

Abstract
Background:
Hearing loss (HL) is a highly prevalent heterogeneous deficiency of sensory-neural system with involvement of several dozen genes. Whole-exome sequencing (WES) is capable of discovering known and novel genes involved with HL.
Materials and Methods:
Two pedigrees with HL background from Khuzestan province of Iran were selected. Polymerase chain reaction-sequencing of GJB2 and homozygosity mapping of 16 DFNB loci were performed. One patient of the first and two affected individuals from the second pedigree were subjected to WES. The result files were analyzed using tools on Ubuntu 16.04. Short reads were mapped to reference genome (hg19, NCBI Build 37). Sorting and duplication removals were done. Variants were obtained and annotated by an online software tool. Variant filtration was performed. In the first family, ENDEAVOUR was applied to prioritize candidate genes. In the second family, a combination of shared variants, homozygosity mapping, and gene expression were implemented to launch the disease-causing gene.
Results:
GJB2 sequencing and linkage analysis established no homozygosity-by-descent at any DFNB loci. Utilizing ENDEAVOUR, BBX: C.C857G (P.A286G), and MYH15: C.C5557T (P.R1853C) were put forward, but none of the variants co-segregated with the phenotype. Two genes, UNC13B and TRAK1, were prioritized in the homozygous regions detected by HomozygosityMapper.
Conclusion:
WES is regarded a powerful approach to discover molecular etiology of Mendelian inherited disorders, but as it fails to enrich GC-rich regions, incapability of capturing noncoding regulatory regions and limited specificity and accuracy of copy number variations detection tools from exome data, it is assumed an insufficient procedure.
Keywords: ENDEAVOUR, hearing loss, homozygosity mapping, Iran, whole-exome sequencing
Introduction
Hearing loss (HL) accounts for the most common sensory-neural defect worldwide with an incidence of 1–2 new cases/1000 newborns.[1] The disease incidence rises during childhood and adolescence lifetime to 2.83 and 3.5/1000 individuals, respectively.[2,3] According to the World Health Organization estimations, there are about 360 million deaf individuals throughout the world.[4] Due to the high rate of consanguineous marriages in Iran,[5] the disease frequency is much higher and stands second among disabilities in the country.[6] Patients with profound hearing difficulties suffer from poor linguistic communication, social engagement, cognitive activity, and low life quality.[7]
More than 50% of the cases have a genetic background. Hereditary HL is divided into two main categories, syndromic and nonsyndromic. Syndromic HL manifests with involvement of other body organs, while deafness is the only symptom in nonsyndromic form. About 75%–85% of nonsyndromic cases have autosomal recessive mode of inheritance. Autosomal dominant inheritance manner comprises 15%–24% of the patients. Rare causes, X-linked recessive and mitochondrial patterns, include 1%–2% of the affected individuals.[8] Lesions of outer ear cause conductive HL, while defects of the middle and inner ear lead to sensory-neural HL. According to age of onset HL is divided into prelingual and postlingual. The severity of disease spectrum ranges from mild to profound based on audiogram profile.[9]
More than 80 distinct loci and 60 genes have been discovered associated with autosomal recessive nonsyndromic HL (ARNSHL).[10] GJB2 mutations at DFNB1 locus are the most common cause of the disease in most part of the world.[11,12] However, the role of DFNB1 locus is variable because of existence of various ethnic groups in Iran.[13,14,15,16,17]
Whole-exome sequencing (WES) is a high-throughput technique which gives the opportunity to detect single-nucleotide variants mainly in the coding regions of the genome.[18,19] To some extent, it has the capability of finding copy number variations (CNVs) through special approaches.[20] Since the development of this technology, several novel genes have been introduced involved with ARNSHL.[21]
Various genes associated with the same heritable disorder usually play a role in certain pathways or have structural and functional similarities. Hence, based on previous knowledge about HL-associated genes, discovering novel genes could be an appropriate selection. Obtaining exome data from different individuals in a family would help to detect homozygous haplotypes and common variants, with the same pattern of inheritance, among affected individuals.
In this study, we selected two different approaches to discover novel disease-causing genes in two large Iranian families affected with ARNSHL. In one family, WES was performed for the proband and gene discovery was based on variant filtering and similarity searching using ENDEAVOR online software tool. In the second family, homozygosity mapping was performed by exome data of two affected individuals. Common disruptive variants, expression pattern and gene function were considered to prioritize candidate genes.
Materials and Methods
Subjects
Two large pedigrees (Ahv-17 and Sho-4) from Khuzestan province of Iran were ascertained with the history of several HL patients. Clinical investigations were accomplished including skeletal, eye, kidney, heart, and cutaneous examinations. In addition, magnetic resonance imaging and computed tomography scan were performed to detect inner ear malformations. Severity of deafness was assessed through pure-tone audiometry from 250 Hz to 8000 Hz.
The study was approved by ethics committee and review boards of Isfahan University of Medical Sciences. About 5–10 mL of peripheral blood from healthy and affected members of the pedigrees were collected in ethylenediaminetetraacetic acid-containing tubes following getting informed written consent from the participants.
Genetic linkage analysis of common DFNB loci
Genomic DNA was extracted from peripheral lymphocytes using Prime Prep Genomic DNA Extraction kit from blood (GeNet Bio, Korea) according to the manufacturer's instruction.
Coding mutations of the GJB2 gene were screened by polymerase chain reaction (PCR)-sequencing as described previously.[17]
More than 60 Short Tandem Repeat Polymorphic (STRP) markers linked to DFNB1A/B, DFNB2, DFNB3, DFNB4, DFNB7/11, DFNB9, DFNB15, DFNB21, DFNB22, DFNB24, DFNB42, DFNB59, DFNB63, DFNB93, and DFNB101 were selected from NCBI Map Viewer and UCSC genome browser. Primers were designed using Primer3 v. 0.4.0 (http://bioinfo.ut.ee/primer3–0.4.0/) [Table 1] or selected through NCBI Probe. Specificity of the primers was checked through NCBI Primer-BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Touchdown PCR was carried out to amplify STRP markers by 2X Master Mix (Ampliqon®, Denmark). To genotype STRP markers, PCR products were resolved on a nondenaturing 12%–15% polyacrylamide gel and stained under standard silver nitrate protocol. Genotypes were determined by visual inspection. Haplotype reconstruction was done by HaploPainter v. 1.043.
Table 1.
Primers used for co-segregation of the variants in the BBX and MYH15 genes

Whole-exome sequencing experiment, bioinformatics analysis, and variant prioritization
One affected individual from Sho-4 pedigree and two affected individuals from Ahv-17 pedigree were subjected to high-throughput sequencing. Genomic DNA was isolated from peripheral blood lymphocytes according to phenol–chloroform protocol. Using 1% agarose gel and Nanospec Cube Biophotometer (Nanolytik®, Dusseldorf, Germany) DNA concentration, purity and integrity were checked. About 1 μg of genomic DNA of each individual was utilized to prepare library sequencing in a solution-phase targeted genomic enrichment by Agilent SureSelect Human All Exon kit v6 (Agilent Technologies, CA, USA) according to the manufacturer's instruction. Sequencing was carried out by Illumina HiSeq 2000 (Illumina, San Diego, California, USA) to generate 105 bp short reads.
Sequence reads were aligned against human reference genome (hg19, NCBI Build 37) by BWA v0.7.8-r455 software. Samtools v1.0 was used to sort BAM files and Picard v1.111 was utilized to remark duplicates. Variant calling was accomplished by GATK v3.1 and variant annotation was done by wANNOVAR online software tool (http://wannovar.wglab.org/).
Missense, nonsense, stop loss, initiation codon change, splice site, small in-frame, and frameshift indels with CAAD score of >10 and global minor allele frequency of <1% in NCBI dbSNP human build 147, 1000 genomes project phase3, NHLBI GO exome sequencing project, exome aggregation consortium version 0.3.1 were retained for further analysis. Pathogenic effect of the variants was evaluated by SIFT, PolyPhen-2 v2.2.2r398, and MutationTaster2.
ENDEAVOUR gene prioritization and conformation
For the Sho-4 family, ENDEAVOUR (https://endeavour.esat.kuleuven.be/Endeavour.aspx) was applied for novel gene discovery. Human was selected as species. Known deafness-associated genes with various patterns of inheritance were utilized for training. Genes from exome data were used as data source to build models. Prioritized candidate genes were fully investigated for their function and probable association with HL by means of comprehensive literature review and online sources including UniProt (http://www.uniprot.org/) and GeneCards (www.genecards.org).
Primers were designed encompassing exon 10 of the BBX gene and exon 39 of the MYH15 gene [Table 1]. PCR-sequencing was carried out among healthy and affected family members.
Running autozygosity mapping by HomozygosityMapper web application
A Perl-based tool was accomplished to compare variant call format (VCF) files and pinpoint shared positions which variants are located. VCF tools v0.1.13 were utilized to find shared variants causing HL between candidates. The tool merges two output file and results in a VCF file with information about shared variants and facilitates decision-making. Using HomozygosityMapper web application (http://www.homozygositymapper.org/), homozygous chromosomal segments were detected. In the following, we focused on genes with expression in the inner and outer hair cells and auditory and vestibular ear ganglion neurons.[22,23,24]
Results
Clinical findings
Two large inbred pedigrees, each with 4 HL patients, from Khuzestan province of Iran were recruited for molecular investigations. Pedigree analysis revealed autosomal recessive mode of inheritance in both of the pedigrees [Figure 1]. Affected individuals in pedigree Sho-4 showed mixed progressive severe-to-profound HL when they were teenager. At the age of 20, all individuals suffered from mixed profound HL. Affected individuals in pedigree Ahv-17 had severe sensory-neural HL. Involvement of other body organs were not observed in any of the pedigrees.
Figure 1.

Pedigrees. There are four affected individuals in each pedigree due to consanguineous marriage that suggests autosomal recessive mode of inheritance
DFNB loci screening and whole-exome sequencing results
Direct sequencing showed no mutation in the coding exon of the GJB2 gene. DFNB1A/B linkage analysis ruled out the role of the most common cause of ARNSHL. After genotyping the STRP markers and haplotype reconstruction, we could not establish homozygosity-by-descent at any DFNB loci.
WES was implemented to recognize the molecular etiology of the disease among known deafness-associated genes or finding novel genes. The mean depth of coverage was 100X and more than 99% of targeted regions were covered. No variant was found in known deafness-associated genes after proper variant filtering.
ENDEAVOUR gene prioritization results
Four candidate genes according to their function, domain, and expression pattern were prioritized by ENDEAVOR [Table 2]. Genes and variants include BBX: C.C857G (P.A286G) and MYH15: C.C5557T (P.R1853C) were evaluated through PCR-sequencing but none of the variants to be co-segregating with the phenotype in the pedigree [Figures 2 and 3]. Primer sequences are available in Table 2.
Table 2.
List of candidate genes for families Sho-4 and Ahv-17
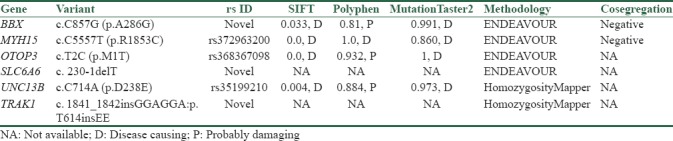
Figure 2.
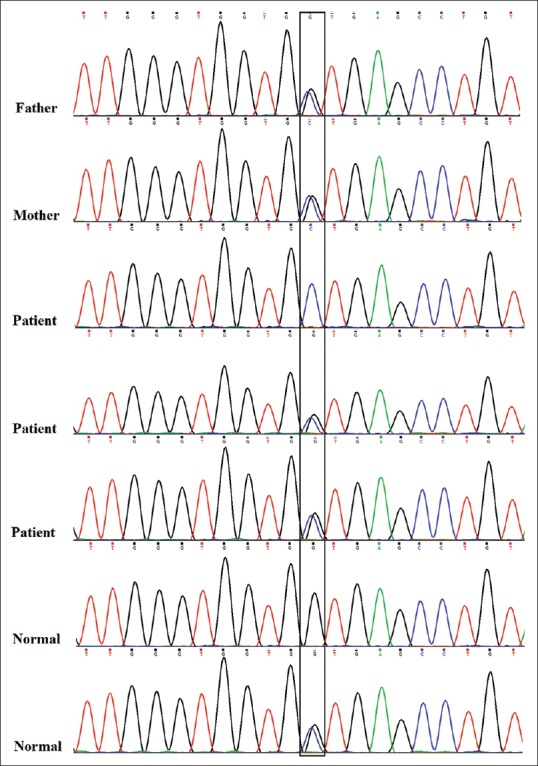
Sequence electropherogram of BBX: c.C857G (P.A286G) variant. The variant does not co-segregates with ARNSHL in the family as other affected members are heterozygous and one healthy sibling is homozygous for mutant allele
Figure 3.
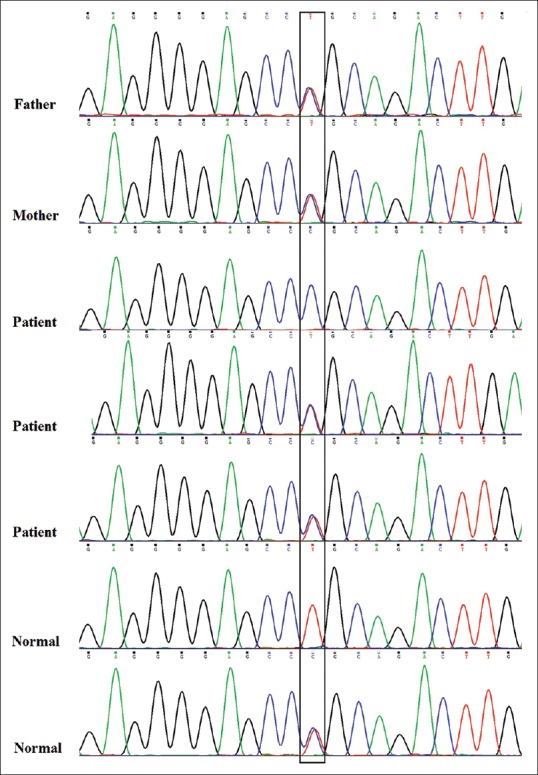
Sequence electropherogram of MYH15: c.C5557T (P.R1853C) variant. As one of the healthy individuals is homozygous for mutant allele and other affected individuals do not have homozygous mutant allele, this variant cannot be molecular etiology of the disease
Homozygosity Mapper results
Two variants, UNC13B: C.C714A (P.D238E) and TRAK1: C.1841-1842insGGAGGA (P.T614insEE), remained after autozygosity mapping, expression in the inner and outer hair cells, auditory and vestibular ear ganglion neurons, and shared in both affected individuals as novel candidate genes for ARNSHL [Table 2].
Discussion
By applying two different approaches, ENDEAVOUR gene prioritization and homozygosity mapping using WES data, we aimed to introduce novel genes related to ARNSHL in two large families from Southwest of Iran. Although ENDEAVOUR prioritized four interesting genes, co-segregation analysis of the first two candidates ruled them out. In the second family, a combination of homozygosity mapping by exome data of two affected individuals, ear gene expression pattern, and shared variants in both individuals suggested the UNC13B and TRAK1 genes as potential candidate genes for ARNSHL.
Although WES is a high-throughput sequencing method, some practical and analysis limitations prevent finding responsible variants for a single-gene disorder. First of all, this method is poor to enrich GC-rich regions of the genome.[25] Second, noncoding regulatory regions are not routinely captured by this methodology. Using whole-genome sequencing (WGS) would help us to uncover such pathogenic alterations, although variant interpretation and prioritization occurring in these regions is problematic.[26] The last limitation is detecting CNVs from exome data. Although several software and algorithms have been developed to overcome this problem, they have limited specificity and accuracy.[27] There are two disadvantages in using exome data for linkage analysis. First, only single-nucleotide polymorphisms (SNPs) at exonic regions will be captured and second, error rate is higher in comparison with array-based technologies.[28] Smith et al. implemented linkage analysis through exome data successfully to uncover genetic causes of recessive and dominant inherited diseases in three families and the results were comparable with SNP array-based linkage analysis.[28] Biallelic mutations in the WEE2 gene have been reported to cause female infertility using the same strategy.[29] ENDEAVOUR has been introduced as a powerful tool for gene prioritization through knowledge from former identified genes and genomics data to integrate them for global ranking.[30] Recent publications have verified the efficiency of ENDEAVOUR for discovering novel genes associated with neurodevelopmental disorder and congenital anomalies.[31,32,33,34]
As the families are large enough for genome-wide linkage analysis, a dense SNP array and using STRP markers could help us to limit the disease gene to an interval. In addition, WGS would be a method of choice to detect CNVs, noncoding regulatory region variants, and mutations in GC-rich regions.
Conclusion
WES has revolutionized molecular investigations in medical genetics practice. However the procedure has wet-lab and dry-lab limitations. It seems that genomic approaches like SNP arrays could help us for fine mapping of the disease interval. In addition, WGS could be method of choice for scanning CNVs, noncoding regulatory regions and GC-rich content of the identified interval.
Financial support and sponsorship
This work was financially supported by Isfahan University of Medical Sciences grant NO.394805, NO.394531 and NO.194068.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
We kindly appreciate our patients and their family for their participation in the project. This paper is published as part of thesis mainly done by Mohammad Reza Pourreza.
References
- 1.Morton CC, Nance WE. Newborn hearing screening – A silent revolution. N Engl J Med. 2006;354:2151–64. doi: 10.1056/NEJMra050700. [DOI] [PubMed] [Google Scholar]
- 2.Tucci D, Merson MH, Wilson BS. A summary of the literature on global hearing impairment: Current status and priorities for action. Otol Neurotol. 2010;31:31–41. doi: 10.1097/mao.0b013e3181c0eaec. [DOI] [PubMed] [Google Scholar]
- 3.LaSasso C, Lollis J. Survey of residential and day schools for deaf students in the United States that identify themselves as bilingual-bicultural programs. J Deaf Stud Deaf Educ. 2003;8:79–91. doi: 10.1093/deafed/8.1.79. [DOI] [PubMed] [Google Scholar]
- 4.Deafness and Hearing Loss. [Last accessed on 2018 Apr 11]. Available from: http://www.who.int/mediacentre/factsheets/fs300/en .
- 5.Saadat M, Ansari-Lari M, Farhud DD. Consanguineous marriage in Iran. Ann Hum Biol. 2004;31:263–9. doi: 10.1080/03014460310001652211. [DOI] [PubMed] [Google Scholar]
- 6.Mahdieh N, Rabbani B, Wiley S, Akbari MT, Zeinali S. Genetic causes of nonsyndromic hearing loss in Iran in comparison with other populations. J Hum Genet. 2010;55:639–48. doi: 10.1038/jhg.2010.96. [DOI] [PubMed] [Google Scholar]
- 7.Brink P, Stones M. Examination of the relationship among hearing impairment, linguistic communication, mood, and social engagement of residents in complex continuing-care facilities. Gerontologist. 2007;47:633–41. doi: 10.1093/geront/47.5.633. [DOI] [PubMed] [Google Scholar]
- 8.Oonk AM, Huygen PL, Kunst HP, Kremer H, Pennings RJ. Features of autosomal recessive non-syndromic hearing impairment: A review to serve as a reference. Clin Otolaryngol. 2016;41:487–97. doi: 10.1111/coa.12567. [DOI] [PubMed] [Google Scholar]
- 9.Schrijver I. Hereditary non-syndromic sensorineural hearing loss: Transforming silence to sound. J Mol Diagn. 2004;6:275–84. doi: 10.1016/S1525-1578(10)60522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang KW. Genetics of hearing loss – Nonsyndromic. Otolaryngol Clin North Am. 2015;48:1063–72. doi: 10.1016/j.otc.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 11.Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature. 1997;387:80–3. doi: 10.1038/387080a0. [DOI] [PubMed] [Google Scholar]
- 12.Morell RJ, Kim HJ, Hood LJ, Goforth L, Friderici K, Fisher R, et al. Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N Engl J Med. 1998;339:1500–5. doi: 10.1056/NEJM199811193392103. [DOI] [PubMed] [Google Scholar]
- 13.Bazazzadegan N, Nikzat N, Fattahi Z, Nishimura C, Meyer N, Sahraian S, et al. The spectrum of GJB2 mutations in the Iranian population with non-syndromic hearing loss – A twelve year study. Int J Pediatr Otorhinolaryngol. 2012;76:1164–74. doi: 10.1016/j.ijporl.2012.04.026. [DOI] [PubMed] [Google Scholar]
- 14.Koohiyan M, Hashemzadeh-Chaleshtori M, Salehi M, Abtahi H, Reiisi S, Pourreza MR, et al. GJB2 mutations causing autosomal recessive non-syndromic hearing loss (ARNSHL) in two Iranian populations: Report of two novel variants. Int J Pediatr Otorhinolaryngol. 2018;107:121–6. doi: 10.1016/j.ijporl.2018.01.012. [DOI] [PubMed] [Google Scholar]
- 15.Najmabadi H, Cucci RA, Sahebjam S, Kouchakian N, Farhadi M, Kahrizi K, et al. GJB2 mutations in Iranians with autosomal recessive non-syndromic sensorineural hearing loss. Hum Mutat. 2002;19:572. doi: 10.1002/humu.9033. [DOI] [PubMed] [Google Scholar]
- 16.Najmabadi H, Nishimura C, Kahrizi K, Riazalhosseini Y, Malekpour M, Daneshi A, et al. GJB2 mutations: Passage through Iran. Am J Med Genet A. 2005;133A:132–7. doi: 10.1002/ajmg.a.30576. [DOI] [PubMed] [Google Scholar]
- 17.Tabatabaiefar M, Alasti F, Zohour MM, Shariati L, Farrokhi E, Farhud D, et al. Genetic linkage analysis of 15 DFNB loci in a group of Iranian families with autosomal recessive hearing loss. Iran J Public Health. 2011;40:34–48. [PMC free article] [PubMed] [Google Scholar]
- 18.De Keulenaer S, Hellemans J, Lefever S, Renard JP, De Schrijver J, Van de Voorde H, et al. Molecular diagnostics for congenital hearing loss including 15 deafness genes using a next generation sequencing platform. BMC Med Genomics. 2012;5:17. doi: 10.1186/1755-8794-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tabatabaiefar MA, Alipour P, Pourahmadiyan A, Fattahi N, Shariati L, Golchin N, et al. A novel pathogenic variant in an Iranian ataxia telangiectasia family revealed by next-generation sequencing followed by in silico analysis. J Neurol Sci. 2017;379:212–6. doi: 10.1016/j.jns.2017.06.012. [DOI] [PubMed] [Google Scholar]
- 20.Poultney CS, Goldberg AP, Drapeau E, Kou Y, Harony-Nicolas H, Kajiwara Y, et al. Identification of small exonic CNV from whole-exome sequence data and application to autism spectrum disorder. Am J Hum Genet. 2013;93:607–19. doi: 10.1016/j.ajhg.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Atik T, Bademci G, Diaz-Horta O, Blanton SH, Tekin M. Whole-exome sequencing and its impact in hereditary hearing loss. Genet Res (Camb) 2015;97:e4. doi: 10.1017/S001667231500004X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scheffer DI, Shen J, Corey DP, Chen ZY. Gene expression by mouse inner ear hair cells during development. J Neurosci. 2015;35:6366–80. doi: 10.1523/JNEUROSCI.5126-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu H, Pecka JL, Zhang Q, Soukup GA, Beisel KW, He DZ, et al. Characterization of transcriptomes of cochlear inner and outer hair cells. J Neurosci. 2014;34:11085–95. doi: 10.1523/JNEUROSCI.1690-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu CC, Appler JM, Houseman EA, Goodrich LV. Developmental profiling of spiral ganglion neurons reveals insights into auditory circuit assembly. J Neurosci. 2011;31:10903–18. doi: 10.1523/JNEUROSCI.2358-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Metzker ML. Sequencing technologies – The next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 26.Smedley D, Schubach M, Jacobsen JO, Köhler S, Zemojtel T, Spielmann M, et al. A whole-genome analysis framework for effective identification of pathogenic regulatory variants in mendelian disease. Am J Hum Genet. 2016;99:595–606. doi: 10.1016/j.ajhg.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abel HJ, Duncavage EJ. Detection of structural DNA variation from next generation sequencing data: A review of informatic approaches. Cancer Genet. 2013;206:432–40. doi: 10.1016/j.cancergen.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith KR, Bromhead CJ, Hildebrand MS, Shearer AE, Lockhart PJ, Najmabadi H, et al. Reducing the exome search space for mendelian diseases using genetic linkage analysis of exome genotypes. Genome Biol. 2011;12:R85. doi: 10.1186/gb-2011-12-9-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sang Q, Li B, Kuang Y, Wang X, Zhang Z, Chen B, et al. Homozygous mutations in WEE2 cause fertilization failure and female infertility. Am J Hum Genet. 2018;102:649–57. doi: 10.1016/j.ajhg.2018.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tranchevent LC, Ardeshirdavani A, ElShal S, Alcaide D, Aerts J, Auboeuf D, et al. Candidate gene prioritization with endeavour. Nucleic Acids Res. 2016;44:W117–21. doi: 10.1093/nar/gkw365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hussain MS, Baig SM, Neumann S, Nürnberg G, Farooq M, Ahmad I, et al. A truncating mutation of CEP135 causes primary microcephaly and disturbed centrosomal function. Am J Hum Genet. 2012;90:871–8. doi: 10.1016/j.ajhg.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thienpont B, Zhang L, Postma AV, Breckpot J, Tranchevent LC, Van Loo P, et al. Haploinsufficiency of TAB2 causes congenital heart defects in humans. Am J Hum Genet. 2010;86:839–49. doi: 10.1016/j.ajhg.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Erlich Y, Edvardson S, Hodges E, Zenvirt S, Thekkat P, Shaag A, et al. Exome sequencing and disease-network analysis of a single family implicate a mutation in KIF1A in hereditary spastic paraparesis. Genome Res. 2011;21:658–64. doi: 10.1101/gr.117143.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thiel C, Kessler K, Giessl A, Dimmler A, Shalev SA, von der Haar S, et al. NEK1 mutations cause short-rib polydactyly syndrome type majewski. Am J Hum Genet. 2011;88:106–14. doi: 10.1016/j.ajhg.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]