

Abstract
Misfolded secretory proteins are retained by endoplasmic reticulum quality control (ERQC) and degraded in the proteasome by ER-associated degradation (ERAD). However, in yeast and mammals, misfolded glycosylphosphatidylinositol (GPI)-anchored proteins are preferentially degraded in the vacuole/lysosome. We investigate this process in the divergent eukaryotic pathogen Trypanosoma brucei using a misfolded GPI-anchored subunit (HA:E6) of the trypanosome transferrin receptor. HA:E6 is N-glycosylated and GPI-anchored and accumulates in the ER as aggregates. Treatment with MG132, a proteasome inhibitor, generates a smaller protected polypeptide (HA:E6*), consistent with turnover in the proteasome. HA:E6* partitions between membrane and cytosol fractions, and both pools are proteinase K-sensitive, indicating cytosolic disposition of membrane-associated HA:E6*. HA:E6* is de-N-glycosylated and has a full GPI-glycan structure from which dimyristoylglycerol has been removed, indicating that complete GPI removal is not a prerequisite for proteasomal degradation. However, HA:E6* is apparently not ubiquitin-modified. The trypanosome GPI anchor is a forward trafficking signal; thus the dynamic tension between ERQC and ER exit favors degradation by ERAD. These results differ markedly from the standard eukaryotic model systems and may indicate an evolutionary advantage related to pathogenesis.
INTRODUCTION
The initial site for the biosynthesis, folding, and assembly of proteins destined for the secretory pathway is the endoplasmic reticulum (ER). Within the ER, an elaborate quality control system (ERQC) ensures that only completely folded and assembled proteins are exported to distal compartments of the secretory pathway. Terminally misfolded or incompletely assembled proteins are retained by the ERQC, retrotranslocated to the cytosol, and ultimately degraded by the proteasome system. This process is termed ER-associated degradation (ERAD; Määttänen et al., 2010; Brodsky, 2012; Hampton and Sommer, 2012). Despite the efficiency of ERQC, levels of misfolded proteins can sometimes exceed ER processing capacity, leading to stress within the ER lumen. When this occurs, an adaptive signaling response known as the unfolded protein response (UPR) is activated (Ron and Walter, 2007). UPR occurs in both yeast and metazoan cells and in each case promotes cell survival, since chronic ER stress can ultimately trigger apoptosis.
A special case for ERAD is disposal of misfolded glycosylphosphatidylinositol-anchored proteins (GPI-APs), in which the mature GPI anchor forms a positive forward trafficking signal for ER export. Recent work has suggested that two misfolded GPI-APs, PrP* and Gas1*, are poor ERAD substrates (Ashok and Hegde, 2008; Satpute-Krishan et al., 2014; Sikorska et al., 2016). In mammalian cells, under acute pharmacologically induced ER stress, misfolded PrP* is exported from the ER, transiently delivered to the plasma membrane via the Golgi, and eventually targeted to lysosomes for degradation (Satpute-Krishan et al., 2014). This stress-responsive pathway, termed “rapid ER stress-induced export” (RESET), has been proposed as a mechanism for alleviating ER overload for diverse misfolded GPI-APs in multiple mammalian cell types, but it also operates constitutively in unstressed cells (Satpute-Krishan et al., 2014). In yeast, misfolded GPI-anchored Gas1*, a cell wall protein, is degraded by both Pep4-dependent (vacuolar) and Hrd1-dependent (ERAD) pathways, albeit predominantly by the former pathway (Sikorska et al., 2016). It is worth noting that both Gas1*-TMD and soluble PrP* variants lacking GPI anchors are mainly degraded by ERAD (Ashok and Hegde, 2008; Sikorska et al., 2016). Collectively, these studies suggest that ERQC retention signals are in an opposing dynamic with functional ER export signals of GPI-APs, that is, mature remodeled GPI anchors, such that during states of overload, GPI recognition overrides ERQC retention mechanisms, resulting in ER export. Furthermore, because these studies were carried out in the conventional model systems of yeast and mammalian cells, this concept has emerged as a paradigm of protein folding and trafficking in the early secretory pathways of all eukaryotes (Sikorska et al., 2016).
Trypanosoma brucei is a unicellular eukaryotic protozoan parasite that causes African sleeping sickness in humans and nagana in cattle. The parasite resides extracellularly in the bloodstream and tissues of the mammalian host and relies on host-derived iron for survival, acquired via the high-affinity transferrin receptor (TfR; Ligtenberg et al., 1994; Salmon et al., 1994; Steverding et al., 1995; Tiengwe et al., 2017). TfR is essential for viability and is a heterodimer composed of two subunits, ESAG6 (E6, GPI-anchored) and ESAG7 (E7, non-GPI). At steady state, TfR localizes in endosomal compartments and in the flagellar pocket, where it binds and internalizes Tf. The receptor is continuously recycled to the flagellar pocket, but eventually TfR is diverted to the single terminal lysosome and degraded (t1/2 ∼ 1.5 h) (Schwartz et al., 2005; Tiengwe et al., 2017). Both subunits genes are located proximal to the promoter in ∼15 telomeric polycistronic loci known as expression sites (ES) (Hertz-Fowler et al., 2008). At any one time, transcription is restricted to a single active ES, but ∼20% of all TfR transcripts derive from limited transcription of the other “silent” ESs (Ansorge et al., 1999). Thus, targeted genetic manipulation of TfR subunits has been experimentally challenging. Consequently, we have developed an RNA interference (RNAi)-based system for manipulating expression of native and modified TfR subunits to unambiguously study its folding, intracellular trafficking, and mode of turnover in bloodstream-form trypanosomes (Tiengwe et al., 2016, 2017). Coincidentally, during these studies, we found that a misfolded non–GPI anchored Ty-tagged E7 reporter (E7:Ty) accumulates in the ER, and that its turnover is selectively rescued by the proteasomal inhibitor MG132, providing the first functional evidence for the ERAD process in T. brucei (Tiengwe et al., 2016).
The central virulence process in trypanosome infection is antigenic variation, the lynchpin of which is the variant surface glycoprotein (VSG; Schwede and Carrington, 2010; Rudenko, 2011). VSGs are abundant homodimeric GPI-anchored proteins that are structurally related to TfR, which make up a dense coat covering the entire cell surface. There are hundreds of VSG genes in the genome, but only one is expressed at a time from the same expression sites as TfR. The main mode of antigenic variation is gene conversion of the active gene from the repertoire of silent VSG genes, and this process is often segmental, leading to creation of new chimeric VSGs (Robinson et al., 1999; Mugnier et al., 2015). We argued previously (Tiengwe et al., 2016) that this process will inevitably generate “failed” VSGs that are incapable of folding, leading to catastrophic overexpression—10% of total cell protein—of misfolded GPI-anchored VSG. This situation would likely be lethal without rapid clearance, and we proposed ERAD as the mechanism for maintaining short-term viability. However, given that ERAD is not favored for disposal of misfolded GPI-APs in yeast and mammals, and that GPI anchors are also forward trafficking signals for ER exit in trypanosomes (Triggs and Bangs, 2003; Sevova and Bangs, 2009; Kruzel et al., 2017), it is not certain that ERAD can actually serve this purpose. We now ask whether a misfolded HA-tagged E6 reporter (HA:E6) is also degraded by ERAD, or by preferential transport to the lysosome. We perform detailed analyses of the functionality and fate of HA:E6 and its GPI-minus variant (HA:E6ΔG) in bloodstream-form trypanosomes. Our results indicate that ERAD is the main mode of disposal of misfolded GPI-APs, a finding that differs markedly from those in yeast and mammalian cells.
RESULTS
Expression of misfolded HA:E6 and HA:E6ΔG reporters
We initially created the HA:E6 reporters to study the behavior of TfR subunits without background interference of native TfR from “silent” ESs. We inserted a 3xHA epitope tag downstream of the EP1 Procyclin signal sequence and fused it in frame with RNAi-resistant E6 (Figure 1, HA:E6). This reporter failed in trafficking studies, as it did not efficiently bind Tf ligand and it localized to the ER (see below), suggesting a misfolded protein that is retained by ERQC. To further characterize this reporter and the role of GPI anchors in proteostasis, we generated a control GPI-minus variant by deleting the GPI-anchoring signal from the C-terminus of HA:E6 to generate HA:E6ΔG (Figure 1). The two constructs were independently expressed in situ from the active expression site in an inducible TfR RNAi cell line (Tiengwe et al., 2016). Without RNA silencing, constitutive expression of the HA:E6 or HA:E6ΔG reporters did not impact the viability of the cell lines (Figure 2A). However, upon silencing of wild-type TfR, both reporters were unable to rescue growth, as there are no native E6 and E7 subunits to form functional heterodimers. Without silencing, these partners are provided by low-level transcription from the other expression sites (Ansorge et al., 1999), thereby maintaining viability. As seen previously with a misfolded E7:Ty reporter (Tiengwe et al., 2016), silencing of native E6 (∼90% reduction) in both cell lines led to an ∼10-fold increase in the steady state level of the RNAiR reporter transcripts (Figure 2B). Immunoblotting with anti-TfR confirmed the depletion of native E6 protein (Figure 2C, αTfR) and up-regulation of RNAiR HA:E6 and HA:E6ΔG proteins (Figure 2C, αHA), mimicking the observed increase in RNAiR transcripts. These results are consistent with changes in native TfR transcript and protein levels seen under iron-starvation conditions (Mussman et al., 2003, 2004; Tiengwe et al., 2016, 2017). Levels of the cytoplasmic marker Hsp70 remained unaffected by TfR silencing, validating the specificity of loss of native E6 and up-regulation of the tagged RNAiR subunits. Importantly, the steady state level of the ER marker BiP was also unaffected, indicating that accumulation of both misfolded HA:E6 and HA:E6ΔG in the ER (see below) did not induce a UPR-like response. A similar lack of UPR was also seen previously with misfolded E7:Ty reporter (Tiengwe et al., 2016). Finally, TfR silencing specifically abrogated endocytosis of Tf in both cell lines (Figure 2D), but did not affect uptake of tomato lectin (TL), a surrogate for receptor-mediated endocytosis, indicating that general endocytosis remained unimpaired.
FIGURE 1:
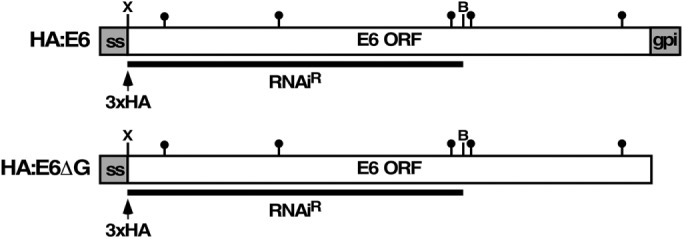
Schematic representation of E6 reporters. Diagrams depict RNAiR GPI-anchored (HA:E6) and GPI-minus (HA:E6ΔG) HA-tagged ESAG6 reporters. N-terminal gray boxes (ss) denote the EP1 Procyclin signal sequence (not to scale); arrowhead indicates position of HA epitope tag (3xHA); filled lollipops indicate N-linked glycan sites; C-terminal gray box (gpi) signifies the GPI attachment peptide (not to scale); underbar indicates the E6 RNAiR insert with flanking XhoI (X) and BamHI sites (B).
FIGURE 2:

Expression of E6 reporters. TfR RNAi cell lines containing RNAiR HA:E6 or HA:E6ΔG were cultured without or with tetracycline. (A) Cell density was enumerated by hemocytometer and cultures were adjusted to starting density daily. Data are presented for three biological replicates (mean ± SD). All subsequent analyses were performed after 24 h of silencing. (B) Wild-type RNAi-sensitive E6 (E6wt) and RNAiR HA:E6 or HA:E6ΔG (E6R) transcript levels were assessed by qRT-PCR. Results are normalized to uninduced controls and are presented as fold changes for three biological replicates (mean ± SD). (C) Cell extracts were subjected to immunoblotting with rabbit anti-TfR (αTfR), mAb anti-HA (αHA), rabbit anti-Hsp70 (αH70), or anti-BiP (αBiP). All lanes have 107 cell equivalents. Mobilities of molecular mass markers are indicated. Star indicates a nonspecific background polypeptide consistently observed with anti-TfR. (D) Receptor-mediated uptake of fluorescent transferrin (Tf) and tomato lectin (TL) was measured by flow cytometry. Data are presented as median fluorescent intensity (MFI ± SD) for three biological replicates and are normalized to unsilenced control cells.
Localization of HA:E6 and HA:E6ΔG
Native TfR heterodimer normally localizes to the flagellar pocket and endosomal compartments, consistent with its role as a recycling nutrient receptor. However, when expressed alone, without the E7 subunit, wild-type E6 homodimerizes and relocalizes to the cell surface (Tiengwe et al., 2017). To determine the location of HA:E6 and HA:E6ΔG, we performed immunofluorescent costaining with the ER molecular chaperone BiP and variant surface glycoprotein (VSG) in the absence of endogenous TfR subunits (Figure 3). Under TfR silencing, both HA:E6 and HA:E6ΔG staining markedly overlapped with BiP indicating that at steady state both reporters localize to the ER. The same pattern was seen previously for misfolded GPI-minus E7:Ty (Tiengwe et al., 2016). No colocalization of HA:E6 with VSG was observed, suggesting that cell surface trafficking, if it does occur, is below the level of detection. Without TfR silencing, no anti-HA staining was seen with either reporter, consistent with the observed low levels of expression in the presence of native TfR (Figure 2C).
FIGURE 3:

Location of E6 reporters under TfR silencing. IFA was performed post–TfR RNAi induction with the HA:E6 (A)- or HA:E6ΔG (B)-expressing cell lines. Staining of fixed permeabilized cells was performed with mAb anti-HA (left, red), and with rabbit anti-BiP or rabbit anti-VSG (middle, green). Three-channel merged images with DAPI staining (blue) are presented (right). In each case, representative deconvolved summed stacked projections are presented. Cell outlines were traced from matched DIC images.
GPI anchoring of HA:E6 and HA:E6ΔG
To assess the GPI-anchor status of HA:E6 and HA:E6ΔG, we utilized the fact that bloodstream-form trypanosomes have a tightly regulated endogenous GPI-specific phospholipase C (GPI-PLC) that is activated by nondenaturing lysis and that cleaves the phosphodiester bond between inositol and dimyristoylglycerol in the GPI anchor (Bülow and Overath, 1986; Hereld et al., 1986). GPI-PLC cleavage exposes the residual GPI glycan structure (EtN-P-Man3-GlcN-cyclic Inos-P), which forms a cross-reacting determinant (CRD) epitope reactive with specific anti-CRD antibodies (Barbet and McQuire, 1978; Zamze et al., 1988). Thus, a positive cross-reaction under such conditions is diagnostic for the presence of a GPI anchor.
Following native TfR silencing, detergent lysis, and GPI-PLC–mediated cleavage, HA:E6 and HA:E6ΔG cell lysates were immunoprecipitated with either anti-TfR or anti-VSG and then subjected to immunoblotting with anti-CRD (Figure 4). Strong reactivity was seen with HA:E6 but not HA:E6ΔG (αCRD, top left). The weak signal in HA:E6ΔG pull downs likely represents contaminating native VSG, which is superabundant and has the same electrophoretic mobility as HA:E6 (αHA, top right). Note that the smaller size of HA:E6ΔG in the anti-HA blot is consistent with the absence of a GPI anchor. As an internal control, GPI-anchored native VSG showed positive anti-CRD reactivity in both cell lines (αCRD, bottom left), confirming GPI-PLC activity, validating functionality of the assay, and confirming equal recovery and loading in both cell lines (αVSG, bottom right). These results demonstrate that the HA:E6 reporter, but not HA:E6ΔG, is efficiently attached to a GPI anchor.
FIGURE 4:
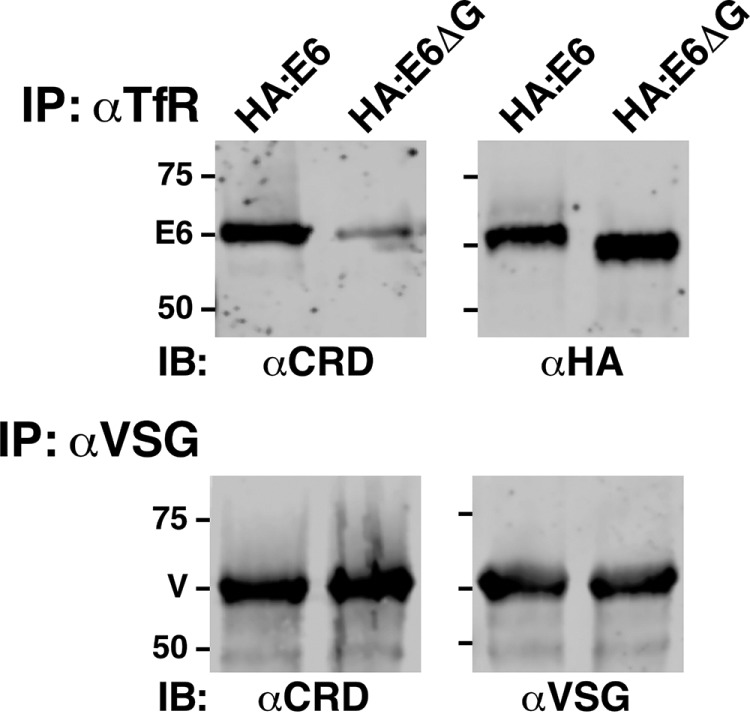
GPI anchoring of E6 reporters. Following TfR silencing, HA:E6- or HA:E6ΔG-expressing cells were lysed at 108 cells/ml in TEN buffer containing 1% NP40. Lysates were incubated at 37°C for 5 min to allow complete hydrolysis of all GPI anchors by the activity of endogenous GPI-specific phospholipase C. Lysates were then immunoprecipitated (IP) as indicated with anti-TfR (αTfR, 107 cell equivalents/precipitate) or anti-VSG (αVSG, 5 × 105 cell equivalents/precipitate) antibodies covalently cross-linked to protein A sepharose. One set (top) of matched anti-TfR precipitates was immunoblotted (IB) with anti-CRD (αCRD), and the other with anti-HA (αHA). Likewise, one set (bottom) of matched anti-VSG precipitates was immunoblotted with anti-CRD (αCRD), and the other with anti-VSG. Images from a representative experiment are presented. Mobilities of E6 subunits, VSG, and molecular mass markers (kDa) are indicated.
HA-E6 and HA:E6ΔG are nonfunctional
To address the activity of the HA:E6 and HA:E6ΔG reporters, we assessed the ability of each to form functional TfR heterodimers. These experiments were performed without TfR silencing to allow the presence of native E7 subunits for heterodimerization. Sequential pull downs with lysates from both cell lines were performed with anti-HA and transferrin-conjugated beads (Tf beads), followed by immunoblotting with anti-HA or anti-TfR. 1o and 2o pull down with anti-HA depleted essentially all HA:E6 from cell lysates, leaving no reporter detectable in 3o pull down with Tf beads (Figure 5A, top, lanes 1–3, αHA blot). However, abundant functional heterodimer formed by native E6 and E7 remained (Figure 5A, bottom, lane 3, αTfR blot). Conversely, 1o and 2o Tf-bead pull downs, despite detecting large amounts of functional native TfR heterodimer (Figure 5A, bottom, lanes 4 and 5, αTfR blot), detected very little HA:E6, most of which was recovered in 3o pull down with anti-HA (Figure 5A, top, lanes 4–6, αHA blot). These results indicate that HA:E6 is severely impaired in forming functional TfR. However, HA:E6 apparently heterodimerizes, as seen by stoichiometric recovery of E7 polypeptide in anti-HA pull downs (Figure 5A, bottom, lanes 1 and 6, αTfR blot). This was confirmed by Native Blue Gel electrophoresis of whole lysates followed by immunoblotting with anti-HA. HA:E6 migrated as a single dominant species of a size consistent with heterodimerization with endogenous E7, and urea treatment partly reduced this to a monomer (Figure 5B, lanes 1 and 2). As a loading control, VSG was detected as a larger dimer that was almost completely converted to monomer by denaturation (Figure 5B, lanes 5 and 6). Note that the relative resistance of HA:E6 heterodimers to urea denaturation versus native VSG was seen previously with native TfR (Tiengwe et al., 2017). In the absence of native E7, however, HA:E6 was largely detected as a monomer, along with a considerable high–molecular weight smear that is partially converted to monomer by urea treatment, consistent with aggregation (Figure 5B, lanes 3 and 4). In contrast, we have previously shown that, when expressed without E7, wild-type E6 efficiently homodimerizes and is exported from the ER (Tiengwe et al., 2017). Collectively, these results indicate that 1) HA:E6 is capable of heterodimerization when endogenous E7 is available; 2) the resultant TfR heterodimers are largely nonfunctional for Tf binding; and 3) in the absence of E7, the reporter is incapable of homodimerization. In every critical respect, the GPI-minus HA:E6ΔG reporter behaves identically to HA:E6 (Figure 5, C and D). Thus, both reporters are severely impaired in folding and assembly in the absence of partner E7 subunit.
FIGURE 5:

Misfolding of E6 reporters. (A, C) Cell extracts were prepared from HA:E6- or HA:E6ΔG-expressing cell lines without silencing of native TfR. Aliquots (107 cell equivalents) were sequentially immuno/affinity precipitated in two sets [1° and 2° αHA, 3° Tf-beads (Tfb), lanes 1–3; and 1° and 2° Tfb, 3° αHA, lanes 4–6). Pull downs were subjected to simultaneous immunoblotting with mAb anti-HA and rabbit anti-TfR using Licor IR-fluorescent secondary antibodies with different emission wavelengths. The anti-HA (top) and anti-TfR (bottom) signals were digitally separated for presentation. Mobilities of E6/E7 subunits and molecular mass markers (kDa) are indicated. (B, D) Cell extracts were prepared with and without RNAi silencing (tet +/-) and fractionated by Blue Native gel electrophoresis (106 cell equivalents/lane) with and without denaturation (urea +/-). Gels were transblotted and probed simultaneously with mAb anti-HA and rabbit anti-VSG and specific Licor secondary reagents. Anti-HA and anti-VSG signals were digitally separated for presentation. Mobilities of E6 dimers (dE6), E6 monomers (mE6), VSG dimers (dVSG), VSG monomers (mVSG), and molecular mass markers (kDa) are indicated.
Fate of misfolded HA:E6 and HA:E6ΔG reporters
Studies in yeast and mammalian cells found that misfolded GPI-APs are typically targeted for vacuolar/lysosomal degradation (Ashok and Hegde, 2008; Satpute-Krishan et al., 2014; Sikorska et al., 2016), while those lacking GPI anchors are eliminated by ERAD (Ashok and Hegde, 2008). Having validated GPI attachment for HA-E6, we investigated the cellular degradation pathway(s) involved in its disposal. First, we measured turnover of steady state E6 reporters in TfR silenced cells following inhibition of protein synthesis with cycloheximide. Cells were simultaneously treated with the proteasome inhibitor MG132 (ERAD pathway) or with the cathepsin-L inhibitor FMK024 (lysosomal pathway). Control HA:E6 cells showed continuous loss of HA:E6 over 4 h, and treatment with FMK024 had little effect on this loss (Figure 6A, lanes 1–3 and 4–6). However, treatment with MG132 markedly delayed loss of HA:E6 and led to accumulation of a protease-protected species of ∼50 kDa (Figure 6A, lanes 7–9, HA:E6*), which must have an intact N-terminus, since it is reactive with anti-HA antibody. No additional rescue was seen by combined treatment with FMK024 and MG132 (Figure 6A, lanes 10–12). Identical results were obtained with the GPI-minus HA:E6ΔG reporter (Figure 6B). In contrast, native E6, which is normally degraded in the lysosome as part of the TfR heterodimer (Schwartz et al., 2005; Tiengwe et al., 2017), was completely rescued by FMK024 (Figure 6C, lanes 1–3 vs. lanes 4–6). MG132 also partially rescued turnover (Figure 6C, lanes 7–9), albeit less effectively—a not unexpected result, as we have previously demonstrated that MG132 cross-inhibits lysosomal thiol protease activity (Tiengwe et al., 2016). Treatment with FMK024 and MG132 gave the same maximal rescue seen with FMK024 alone (Figure 6C, lanes 10–12).
FIGURE 6:

Turnover of E6 reporters. (A, B) HA:E6- and HA:E6ΔG-expressing cell lines were harvested after TfR RNAi induction (24 h) and cultured with cycloheximide (CHX, 100 μg/ml) to stop protein synthesis. The cells were also treated with FMK024 (FMK, 20 μM) or MG132 (MG, 25 μM) as indicated. Samples (107 cell equivalents) were collected at 0, 2, or 4 h and immunoblotted with anti-HA (αHA) or anti-Hsp70 (αH70). Assay measures loss of steady state reporter as a function of time. The mobility of the MG132-protected species is indicated by a star. (C) Turnover of native E6 was assessed in the HA:E6 cell line without silencing exactly as in panels A and B. (D) Turnover of HA:E6 and HA:E6ΔG reporters was assayed by pulse/chase (15 min/4 h) radiolabeling in cells post–TfR RNAi (24 h). As indicated, cells were either untreated (Ct) or treated with MG132 (MG, 25 μM). Typical phosphor images of anti-HA immunoprecipitates at the indicated chase times are presented (107 cell equivalents/lane). The mobilities of the MG132-protected E6 species are indicated by a star. Loss of the HA:E6 or HA:E6ΔG reporter during the chase period was quantified (mean ± SD, n = 5). (E) IFA was performed post–TfR RNAi induction with the HA:E6-expressing cell line, with and without FMK024 treatment (20 μM, 4 h). Staining of fixed permeabilized cells was performed with mAb anti-HA (green) and with rabbit anti-TbCatL (red). Deconvolved three-channel summed stack images with DAPI staining (blue) are presented. Insets are the single channel anti-HA images in the region of the lysosome. Bar, 2 μm.
To independently validate the mode of degradation, we examined the biosynthesis and turnover of the HA:E6 and HA:E6ΔG reporters by quantitative pulse-chase radiolabeling experiments in TfR silenced cells (Figure 6D). Newly synthesized HA:E6 reporter again rapidly disappeared, but without the appearance of the larger mature glycoform indicative of transport to the Golgi (Schwartz et al., 2005; Tiengwe et al., 2017) (Figure 6D, left top), suggesting that there is little if any anterograde transport of this reporter from the ER. As in the cycloheximide chase, MG132 treatment dramatically reduced the rate of degradation and generated the smaller HA:E6* species. Essentially identical behavior was observed with the HA:E6ΔG reporter (Figure 6D, left bottom). These results strongly indicate that in the absence of E7 both reporters fail to leave the ER by anterograde secretory transport.
To confirm that HA:E6 does not access the lysosome, we performed IFA of control and FMK024-treated cells imaging reporter versus the lysosomal marker TbCatL (Figure 6E). In control cells, HA:E6 was seen with prominent ER localization as in Figure 3, and the lysosome presented as a single locus in the postnuclear region. No lysosomal localization was seen with the reporter. FMK024 treatment led to typically enlarged lysosomes, but again, no colocalization with HA:E6 was seen. Had the reporter been delivered to the lysosome in significant amounts, we would have expected FMK024 to prevent degradation, thereby generating a lysosomal signal, as we have seen for other mistargeted reporters (Triggs and Bangs, 2003; Schwartz et al., 2005; Silverman et al., 2013).
Along with the pull down and Native Blue Gel findings (Figure 5), these data suggest that E7 can rescue the folding defect of HA:E6 and HA:E6ΔG, leading to production of heterodimers, albeit nonfunctional for Tf binding. However, in the absence of a dimerization partner, both apparently misfold catastrophically and are subject to degradation by the ERAD pathway. Collectively, and in contrast to yeast and mammals, these results suggest that ERAD is the preferential mode of degradation for misfolded GPI-APs in trypanosomes.
Characterization of the MG132 protected species
During ERAD, misfolded glycoproteins are retrotranslocated to the cytosol and deglycosylated by a peptide:N-glycanase prior to proteasomal degradation (Carvalho et al., 2010; Suzuki et al., 2016). To further investigate the involvement of the ERAD pathway in turnover of HA:E6 and HA:E6ΔG, we tested whether these reporters are extracted from the ER to the cytosol. TfR silenced cells expressing HA:E6 or HA:E6ΔG were treated with MG132, and cell lysates were separated by centrifugation into membrane-associated and cytosolic fractions, as indicated by the distribution of endogenous BiP (membrane) and Hsp70 (cytosol), respectively. Again, MG132 treatment led to accumulation of HA:E6* in HA:E6 cells (Figure 7A, left, lane 1 vs. lane 4). While HA:E6 was recovered exclusively in the membrane fraction (Figure 7A, left, lanes 3 and 6), HA:E6* was distributed evenly between cytosol and membranes (Figure 7A, left, lanes 5 and 6). Identical results were obtained with the soluble HA:E6ΔG reporter (Figure 7A, right). These results suggest that generation of the protected species is related to retrotranslocation to the cytosol, consistent with the ERAD pathway.
FIGURE 7:

Characterization of MG132 protected species. All analyses were performed after RNAi silencing (24 h) of endogenous TfR. All MG132 treatments were for 2 h at 25 μM. (A) HA:E6- and HA:E6ΔG-expressing cells were incubated without (Cont) or with MG132 (MG). The cells were hypotonically lysed and total (T), cytoplasmic (C), and membrane (M) fractions were prepared. All fractions (107 cell equivalents/lane) were analyzed by immunoblotting with anti-HA (αHA), anti-Hsp70 (αH70, cytoplasmic marker), or anti-BiP (αBiP, ER marker). (B) Cell fractions prepared from MG132-treated HA:E6 cells were treated with proteinase K (Prot-K) as indicated in the absence or presence of NP40. Samples (107 cell equivalents/lane) were immunoblotted with anti-HA, anti-Hsp70, and anti-BiP. (C–E) HA:E6- or HA:E6ΔG-expressing cells were incubated without (-) or with (+) MG132 treatment. (C) Cells were solubilized under denaturing conditions and each was split into two equal fractions (107 cell equivalents). One set was mock-treated (-) and the other digested (+) with PNGase F (PNG). Samples were analyzed by immunoblotting with anti-HA. (D) Lysates were immunoprecipitated with anti-TfR antibodies (107 cell equivalents/precipitate) covalently cross-linked to Protein A sepharose. One set (right) was immunoblotted with anti-HA (αHA) and the other blotted with biotinylated ConA (left). Mobilities of E6 and molecular mass markers (kDa) are indicated on the left. White strip indicates digital reordering of lanes after image processing. Stars in A–D indicate mobility of MG132-protected E6 or fully deglycosylated species. (E) Total extracts of HA:E6-expressing cells were prepared in SDS sample buffer, and lysates for immunoprecipitation were prepared identically to GPI-PLC-lysates in Figure 8 to minimize deubiquitinating activities. Total cell extracts (lanes 1–4) and anti-HA immunoprecipitates (lanes 5–8) were immunoblotted (107 cell equivalents/lane) sequentially with anti-ubiquitin and anti-HA on the same membrane and the individual signals were processed prior to digital separation for presentation.
To confirm this possibility, we performed exogenous protease protection experiments with fractions from MG132-treated cells (Figure 7B, HA:E6 cell line only). Proteinase K treatment was performed with and without added detergent to solubilize membranes, and immunoblotting was performed with anti-HA, anti-Hsp70, and anti-BiP. Treatment completely eliminated HA:E6* from the cytosolic fraction without addition of detergent, as it did for the cytosolic marker Hsp70 (Figure 7B, lanes 2 and 3). A small amount of BiP is released from the ER by the fractionation procedure, and this too is protease-sensitive. On the other hand, both HA:E6 and BiP in the membrane fraction were largely resistant to protease in the absence, but not the presence, of detergent (Figure 7B, lanes 6–9). Small amounts of contaminating Hsp70 were fully susceptible to protease. Interestingly, the membrane-associated HA:E6* species was also fully sensitive to proteolysis, even in the absence of detergent. This may represent minor cytosolic contamination of the membrane fraction, but its relative abundance suggests that it represents HA:E6* that is physically associated with the cytosolic side of the ER.
Typically, cytosolic ERAD substrates are deglycosylated as a prerequisite for proteasomal degradation. The wild-type E6 amino acid sequence contains five potential N-glycosylation sites (Figure 1). We investigated the glycosylation status of HA:E6* by 1) treating cell lysates with PNGase F, which removes all N-glycans, and 2) assessing its reactivity with the α-mannose-specific lectin concanavilin A (ConA). PNGase F treatment led to disappearance of HA:E6 and accumulation of a polypeptide identical in size to HA:E6* (Figure 7C, lane 1 vs. lane 3), consistent with full deglycosylation. However, the electrophoretic mobility of HA:E6* itself in MG132-treated cells was unaffected (Figure 7C, lane 2 vs. lane 4), suggesting that this species lacks N-linked glycans. Moreover, HA:E6 gave a positive signal with ConA, but HA:E6* was nonreactive (Figure 7D, ConA), confirming the lack of N-glycans. Equal recovery and loading in both cell lines are shown by anti-HA immunoblotting (Figure 7D, αHA). Identical results were obtained with HA:E6ΔG in both deglycosylation and lectin blotting assays. Collectively, these results indicate that cytosolic MG132-protected HA:E6 and HA:E6ΔG polypeptides undergo complete removal of N-linked glycans (presumably by an endogenous cytosolic glycanase) prior to degradation through the proteasome-dependent pathway.
Typically, ERAD substrates are poly-ubiquitinylated during the retrotranslocation process as a signal for subsequent degradation by the proteasome. However, the sharp electrophoretic mobility of HA:E6* and the lack of upward laddering suggest that the protected species is not ubiquitinylated. To investigate this further, whole cell extracts and anti-HA immunoprecipitates were subjected to blotting with anti-ubiquitin antibodies. A broad range of ubiquitinylated polypeptides was detected in control cell extracts, and these signals were elevated in MG132-treated cells (Figure 7E, lanes 1 and 2), consistent with proteasomal inhibition. However, there was no indication of ubiquitinylated HA:E6*, even though blotting with anti-HA confirmed the typical pattern of HA:E6 and HA:E6* polypeptides in these extracts (Figure 7E, lanes 3 and 4). This was confirmed with anti-HA pull downs, which were negative in anti-ubiquitin blots (Figure 7E, lanes 5 and 6), again despite the presence of the HA:E6 and HA:E6* species in the immunoprecipitates (Figure 7E, lanes 7 and 8). These results indicate that at steady state the HA:E6* species is not ubiquitinylated.
GPI-anchor cleavage precedes proteolysis of misfolded HA:E6
Finally, we asked whether removal of the GPI anchor from misfolded HA:E6 also occurs prior to engagement with the proteasome. Cell extracts were prepared by procedures that either preserve GPI anchors or support removal of dimyristoylglycerol by the endogenous GPI-PLC (Ferguson et al., 1985; Bangs et al., 1986). Samples were immunoprecipitated with anti-HA antibody and then immunoblotted with anti-CRD to assess GPI status, followed by blotting with anti-HA to confirm the presence of the expected polypeptides (Figure 8A). As a control for GPI-PLC activation, parallel pull downs and blots were performed to assess the GPI status of endogenous VSG (Figure 8B). There are three possible outcomes of this experiment: 1) anti-CRD reactivity only following GPI-PLC activation, indicating a fully intact GPI anchor at the time of lysis; 2) anti-CRD reactivity without GPI-PLC activation, indicating cleavage of the GPI anchor prior to lysis; and 3) no reactivity under any conditions, indicating partial or complete removal of the residual GPI glycan. Strong anti-CRD reactivity with HA:E6 was seen, but only after activation of GPI-PLC (Figure 8A, lanes 1 and 2 vs. lanes 3 and 4). Likewise, and as expected, VSG was reactive with anti-CRD only after GPI-PLC activation, validating our experimental conditions (Figure 8B, lanes 1 and 2 vs. lanes 3 and 4). These results indicate that HA:E6 is sequestered from GPI-PLC, which resides on the cytoplasmic face of internal membranes (Bülow et al., 1989; Sunter et al., 2013), consistent with its ER location (Figure 3A) and Proteinase K protected status (Figure 7B). In contrast, HA:E6* was equally reactive with anti-CRD regardless of GPI-PLC activation (Figure 8A, lane 2 vs. lane 4), indicating that an intact GPI glycan remains following in vivo removal of dimyristoylglycerol and prior to proteasomal degradation. It is striking that HA:E6* is apparently fully GPI-hydrolyzed, yet has a strong membrane-associated fraction (Figure 7A). To resolve this issue, we prepared cell fractions and assessed the GPI status of cytosolic and membrane-associated HA:E6* (Figure 8C). Both pools were anti-CRD reactive in the same ratio (∼50:50) as the actual polypeptides (anti-HA blot), strongly suggesting that all of HA:E6*, whether soluble or membrane-bound, has hydrolyzed GPI anchors. The implications of this finding for an ordered pathway for retrotranslocation and proteasomal degradation are discussed below.
FIGURE 8:

GPI-anchor status of HA:E6*. Following TfR silencing (24 h), HA:E6-expressing cells were incubated with MG132 (2 h) to block proteasomal degradation. Cells were then detergent-lysed under conditions that either subject all GPI anchors to hydrolysis by endogenous GPI-PLC (PLC+) or preserved GPI integrity (PLC-). (A) Lysates (107 cell equivalents) were immunoprecipitated with mouse anti-HA cross-linked to Protein G Sepharose and sequentially immunoblotted with rabbit anti-CRD (top panel) followed by mAb anti-HA (bottom panel), generating matched signals from the same membrane. (B) Lysates (5 × 105 cell equivalents) were immunoprecipitated with rabbit anti-VSG cross-linked to Protein A Sepharose and then immunoblotted with rabbit anti-CRD (top panel) or mAb anti-VSG (bottom panel), generating matched signals from the same membrane. Blots from a single representative experiment are presented in A and B. (C) Total (T), cytosolic (C), and membrane (M) fractions were prepared as in Figure 7A. HA:E6 polypeptides were immunoprecipitated (107 cell equivalents) with anti-HA and then blotted with anti-CRD (top) followed by anti-HA (bottom), generating matched signals from the same membrane. Mobilities of intact HA:E6 (E6), protected deglycosylated HA:E6 (star), and VSG (V) are indicated on the left in A–C; mobilities of molecular mass markers on the right.
DISCUSSION
The ERQC and ERAD machineries are found broadly across the eukaryotic domain, but the actual processes have been defined mainly in the standard model systems of mammalian cells and yeast (Määttänen et al., 2010; Brodsky, 2012; Hampton and Sommer, 2012). For misfolded luminal proteins, as well as transmembrane proteins with misfolded luminal domains, a similar pathway is followed. Unfolded proteins are retained in the ER by calreticulin and/or calnexin, which recognize monoglucosylated N-glycans. These proteins go through cycles of glucosylation/binding and deglucosylation/release, mediated by UDP-glucose:glycoprotein glucosyltransferase and ER glucosidase II, during which they are acted upon by resident ER molecular chaperones. Once folded, proteins are terminally deglucosylated and packaged in COPII-coated vesicles for export to the Golgi (Hughes and Stephens, 2008). Proteins that fail to achieve proper folding are eventually subject to turnover by ERAD. First, terminal α1-2–linked mannose residues are removed from one or more N-glycans by ER α-mannosidase I and related mannosidase-like lectins, EDEMs, thereby terminating the glucosylation cycle. A single terminal α1-6–linked mannose on the residual C branch is recognized by the OS-9 lectin, which delivers the misfolded protein to a membrane complex for retrotranslocation to the cytosol. The ubiquitin ligase (Hrd1) assists in that process and mediates subsequent ubiquitinylation. The tagged protein is then extracted from ER membranes by an AAA ATPase, p97/CDC48 (aka, VCP), and prior to degradation by the proteasome, N-glycans are removed by a cytosolic peptide:N-glycanase (Suzuki et al., 2016).
The T. brucei genome (http://tritrypdb.org) has obvious orthologues of all the critical components of the ERAD pathway (Table 1), except ER α-mannosidase I and N-glycanase. Presumably TbEDEMs mediate α1-2 mannose removal, and there is a strong orthologue of cytosolic endo-β-N-acetylglucosaminidase, which can substitute for N-glycanase in deficient mammalian cells (Suzuki et al., 2016). Not surprisingly then, we have demonstrated previously that disposal of a soluble misfolded E7 subunit (E7:Ty) proceeds via the ERAD pathway (Tiengwe et al., 2016). Most notably, turnover of E7:Ty was rescued by MG132, resulting in accumulation of a smaller protected species.
TABLE 1:
T. brucei ERAD orthologues.
| Protein | GENE IDa | E valueb | Reference |
|---|---|---|---|
| TbEDEM1-3 | Tb927.8.2910-2940 | 4.8 × 10-71 | Engstler et al. (2006); Field et al. (2010) |
| TbHrd1 | Tb927.9.5260 | 2.0 × 10-7 | This work |
| TbOS-9 | Tb927.11.10700 | 2 × 10-12 | Field et al. (2010); this work |
| TbVCP | Tb927.10.5770 | 0.0 | Roggy and Bangs (1999) |
| TbENGasec | Tb927.9.3400 | 2 × 10-55 | This work |
aTriTryp Database (http://tritrypdb.org).
bBLASTP query with human orthologue.
cCytosolic endo-β-N-acetylglucosaminidase.
We have now extended this work to include the misfolded GPI-anchored reporter HA:E6. A matched GPI-minus reporter (HA:E6ΔG) was used as a control for most experiments; it behaved in all ways identically to HA:E6 and E7:Ty. HA:E6 was capable of forming heterodimers with endogenous native E7, but they were nonfunctional for Tf binding, suggesting that native E7 is able to act cooperatively to assist in the folding of HA:E6. Whether the inability to bind Tf is due to residual localized misfolding of HA:E6 within the heterodimer, or to occlusion of the binding site by the epitope tag, is not clear. In the absence of E7, the misfolding phenotype of HA:E6 was much more severe. Native E6, when expressed alone, forms folded homodimers that do not bind Tf, but traffic freely to the flagellar pocket and onto the cell surface (Tiengwe et al., 2017). In contrast, when expressed without a dimerization partner, misfolded HA:E6 1) accumulated in the ER, 2) was not subject to normal glycan processing characteristic of transit to the Golgi, and 3) presented as a mixture of monomer and high–molecular weight aggregates. These findings are all consistent with a severe misfolding defect with consequent retention in the ER by ERQC. Despite this, HA:E6 was fully N-glycosylated and received a GPI anchor. As seen in other reports (Izquierdo et al., 2009a; Tiengwe et al., 2015, 2016), accumulation of misfolded aggregated HA:E6 in the ER did not induce a UPR-like response, as the levels of endogenous BiP remained unaltered.
Degradation of HA:E6 was specifically retarded by the proteasomal inhibitor MG132, but not the lysosomal thiol protease inhibitor FMK024, resulting in accumulation of a smaller protected species, HA:E6*. The physical characteristics of HA:E6* are consistent with a preproteasomal substrate—a full-length de-N-glycosylated cytoplasmically disposed polypeptide with intact N (HA tag)- and C (GPI glycan)-termini. Strikingly, although HA:E6* is fully sensitive in protease protection assays, it is distributed fairly evenly between cytosol and membrane fractions. This raised the possibility that following retrotranslocation, the polypeptide associates with ER membranes via an intact GPI anchor, and that release to the cytosolic preproteasomal pool is by GPI-PLC hydrolysis. However, using reactivity with anti-CRD antibody, we found that both pools of HA:E6* retain the delipidated soluble GPI glycan structure. Trypanosomes have an enigmatic GPI-PLC activity that is associated with the cytosolic face of internal membranes (Bülow et al., 1989; Sunter et al., 2013); presumably this enzyme is responsible for cleavage. Two observations argue that most, if not all, of the steady state HA:E6* has a hydrolyzed GPI anchor: 1) reactivity with anti-CRD is the same whether GPI-PLC is active or inactive during lysis (Figure 8A, lane 2 vs. lane 4), indicating that cleavage occurs in vivo prior to lysis, and 2) the ratio of CRD-positive HA:E6* in cytosol and membrane fractions is the same as the ratio of total HA:E6* (Figure 8C, lane 2 vs. lane 3). Collectively, these data indicate that both N-glycan removal and GPI hydrolysis occur prior to extraction from the ER membrane.
These results allow a working model for misfolded GPI-APs in trypanosomes (Figure 9). First, the target protein is retained in the ER by the calreticulin cycle (Conte et al., 2003; Izquierdo et al., 2009a; trypanosomes do not have a calnexin orthologue). The protein fails to achieve final folding, but the forward trafficking properties of the GPI anchor (see below) are insufficient to overcome ERQC-mediated retention. Trimming of oligomannose N-linked oligosaccharides eventually terminates the glucosylation/deglucosylation cycle and engages the luminal ERAD machinery for delivery to the Hrd1-containing retrotranslocation complex. Following export, the target protein remains membrane-associated, either with the retrotranslocon or with some other membrane-associated chaperone. We presume that the polypeptide is ubiquitinylated by Hrd1 at this time (but see below). While still membrane-associated, the target protein is subject to deglycosylation and GPI hydrolysis by cytoplasmic endo-β-N-acetylglucosaminidase and membrane-associated GPI-PLC, respectively. The GPI anchor could initially contribute to membrane association, but continued retention must involve some other protein:protein interaction following GPI hydrolysis, consistent with the fact that the GPI-minus HA:E6ΔG* reporter also distributes equally between cytosol and membrane fractions (Figure 7A). Finally, the target protein is extracted from the membrane to the soluble pool by the trypanosomal p97 orthologue, TbVCP, for delivery to the proteasome. An important aspect of this model, which is supported by our data, is that complete degradation of the GPI anchor is not necessary for proteasomal degradation, only the removal of dimyristoylglycerol. Proteasomal degradation can begin at either terminus or from internal sites of target proteins (Berko et al., 2012). It is possible, then, that degradation of misfolded GPI-APs could proceed to the ultimate C-terminal residue with attached GPI glycan. The remaining linkage is essentially a peptide bond between the amino acid α-carboxyl and the amine of the GPI phosphoethanolamine, which might be a substrate for proteasomal cleavage, following which the residual glycan would be degraded by some other mechanism. In this regard, it is worth noting that in mammals the residual cytosolic N-glycans that are removed from ERAD substrates are delivered to the lysosome for ultimate catabolism (Suzuki, 2016). Perhaps a similar process disposes of the residual GPI glycan.
FIGURE 9:

ERAD model in trypanosomes. 1) Newly synthesized GPI-anchored glycoprotein enters into the calnexin cycle of de-/reglucosylation. 2) The protein fails to achieve final folding status, but the GPI forward trafficking signal is unable to overcome retention by ER quality control. 3) The calnexin cycle is broken by TbEDEM-mediated demannosylation. 4) The trimmed glycoprotein is delivered to the TbHrd1-containing retrotranslocation complex by the lectin TbOS-9. 5) Translocation occurs, presumably with concomitant ubiquitinylation by TbHrd1, but this is not certain, based on data presented here (indicated by “?”). Dimyristoylglycerol is removed by membrane-bound GPI-PLC. N-glycans are removed by cytosolic endo-β-N-acetylglucosaminidase (TbENG). 6) The substrate is extracted from the membrane by TbVCP and delivered to the proteasome for degradation.
There are two somewhat confounding issues with this working model. First, while we presume that HA:E6* is poly-ubiquitinlyated, based on work in other systems, we were unable to detect this modification by immunoblotting. Whether this is because HA:E6* ubiquitinlyation does not happen at all, or because it is subject to deubiquitinylation when subsequent proteolysis is blocked (or even during lysis and sample preparation) is not clear. The second is presumed de-N-glycosylation by the putative cytosolic endo-β-N-acetylglucosaminidase, TbENG. BSF trypanosomes transfer two distinct N-glycan structures to nascent secretory proteins in the ER—standard triantennary Man9GlcNAc2 and abbreviated biantennary Man5GlcNAc2 (Izquierdo et al., 2009b, 2012). The former structure is susceptible to cleavage by commercially available endo-β-N-acetylglucosaminidase H (EndoH), which is of bacterial origin, but the latter is not. E6, which has 5 N-glycan sites, has a mixture of both structures (Mehlert et al., 2012). Thus, if TbENG is responsible for removal of all N-glycans, it requires a broader substrate specificity than EndoH. This issue does not arise in other eukaryotic systems because only triantennary Man9GlcNAc2 is transferred to secretory proteins.
Our findings contrast starkly with similar work in yeast and mammalian cells (Ashok and Hegde, 2008; Satpute-Krishan et al., 2014; Sikorska et al., 2016). In these systems, misfolded GPI-APs, such as Gas1p* in yeast and PrP* in mammals, are preferentially degraded by secretory export from the ER and delivery to the vacuole/lysosome. Initially, it was thought that the presence of a GPI anchor sterically blocked entry into the ERAD pathway, since non–GPI anchored versions of PrP* were robust ERAD substrates (Ashok and Hegde, 2008; Satpute-Krishan et al., 2014). More recently, however, it has been shown with the yeast/Gas1p* system that misfolded GPI-APs are facile ERAD substrates if forward trafficking from the ER is blocked (Sikorska et al., 2016), suggesting that a dynamic tension exists between GPI-mediated forward trafficking and ERQC-mediated retention of misfolded GPI-APs. GPI anchors function as forward trafficking signals in both yeast and mammalian cells. In each case, the final step of postattachment GPI remodeling before ER exit is removal of phosphoethanolamine from the second mannose of the GPI core, allowing GPI cargo to be recognized by transmembrane p24 receptor complexes for inclusion in COP II vesicles (Castillon et al., 2011; Fujita et al., 2011). Under normal conditions, this process is apparently able to override the ERQC/ERAD pathway.
We have generated considerable evidence that GPI anchors also act as forward trafficking signals for ER exit in trypanosomes: 1) deletion of the GPI signal sequence reduces rates of ER exit (Triggs and Bangs, 2003); 2) conversely, addition of a GPI signal accelerates forward trafficking of soluble cargo (Kruzel et al., 2017); and 3) GPI-dependent ER exit is mediated by a specific subset of COPII Sec23:Sec24 heterodimers (Sevova and Bangs, 2009) and 4) also by a subset of p24 orthologues (Kruzel et al., 2017). One difference from yeast and mammals is that the trimannosyl core of the newly attached GPI anchor is undecorated with phosphoethanolamine (Krakow et al., 1986; Menon et al., 1988). The core can be modified with variable side-chain galactose residues, but this occurs primarily after transport to the Golgi (Bangs et al., 1986, 1988; Mayor et al., 1992). Thus, it is possible that the same exposed second mannose as seen in yeast and mammals serves as the recognition signal for capture by p24 complexes. Whatever the signal, it is clear that in trypanosomes the dynamic tension between ERQC and ER exit tilts toward retention and destruction by ERAD.
In summary, this work extends our previous efforts demonstrating that the processes of ERQC and ERAD span the broad range of eukaryotic phylogeny (Tiengwe et al., 2016). However, in contrast to yeast and mammals, ERAD is the primary mode of eliminating misfolded GPI-anchored proteins in trypanosomes. These results confirm that ERAD could provide critical disposal capacity should antigenic variation produce a “failed” misfolded VSG. We suggest that trypanosomes have evolved to rely on ERAD as the most efficient method of coping with such a catastrophic event. Future efforts will focus on testing whether ERAD can also function for disposal of deliberately misfolded VSGs.
MATERIALS AND METHODS
Cell lines and culture
All experiments were carried out with the bloodstream-form Lister 427 strain of T. brucei brucei (MITat1.2 expressing VSG221) grown at 37°C in HMI9 medium (Hirumi and Hirumi, 1994). The tetracycline-responsive single-marker (SM) MITat1.2 BSF cell line (Wirtz et al., 1999) was used for all experiments involving inducible RNAi silencing of endogenous TfR expression. For experiments, cells were harvested at mid-late log phase (0.5 × 106–1 × 106). Generation of the TfR RNAi cell line using SM cells as the parental cell line has been extensively described in (Tiengwe et al., 2016). Induction of anti-TfR double-stranded RNA was achieved by addition of 1 μg/ml tetracycline.
Construction of epitope-tagged RNAi-resistant ESAG6
We generated misfolded HA-tagged E6 reporters (Figure 1) for unrelated studies on TfR trafficking based on constructs available in the laboratory and described in Tiengwe et al. (2016). Briefly, using our pXS6 vector (Silverman et al., 2011) as the backbone, the GPI-anchored construct was assembled as follows (5′-3′): 5′ UTR targeting region (nts –484 to +1 relative to the E6 ORF); hygromycin resistance cassette; βα-tubulin intergenic region; EP1 Procyclin signal sequence (Tb927.10.20160; nts 1-111, codons 1–37, cleavage site at 27/28) fused in frame to a 3xHA epitope tag (1x: YPYDVPDYA); the in-frame E6 ORF minus the native signal sequence (nts 58–1206, codons 20–402); 3′ UTR targeting region (nts 1–601 relative to E6 stop codon). All E6 segments were PCR-amplified from H25N7 BAC DNA containing the BES1 telomeric expression site (Berriman et al., 2002; gift of Gloria Rudenko, Imperial College). Finally, the 5′ end of the E6 ORF (nts 54–742, codons 20–247, XhoI-BamHI) was replaced with a synthetic recoded RNAi-resistant E6 segment (Integrated DNA Technologies) to generate the N-terminally 3xHA tagged RNAi-resistant E6 reporter (referred to as HA:E6). To generate the GPI-minus variant of HA:E6, nts 58–1125 was PCR-amplified from the HA:E6 construct, digested with XhoI/MfeI, and cloned into the HA:E6 construct with the same restriction enzymes creating HA:E6ΔG, without a GPI attachment signal (Figure 1). All segments were confirmed by sequencing. The resultant RNAiR reporters were linearized with ClaI/FseI for homologous replacement of the endogenous E6 gene in the active ES1 of the TfR RNAi cell line as described previously in Tiengwe et al. (2016, 2017).
Antibodies and blotting reagents
The following antibodies have been described in our prior publications (Silverman et al., 2011; Tiengwe et al., 2016): mouse and rabbit anti-VSG221, rabbit anti-BiP, rabbit anti-HSP70, rabbit anti-TbCatL, and mouse mAb and rabbit polyclonal anti-HA (Sigma). Anti–cross reacting determinant (CRD) is described in Bangs et al. (1985). Rabbit anti-TfR (BES1 specific) was a generous gift of Piet Borst and Henri Luenen (Netherlands Cancer Institute), and biotinylated concanavalin A (ConA) was from Vector Laboratories. P4D1 monoclonal anti-ubiquitin was from Thermo Fisher. Secondary reagents for Western blotting were IRDye800CW streptavidin and IRDye680- and IRDye800-conjugated goat anti-rabbit and anti-mouse immunoglobulin G (IgG) (Li-Cor, Lincoln NB). Secondary reagents for immunofluorescent imaging were species-specific Alexa-conjugated goat anti-IgG as appropriate (Molecular Probes).
Endocytosis assay
Endocytosis was assayed by flow cytometry as described in Silverman et al. (2011). Ligands were Alexa488 conjugated bovine transferrin and tomato lectin (Molecular Probes).
Immunoblotting
Gels were transferred to Immobilon-P membranes (Millipore) using a Trans-Blot Turbo apparatus (BioRad). Membranes were blocked and probed with appropriate dilutions of primary and secondary antibodies in Odyssey Blocking Buffer (LI-COR Biosciences). All washes were with PBS, 0.5% Tween20. Quantitative fluorescent signals were scanned on an Odyssey CLx Imager (LI-COR Biosciences).
qRT-PCR
Specific transcript levels were determined using quantitative reverse transcription-PCR (qRT-PCR). Total RNA was isolated from log phase cultures using RNeasy Mini kit (Qiagen). RNA was treated with DNase1 on column using an RNase-Free DNase Set (Qiagen) and cDNA was synthesized using an iScript cDNA synthesis kit (BioRad). qPCR was performed using diluted cDNAs and Power SYBR green PCR Master Mix (Life Technologies) with oligonucleotide pairs specifically targeting transcripts for wild-type E6 and RNAiR HA:E6 and HA:E6ΔG. The positions of these primers are described in Tiengwe et al. (2017). TbZFP3 (Tb927.3.720, nts 241–301) was used as the control amplicon. Amplification was performed using an Applied Biosystems StepOne real-time PCR system (Life Technologies). For each transcript, postamplification melting curves indicated a single dominant product. All calculations and normalizations were done using StepOne software, version 2.2.2. Reactions were performed in triplicate, and means ± standard deviation (SD) for three biological replicates are presented.
Epifluorescence microscopy
Immunofluorescence (IFA) microscopy was performed with formaldehyde-fixed/detergent-permeabilized cells as described in Bangs (2011). Cells were also stained with 4’,6’-diamidino-2-phenylindole (DAPI) (0.5 μg ml-1) to reveal nuclei and kinetoplasts. Serial image stacks (Z-increment 0.2 μm) were collected with capture times from 100 to 500 ms (100× PlanApo, oil immersion, 1.46 na) on a motorized Zeiss Axioimager M2 stand equipped with a rear-mounted excitation filter wheel, a triple-pass (DAPI/FITC/Texas Red) emission cube, differential interference contrast (DIC) optics, and an Orca ER CCD camera (Hamamatsu). Images were collected with a Volocity 6.1 Acquisition Module (Improvision) and individual channel stacks were deconvolved by a constrained iterative algorithm, pseudocolored, and merged using a Volocity 6.1 Restoration Module. All images presented are summed stack projections of merged channels. The xyz pixel precision of this arrangement has been validated in Sevova and Bangs (2009; see Figure S1 therein).
Assessing GPI-anchor status of reporters
Cell lines expressing reporters were lysed at 108 cells/ml in TEN buffer (50 mM Tris HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA) containing 1% NP40 and protease inhibitor cocktail (PIC) (Bangs et al., 1986). Lysates were incubated at 37°C for 5 min to allow complete hydrolysis of all GPI anchors by the specific activity of endogenous GPI-specific phospholipase C (GPI-PLC) and were then adjusted to RIPA detergent conditions for immunoprecipitation (107 cells/ml in TEN containing 1% NP40, 0.5% deoxycholate, 0.1% SDS, PIC). All lysates were clarified by centrifugation prior to immunoprecipitation, SDS–PAGE, and immunoblotting. Alternatively, to generate matched lysates without activation of GPI-PLC, cells were lysed in 1% SDS at room temperature (10 min) and then adjusted to final RIPA conditions on ice. This same procedure was used to minimize deubiquitinylation while generating lysates for immunoprecipitation and anti-ubiquitin blotting.
Blue native PAGE (BN-PAGE)
BN-PAGE was performed using the NativePAGE Bis-Tris Gel System (Thermo Fisher Scientific). Briefly, cells expressing both reporters were harvested without TfR silencing, washed with HBS, and solubilized in NativePAGE sample buffer supplemented with 10% glycerol, 1% DDM (n-dodecyl-β-d-maltoside), 1× protease inhibitor cocktail, and 100 μg/ml DNaseI. The samples were incubated in the solubilization buffer on ice for 30 min and centrifuged (13,000 × g at 4°C, 1 h), and the resulting supernatants were either untreated or treated with 4 M urea to denature protein complexes. Samples were then fractionated on precast 4–16% BN gradient gels (Thermo Fisher Scientific). After electrophoresis, proteins were transferred to polyvinylidene membranes (Millipore) and detected by our standard immunoblotting protocol with anti-HA or anti-VSG221.
Cycloheximide chase experiments
Cell lines expressing reporters were harvested after TfR RNAi induction (24 h) and cultured (5 × 106/ml) with cycloheximide (CHX; 100 μg/ml) to stop protein synthesis. The cells were also treated with FMK024 (FMK; 20 μM) or MG132 (MG; 25 μM) as indicated. Samples (1 × 107) were collected at 0, 2, or 4 h and immunoblotted with anti-HA (αHA) or anti-Hsp70 (αH70). Assay measures loss of steady state reporter as a function of time.
Metabolic labeling and immunoprecipitation
Log-phase cells expressing reporters were pulse-chase radiolabeled with [35S]methionine/cysteine, and subsequent immunoprecipitation of labeled polypeptides was performed as described previously (Tazeh and Bangs, 2007; Peck et al., 2008). Pulse and chase times are indicated in the figure legends. All immunoprecipitates were fractionated by SDS–PAGE, and gels were analyzed by phosphor imaging using a Molecular Dynamics Typhoon FLA 9000 system with native ImageQuant Software (GE Healthcare).
Cell fractionation and protease protection
Cell lines expressing reporters were cultured (1 × 106 cells/ml) with dimethyl sulfoxide alone or with MG132 (Sigma-Aldrich; 25 μM) for 2 h. The cells were harvested, hypotonically lysed (109 cells/ml) in distilled H2O plus protease inhibitor cocktail and then adjusted to 1× TEN buffer conditions. Cytosolic and membrane fractions were separated by centrifugation (17,000 × g for 10 min at 4°C). Total, cytosolic, and membrane fractions (107 cells equivalents) were processed for standard SDS–PAGE and immunoblotting. For protease protection, cytosolic and membrane fractions were prepared in the same manner, substituting TLCK (tosyl-lysyl-chloromethyl-ketone; Sigma-Aldrich). Samples (107 cells equivalents, 25 μl) were treated (37°C, 30 min) with Proteinase K (Promega; 50 µg/ml final) in 1× TEN buffer with 10 mM CaCl2 and in the presence or absence of 0.1% NP40 (final volume 40 μl). The enzyme was inhibited by addition of phenylmethylsulfonyl fluoride (PMSF, 5 mM final; Sigma-Aldrich) on ice for 5 min. Samples were then processed for standard SDS–PAGE and immunoblotting.
Deglycosylation and lectin blotting
Cell lines expressing reporters were cultured in HMI-9 growth media (1 × 106 cells/ml) with DMSO alone or with MG132 for 2 h. The cells were harvested, solubilized under denaturing conditions with PIC, and then treated with peptide N-glycanase (PNGase F) according to the manufacturer’s specifications (New England BioLabs). Samples were then fractionated by SDS–PAGE for standard immunoblotting or immunoprecipitated in RIPA buffer (as described above) and subsequent blotting with biotinylated ConA (1:3000) followed by IRDye800CW streptavidin.
Acknowledgments
We are grateful to Piet Borst and Henri Luenen (Netherlands Cancer Institute) for anti-TfR antibody. This work was supported by U.S. Public Health Service Grant R01 AI035739 and funds from the Jacobs School of Medicine and Biomedical Sciences to J.D.B.
Abbreviations used:
- ConA
concanavalin A
- CRD
cross-reacting determinant
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- ERQC
ER quality control
- ES
expression site
- ESAG6&7
expression site–associated genes 6 and 7
- GPI
glycosylphosphatidylinositol
- GPI-PLC
GPI-specific phospholipase C
- Tf
transferrin
- TfR
transferrin receptor
- VSG
variant surface glycoprotein.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-06-0380) on August 9, 2018.
REFERENCES
- Ansorge I, Steverding D, Melville S, Hartmann C, Clayton C. (1999). Transcription of “inactive’” expression sites in African trypanosomes leads to expression of multiple transferrin receptor RNAs in bloodstream forms. Mol Biochem Parasitol , 81–94. [DOI] [PubMed] [Google Scholar]
- Ashok A, Hegde RS. (2008). Retrotranslocation of prion proteins from the endoplasmic reticulum by preventing GPI signal transamidation. Mol Biol Cell , 3463–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangs JD. (2011). Replication of the ERES:Golgi junction in bloodstream form African trypanosomes. Mol Microbiol , 1433–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangs JD, Andrews N, Hart GW, Englund PT. (1986). Posttranslational modification and intracellular transport of a trypanosome variant surface glycoprotein. J Cell Biol , 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangs JD, Doering DL, Englund PT, Hart GW. (1988). Biosynthesis of a variant surface glycoprotein of Trypanosoma brucei: processing of the glycolipid membrane anchor and N-linked oligosaccharides. J Biol Chem , 17697–17705. [PubMed] [Google Scholar]
- Bangs JD, Herald D, Krakow JL, Hart GW, Englund PT. (1985). Rapid processing of the carboxyl terminus of a trypanosome variant surface glycoprotein. Proc Natl Acad Sci USA , 3207–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbet AF, McQuire TC. (1978). Crossreacting determinants in variant-specific surface antigens of African trypanosomes. Proc Natl Acad Sci USA , 1989–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berko D, Tabachnick-Cherny S, Shental-Bechor D, Cascio P, Mioletti S, Levy Y, Admon A, Ziv T, Tirosh B, Goldberg AL, Navon A. (2012). The direction of protein entry into the proteasome determines the variety of products and depends on the force needed to unfold its two termini. Mol Cell , 601–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berriman M, Hall N, Sheader K, Bringaud F, Tiwari B, Isobe T, Bowman S, Corton C, Clark L, Cross GAM, et al (2002). The architecture of variant surface glycoprotein gene expression sites in Trypanosoma brucei . Mol Biochem Parasitol , 131–140. [DOI] [PubMed] [Google Scholar]
- Brodsky JL. (2012). Cleaning up: ER-associated degradation to the rescue. Cell , 1163–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bülow R, Griffiths G, Webster P, Stierhof Y-D, Opperdoes FR, Overath P. (1989). Intracellular localization of the glycosyl-phosphatidylinositol-specific phospholipase C of Trypanosoma brucei . J Cell Sci , 233–240. [DOI] [PubMed] [Google Scholar]
- Bülow R, Overath P. (1986). Purification and characterization of the membrane-form variant surface glycoprotein hydrolase. J Biol Chem , 11918–11923. [PubMed] [Google Scholar]
- Carvalho P, Stanley AM, Rapoport TA. (2010). Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell , 579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillon GA, Aguilera-Romero A, Manzano-Lopez J, Epstein S, Kajiwara K, Funato K, Watanabe R, Reizman H, Muñiz M. (2011). The yeast p24 complex regulates GPI-anchored protein transport and quality control by monitoring anchor remodeling. Mol Biol Cell , 2924–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte I, Labriola C, Cazzulo JJ, Docampo R, Parodi AJ. (2003). The interplay between folding–facilitating mechanisms in Trypanosoma cruzi endoplasmic reticulum. Mol Biol Cell , 3529–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstler M, Bangs JD, Field MC. (2006). Intracellular transport systems in trypanosomes: function, evolution and virulence. In: Trypanosomes—After the Genome, ed. Barry JD, Mottram JC, McCulloch R, Acosta-Serrano A, Wymondham, UK: Horizon Scientific Press, 281–317. [Google Scholar]
- Ferguson MAJ, Haldar K, Cross GAM. (1985). Trypanosoma brucei variant surface glycoprotein has an sn-1,2-dimyristyl glycerol membrane anchor at its COOH terminus. J Biol Chem , 4963–4968. [PubMed] [Google Scholar]
- Field MC, Sergeenko T, Wang Y-N, Böhm S, Carrington M. (2010). Chaperone requirements for biosynthesis of the trypanosome variant surface glycoprotein. PLoS One , e8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita M, Watanabe R, Jaensch N, Romanova-Michaelides M, Satoh T, Kato M, Riezman H, Yamaguchi Y, Maeda Y, Kinoshita T. (2011). Sorting of GPI-anchored proteins into ER exit sites by p24 proteins is dependent on remodeled GPI. J Cell Biol , 61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Sommer T. (2012). Finding the will and the way of ERAD substrate retrotranslocation. Cur Opin Cell Biol , 460–466. [DOI] [PubMed] [Google Scholar]
- Hereld D, Krakow JL, Bangs JD, Hart GW, Englund PT. (1986). A phospholipase C from Trypanosoma brucei which selectively cleaves the glycolipid on the variant surface glycoprotein. J Biol Chem , 13813–13819. [PubMed] [Google Scholar]
- Hertz-Fowler C, Figueiredo LM, Quail MA, Becker M, Jackson A, Bason N, Brooks K, Churcher C, Fahkro S, Goodhead I, et al (2008). Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS One , e3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirumi H, Hirumi K. (1994). Axenic culture of African trypanosome bloodstream forms. Parasitol Today , 81–84. [DOI] [PubMed] [Google Scholar]
- Hughes H, Stephens DJ. (2008). Assembly, organization, and function of the COPII coat. Histochem Cell Biol , 129–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo L, Atrih A, Rodriques JA, Jones DC, Ferguson MAJ. (2009a). Trypanosoma brucei UDP-glucose:glycoprotein glucosyltransferase has unusual substrate specificity and protects the parasite from stress. Eukaryot Cell , 230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo L, Mehlert A, Ferguson MAJ. (2012). The lipid-linked oligosaccharide donor specificities of Trypanosoma brucei oligosaccharyltransferases. Glycobiol , 696–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo L, Schulz BL, Rodriques J, Guther ML, Procter JB, Barton GJ, Aebi M, Ferguson MAJ. (2009b). Distinct donor and acceptor specificities of Trypanosoma brucei oligosaccharyltransferases. EMBO J , 2650–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakow JL, Hereld D, Bangs JD, Hart GW, Englund PT. (1986). Identification of a glycolipid precursor of the Trypanosoma brucei variant surface glycoprotein. J Biol Chem , 12147–12153. [PubMed] [Google Scholar]
- Kruzel EK, Zimmett GP, III, Bangs JD. (2017). Life stage-specific cargo receptors facilitate glycosylphosphatidylinositol-anchored surface coat protein transport in Trypanosoma brucei. mSphere , e00282–00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligtenberg MJL, Bitter W, Kieft R, Sterverding D, Janssen H, Calafat J, Borst P. (1994). Reconstitution of a surface transferrin binding complex in insect form Trypanosoma brucei . EMBO J , 2565–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Määttänen P, Gehring K, Bergeron JJM, Thomas DY. (2010). Protein quality control in the ER: The recognition of misfolded proteins. Sem Cell Dev Bio , 500–511. [DOI] [PubMed] [Google Scholar]
- Mayor S, Menon AK, Cross GAM. (1992). Galactose-containing glycosylphosphatidylinositols in Trypanosoma brucei . J Biol Chem , 754–761. [PubMed] [Google Scholar]
- Mehlert A, Wormald MR, Ferguson MAJ. (2012). Modeling of the N-glycosylated transferrin receptor suggests how transferrin binding can occur within the surface coat of Trypanosoma brucei . PLoS Pathog , e1002618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon AK, Mayor S, Ferguson MAJ, Duszenko M, Cross GAM. (1988). Candidate glycophospholipid precursor fo the glycosylphosphatidylinositol membrane anchor of Trypanosoma brucei variant surface glycoprotein. J Biol Chem , 1970–1977. [PubMed] [Google Scholar]
- Mugnier MR, Cross GA, Papavasiliou FN. (2015). The in vivo dynamics of antigenic variation in Trypanosoma brucei. Science , 1470–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussman R, Engstler M, Gerrits H, Kieft R, Toaldo CB, Onderwater J, Koerten H, van Luenen HGAM, Borst P. (2004). Factors affecting the level and localization of the transferrin receptor in Trypanosoma brucei . J Biol Chem , 40690–40698. [DOI] [PubMed] [Google Scholar]
- Mussman R, Hanssen H, Calafat J, Engstler M, Ansorge I, Clayton C, Borst P. (2003). The expression level determines the surface distribution of the transferrin receptor in Trypanosoma brucei . Mol Microbiol , 23–35. [DOI] [PubMed] [Google Scholar]
- Peck RF, Shiflett AM, Schwartz KJ, McCann A, Hajduk SL, Bangs JD. (2008). The LAMP-like protein p67 plays an essential role in the lysosome of African trypanosomes. Mol Microbiol , 933–946. [DOI] [PubMed] [Google Scholar]
- Robinson NP, Burman N, Melville SE, Barry JD. (1999). Predominance of duplicative VSG gene conversion in antigenic variation in African trypanosomes. Mol Cell Biol , 5839–5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roggy JL, Bangs JD. (1999). Molecular cloning and biochemical characterization of a VCP homolog in African trypanosomes. Mol Biochem Parasitol , 1–15. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol , 519–529. [DOI] [PubMed] [Google Scholar]
- Rudenko G. (2011). African trypanosomes: the genome and adaptations for immune evasion. Essays Biochem , 47–62. [DOI] [PubMed] [Google Scholar]
- Salmon D, Geuskens M, Hanocq F, Hanocq-Quertier J, Nolan D, Ruben L, Pays E. (1994). A novel heterodimeric transferrin receptor encoded by a pair of VSG expression site-associated genes in T. brucei. Cell , 75–86. [DOI] [PubMed] [Google Scholar]
- Satpute-Krishan P, Ajinkya M, Bhat S, Itakura E, Hegde RS, Lippincott-Schwartz J. (2014). ER stress-induced clearance of misfolded GPI-anchored proteins via the secretory pathway. Cell , 522–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz KJ, Peck RF, Tazeh NN, Bangs JD. (2005). GPI valence and the fate of secretory membrane proteins in African trypanosomes. J Cell Sci , 5499–5511. [DOI] [PubMed] [Google Scholar]
- Schwede A, Carrington M. (2010). Bloodstream form trypanosome plasma membrane proteins: antigenic variation and invariant antigens. Parasitol , 2029–2039. [DOI] [PubMed] [Google Scholar]
- Sevova ES, Bangs JD. (2009). Streamlined architecture and GPI-dependent trafficking in the early secretory pathway of African trypanosomes. Mol Biol Cell , 4739–4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorska N, Lemus L, Aguilera-Romero A, Manzano-Lopez J, Riezman H, Muniz M, Goder V. (2016). Limited ER quality control for GPI-anchored proteins. J Cell Biol , 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JS, Muratore KA, Bangs JD. (2013). Characterization of the late endosomal ESCRT machinery in Trypanosoma brucei . Traffic , 1078–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JS, Schwartz KJ, Hajduk SL, Bangs JD. (2011). Late endosomal Rab7 regulates lysosomal trafficking of endocytic but not biosynthetic cargo in Trypanosoma brucei . Mol Microbiol , 664–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steverding D, Stierhof YD, Fuchs H, Tauber R, Overath P. (1995). Transferrin-binding protein complex is the receptor for transferrin uptake in Trypanosoma brucei . J Cell Biol , 1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunter J, Webb H, Carrington M. (2013). Determinants of GPI-PLC localisation to the flagellum and access to GPI-anchored substrates in trypanosomes. PLoS Pathog , e1003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T. (2016). Catabolism of N-glycans in mammalian cells: Molecular mechanisms and genetic disorders related to the processes. Mol Aspects Med , 89–103. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Huang C, Fujihira H. (2016). The cytoplasmic peptide:N-glycanase (NGLY1)—structure, expression and cellular functions. Gene , 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tazeh NN, Bangs JD. (2007). Multiple signals regulate trafficking of the lysosomal membrane protein p67 in African trypanosomes. Traffic , 1007–1017. [DOI] [PubMed] [Google Scholar]
- Tiengwe C, Brown DNA, Bangs JD. (2015). Unfolded protein response pathways in bloodstream form Trypanosoma brucei? Eukaryot Cell , 1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiengwe C, Bush PJ, Bangs JD. (2017). Controlling transferrin receptor trafficking with GPI-valence in bloodstream stage African trypanosomes. PLoS Pathog , e1006366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiengwe C, Muratore KA, Bangs JD. (2016). Surface proteins, ERAD and antigenic variation in Trypanosoma brucei. Cell Microbiol , 1673–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triggs VP, Bangs JD. (2003). Glycosylphosphatidylinositol-dependent protein trafficking in bloodstream stage Trypanosoma brucei . Eukaryot Cell , 76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz E, Leal S, Ochatt C, Cross G. (1999). A tightly regulated inducible expression system for conditional gene knockouts and dominant-negative genetics in Trypanosoma brucei . Mol Biochem Parasitol , 89–101. [DOI] [PubMed] [Google Scholar]
- Zamze SE, Ferguson MAJ, Collins R, Dwek RA, Rademacher TW. (1988). Characterization of the cross-reacting determinant (CRD) of the glycosyl-phosphatidylinositol membrane anchor of Trypanosoma brucei variant surface glycaoprotein. Eur J Biochem , 527–534. [DOI] [PubMed] [Google Scholar]