

Sir,
A 66-year-old female presented with multiple brown reticulate pigmentary plaques on perineum for decades [Figures 1a and b]. A biopsy specimen showed irregular, filiform epidermal elongations of hyperpigmented rete ridges [Figure 1c]. Multiple subcutaneous nodules on groins were also noted [Figure 1a] and the biopsy proved that those were epidermal inclusion cysts. According to the above findings, Dowling–Degos disease (DDD) associated with multiple epidermal inclusion cysts were diagnosed. Her younger sister also had the same lesions. On the other hand, her brothers and nieces had multiple epidermal inclusion cysts on the trunk. Because of the benign prognosis of DDD, the patient preferred long-term observation.
Figure 1.
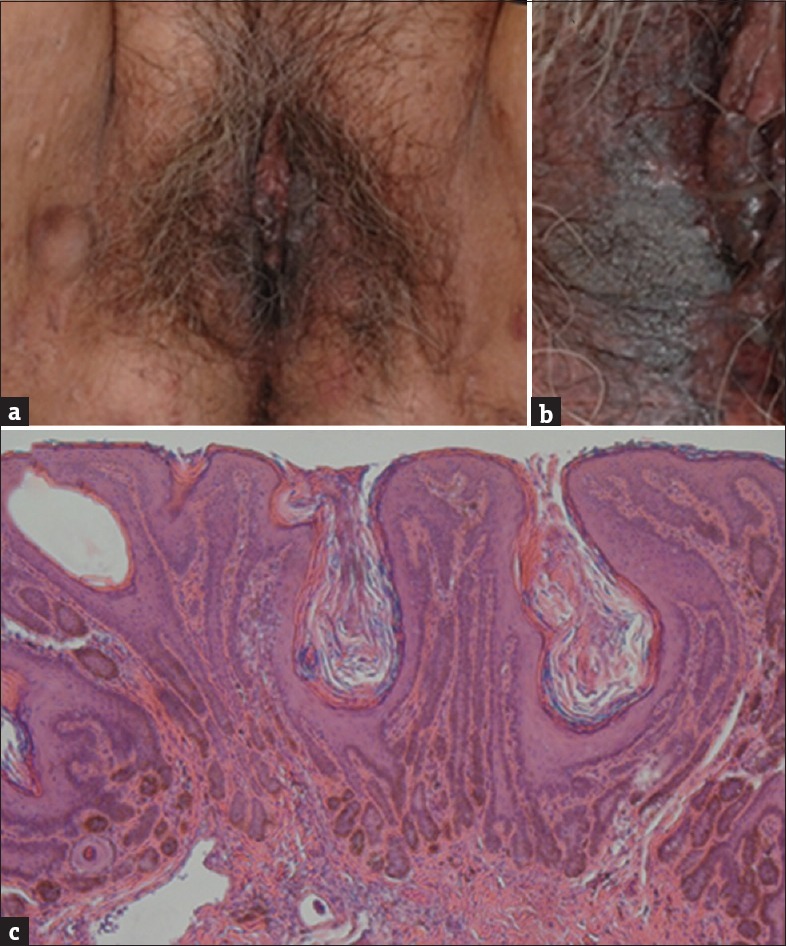
(a and b) Multiple brown reticulate pigmentary macules on labia minora and confluent brown plaques on labia majora with multiple epidermal inclusion cysts on inguinal area. (c) Typical filiform epidermal elongations of hyperpigmented rete ridges with a concentration of melanin at the tips (H and E, ×100)
As the genetic basis of DDD was recently reported, we focused our genetic analysis on exon 1 of the KRT5 gene. Direct DNA sequencing of PCR products derived from the patient did not reveal any mutation compared to the control sequence. We identified c.-73 T>C (negative strand), which was a single-nucleotide polymorphism (SNP) 4rs371725 of keratin 5 (K5) located in chromosome 12 52914153 [Figure 2]. This SNP was located at 5' end of exon 1 but not translated.
Figure 2.
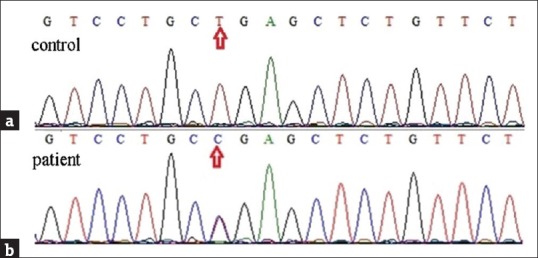
Mutation analysis. (a) Control from a normal person. (b) Direct DNA sequencing of exon 1 of KRT5 from the proband. We identified c.-73 T>C (complement strain), which was a single-nucleotide polymorphism of KRT5 gene located in chromosome 12
DDD is characterized by acquired, progressive pigmented lesions that primarily involve the large flexural areas. It is common that DDD involved the genitalia;[1] however, DDD resulted from SNP and associated with family members had not been reported. In addition, the distinct clinical presentation might be due to long-standing lesions with local stimuli by mechanical rubbing in daily life and it should be differentiated from condyloma acuminata, bowenoid papulosis, acanthosis nigricans, and seborrheic keratosis.
DDD may present as either sporadic or autosomal dominant genodermatosis with a strong family history. However, the genetic defect of DDD has not yet been well defined. A recently reported series mapped DDD to chromosome 12q and described the loss of function mutations in the exon 1 of the KRT5 gene encoding K5 in two German pedigrees[2] and heterozygous frameshift mutation in the V1 Domain of K5 in the proband from an extended Spanish DDD kindred.[3] It has been established that haploinsufficiency for K5 appears to be a common genetic mechanism underlying DDD.[4] A variety of follicular abnormalities, such as facial acne-like pitted scars, hyperkeratotic follicular papules, and comedones,[1] is associated with DDD and K5 may also play a role in follicle development. However, it failed to identify anomalies of the KRT5 gene in other cases with DDD.[5] We also failed to identify anomalies of the KRT5 gene. Another larger linkage analysis from a 4-generation Chinese family showed a possible DDD-related gene locus mapping to chromosome 17p13.3.[6]
Taken together, DDD is easily misdiagnosed as other sexual transmitted diseases, such as condyloma acuminata. It is crucial to have a detailed family history and physical examination. According to our result and those of other investigators, DDD is a heterogeneous disease and patients with DDD do not necessarily have KRT5 gene haploinsufficiency.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Kang HS, Hur J, Lee JW, Oh DH, Yeo KY, Kim JS, et al. A case of Dowling-Degos disease on the vulva. Ann Dermatol. 2011;23:205–8. doi: 10.5021/ad.2011.23.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Betz RC, Planko L, Eigelshoven S, Hanneken S, Pasternack SM, Bussow H, et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet. 2006;78:510–9. doi: 10.1086/500850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liao H, Zhao Y, Baty DU, McGrath JA, Mellerio JE, McLean WH, et al. A heterozygous frameshift mutation in the V1 domain of keratin 5 in a family with Dowling-Degos disease. J Invest Dermatol. 2007;127:298–300. doi: 10.1038/sj.jid.5700523. [DOI] [PubMed] [Google Scholar]
- 4.Guo L, Luo X, Zhao A, Huang H, Wei Z, Chen L, et al. A novel heterozygous nonsense mutation of keratin 5 in a Chinese family with Dowling-Degos disease. J Eur Acad Dermatol Venereol. 2012;26:908–10. doi: 10.1111/j.1468-3083.2011.04115.x. [DOI] [PubMed] [Google Scholar]
- 5.Asahina A, Ishii N, Kai H, Yamamoto M, Fujita H. Dowling-Degos disease with asymmetrical axillary distribution and no KRT 5 exon 1 mutation. Acta Derm Venereol. 2007;87:556–7. doi: 10.2340/00015555-0313. [DOI] [PubMed] [Google Scholar]
- 6.Li CR, Xing QH, Li M, Qin W, Yue XZ, Zhang XJ, et al. A gene locus responsible for reticulate pigmented anomaly of the flexures maps to chromosome 17p13.3. J Invest Dermatol. 2006;126:1297–301. doi: 10.1038/sj.jid.5700271. [DOI] [PubMed] [Google Scholar]