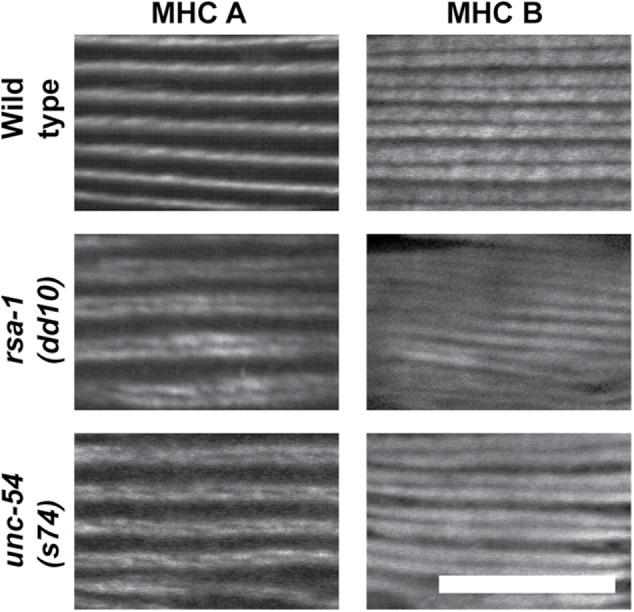
MHC B localization in wild type, rsa-1, and unc-54 mutants. Nematodes of the indicated genotypes were immunostained with anti-MHC A and anti-MHC B and imaged by confocal microscopy. Because both antibodies were mouse monoclonals, it was not practical to conduct costaining. MHC B localization broadly to the A-bands, with a gap in the middle, is the same in the wild type and the mutants. In the two mutants, the double-line MHC A localization is narrower than the localization of MHC B. Scale bar, 10 μm.
The MHC B mutation found in unc-54(s74) results in increased phosphorylation of MHC A
To determine whether rsa-1 affects the phosphorylation of myosin, we developed a method for specifically immunoprecipitating (IP) the major myosin heavy chains of body-wall muscle, MHC A and MHC B, from nematode samples. After establishing the technique for IP these myosins from wild-type animals, we tried to immunoprecipitate myosins from rsa-1(dd10) homozygotes. Owing to maternal-effect lethality of rsa-1(dd10), it was not possible to obtain sufficient quantities of protein from this mutant to determine the phosphorylation status of myosin. Nevertheless, we were able to determine the state of myosin phosphorylation of wild-type and adult-viable unc-54(s74) mutant animals, which show the same split A-band phenotype as rsa-1 mutants. As shown in Table 2, we detected phosphorylation of serines primarily in the nonhelical ∼30-residue C-termini of both MHC A and MHC B. These sites primarily overlap with sites predicted as phosphorylation sites by Schriefer and Waterston (1989; Supplemental Figure S6). Interestingly, for MHC A, two additional sites are phosphorylated (S1940 and S1942) in unc-54(s74) mutants as compared with wild type. Moreover, for MHC B, two additional sites are phosphorylated (T1426 and S1428) in wild type as compared with unc-54(s74). As noted below, RSA-1 is located at the M-line. Given the similar split A-band phenotype for unc-54(s74) and rsa-1, we would expect that in the rsa-1 mutant, there would be a similar increase in the number of phosphorylated residues in MHC A, consistent with loss of RSA-1 directed PP2A activity at the M-line.
TABLE 2:
Phosphorylation sites in MHC A and MHC B.

Knockdown of either the catalytic or scaffold subunit of PP2A results in a severe muscle phenotype
Loss of function mutations for either let-92, which encodes the unique catalytic subunit (C), or paa-1, which encodes the unique scaffolding subunit (A), are embryonic lethal (Ogura et al., 2003, 2010; Kao et al., 2004). Therefore, we performed RNAi for each gene after the lethal period, that is, from the L1 larval stage. For each gene, this resulted in adults that are small, paralyzed, and in a “jack-knifed” posture (Figure 11A; Table 1). Examination of muscle from these let-92(RNAi) or paa-1(RNAi) animals revealed that the sarcomeres are highly disorganized and some muscle cells have detached from their basement membranes (Figure 11B; Table 1).
FIGURE 11:
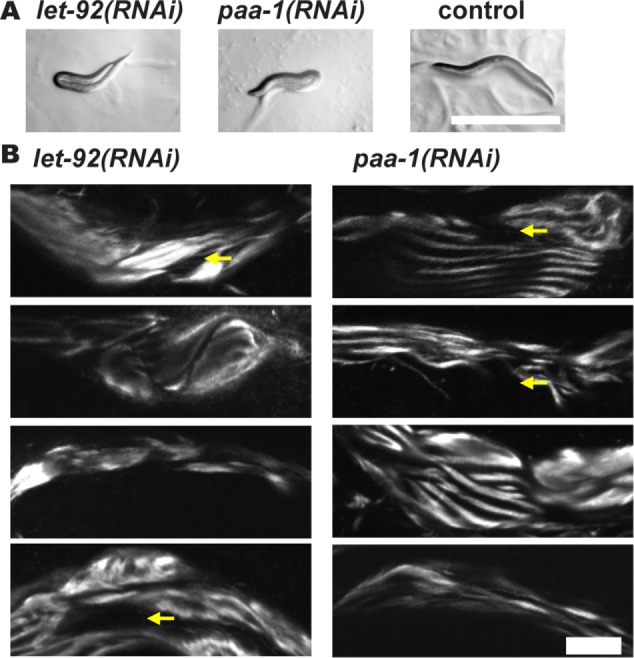
Muscle phenotypes for loss of function for the PP2A catalytic subunit (LET-92) and for the PP2A scaffolding subunit (PAA-1). RNAi was performed beginning from the L1 stage and continuing into adulthood. (A) RNAi for let-92 or paa-1 results in paralyzed folded-over or “jack-knifed” adults. Scale bar, 500 μm. (B) RNAi for let-92 or paa-1 results in highly disorganized A-bands and in some cells, detachment of the myofilament lattice from the muscle cell/basement membrane (indicated by yellow arrows). Four representative examples are shown for each gene. Scale bar, 20 μm.
The PP2A scaffolding subunit PAA-1 and two additional regulatory subunits, RSA-1 and SUR-6, also localize in sarcomeres
Because of the genetic evidence we found for a role for these PP2A subunits in striated muscle, we next wondered whether these proteins, like PPTR-1 and PPTR-2, are localized in the sarcomere. Fortunately, we were able to obtain antibodies from colleagues for PAA-1, SUR-6, and RSA-1. We verified by Western blot (Supplemental Figure S7) that each antibody recognizes the expected-size polypeptides. When these antibodies were reacted to adult animals, we found that all three are localized in the sarcomere (Figure 12; Table 1). PAA-1 and SUR-6 localize to I-bands, and RSA-1 localizes to both I-bands and M-lines. The M-line localization of RSA-1 is compatible with the split–MHC A localization phenotype, and since MHC A is located in the M-line region, this is also compatible with the idea that RSA-1-containing PP2A complexes are involved in dephosphorylating MHC A or other M-line proteins.
FIGURE 12:
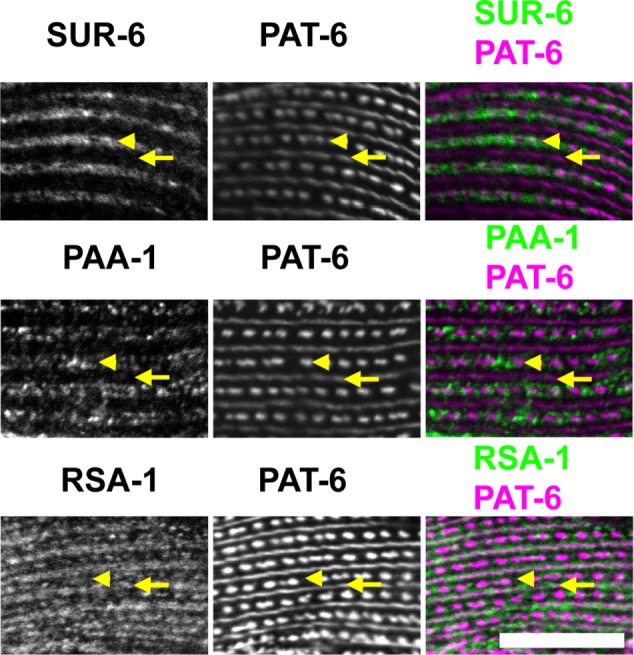
Sarcomeric localization of additional PP2A components. Wild-type animals were immunostained with antibodies to SUR-6, PAA-1, or RSA-1, together with anti–PAT-6 (α-parvin) to mark M-lines (arrows) and dense bodies (arrowheads). As shown, SUR-6 localizes to I-bands, PAA-1 localizes to I-bands, and RSA-1 localizes to M-lines and I-bands. Scale bar, 10 μm.
DISCUSSION
We have shown that a portion of the giant sarcomeric protein UNC-89 interacts with a component of PP2A. The interaction was found by a yeast two-hybrid screen (Figure 1) and verified by in vitro binding experiments using recombinant proteins (Figure 2). An interaction in vivo is supported by two results: 1) by antibody staining, PPTR-2 and UNC-89 colocalize to the M-line (Figure 4C), and 2) in an unc-89 mutant that lacks expression of UNC-89 isoforms containing the PPTR-2 binding region, PPTR-2 shows reduced levels at the M-line (Figure 5). The paralogue PPTR-1 is 58% identical in protein sequence (Supplemental Figure S2). Yeast two-hybrid clones representing PPTR-1 were also isolated using the same bait as was used to isolate PPTR-2. However, antibody staining shows that PPTR-1 is located at the I-band (Figure 4C), a region of the sarcomere that does not contain UNC-89. Therefore, we conclude that the interaction found between UNC-89 and PPTR-1 is not relevant in vivo and was identified because of the high sequence homology between PPTR-2 and PPTR-1. Nevertheless, PPTR-1 has a muscle function, as the loss-of-function mutant for pptr-1 shows disorganized sarcomeres. There is also a biological function for PPTR-2 in muscle: although certain loss-of-function alleles of pptr-1 and pptr-2 do not by themselves show a defect in myofibril organization, when they are combined in a double mutant, a defect in myofibril organization is revealed (Figure 3B). This shows that reducing the function of pptr-1 uncovers a function for pptr-2 in muscle. Either pptr-2 is partially redundant with pptr-1 in muscle, or more likely, the additive phenotype of the double mutant indicates that the two PPTR proteins disrupt separate processes in muscle. The latter possibility is supported by the distinctly different localizations of the two proteins (Figure 4C).
This is the first time that a UNC-89/obscurin family member has been shown to interact directly with PP2A. The interaction is with a regulatory component of the PP2A complex, PPTR-2, which is highly homologous to human regulatory B56 subunits, ranging from 64 to 74% identities, with the highest identity to B56gamma (74%; Supplemental Figure S2). Nevertheless, in mouse heart muscle, such an association occurs via an intermediary protein, ankyrin B. Bhasin et al. (2007) demonstrated that a portion of ankyrin B binds to B56α, that ankyrin B coIPs the entire PP2A complex from heart lysates, and that ankyrin B and B56α colocalize at M-lines in neonatal and adult cardiomyocytes. Cunha and Mohler (2008) reported that obscurin (the mammalian UNC-89 homologue) interacts with ankyrin B and that ankyrin B and obscurin can be coIPed from a heart lysate. Most importantly, they show that the obscurin-to-ankyrin B interaction is required for the recruitment of B56α to the M-line. This last result is consistent with our finding that, in an unc-89 mutant lacking the PPTR-2 binding region, there are reduced levels of PPTR-2 at the M-line. However, in this unc-89 mutant, PPTR-2 is not completely absent from the M-line. Perhaps PPTR-2, like B56α in mammals, interacts with an ankryin and this helps localize PPTR-2 to the M-line in nematode muscle. Further experiments are required to address this question.
Interestingly, Yin et al. (2010) report that in rat or mouse cardiomyocytes, B56α is normally located at both M-lines and Z-disks. Again, this is consistent with our finding that PPTR-2 is located both at M-lines and at dense bodies (the nematode analogs of Z-disks). Although our studies indicate that a major determinant of the M-line localization of PPTR-2 is its interaction with UNC-89, we do not yet know which protein interactions determine the dense-body localization of PPTR-2. It is also not clear why the localization results of these several studies on mammalian B56α do not agree (M-line vs. M-line and Z-disk), but it may be due to differences in the antibodies used or the fixation methods employed. Yin et al. (2010) report another fascinating result: treatment with isoproterenol, a β-adrenergic agonist, which induces the phosphorylation of several myofilament proteins including TnI and MyBP-C, results in translocation of B56α from M-lines and Z-disks, to the cytosol. It will be interesting to determine whether the location of PPTR-2 is also dynamic in nematode muscle, which can be addressed by the use of GFP-tagged PPTR-2 in live animals.
Our results with PPTR-1 and PPTR-2 led us to explore the function and localization of all the currently reported C. elegans components of PP2A complexes (Figure 6). We found that loss-of-function mutation or RNAi for any of the seven components results in defects in sarcomere organization (summarized in Table 1). Using antibodies, we were able to show that three of these components, in addition to PPTR-1 and PPTR-2, are localized in the sarcomere (Figure 12). Because PP2A complexes are likely to be expressed in every tissue, it is not surprising that loss of function for several components (the catalytic subunit LET-92, the scaffolding component PAA-1, and the regulatory B subunit SUR-6) was embryonic lethal, and we had to examine adult muscle phenotypes either in a temperature-sensitive mutant or by RNAi from the L1 larval period onward. RNAi of sur-6 from the L1 stage resulted in adults with normal sarcomere organization (unpublished data). In contrast, RNAi of either let-92 or paa-1 from the L1 stage resulted in larvae with severely disorganized muscle—disorganization of sarcomeres and detachment of sarcomeres from the muscle-cell membrane (Figure 11B). Thus, a general conclusion is that loss of function in PP2A components encoded by single genes (let-92 encoding the single catalytic PP2A subunit, and paa-1 encoding the single scaffolding PP2A subunit) results in the most severe muscle sarcomere phenotype. A corollary is that loss of function of any one of the five genes encoding regulatory PP2A subunits results in less severe muscle phenotypes (Table 1; Figures 3 and 7).
Perhaps one of the most fascinating phenotypes revealed was for loss of function of the regulatory subunit B”, RSA-1 (Figures 7–9): the “split” or “double-line” localization of MHC A. The thick filaments of nematode body-wall muscle contain two myosin heavy chain isoforms that primarily form homodimers (Schachat et al., 1978), and are differentially localized, with MHC A located in the central region and MHC B lying in the outer regions of the thick filament (Miller et al., 1983). Curiously, we found the “double-line” localization of MHC A also in rare, missense mutations in the myosin head region of MHC B, encoded by the unc-54 gene (Moerman et al., 1982; Moerman and Fire, 1997; Figures 8 and 9). RSA-1 is located at both M-lines and I-bands (Figure 12). The M-line localization of RSA-1 suggests that PP2A targeted by RSA-1 could influence the phosphorylation of MHC A, which is located near the M-line. Because rsa-1 mutants are maternal-effect embryonic lethal, we could not obtain sufficient material to examine the phosphorylation status of MHC A in rsa-1 mutants. However, we could obtain enough material from unc-54(s74) and determine that in this mutant, as compared with wild type, two additional serines (S1940 and S1942) in the nonhelical tailpiece of MHC A are phosphorylated (Table 2). Given the similarity of the MHC A double-line phenotype of unc-54(s74) and rsa-1 mutants, we speculate that in rsa-1 mutants, which may have reduced PP2A activity at M-lines, a similar increase in phosphorylation of MHC A would be found. To test this question about rsa-1 mutants in the future, we could develop antibodies that recognize phosphoserines S1940 and S1942 in MHC A. We also found that in unc-54(s74), two residues (T1426 and S1428) are not phosphorylated, in contrast to wild type (Table 2). How a mutation in the myosin head domain of UNC-54 (MHC B) leads to a change in the phosphorylation status of both MHC A and MHC B rods is unknown. However, it is known that 1) there are a number of unc-54 mutants that display disrupted thick-filament assembly and organization, and yet the mutations all reside in the myosin head region (Bejsovec and Anderson, 1988, 1990); 2) the end of the rod and the nonhelical tailpiece of these myosin heavy chains influences the organization of thick filaments (Hoppe et al., 2003); 3) by mutational analysis, S1940 and S1942 are crucial for proper assembly of MHC A into the myofilament lattice (Hoppe et al., 2010b); and 4) the nonhelical tail of MHC A contributes to binding to the M-line protein UNC-98 (Miller et al., 2006). Finally, it should be noted that all the residues we determined to be phosphorylated in MHC A and MHC B were predicted as phosphorylation sites by Schriefer and Waterston (1989), by homology to sites that they determined to be phosphorylated in the N-terminal nonhelical region of paramyosin (Supplemental Figure 6).
Our studies are the first to demonstrate the importance of PP2A for myofibril organization in any system. However, they are not the first to demonstrate the importance of protein phosphatases in myofibril organization or formation. Terry et al. (2006), using a Xenopus model of vertebrate skeletal myofibrillogenesis and a set of chemical inhibitors of protein phosphatases, reported that PP1, but interestingly not PP2A, is required for myofibril assembly. In addition, morpholino-induced knockdown of a myosin-targeting subunit of PP1 resulted in fewer somites and less myofibril organization. Whether this reflects a true difference between nematodes and vertebrates in the requirements of various protein phosphatases is not known.
Our finding that UNC-89 interacts with a regulatory subunit of PP2A suggests that perhaps UNC-89 is a substrate for PP2A. Unfortunately, despite multiple attempts with several antibodies, we were not able to IP UNC-89 in sufficient quantity to subject it to mass spectrometry analysis. Perhaps in the future we can use CRISPR/Cas9 to introduce an HA or FLAG tag that will permit us to IP UNC-89 in an informative manner.
It should be noted that a previous study from our laboratory (Qadota et al., 2008b) demonstrated that the kinase region of UNC-89 interacts with another protein phosphatase, SCPL-1, a member of the CTD phosphatase family. Interaction occurs via Fn1-Ig52-PK1, and via Ig53-Fn2-PK2. One of the SCPL-1 interaction sites, overlaps with the interaction site of PPTR-2, namely Ig53-Fn2 (Figure 1B). Whether this region just N-terminal of PK2 interacts with PPTR-2 and SCPL-1 simultaneously, or mutually exclusively, is not known. Homology modeling (Small et al., 2004) and sequence considerations (Mayans et al., 2013) suggest that PK1 is an inactive pseudokinase, whereas PK2 is an active kinase. In fact, we have preliminary evidence that indeed PK2 has catalytic activity in vitro (T. Ferrara and G.M. Benian, unpublished data). Although we do not yet know the substrates for UNC-89 PK2, SCPL-1, and PPTR-2, perhaps some of the substrates are shared. Perhaps the kinase activity of UNC-89 PK2 needs to be under tight control; soon after a substrate is phosphorylated by PK2, it needs to be dephosphorylated by the nearby SCPL-1 or PPTR-2-directed PP2A. Hopefully, this question can be addressed when we finally identify the substrates for these enzymes. It should also be noted that the nematode M-line is the fixed location of at least two other protein kinases, PKN-1 (Qadota et al., 2011), and UNC-82 (Hoppe et al., 2010a). This is perhaps another reason for PPTR-2-directed PP2A and SCPL-1 phosphatases being “solid state” components of the M-line. Another possibility is that at the M-line PPTR-2-directed PP2A might regulate the kinase activity of UNC-89 PK2, PKN-1 or UNC-82.
MATERIALS AND METHODS
Screening of yeast two-hybrid library and domain mapping
Yeast two-hybrid screening of a C. elegans cDNA library was performed as previously described (Miller et al., 2006). Purification of plasmids from yeast cells was done using Zymoprep Yeast Plasmid Miniprep II (Zymo Research). Bait plasmids of UNC-89 were described previously (Wilson et al., 2012a; Warner et al., 2013). Some bait plasmids used for domain mapping (Figure 2B) were also described before, including 1/3IK-Ig53-Fn2 (Wilson et al., 2012a), Ig53-Fn2-PK2 (Qadota et al., 2008b), 1/3IK-Ig53 (Wilson et al., 2012a), 1/3IK (Xiong et al, 2009), and Ig53-Fn2 (Wilson et al., 2012a). Bait plasmids of UNC-89 Ig53 and UNC-89 Fn2 were created by cloning of PCR-amplified fragments using primers shown in Supplemental Table S3 and inserted into pGBDU-C1. Purification of plasmids from bacterial cells was done using ZR Plasmid Miniprep-Classic (Zymo Research).
Domain mapping by far-Western assay
The minimum regions of PPTR-1 (75-543) and PPTR-2 (10-558) for interaction with UNC-89, as determined by yeast two-hybrid analysis, were expressed as His-tagged proteins in Escherichia coli. cDNAs encoding these two proteins were PCR-amplified using primers shown in Supplemental Table S3 from yeast two-hybrid clones, and their sequences were confirmed to be error-free. These cDNAs were cloned via the NcoI and KpnI sites into pETM-11 and expressed at 15°C for 17 h after induction with 750 μM Isopropyl β-d-1-thiogalactopyranoside (IPTG). Bacterial cells from a 500-ml culture were suspended in 30 ml of His Lysis Buffer (50 mM Tris, pH 8.0, 50 mM NaCl, 40 mM imidazole, 10 mM β-mercaptoethanol, 10% glycerol [all adjusted to pH 8.0], 1 mM phenyl methane sulfonate (PMSF), containing three tablets of cOmplete Mini, EDTA-free protease inhibitors [Roche]) and broken by one passage through a French pressure cell at 1000 psi. To this lysate was added 80 μl of 20% Triton X-100 and 20 μl of Benzonase Nuclease (25 U/μl; Novagen), and then it was centrifuged at 39,000 × g for 45 min at 4°C. The resulting supernatant was added to 2.7 ml of a 50:50 slurry of Ni-NTA Agarose (Qiagen) that had been equilibrated in His Lysis Buffer and incubated with mixing at 4°C for 30 min. The beads were pelleted by low-speed centrifugation and washed five times with His Wash Buffer (50 mM Tris, pH, 8.0, 50 mM NaCl, 60 mM imidazole, 10 mM β-mercaptoethanol, 10% glycerol [all adjusted to pH 8.0], 1 mM PMSF). After being poured into a small (∼12-ml) disposable column and the beads being allowed to settle, the 6His-tagged protein was eluted with His Elution Buffer (50 mM Tris, pH 8.0, 50 mM NaCl, 200 mM imidazole, 10 mM β-mercaptoethanol, 10% glycerol [all adjusted to pH 8.0]).
The plasmid used for expressing a maltose-binding protein (MBP) fusion of UNC-89-Ig53-Fn2 was described previously (Wilson et al., 2012a). MBP fusions of UNC-89 1/3 IK-Ig53-Fn2 were created by cloning the corresponding fragment excised from pGDBU-UNC-89 1/3IK-Ig53-Fn2 (Wilson et al., 2012a) into pMAL-KK1. MBP, MBP-UNC-89-Ig53-Fn2, and MBP-UNC-89 1/3 IK-Ig53-Fn2 were expressed and purified as follows: expression was conducted at 20°C for 5 h after induction with 750 μM IPTG. Bacterial cells from a 500-ml culture were suspended in 30 ml of MBP Lysis Buffer (50 mM Tris, pH 8.0, 1 mM PMSF) plus three tablets of cOmplete Mini protease inhibitors (Roche) and broken by one passage through a French pressure cell at 1000 psi. After addition of Triton X-100 to a final concentration of 1% and thorough mixing, the lysate was centrifuged at 12,000 × g for 20 min. The resulting supernatant was added to 2.7 ml of a 50:50 slurry of Amylose Resin beads (New England BioLabs) and incubated with mixing for 30 min at 4°C. The beads were pelleted by low-speed centrifugation and washed five times in MBP Lysis Buffer plus Triton (50 mM Tris, pH 8.0, 1% Triton X-100, 1 mM PMSF) and three times in MBP Lysis Buffer. After the mixture was poured into a small column and the beads were allowed to settle, the MBP fusion proteins were eluted with 10 mM maltose.
A far-Western assay was conducted as follows: 2 μg of His-PPTR-1 and His-PPTR-2, in triplicate was resolved by SDS–PAGE, transferred to a nitrocellulose membrane, and blocked overnight at room temperature in 5% milk and Tris-buffered saline (TBS)/Tween 20 (TBS-T). Blot strips containing His-PPTR-1 and His-PPTR-2 were incubated with either MBP, MBP-UNC-89 Ig53-Fn2, or MBP-UNC-89 1/3 IK-Ig53-Fn2 at 5 μg/ml at room temperature for 1 h, washed multiple times in TBS-T, reacted with anti–MBP-horseradish peroxidase (HRP) (New England BioLabs) at 1:5000 dilution, and washed multiple times, and reactions were visualized by enhanced chemiluminescence (ECL Pierce, Thermo Fisher Scientific).
In-solution pull-down binding assay
Pairs of proteins were tested for in-solution binding by diluting the proteins approximately 10-fold in Binding/Washing Buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 0.25% gelatin, 0.1% NP40, cOmplete Mini protease inhibitors [Roche]) and incubated with mixing for 3 h at 4°C. These reactions were carried out in a total volume of 400 μl and included 5 μg of either His-PPTR-1 or His-PPTR-2 and 10 μg of either MBP or MBP-UNC-89-Ig-Fn. Each solution was mixed with 50 μl of a 1:1 slurry of Anti–His-tag mAb-Agarose beads (Medical and Biological Laboratories) and incubated for 1 h at 4°C. The beads were pelleted by a brief centrifugation and then washed four times with 1 ml each of the Binding/Washing Buffer. Proteins from the beads were eluted using Laemmli buffer at 95°C for 5 min. The entire eluted proteins from each binding reaction were separated by SDS–PAGE, transferred to a nitrocellulose membrane, stained with Ponceau S, blocked with 5% milk TBS-T, reacted with anti–MBP-HRP (New England BioLabs) at 1:5000 dilution, and washed multiple times, and reactions were visualized by ECL (Pierce, Thermo Fisher Scientific). A Coomassie stained gel of all the proteins used in the far-Western and in-solution binding experiments is shown in Supplemental Figure S4.
Caenorhabditis elegans strains and RNAi
Standard growth conditions for C. elegans were used (Brenner, 1974). Wild-type nematodes were the N2 (Bristol) strain. The following strains were used in this study: JH2787 pptr-1(tm3103)V (outcrossed 6X with N2), MAS268 pptr-2(ok1467)V (outcrossed 4X with N2), MAS264 pptr-1(abc19)V, MAS294 pptr-1(abc19) pptr-2(ok1467)V, GB283 unc-89(tm752)I (outcrossed 5X with N2), EU1062 sur-6(or550)I, TH322 unc-13(e51) rsa-1(dd10) I/hT2 [bli-4(e937) let-?(q782) qIs48] (I;III), TH323 unc-13(e51) rsa-1(dd13) I/hT2 [bli-4(e937) let-?(q782) qIs48] (I;III), BC347 unc-54(s74)I, RW5008 unc-54(s95)I, and RW134 unc-54(st134)I. Some strains were obtained from the Caenorhabditis Genetics Center. The pptr-1(abc19) missense mutant was identified in an EMS mutagenesis screen (Lange et al., 2013). Plasmids for RNAi of let-92 and paa-1 were created by cloning of PCR-amplified cDNA using primers shown in Supplemental Table S3 and inserted into pPD129.36. The plasmids for RNAi of cash-1 were created by cloning of a BamHI fragment of yk294d9 or an XbaI-XhoI fragment of yk297d9 into pPD129.36.
Generation of antibodies to PPTR-1 and PPTR-2 and Western blots
Glutathione S-transferase (GST) and MBP fusions of the 80 C-terminal residues of PPTR-1 and the 116 C-terminal residues of PPTR-2 were expressed in E. coli (Mercer et al., 2006) after cloning into pGEX-KK1 and pMAL-KK1, using primers listed in Supplemental Table S3, and SmaI and SalI sites. GST fusions were supplied to Spring Valley Laboratories for production of rabbit antibodies. Anti–PPTR-1 and anti–PPTR-2 were affinity-purified using Affigel (BioRad)-conjugated MBP fusions, as described previously (Mercer et al., 2003). We used the procedure of Hannak et al. (2002) to prepare total protein lysates from wild-type, pptr-1(tm3103), and pptr-2(ok1467) mixed-staged animals. Equal amounts of total protein from these strains were separated by 10% polyacrylamide-SDS Laemmli gels, transferred to nitrocellulose membranes, reacted with affinity-purified anti–PPTR-1 at 1:70,000 dilution or affinity-purified anti–PPTR-2 at 1:10,000 dilution, reacted with goat anti-rabbit immunoglobulin G conjugated to HRP at 1:10,000 dilution, and visualized by ECL.
Immunolocalization in adult body-wall muscle
Adult nematodes were fixed and immunostained according to the method described in Nonet et al. (1993) and described in further detail in Wilson et al. (2012b). The following primary antibodies were used at 1:200 dilution: anti–UNC-89 (mouse monoclonal MH42; Benian et al., 1996; Hresko et al., 1994), anti–myosin heavy chain A (MHC A; mouse monoclonal 5-6; Miller et al., 1983), anti–myosin heavy chain B (MHC B; mouse monoclonal 5-8; Miller et al., 1983); at 1:100 dilution: anti–UNC-95 (rabbit polyclonal Benian-13; Qadota et al., 2007), anti–PAT-6 (rat polyclonal; Warner et al., 2013), anti–PPTR-1 (rabbit polyclonal; this study), anti–PPTR-2 (rabbit polyclonal; this study), anti–SUR-6 (rabbit polyclonal; Song et al., 2011), anti–PAA-1 (affinity-purified rabbit polyclonal Sr3-4 generated to a C-terminal region of PAA-1; residues 288–590; Lange et al., 2013), and anti–RSA-1 (rabbit polyclonal; Schlaitz et al., 2007). MH42 was kindly provided by Pamela Hoppe (Western Michigan University); 5-6 was obtained from the Developmental Studies Hybridoma Bank, created by the National Institute of Child Health and Human Development of the National Institutes of Health and maintained at the University of Iowa, Iowa City. Secondary antibodies, also used at 1:200 dilution, including anti-rabbit Alexa 488 (Invitrogen), anti-rat Alexa 594 (Invitrogen), and anti-mouse Alexa 594 (Invitrogen). Images, except Figure 9, were captured at room temperature with a Zeiss confocal system (LSM510) equipped with an Axiovert 100M microscope and an Apochromat ×63/1.4 numerical aperture oil immersion objective, in ×2.5 zoom mode. The color balances of the images were adjusted by using Adobe Photoshop. To obtain the intensity profiles presented in Figure 5B, confocal images were processed using Zeiss ZEN 2009 software. Using this software, the mode was changed from 2D to 2.5D to show an “intensity profile.” After the angles were adjusted by rotation of 2.5D images, snapshots of intensity profiles were obtained.
N-SIM microscopy and 3D image reconstruction
Superresolution microscopy was performed with a Nikon N-SIM system in 3D structured illumination mode on an Eclipse Ti-E microscope equipped with a 100×/1.49 NA oil immersion objective, 488- and 561-nm solid-state lasers, and an EM-CCD camera (DU-897, Andor Technology). Superresolution images were reconstructed using the N-SIM module in NIS-Elements software. The color balances of the images were adjusted using Adobe Photoshop. A Z-series every 0.2 μm was taken from the outer muscle cell membrane deeper into the muscle cell. This Z stack was then reconstructed into a 3D image using software on the N-SIM microscope. The reconstructed 3D z-stacks were analyzed using Imaris 8.3 (Bitplane).
Immunopreciptation of MHC A and MHC B
Large quantities (∼3 ml of packed worms) of wild type and unc-54(s74) were grown from 30 15-cm-high peptone NGM plates, and “worm powders” were obtained by grinding them extensively in a mortar and pestle in liquid nitrogen. Lysates (∼1 ml) were prepared by adding worm powder to “IP buffer” (∼20% vol/vol) consisting of 25 mM Tris, pH 7.5, 600 mM KCl, 0.1 mM ethylene glycol-bis (β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid (EGTA), 0.5% Nonidet P-40, 5% glycerol, 5 mM MgCl2, 5 mM ATP, cOmplete Mini protease inhibitors tablets (–EDTA) (Roche), and phosphatase inhibitor mini tablets (Pierce), vortexing for 1 min, incubating on ice for 30 min, vortexing for 1 min, spinning at top speed in a microcentrifuge for 10 min at 4°C, and saving the supernatant. To 500 μl of supernatant was added 25 μl of monoclonal antibodies to MHC A or MHC B (5-6 ascites, or 5-13 ascites, respectively), followed by mixing and incubating at 4°C for 4 h. Next, each sample was diluted by adding 500 μl of IP buffer lacking KCl, mixing, and then adding 160 μl of a 50:50 slurry of protein A sepharose beads (Sigma) and mixing and incubating at 4°C for 1 h. The beads were pelleted using a microcentrifuge and washed three times with IP buffer containing 300 mM KCl and three times with phosphate-buffered saline (PBS). Both wash buffers contained protease and phosphatase inhibitors. The pellets were delivered to Emory’s Proteomics Core Facility.
On-bead digestion
A published protocol was followed for on-bead digestion (Soucek et al., 2016). The IP beads were spun down and residual PBS was removed. Digestion buffer (50 mM NH4HCO3, three times the bead volume) was added and the bead solution was then treated with 1 mM dithiothreitol (DTT) at room temperature for 30 min, followed by 5 mM iodoacetimide (IAA) at room temperature for 30 min in the dark. Proteins were digested with 1 µg of lysyl endopeptidase (Wako) at room temperature for 4 h and further digested overnight with 2 µg trypsin (Promega) at room temperature. Resulting peptides were desalted with an Oasis HLB column (Waters) and dried under vacuum.
LC-MS/MS
Peptides were analyzed with nano–high pressure liquid chromatography–tandem mass spectrometry (nano-LC-MS/MS). Briefly, the peptides were loaded onto an in-house packed column (40 cm long × 75 μm ID × 360 OD, Dr. Maisch GmbH ReproSil-Pur 120 C18-AQ 3.0-µm beads) analytical column (Thermo Scientific) using a Dionex nanoLC system (Thermo Scientific). A flow rate of 0.300 μl/min with a linear acetonitrile gradient from 8 to 27% in 0.1% formic acid for 120 min was used. The column output was connected to a Q Exactive Plus mass spectrometer (Thermo Scientific) through a nanoelectrospray ion source. The mass spectrometer was controlled by Xcalibur software (Thermo, 4.0.27.19) and operated in the data-dependent mode in which the initial MS scan recorded the mass-to-charge ratios (m/z) of ions over the range 350–1750 at a resolution of 70,000 with a target value of 1 × 106 ions and a maximum injection time of 100 ms. The 10 most abundant ions were automatically selected for subsequent higher-energy collision dissociation (HCD) with the energy set at 28 NCE. The MS/MS settings included a resolution of 35,000, a target value of 5 × 105 ions, a maximum integration time of 108 ms, and an isolation window set at 3.0 m/z. Ions with undetermined charge, z = 1, and z > 7 were excluded.
Mass spectrometry data analysis
Mass spectrometry data were analyzed as reported in Wingo et al. (2017). Spectra were searched using Proteome Discoverer 2.1 against C. elegans Uniprot database (27,194 target sequences). Searching parameters included fully tryptic restriction and a parent ion mass tolerance (±20 ppm). Methionine oxidation (+15.99492 Da), asparagine and glutamine deamidation (+0.98402 Da), serine, threonine, tyrosine phosphorylation (+79.96633 Da), and protein N-terminal acetylation (+42.03670) were variable modifications (up to three allowed per peptide); cysteine was assigned a fixed carbamidomethyl modification (+57.021465 Da). Percolator was used to filter the peptide spectrum matches to a false discovery rate of 1%.
Sequence analysis
Multiple polypeptide sequences were aligned using CLUSTALW (Supplemental Figures S2A and S6). The amino acid sequence identities shown in Supplemental Figure S2B were determined by BLAST.
Supplementary Material
Acknowledgments
We thank Anne Schwager and Tony Hyman (Max-Planck Institute, Dresden, Germany) for antibodies to RSA-1 and Kevin O’Connell (National Institutes of Health) for antibodies to SUR-6. We thank Robert Barstead (Oklahoma Medical Research Foundation) for the C. elegans yeast two-hybrid library RB2 and Yuji Kohara (National Institute of Genetics, Japan) for “yk” cDNA clones. This study was supported in part by the Emory Integrated Proteomics Core (EIPC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. This work was also supported by the Georgia Institute of Technology’s Parker H. Petit Institute for Bioengineering and Bioscience, including the Systems Mass Spectrometry Core Facility. This study was supported by grants from the National Institutes of Health (R01 AR064307 to G.M.B. and R01 CA57327 to D.C.P.). Many of the nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440).
Abbreviations used:
- GST
glutathione S-transferase
- HRP
horseradish peroxidase
- IPTG
Isopropyl β- d-1-thiogalactopyranoside
- MBP
maltose-binding protein
- MHC
myosin heavy chain
- PMSF
phenyl methane sulfonate
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-03-0192) on June 27, 2018.
REFERENCES
- Aoki H, Sadoshima J, Izumo S. (2000). Myosin light chain kinase mediates sarcomere organization during cardiac hypertrophy in vitro. Nat Med , 183–188. [DOI] [PubMed] [Google Scholar]
- Barefield D, Sadayappan S. (2010). Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J Mol Cell Cardiol , 866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejsovec A, Anderson P. (1988). Myosin heavy-chain mutations that disrupt Caenorhabditis elegans thick filament assembly. Genes Dev , 1307–1317. [DOI] [PubMed] [Google Scholar]
- Bejsovec A, Anderson P. (1990). Functions of the myosin ATP and actin binding sites are required for C. elegans thick filament assembly. Cell , 133–140. [DOI] [PubMed] [Google Scholar]
- Benian GM, Tinley TL, Tang X, Borodovsky M. (1996). The Caenorhabditis elegans gene unc-89, required for muscle M-line assembly, encodes a giant modular protein composed of Ig and signal transduction domains. J Cell Biol , 835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhasin N, Cunha SR, Mudannayake M, Gigena MS, Rogers TB, Mohler PJ. (2007). Molecular basis of PP2A regulatory subunit B56alpha targeting in cardiomyocytes. Am J Physiol Heart Circ Physiol , H109–H119. [DOI] [PubMed] [Google Scholar]
- Brenner S. (1974). The genetics of Caenorhabditis elegans . Genetics , 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha SR, Mohler PJ. (2008). Obscurin targets ankyrin-B and protein phosphatase 2A to the cardiac M-line. J Biol Chem , 31968–31980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey CS, Deitiker PR, Epstein HF. (1992). Assembly-dependent phosphorylation of myosin and paramyosin of native thick filaments in Caenorhabditis elegans. Biochem Biophys Res Commun , 1528–1532. [DOI] [PubMed] [Google Scholar]
- Du A, Sanger JM, Linask KK, Sanger JW. (2003). Myofibrillogenesis in the first cardiomyocytes formed from isolated quail precardiac mesoderm. Dev Biol , 382–394. [DOI] [PubMed] [Google Scholar]
- Ferrara TM, Flaherty DB, Benian GM. (2005). Titin/connectin-related proteins in C. elegans: a review and new findings. J Musc Res Cell Motil , 435–447. [DOI] [PubMed] [Google Scholar]
- Gieseler K, Qadota H, Benian GM. (2017). Development, structure and maintenance of C.elegans body wall muscle. In: WormBook, ed. The C. elegans Research Community, WormBook, / 10.1895/wormbook.1.81.2, www.wormbook.org [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannak E, Oegema K, Kirkham M, Gonczy P, Habermann B, Hyman AA. (2002). The kinetically dominant assembly pathway for centrosomal asters in Caenorhabditis elegans is gamma-tubulin dependent. J Cell Biol , 591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe PE, Andrews RC, Parikh PD. (2003). Differential requirement for the nonhelical tailpiece and the C terminus of the myosin rod in Caenorhabditis elegans muscle. Mol Biol Cell , 1677–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe PE, Chau J, Flanagan KA, Reedy AR, Schriefer LA. (2010a). Caenorhabditis elegans unc-82 encodes a serine-threonine kinase important for myosin filament organization in muscle during growth. Genetics , 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe PE, Heustis RJ, Flanagan KA, Reedy AR. (2010b). Phosphorylation motifs in the nonhelical domains of myosin heavy chain and paramyosin may negatively regulate assembly in Caenorhabditis elegans striated muscle. Cytoskel , 309–321. [DOI] [PubMed] [Google Scholar]
- Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, (2012). PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res , D261–D270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hresko MC, Williams BD, Waterston RH. (1994). Assembly of body wall muscle and muscle cell attachment structures in Caenorhabditis elegans. J Cell Biol , 491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao G, Tuck S, Baillie D, Sundaram MV. (2004). C.elegans SUR-6/PR55 cooperates with LET-92/protein phosphatase 2A and promotes Raf activity independently of inhibitory Akt phosphorylation sites. Development , 755–765. [DOI] [PubMed] [Google Scholar]
- Kostich M, English J, Madison V, Gheyas F, Wang L, Qiu P, Greene J, Laz TM. (2002). Human members of the eukaryotic protein kinase family. Genome Biol , research0043.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange KI, Heinrichs J, Cheung K, Srayko M. (2013). Suppressor mutations identify amino acids in PAA-1/PR65 that facilitate regulatory RSA-1/B” subunit targeting of PP2A to centrosomes in C. elegans . Biol Open , 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manring HR, Carter OA, Ackermann MA. (2017). Obscure functions: the location–function relationship of obscurins. Biophys Rev , 245–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayans O, Benian GM, Simkovic F, Rigden DJ. (2013). Mechanistic and functional diversity in the mechanosensory kinases of the titin-like family. Biochem Soc Trans , 1066–1071. [DOI] [PubMed] [Google Scholar]
- Meissner B, Warner A, Wong K, Dube N, Lorch A, McKay SJ, Khattra J, Rogalski T, Somasiri A, Chaudhry I, et al (2009). An integrated strategy to study muscle development and myofilament structure in Caenorhabditis elegans . PLoS Genet , e1000537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer KB, Flaherty DB, Miller RK, Qadota H, Tinley TL, Moerman DG, Benian GM. (2003). Caenorhabditis elegans UNC-98, a C2H2 Zn finger protein, is a novel partner of UNC-97/PINCH in muscle adhesion complexes. Mol Biol Cell , 2492–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer KB, Miller RK, Tinley TL, Sheth S, Qadota H, Benian GM. (2006). Caenorhabditis elegans UNC-96 is a new component of M-lines that interacts with UNC-98 and paramyosin and is required in adult muscle for assembly and/or maintenance of thick filaments. Mol Biol Cell , 3832–3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DM, Ortiz I, Berliner GC, Epstein HF. (1983). Differential localization of two myosins within nematode thick filaments. Cell , 477–490. [DOI] [PubMed] [Google Scholar]
- Miller RK, Qadota H, Landsverk ML, Mercer KB, Epstein HF, Benian GM. (2006). UNC-98 links an integrin-associated complex to thick filaments in Caenorhabditis elegans muscle. J Cell Biol , 853–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerman DG, Fire A. (1997). Muscle: structure, function, and development. In: C. elegans II, ed. Riddle DL, Blumenthal T, Meyer BJ, Priess JR, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 417–470. [PubMed] [Google Scholar]
- Moerman DG, Plurad S, Waterston RH, Baillie DL. (1982). Mutations in the unc-54 myosin heavy chain gene of Caenorhabditis elegans that alter contractility but not muscle structure. Cell , 773–781. [DOI] [PubMed] [Google Scholar]
- Nonet ML, Grundahl K, Meyer BJ, Rand JB. (1993). Synaptic function is impaired but not eliminated in C. elegans mutants lacking synaptotagmin. Cell , 1291–1305. [DOI] [PubMed] [Google Scholar]
- Ogura K, Kishimoto N, Mitani S, Gengyo-Ando K, Kohara Y. (2003). Translational control of maternal glp-1 mRNA by POS-1 and its interacting protein SPN-4 in C. elegans . Development , 2495–2503. [DOI] [PubMed] [Google Scholar]
- Ogura K, Okado T, Mitani S, Gengyo-Ando K, Baillie DL, Kohara Y, Goshima Y. (2010). Protein phosphatase 2A cooperates with the autophagy-related kinase UNC-51 to regulate axon guidance in Caenorhabditis elegans. Development , 1657–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan S, Mukhopadhyay A, Narasimhan SD, Tesz G, Czech MP, Tissenbaum HA. (2009). A PP2A regulatory subunit regulates C. elegans insulin/IGF-1 signaling by modulating AKT-1 phosphorylation. Cell , 939–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Lant B, Yu B, Tian R, Tong J, Krieger JR, Moran MF, Gingras A-C, Derry WB. (2017). CCM-3 promotes C.elegans germline development by regulating vesicle trafficking cytokinesis and polarity. Curr Biol , 868–876. [DOI] [PubMed] [Google Scholar]
- Qadota H, Blangy A, Xiong G, Benian GM. (2008a). The DH-PH region of the giant protein UNC-89 activates RHO-1 GTPase in Caenorhabditis elegans body wall muscle. J Mol Biol , 747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qadota H, Mayans O, Matsunaga Y, McMurry JL, Wilson KJ, Kwon GE, Stanford R, Deehan K, Tinley TL, Ngwa VM, Benian GM. (2016). The SH3 domain of UNC-89 (obscurin) interacts with paramyosin, a coiled-coil protein, in Caenorhabditis elegans muscle. Mol Biol Cell , 1606–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qadota H, McGaha LA, Mercer KB, Stark TJ, Ferrara TM, Benian GM. (2008b). A novel protein phosphatase is a binding partner for the protein kinase domains of UNC-89 (Obscurin) in Caenorhabditis elegans . Mol Biol Cell , 2424–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qadota H, Mercer KB, Miller RK, Kaibuchi K, Benian GM. (2007). Two LIM domain proteins and UNC-96 link UNC-97/pinch to myosin thick filaments in Caenorhabditis elegans muscle. Mol Biol Cell , 4317–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qadota H, Miyauchi T, Nahabedian JF, Stirman JN, Lu H, Amano M, Benian GM, Kaibuchi K. (2011). PKN-1, a homologue of mammalian PKN, is involved in the regulation of muscle contraction and force transmission in C. elegans. J Mol Biol , 222–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachat F, Garcea RL, Epstein HF. (1978). Myosins exist as homodimers of heavy chains: demonstration with specific antibody purified by nematode mutant myosin affinity chromatography. Cell , 405–411. [DOI] [PubMed] [Google Scholar]
- Schlaitz AL, Srayko M, Dammermann A, Quintin S, Wielsch N, MacLeod I, de Robillard Q, Zinke A, Yates JR, 3rd, Müller-Reichert T, et al (2007). The C. elegans RSA complex localizes protein phosphatase 2A to centrosomes and regulates mitotic spindle assembly. Cell , 115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schriefer LA, Waterston RH. (1989). Phosphorylation of the N-terminal region of Caenorhabditis elegans paramyosin. J Mol Biol , 451–454. [DOI] [PubMed] [Google Scholar]
- Shi Y. 2009. Serine/threonine phosphatases: mechanism through structure. Cell , 468–484. [DOI] [PubMed] [Google Scholar]
- Sieburth DS, Sundaram M, Howard RM, Han M. (1999). A PP2A regulatory subunit positively regulates Ras-mediated signaling during Caenorhabditis elegans vulval induction. Genes Dev , 2562–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small TM, Gernert KM, Flaherty DB, Mercer KB, Borodovsky M, Benian GM. (2004). Three new isoforms of Caenorhabditis elegans UNC-89 containing MLCK-like protein kinase domains. J Mol Biol , 91–108. [DOI] [PubMed] [Google Scholar]
- Solaro RJ, Kobayashi T. (2011). Protein phosphorylation and signal transduction in cardiac thin filaments. J Biol Chem , 9935–9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MH, Liu Y, Anderson DE, Jahng WJ, O’Connell KF. (2011). Protein phosphatase 2A-SUR-6/B55 regulates centriole duplication in C. elegans by controlling the levels of centriole assembly factors. Dev Cell , 563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek S, Zeng Y, Bellur DL, Bergkessel M, Morris KJ, Deng Q, Duong D, Seyfried NT, Guthrie C, Staley JP, et al (2016). The evolutionarily-conserved polyadenosine RNA binding protein, Nab2, cooperates with splicing machinery to regulate the fate of pre-mRNA. Mol Cell Biol , 2697–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terentyev D, Hamilton S. (2016). Regulation of sarcoplasmic reticulum Ca2+ release by serine-threonine phosphatases in the heart. J Mol Cell Cardiol ,156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry M, Walker DD, Ferrari MB. (2006). Protein phosphatase activity is necessary for myofibrillogenesis. Cell Biochem Biophys , 265–278. [DOI] [PubMed] [Google Scholar]
- Virshup DM, Shenolikar S. (2009). From promiscuity to precision: protein phosphatases get a makeover. Mol Cell , 537–545. [DOI] [PubMed] [Google Scholar]
- Wang JT, Smith J1, Chen BC, Schmidt H, Rasoloson D, Paix A, Lambrus BG, Calidas D, Betzig E, Seydoux G. (2014). Regulation of RNA granule dynamics by phosphorylation of serine-rich, intrinsically disordered proteins in C. elegans . Elife , e04591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner A, Xiong G, Qadota H, Rogalski T, Vogl AW, Moerman DG, Benian GM. (2013). CPNA-1, a copine domain protein, is located at integrin adhesion sites, and is required for myofilament stability in C. elegans . Mol Biol Cell , 601–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber S, Meyer-Roxlau S, Wagner M, Dobrev D, El-Armouche A. (2015). Counteracting protein kinase activity in the heart: the multiple roles of protein phosphatases. Front Pharmacol 6, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson KJ, Qadota H, Benian GM. (2012b). Immunofluorescent localization of proteins in Caenorhabditis elegans muscle. Methods Mol Biol , 171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson KJ, Qadota H, Mains PE, Benian GM. (2012a). UNC-89 (obscurin) binds to MEL-26, a BTB-domain protein, and affects the function of MEI-1 (katanin) in striated muscle of Caenorhabditis elegans . Mol Biol Cell , 2623–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingo TS, Duong DM, Zhou M, Dammer EB, Wu H, Cutler DJ, Lah JJ, Levey AI, Seyfried NT. (2017). Integrating next-generation genomic sequencing and mass spectrometry to estimate allele-specific protein abundance in human brain. J Proteome Res , 3336–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong G, Qadota H, Mercer KB, McGaha LA, Oberhauser AF, Benian GM. (2009). A LIM-9 (FHL)/SCPL-1 (SCP) complex interacts with the C-terminal protein kinase regions of UNC-89 (obscurin) in Caenorhabditis elegans muscle. J Mol Biol , 976–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Cuello F, Mayr U, Hao Z, Hornshaw M, Ehler E, Avkiran M, Mayr M. (2010). Proteomics analysis of the cardiac myofilament subproteome reveals dynamic alterations in phosphatase subunit distribution. Mol Cell Proteom , 497–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.