

SUMMARY
Plasticity of thalamocortical (TC) synapses is robust during early development and becomes limited in the adult brain. We previously reported that a short duration of deafening strengthens TC synapses in the primary visual cortex (V1) of adult mice. Here, we demonstrate that deafening restores NMDA receptor (NMDAR)-dependent long-term potentiation (LTP) of TC synapses onto principal neurons in V1 layer 4 (L4), which is accompanied by an increase in NMDAR function. In contrast, deafening did not recover long-term depression (LTD) at TC synapses. Potentiation of TC synapses by deafening is absent in parvalbumin-positive (PV+) interneurons, resulting in an increase in feedforward excitation to inhibition (E/I) ratio. Furthermore, we found that a brief duration of deafening adult mice recovers rapid ocular dominance plasticity (ODP) mainly by accelerating potentiation of the open-eye responses. Our results suggest that cross-modal sensory deprivation promotes adult cortical plasticity by specifically recovering TC-LTP and increasing the E/I ratio.
In Brief
Plasticity of thalamocortical (TC) synapses is limited in adults. Rodríguez et al. demonstrate that a brief period of deafening adults recovers LTP at TC synapses in visual cortex and accelerates ocular dominance plasticity. These results suggest that cross-modal sensory deprivation may be an effective way to promote adult cortical plasticity.
Graphical Abstract
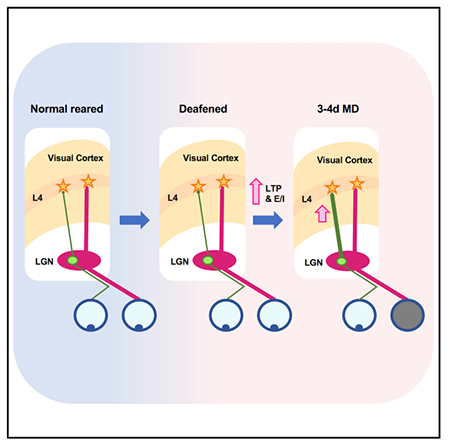
INTRODUCTION
Thalamocortical (TC) inputs convey sensory information to the cortex for further processing and are shaped by sensory experience during a defined early developmental period (Barkat et al., 2011; Crair and Malenka, 1995). Notably, across different sensory cortices, TC synapses in layer 4 (L4) exhibit the earliest and shortest critical period that precedes synaptic plasticity in other layers, which often persists through adulthood (Barkat et al., 2011; Barth and Malenka, 2001; Crair and Malenka, 1995; Desai et al., 2002; Goel and Lee, 2007; Jiang et al., 2007). Therefore, the loss of TC synaptic plasticity may be an essential factor contributing to the limited ability of the adult brain to undergo plasticity. In the mouse visual pathway, TC synapses undergo experience-dependent reorganization and refinement that end between the second and third postnatal week (Gu and Cang, 2016; Jiang et al., 2007). Ascending TC inputs onto L4 neurons express NMDA receptor (NMDAR)-dependent long-term potentiation (LTP) and long-term depression (LTD) during this limited time window, while L4 to L2/3 synapses remain plastic into adulthood in rodent primary visual cortex (V1) (Jiang et al., 2007). The same laminar progression of plasticity has been described in studies of experience-dependent homeostatic synaptic scaling in V1, where L4 plasticity ends earlier while that in L2/3 persists (Desai et al., 2002; Goel and Lee, 2007). Studies performed in vivo also showed that ocular dominance plasticity (ODP) is more robust in L4 early in development, while in adults, it is more evident in L2/3 (Pham et al., 2004). Taken together, these cases support the view that TC synapses onto V1 L4 undergo plasticity early on, and subsequent experience-dependent changes are mainly mediated by plasticity in the superficial layers.
Recent studies have challenged this notion by demonstrating TC plasticity in adults under particular manipulations, such as environmental enrichment (Mainardi et al., 2010), prolonged visual deprivation (Montey and Quinlan, 2011), or peripheral nerve transection (Chung et al., 2017; Yu et al., 2012). Furthermore, we previously reported that visual deprivation results in strengthening of TC synapses to auditory cortex (A1) while deafening leads to TC potentiation in V1 (Petrus et al., 2014). This indicates that cross-modal sensory deprivation in adults readily reactivates TC plasticity, which could represent the cellular basis for cross-modal plasticity that allows enhanced processing in spared cortices after sensory loss (Lee and Whitt, 2015). However, the synaptic mechanisms underlying this change remain unexplored. Here, we used targeted optogenetic activation of TC synapses to demonstrate the reemergence of NMDAR-dependent LTP at V1 TC synapses following a week of deafening adult mice (post-natal day 90 [P90] to P120), which occurred without recovery of TC-LTD. In addition, we found that deafening increases the excitation to inhibition (E/I) ratio of TC inputs onto L4 principal neurons. Such a shift in the E/I ratio has been linked to heightened cortical plasticity (Froemke, 2015; Froemke et al., 2007). Consistent with this notion, we report that restoration of V1 TC plasticity by cross-modal sensory deprivation accelerates ODP in adults. Our findings suggest cross-modal sensory manipulations as a potential method to facilitate plasticity in the adult brain.
RESULTS
Reemergence of LTP at TC Synapses in V1 L4 after Deafening
We previously showed that a brief period of sensory deprivation produces cross-modal strengthening of TC synapses in L4 of the spared sensory cortices of adult mice (Petrus et al., 2014). Importantly, this change in TC synapses was dependent on the spared cortex retaining its own sensory inputs. Hebbian forms of synaptic plasticity have been established as the mechanisms driving experience-dependent changes of TC inputs to L4 in primary sensory cortices during the critical period (Crair and Malenka, 1995; Dudek and Friedlander, 1996; Kirkwood et al., 1995). Therefore, we tested whether the cross-modal potentiation of TC synapses by a period of deafening is due to reengagement of LTP mechanisms in adult V1.
We induced hearing loss in adult animals (P90–P120) via ototoxic lesioning of hair cells by injection of kanamycin (175 mg/mL) into the inner ear coupled with tympanic rupture. We confirmed the efficiency of our approach by measuring auditory brainstem responses (ABRs) in a subset of mice, as well as by verifying cochlear damage in the deafened group by phalloidin staining of hair cells (Figure S1).
In order to selectively activate TC synapses, we expressed ChR2 in the visual thalamus (dLGN) via adeno-associated virus (AAV)-mediated transfection as in our published study (Petrus et al., 2014). We found that ChR2 can only follow stimulation faithfully up to ~40 Hz (Figure S2). Therefore, we induced LTP at TC synapses using a pairing protocol, which consists of pairing low-frequency activation (1 Hz, 200 pulses) of ChR2-expressing TC synapses using light emitting diode (LED) (455 nm) with postsynaptic depolarization (0 mV) of a L4 principal neuron (Figure 1A). This is a modification of a protocol used successfully in cortical slices from young (P8–P17) mice to study TC-LTP using electrical stimulation of the white matter (WM) (Jiang et al., 2007). The location of recorded neurons and expression of ChR2 were verified post hoc (Figure 1B). Consistent with previous studies showing early critical period for WM to L4 LTP, our pairing protocol failed to induce TC-LTP in V1 slices of normal-reared (NR) adults (Figure 1C). In contrast, V1 slices from adult mice that were deafened (DF) a week (7 ± 1 days) prior to the experiment exhibited robust TC-LTP, which was blocked by bath application of 100 μM DL-2-amino-5 phosphonopentanoic acid (APV) (Figure 1C). These results indicate that TC-LTP recovered in adults by cross-modal sensory deprivation is dependent on NMDARs, which is similar to what has been described for TC-LTP in normal young mice (Crair and Malenka, 1995).
Figure 1. Deafening Specifically Recovers TC-LTP in Adult V1 L4.
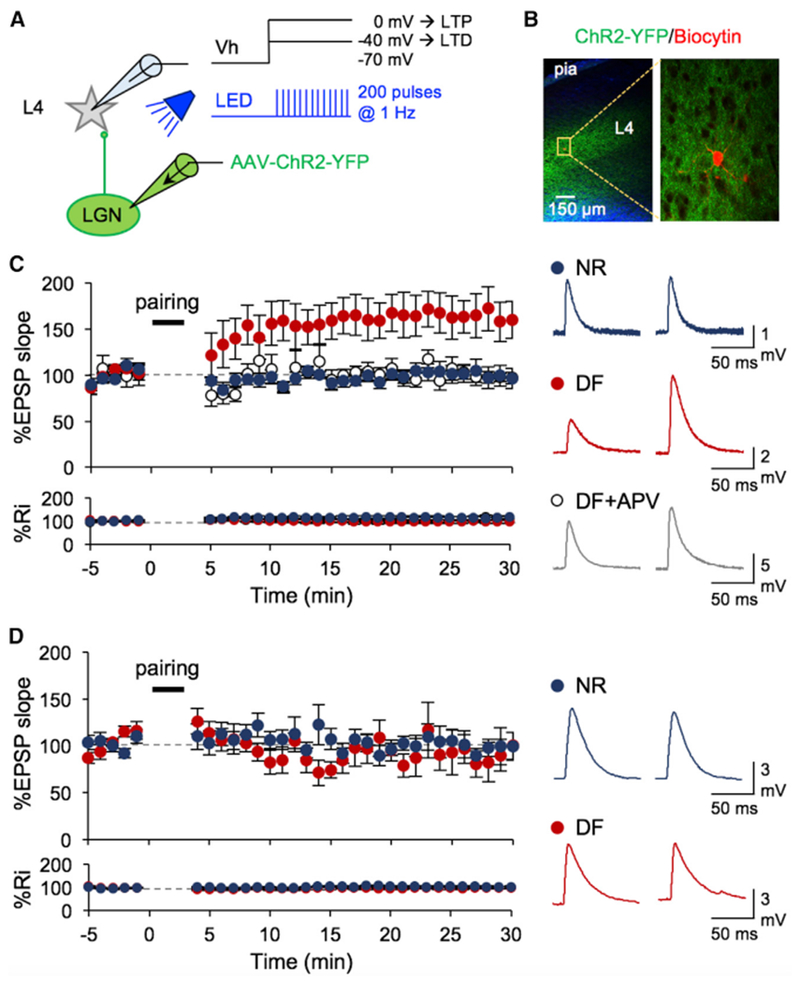
(A) Schematic of V1 recordings where LGN terminals expressed ChR2. LTP and LTD were induced using a pairing protocol, where postsynaptic depolarization to 0 mV and −40 mV, respectively, was paired with presynaptic stimulation with light pulses (455 nm LED; 5 ms pulse duration, 200 pulses at 1 Hz).
(B) Left: low-magnification image of a V1 slice showing ChR2-YFP (green) expression in LGN axons especially in L4. Blue, DAPI. Right: high-magnification image of a recorded L4 neuron filled with biocytin (red) surrounded by ChR2-YFP-expressing LGN axons.
(C) Left: TC-LTP in adult DF mice (red, n = 9 cells/5 mice, *p < 0.02 between baseline and 30 min post-pairing), but not in NR (blue, n = 9 cells/6 mice, not significant [N.S.]p = 0.9934). APV blocked LTP in the DF group (open circles, n = 6 cells/3 mice, N.S. p = 0.7564). Bottom: no significant change in input resistance (Ri). Right: example average EPSPs taken before (left) and 30 min after pairing (right).
(D) Left: lack of TC-LTD in adult control (NR: blue, n = 9 cells/5 mice, N.S. p = 0.7152) and DF (red, n = 9 cells/5 mice, p = 0.3824). Bottom: no significant change in Ri. Right: example average EPSPs taken before (left) and 30 min after pairing (right). Data plotted are mean ± SEM.
Next, we examined whether deafening will also restore LTD at TC synapses in adults. To do this, we repeated the same experimental design, except that the postsynaptic cells were held at −40 mV during pairing protocol (Figure 1A). We did not observe TC-LTD in normal adults, which is consistent with a loss of this form of LTD beyond the third postnatal week (Jiang et al., 2007), and a week of prior deafening did not restore TC-LTD in adults (Figure 1D). Collectively, our results indicate that a brief cross-modal sensory deprivation specifically restores TC-LTP in adults.
Cross-Modal Regulation of NMDARs at TC Synapses
Recovery of NMDAR-dependent TC-LTP in V1 of DF adult mice suggests that deafening may alter NMDAR function. To examine this possibility, we first determined the relative contribution of NMDARs to excitatory postsynaptic currents (EPSCs) evoked from thalamic terminals by measuring the NMDA/AMPA ratio. There was no significant change in the average NMDA/AMPA ratio between the NR and DF groups (Figure 2A). We previously reported that the same duration of DF increases AMPA receptor (AMPAR)-mediated currents at TC synapses (Petrus et al., 2014), which suggests that there is concurrent potentiation of NMDAR-EPSCs to maintain the NMDA/AMPA ratio at TC synapses following DF.
Figure 2. Regulation of NMDAR Function at TC Synapses onto Adult V1 L4 Neurons following Deafening.
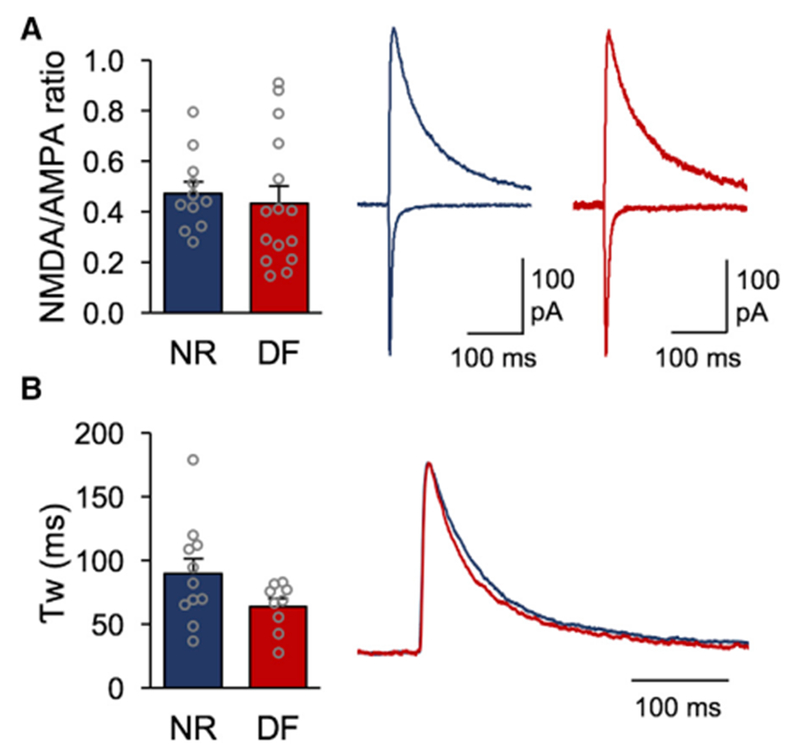
(A). Left: NMDA/AMPA ratio measurements for the NR and DF groups (open circles represent individual cells; NR = 0.49 ± 0.045, 4 mice; DF = 0.43 ± 0.067, 5 mice; t test, N.S. p = 0.5173). Right: averaged example traces for an NR cell (blue) and DF cell (red) normalized to the AMPAR component in NR. The AMPAR component was measured at the peak recorded at −80 mV, while the NMDAR component was measured 70 ms after the onset of the compound EPSC recorded at +40 mV.
(B). Left: weighted decay time constant (Tw) for a pharmacologically isolated NMDAR current measured at +40 mV for the NR and DF groups (open circles represent individual cells; NR = 89.31 ± 11.97 ms, 4 mice; DF = 63.93 ± 6.3 ms, 3 mice; t test, N.S. p = 0.0961). Right: averaged NMDAR responses for the NR (blue) and DF (red) groups normalized to the maximum amplitudes.
Bar graphs: mean ± SEM.
A switch in NMDAR GluN2 subunit composition is known to coincide with decreased synaptic plasticity and the closure of the critical period for cortical plasticity (Barth and Malenka, 2001; Philpot et al., 2001; Quinlan et al., 1999). GluN2B-containing NMDARs predominate at synapses early on but undergo a developmental switch to incorporate GluN2A subunits, which have faster decay kinetics (Monyer et al., 1994; Sheng et al., 1994). Furthermore, changes in GluN2 subunit composition have been shown to alter the threshold for LTP/LTD induction (Philpot et al., 2001, 2003; Quinlan et al., 1999). In orderto determine if changes in NMDAR subunit composition allowed for the recovery of TC-LTP with DF, we measured isolated NMDAR-evoked responses at TC synapses onto L4 in V1 and compared their weighted decay time constants between groups as reported previously (Philpot et al., 2001; Rumbaugh and Vicini, 1999). We observed no significant changes in the NMDAR decay kinetics at TC synapses between the NR and DF groups (Figure 2B). Our results suggest that cross-modal sensory deprivation in adults potentiates the function of synaptic NMDARs without detectable changes in NR2 subunit composition, which contrasts what is observed early in development.
Deafening Increases the E/I Ratio of TC Inputs to V1 L4 Principal Neurons
Thalamic inputs strongly drive feedforward inhibition onto cortical L4 neurons, which serves to regulate coincidence detection by improving the precision of postsynaptic spikes (Chittajallu and Isaac, 2010; Cruikshank et al., 2007; Daw et al., 2007; Gabernet et al., 2005; Kloc and Maffei, 2014). In V1, like other sensory cortices, TC inputs recruit feedforward inhibition mainly by activating parvalbumin-positive (PV+) interneurons (Cruikshank et al., 2007; Daw et al., 2007; Kloc and Maffei, 2014). Maturation of cortical inhibition mediated by PV+ neurons is also implicated in gating cortical plasticity (Jiang et al., 2005). Therefore, we examined if DF could lead to changes in thalamic engagement of cortical inhibition.
We targeted recordings to PV+ cells in L4 V1 by using mice expressing tdTomato in these neurons (PV-tdT) and expressed ChR2 in dLGN using targeted injection of AAV (Figures 3A and 3B). To compare the strength of individual TC synapses regardless of ChR2 expression or stimulation level, we recorded LED-evoked Sr2+ desynchronized (LEv-Sr2+) mEPSCs as described previously (Petrus et al., 2014, 2015) (see STAR Methods). In this scheme, LED-evoked responses are desynchronized such that recorded events represent single-vesicle release and thus, quantal synaptic response size can be determined (Gil et al., 1999). We found no significant differences between the average amplitude of LEv-Sr2+ miniature EPSCs (mEPSCs) recorded from PV+ neurons in the NR and DF groups (Figure 3C). This result contrasts our previous finding that TC synapses on L4 principal neurons potentiate with DF (Petrus et al., 2014), which suggests that a postsynaptic target determines the plasticity of TC synapses with cross-modal deprivation. To test whether the selective potentiation of TC inputs to principal neurons alters the balance of recruiting feedforward excitation and inhibition, we measured the E/I ratio in V1 L4 principal neurons following TC input activation using ChR2 while holding the postsynaptic cell at reversal potential for glutamatergic (Eglu) or GABAergic (EGABA) synaptic transmission (Figure S3). We observed a significant increase in the average E/I ratio values in V1 L4 principal neurons after DF (Figure 3D). Collectively, these data suggest that the increased E/I ratio following DF is driven by selective potentiation of TC inputs onto L4 principal neurons without changes in excitatory drive onto PV+ interneurons.
Figure 3. Deafening Adults Did Not Change the Strength of TC Inputs to PV+ Interneurons and Enhanced E/I Ratio of TC Inputs to V1 L4 Neurons.
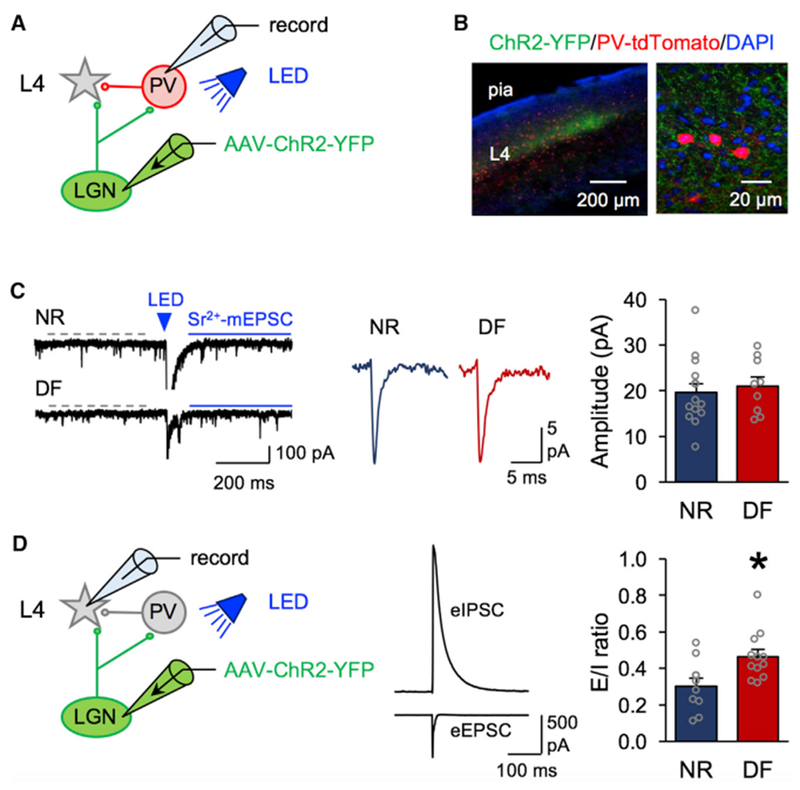
(A). Schematic of a td-Tomato-expressing PV+ neuron (red) targeted in V1 L4 for LEv-Sr+2 mEPSCs recordings to assess the strength of TC synapses.
(B). Confocal image of a V1 slice showing LGN axons expressing ChR2-YFP (green) and PV+ neurons expressing Td-Tomato (red) and counterstained for DAPI (blue).
(C). Left: example traces of LEv-Sr2+ mEPSCs from the NR (top) and DF (bottom) groups. Dashed gray line represents the time window used to measure spontaneous mEPSC events before LED stimulation (blue triangle, 455 nm, 5 ms duration). Solid blue line represents the time window used to measurethe post-LED events. From the measured post-LED events, pre-LED spontaneous events were mathematically subtracted (see STAR Methods) to obtain the events evoked by ChR2 activation of TC axons. Middle: average traces of calculated LEv-Sr2+-mEPSCs (NR, blue; DF, red). Right: comparison of average amplitude of TC LEv-Sr2+-mEPSCs between NR and DF (open circles represent individual cells; NR = 18.9 ± 1.7 pA, 7 mice; DF = 19.6 ± 1.65 pA, 7 mice; t test, N.S. p = 0.7893).
(D). Left: schematic showing targeted V1 L4 principal neurons for E/I ratio recording where thalamic terminals (green) were stimulated with light (455 nm) to evoke a monosynaptic EPSC and a disynaptic inhibitory postsynaptic current (IPSC) responses in L4. Middle: average trace of LED evoked monosynaptic eEPSC (inward current recorded at EGABA = —52 mV) and disynaptic eIPSC (outward current recorded at Eglu = 0 mV) from a L4 cell after light stimulation of ChR2-expressing LGN axons. Right: significant increase in average E/I ratio values after DF (open circles represent each cell; NR, 4 mice; DF, 4 mice; t test, *p = 0.0127).
Bar graphs: mean ± SEM.
Deafening Restores Rapid ODP in Adult V1
Based on our result that short-term deafening of adult mice restores TC-LTP in V1, we next tested whether deafening could restore experience-dependent V1 plasticity. ODP is a classic approach to studying experience-dependent synaptic plasticity in V1, and the mechanisms underlying it in both juveniles and adults have been well characterized (Frenkel and Bear, 2004; Gordon and Stryker, 1996; Sato and Stryker, 2008; Sawtell et al., 2003). It is well established that a short period (3–4 days) of monocular deprivation (MD) elicits quick and robust ocular dominance (OD) shift in V1 during the critical period, which in mice starts at P19 and extends until P35 (Gordon and Stryker, 1996) and mainly involves weakening of the closed-eye inputs (Sato and Stryker, 2008; Sawtell et al., 2003). In adults (P60–P90), however, ODP requires a longer period of MD (5–7 days) and is driven predominantly by a delayed potentiation of the open-eye inputs (Frenkel and Bear, 2004; Sato and Stryker, 2008; Sawtell et al., 2003). Since deafening promotes TC-LTP in adult V1, we reasoned that it may accelerate the open-eye potentiation in adults to allow ODP with a shorter duration of MD, which is normally ineffective.
We measured ODP using optical imaging of V1 intrinsic signals before and after periods of MD (Figures 4A and 4B) and calculated an ocular dominance index value (ODI) at each time point (Figure 4C). First, we verified that brief (3–4 days) MD shifts ODI in young mice within the established critical period (P25–P32) (Figure 4A), which in our hands was due to a combination of weakening of the closed contralateral eye inputs and potentiation of the open ipsilateral eye inputs (Figures 4D and 4E). In adult mice (P90–P110), that were deafened 1 week prior to initiating the brief (3–4 days) MD, we observed a significant shift in the ODI which was driven exclusively by potentiation of the open ipsilateral inputs (Figures 4D and 4E). Deafening by itself did not alter the ODI (Figure S4). Consistent with previous studies, the same brief (3–4 days) duration of MD failed to alter the ODI in normal hearing adults, and consequently there was no change in the strength of either the closed or open eye inputs to V1 (Figures 4D and 4E). Normal adults required a longer duration of MD (5–6 days) to produce a significant shift in the ODI, which was driven by potentiation of the open ipsilateral eye inputs (Figures 4D and 4E) as reported in prior studies (Frenkel and Bear, 2004; Ranson et al., 2012; Sato and Stryker, 2008; Sawtell et al., 2003). Our data indicate that deafening accelerates the emergence of open-eye potentiation in the adults but does not engage the fast depression of the closed-eye inputs, as seen in juveniles.
Figure 4. Deafening Accelerated Ocular Dominance Plasticity in Adult V1 by Promoting Open-Eye Potentiation.
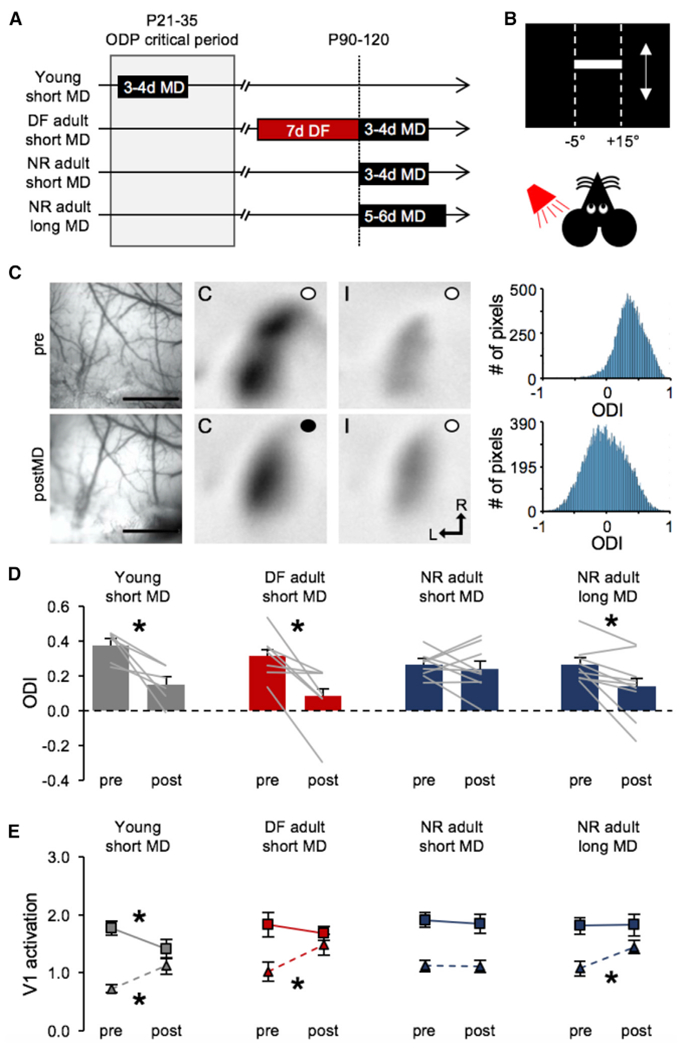
(A). Outline of experimental groups.
(B). Schematic of visual stimulus presented while recording V1 intrinsic signals.
(C). A representative images and data from a young mouse (young short MD). Left: surface vasculature used to guide imaging in the same animal before (top) and after (bottom) MD. Scale bars, 1 mm. Middle two panels: V1 intrinsic signals recorded before (top) and after (bottom) MD for contralateral (C) and ipsilateral (I) eyes (an open circle indicates an open eye, and a closed circle indicates a closed eye; L, lateral, R, rostral). Right: histogram showing the distribution of pixels corresponding to ODI values. Note a left shift in the distribution after MD indicating that more pixels having a lower ODI (ODI = 0 indicates neurons responding equally to both eyes).
(D). Deafening accelerates ODP in the adult V1. Comparison of ocular dominance index [ODI = (C − I)/(C + I)] pre- and post-MD (bars show mean + SEM, gray lines connect ODI values measured pre- and post-MD for each mouse). Short (3–4 days) MD in young mice during the critical period (gray bars) shows a significant shift in ODI toward the open eye (pre = 0.378 ± 0.036, post = 0.1534 ± 0.044; paired t test, *p = 0.0132). A week of deafening adult mice prior to a short (3- to 4-day) MD (red bars) also allows a significant shift in ODI toward the open eye (pre = 0.3164 ± 0.048, post = 0.083 ± 0.068; paired t test *p = 0.0121). The same short-duration MD (3–4 days) in normal adult mice failed to shift ODI (pre = 0.264 ± 0.023, post = 0.2389 ± 0.044; paired t test, N.S. p = 0.5892). However, a longer-duration MD (5–6 days) significantly shifted ODI in normal adults (pre = 0.2682 ± 0.035, post = 0.1411 ± 0.060; paired t test *p = 0.170). Bar graphs: mean ± SEM.
(E). Comparison of average eye-specific V1 activation signal intensity through contralateral eye (square symbols with solid line) and ipsilateral eye (triangles with dashed line) pre- and post-MD. In young mice, short MD (3–4 days) significantly decreased the intensity of signal from the closed contralateral eye while it increased that from the open ipsilateral eye (contralateral: pre = 1.76 ± 0.12, post =1.41 ± 0.16, paired t test *p = 0.0204; ipsilateral: pre = 0.72 ± 0.16, post = 1.21 ± 0.35, paired t test *p = 0.0213). In adults deafened for 1 week prior to short MD (3–4 days), there was only a significant increase in the intensity of signal from the open ipsilateral eye (contralateral: pre = 1.83 ± 0.21, post = 1.68 ± 0.12, paired t test N.S. p = 0.52; ipsilateral: pre =1.02 ± 0.16, post = 1.48 ± 0.18, paired t test *p = 0.0207). In normal adults, short MD (3–4 days) did not alter the intensity of signal from either eye (contralateral: MD = 1.9 ± 0.13, MD = 1.84 ± 0.16, paired t test N.S. p = 0.6782; ipsilateral: pre = 1.12 ± 0.09, post =1.12 ± 0.10, paired t test N.S. p = 0.9168), but a longer-duration MD (5–6 days) significantly increased the intensity of signal from the open ipsilateral eye (contralateral: pre = 1.81 ± 0.14, post =1.82 ± 0.18, paired t test p = 0.9629; ipsilateral: pre =1.08 ± 0.13, post = 1.45 ± 0.11, paired t test *p = 0.0328). Data plotted are mean ± SEM.
DISCUSSION
In this study, we investigated the molecular mechanisms and functional consequences underlying the reactivation of TC plasticity by cross-modal sensory deprivation. In particular, we addressed the impact deafening has on TC plasticity in the spared V1. We observed a reemergence of NMDAR-dependent TC-LTP, which is driven by potentiation of NMDAR currents and an increased E/I ratio. However, deafening did not restore TC-LTD in adults, which is consistent with the expectation that increase in NMDAR function and E/I ratio will promote LTP rather than LTD by providing greater summation of responses and Ca2+ influx. Moreover, we demonstrated that deafening accelerates ODP in adult mice through potentiation of open-eye inputs without any depression of the deprived eye inputs. Notably, the mechanisms described here allow for TC plasticity 10–12 weeks after the defined critical period for these synapses, suggesting that cross-modal sensory deprivation may be an effective means to recover cortical plasticity in adults that are specifically dependent on synaptic potentiation.
Recent studies have begun to highlight the potential for TC synapses to undergo plasticity in adulthood under certain conditions (Chung et al., 2017; Mainardi et al., 2010; Montey and Quinlan, 2011; Petrus et al., 2014; Yu et al., 2012). While our findings are similar to those describing TC-LTP in the spared whisker barrel of adult mice following infraorbital nerve (ION) lesions (Chung et al., 2017; Yu et al., 2012), we extend this by showing that cross-modal sensory deprivation, not just within-sensory modality deprivation, can recover TC-LTP (Figure 1). Furthermore, we show that cross-modal sensory deprivation specifically recovers TC-LTP, but not TC-LTD, in adults. In terms of molecular mechanisms, we found a requirement for NMDARs in adult TC-LTP induction and a potential potentiation of NMDAR function as a result of cross-modal sensory deprivation as deduced by a preserved NMDA/AMPA ratio despite previously reported potentiation of AMPAR current (Petrus et al., 2014) (Figure 2A). However, we did not find observable changes in the kinetics of NMDAR current as would occur if GluN2 subunit composition was altered (Figure 2B). This differs from the findings reported in the ION lesion model, which showed increased ifenprodil sensitivity at spared TC synapses indicative of a switch in NMDAR composition (Chung et al., 2017). Our results suggest that cross-modal NMDAR regulation is likely mediated by a change in the number of functional NMDARs. This is in line with a previous study describing activity-dependent concomitant regulation of AMPAR and NMDAR synaptic components, which is driven mainly by modulation in the number of open NMDAR channels (Watt et al., 2000).
We also found that while TC synapses to L4 principal neurons strengthen (Petrus et al., 2014), those to PV+ inhibitory cells remain unaltered after DF (Figure 3), which suggests that cross-modal reactivation of TC plasticity is specified by the postsynaptic cell type. One functional consequence is an increase in the balance of feedforward excitation and inhibition recruited at TC synapses on V1 L4 principal neurons. Increased E/I ratio is reminiscent of early developmental stages with high potential for plasticity, which becomes restricted as inhibition matures to decrease the E/I ratio (Zhang et al., 2011). Several studies suggested that E/I balance is relatively maintained in the mature cortex (Dehghani et al., 2016; House et al., 2011; Xue et al., 2014), yet our results show that DF causes a deviation from this equilibrium. Changes in E/I balance have been implicated in recruitment of TC plasticity after environmental enrichment (Mainardi et al., 2010) and for learning-induced cortical rearrangement (Froemke, 2015; Froemke et al., 2007). Our result similarly suggests that the E/I ratio is not fixed in adults but can be regulated based on sensory experience to reopen the window for plasticity to sculpt neural circuits in accordance with environmental changes and the need of the cortical network for sensory processing.
While the consequences of cross-modal plasticity on V1 function are still unclear, we demonstrate that a short-term deafening can accelerate ODP in adults. This effect was driven selectively by accelerated potentiation of open-eye inputs without major changes in the deprived-eye inputs. This is qualitatively different from other means of recovering ODP in the adult brain such as dark exposure, which has been shown to recover juvenile-like plasticity that involves weakening of the deprived eye inputs (He et al., 2006, 2007) and a change in NMDAR subunit composition to juvenile form (He et al., 2006). We found that DF specifically recovers TC-LTP, but not TC-LTD, by increasing NMDAR function without significant alterations in subunit composition. In addition, deafening increased the E/I ratio at TC synapses on L4 principal neurons. These cellular level changes correlate with acceleration of open-eye potentiation mechanisms and ODP in the adult V1. In sum, our results suggest that cross-modal sensory deprivation may be an effective way to promote experience-dependent cortical plasticity in adults that are specifically dependent on TC-LTP.
STAR*METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| Alexa Fluor 488-phalloidin conjugate | Molecular Probes | Cat# A-12379; RRID:AB_2315147 | |
| Bacterial and Virus Strains | |||
| AAV5.hSyn.ChR2(H134R)-YFP.WPRE.hGH | Penn Vector Core | Addgene26973P | |
| Chemicals, Peptides, and Recombinant Proteins | |||
| kanamycin | Sigma-Aldrich | 60615; CAS: 70560-51-9 | |
| DAPI | Life technologies | D-1306 | |
| Avidin-Texas red conjugate | Life technologies | A-820 | |
| chlorprothixene | Sigma-Aldrich | C1671; CAS: 6469-93-8 | |
| formalin | Sigma-Aldrich | HT5014; MDL: MFCD00003274 | |
| biocytin | Sigma-Aldrich | B4261 | |
| Isoflurane | Patterson Veterinary | 07-890-8115 | |
| Tetrodotoxin citrate (TTX) | Abcam | ab120055 | |
| DL-APV | Sigma-Aldrich | A5282 | |
| NBQX disodium salt hydrate | Sigma-Aldrich | N183 | |
| glycine | Fisher | BP381-1 | |
| xylazine | Patterson Veterinary | 07-808-1947 | |
| ketamine | Patterson Veterinary | 07-890-8598 | |
| Bicuculline methiodide | Enzo | BML-EA149-0050 | |
| picrotoxin | Tocris | Cat#1128 | |
| Deposited Data | |||
| Dataset for each figure | Mendeley | https://doi.org/10.17632/drgg7v5zjx.1 | |
| Experimental Models: Organisms/Strains | |||
| C57BL/6J | Jackson Laboratories | RRID:IMSR_JAX:000664 | |
| B6.129P2-Pvalbtm1(cre)Arbr/J | Jackson Laboratories | RRID:IMSR_JAX:017320 | |
| B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | Jackson Laboratories | RRID:IMSR_JAX:007914 | |
| Software and Algorithms | |||
| MATLAB | Mathworks | https://www.mathworks.com/products/matlab.html; RRID: SCR_001622 | |
| Prism 7.0 | GraphPad Software | https://www.graphpad.com/scientificsoftware/prism/; RRID: SCR_002798 | |
| Igor Pro | Wavemetrics | https://www.wavemetrics.com/products/igorpro/igorpro.htm; RRID:SCR_000325) | |
| Mini-Analysis | Synaptosoft | http://www.synaptosoft.com/MiniAnalysis/; RRID:SCR_002184 | |
| Other | |||
| gel foam | Pfizer | 0315-08 | |
| CCD camera | Dalsa | DS-1A-01M30-12E | |
| Axon patch-clamp amplifier | Molecular Device | Multiclamp 700B | |
| processor | Tucker-Davis Technologies | RX6 Multifunction Processor | |
| attenuator | Tucker-Davis Technologies | PA5 Programmable Attenuator | |
| monitor | ViewSonic | VX2268WM | |
| 455 nm LED | Thor Labs | M455L3-C5 | |
| Vibratory tissue slicer | Ted Pella | PELCO easiSlicer product#: 11000 | |
| ProLong Gold antifade | Life Technologies | P36930 | |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hey-Kyoung Lee (heykyounglee@jhu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
B6 (C57BL/6J, Jackson Laboratories; RRID: IMSR_JAX:000664) and PV-tdT (offsprings of PV-CrexAi14; PV-Cre: Pvalbtm1(cre)Arbr/J, RRID:IMSR_JAX:017320; Ai14: Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J, RRID:IMSR_JAX:007914) mice were reared in a 12 hours light/dark cycle with water and food pellets ad libitum. All experiments were performed on adult mice (P90-P120) of both sexes where littermates were randomly assigned to experimental groups. All protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at Johns Hopkins University and followed the guidelines established by the Animal Care Act and National Institutes of Health (NIH).
METHOD DETAILS
Thalamic Viral Transfection
Mice (P40-60) of either sex were anesthetized and head fixed in a stereotaxic device (Kopf Instruments, California) under 1.5%–2% isoflurane/oxygen mix. The dorsal lateral geniculate nuclei (dLGN; −2.3 mm lateral: 2 mm from Bregma; 2.42 mm depth from pia) were bilaterally injected with an adeno-associated virus expressing channelrhodopsin (ChR2) under the control of the human synapsin (hSyn) promoter (AAV5.hSyn.ChR2(H134R)-YFP.WPRE.hGH, Penn Vector Core, University of Pennsylvania). Mice recovered on a heated pad and were returned to the animal colony, housed with 2-3 same sex mice until experimental procedures.
Deafening
At P90-110, one week (7 ± 1 day) prior to experiments, male and female mice were deafened as in previous studies (Petrus et al., 2014; Petrus et al., 2015). In brief, animals were anesthetized by exposure to isoflurane vapors in an induction chamber. Following the absence of the toe pinch reflex, the animal was placed in a stereotaxic device (Kopf Instruments, California) under constant administration of 2% isoflurane/oxygen mix. The pinnae were cut and the ventral surface of the ear was slit to aid visualization of the inner ear. The tympanic membrane was punctured with a 30-gauge needle, the ear cavity was injected with 50 μL kanamycin (175 mg/mL), and stuffed with kanamycin soaked gel foam (Pfizer) (Hashimoto et al., 2007). The ventral incision and remainder of pinnae were sutured shut (PSDII; Ethicon) and animals were allowed to recover on a heating pad until movement and drinking behaviors were evident.
Confirmation of deafening
Auditory brainstem responses (ABRs) were measured for a subset of animals as described previously (McGuire et al., 2015). Mice were anesthetized with ketamine/xylazine (i.p., 100 mg/kg and 20 mg/kg respectively), then placed on a heating pad inside a sound-attenuating chamber (IAC) lined with Sonex Acoustic foam. Mice were placed facing a speaker (FT28D; Fostex, Tokyo, Japan) positioned 30 cm from the pinnae. Temperature was monitored via a rectal probe and maintained at 36°C ± 1°. Subcutaneous platinum needle electrodes were placed over the left bulla and at the vertex of the skull, and a ground electrode was inserted into the leg muscle. The electrodes were attached to a preamplifier and amplifier (ISO-80; World Precision Instruments, Sarasota, FL). Stimulus generation, presentation and response acquisition were controlled using custom MATLAB-based software (Mathworks; RRID: SCR_001622), a Tucker Davis processor (RX6; Tucker-Davis Technologies, Alachua, Fl) and programmable attenuator (PA5). Stimuli consisting of clicks and 5-ms tones (0.5 ms onset/offset) at frequencies of 8, 12, 16, 24 and were presented. Stimuli were generated with a sampling frequency of 195 kHz, and presented at a rate of 20/s. Responses were sampled at 9.5 kHz, bandpass filtered from 300-3000 kHz, and averaged over 300 stimulus repetitions. Clicks were tested first to verify electrode placement and the presence of a clearly observable response, and then tones were tested in random order of frequency. We presented a tone of a given frequency at a sound level of 85-105 dB SPL (depending on frequency), and then continued presenting the same tone at lower sound levels until a threshold was reached. Threshold was defined as the sound level at which the peak-to-peak (any peak) amplitude of the response was two standard deviations above the average baseline noise amplitude during a time window when no sound stimulus was present. Testing lasted approximately 40-60 minutes, and mice were returned to their home cages following testing and monitored until recovery.
We also confirmed the effectiveness of deafening by observation of hair cell loss using phalloidin staining. Whole cochleae were dissected from experimental animals and stored in 10% formalin solution (Sigma) at 4°C. Sectioning and staining of cochleae was done in a blinded manner. Before staining cochleae were washed in 0.1 M PB and decalcified in 3% EDTA for 48 hours. Apical, middle and basal turns were dissected. Each turn was permeabilized with 0.2% Triton-X for 1 hour and subsequently incubated with Alexa Fluor 488-phalloidin (Invitrogen; 1:200) and DAPI (1:5000) for 2 hours. Cochlear turns were whole mounted on glass slides using ProLong Gold antifade mounting medium (Life Technologies). Confocal images of NR and DF cochleae were taken using the same settings.
Cortical slice preparation
Each mouse was deeply anesthetized using isoflurane vapors until absence of corneal reflex and toe pinch response, then transcardially perfused with ice cold artificial cerebrospinal fluid (ACSF, in mM: 124 NaCl, 5 KCl, 1.25 NaH2PO4·H2O, 26 NaHCO3, 10 dextrose, 2.5 CaCl2 1.5 MgCl2, bubbled with 95% O2/5% CO2) and immediately decapitated. The brain was removed and immersed in ice-cold dissection buffer (in mM: 212.7 sucrose, 10 dextrose, 3 MgCl2, 1 CaCl2, 2.6 KCl, 1.23 NaH2PO4·H2O, 26 NaHCO3, bubbled with 95% O2/5% CO2). Blocks containing V1 were isolated and sectioned coronally into 300-μm thick slices using a vibratome (PELCO easiSlicer, Ted Pella). Slices were incubated in a light-tight holding chamber filled with ACSF at 30°C for 30 minutes and then allowed to recover at room temperature for at least 30 minutes. The slices were then transferred to a submersion-type recording chamber mounted on an upright microscope (Nikon, E600FN) with oblique infrared illumination.
Electrophysiology
Whole-cell current clamp recordings for LTP and LTD
Visually identified neurons were targeted at a 40%–50% depth from the pia corresponding to L4. The location of the majority of recorded cells was confirmed post hoc by biocytin labeling. Recording pipettes (3-5 MΩ) were filled with an internal solution consisting of (in mM): 130 K-gluconate, 8 NaCl, 0.2 EGTA, 10 HEPES, 3 ATP, 10 Na-phosphocreatine, and 0.5 GTP, pH 7.4, 275–285 mOsm. Excitatory postsynaptic potential (EPSPs) were evoked by shining blue light (Thorlabs 455-nm LED, 5-ms duration) through a 40× objective lens to activate thalamic terminals. Stimulus intensity was adjusted to the minimum required to reliably produce single-peaked, short-onset latency (< 4-ms) EPSPs. Recording configuration switched from current-clamp to voltage-clamp for the plasticity induction protocol. The paired stimulus protocol entailed 200 pulses of presynaptic stimulation at 1-Hz coupled with postsynaptic depolarization to 0 mV or −40 mV (Jiang et al., 2007) to induce LTP or LTD, respectively. An Axon patch-clamp amplifier 700B (Molecular Devices) was used for whole cell recordings and data were acquired through Igor Pro software (WaveMetrics; RRID: SCR_000325). Changes in synaptic strength were quantified as the initial slope of the EPSP normalized by the average baseline slope obtained during the first 5 minutes of stable recordings. Cells were discarded if Vm > −65 mV or if Ri changed more than 30% during the experiment.
NMDAR/AMPAR ratio
Isolated monosynaptic glutamatergic currents were recorded in the presence of bicuculline (20 μM) in ACSF with 4 mM Ca 2+ and 4 mM Mg2+ using internal solution consisted of (in mM): 102 Cs-gluconate, 5 TEA-chloride, 3.7 NaCl, 20 HEPES, 0.3 Na·GTP, 4 Mg·ATP, 0.2 EGTA, 10 BAPTA, and 5 QX-314 chloride, pH 7.2, 300 mOsm. The LED intensity was adjusted to twice the minimum value required to consistently produce single-peaked, short-onset latency (< 4 ms) AMPAR-EPSCs. NMDAR and AMPAR responses were distinguished based on their kinetics and voltage dependence. The NMDAR component was measured as the amplitude 70-ms after the response onset at +40 mV, whereas the AMPAR component was measured as the peak amplitude recorded at −80 mV.
NMDAR kinetics
NMDAR-EPSCs were pharmacologically isolated in the presence of 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX, 10 μM), picrotoxin (50 μM) and glycine (1 μM). Responses were recorded at +40 mV, with 4 mM Ca2+ and 4 mM Mg2+ in ACSF to reduce polysynaptic responses and using the same Cs-gluconate internal solution as mentioned above. A minimum of 15 traces (15-30) were averaged per cell and the decay was fitted with a double exponential equation. Slow (s) and fast (f) exponential values were then used to calculate a weighted time constant (τw) as published(Philpot et al., 2001; Rumbaugh and Vicini, 1999): τw = τf [If/(If + Is)] + τs [Is/(If + Is)] where If and Is are the amplitudes for fast and slow components respectively.
Light evoked Sr2+ mEPSCs (LEv-Sr mEPSCs)
Slices were incubated in the recording chamber with Ca2+-free ACSF containing 4 mM Sr2+ and 4 mM Mg2+ (95% O2/5% CO2) for 20 minutes to equilibrate before initiation of recordings. AMPAR-mediated responses were recorded under two conditions: (i) pharmacologically isolated with 20 μM bicuculline and 100 μM DL-2-amino-5 phosphonopentanoic acid (APV) and (ii) isolated with the same drugs with 1 μM tetrodotoxin (TTX) to confirm monosynaptic responses resulting from ChR2 activation of presynaptic inputs. Recording pipettes (3–5 MΩ) were filled with internal solution containing (in mM): 130 Cs-gluconate, 8 KCl, 1 EGTA, 10 HEPES, 4 ATP, and 5 QX-314, pH 7.4,285–295 mOsm. Visually identified neurons were voltage clamped at −80mV. Only recordings with Rs < 25 MΩ and Ri > 150MΩ that changed less than 15% were included in the analysis. The responses were evoked using a 455-nm LED (Thor labs) illuminated through a 40x objective lens (Nikon). Stimulation intensity was set to the minimal light required to produce a reliable response with 5-ms stimulus duration delivered every 10 s. Spontaneous events were quantified during a 400-ms time window before LED illumination (preLED) and LED-evoked Sr2+ desynchronized (LEv-Sr) events were quantified in a 400-ms window that started 50-ms from LED onset (postLED). Events were selected using MiniAnalysis (Synaptosoft; RRID: SCR_002184) with threshold set to 3 times the root mean square (RMS) noise. Cells with an RMS noise > 2 were excluded. To calculate the mean amplitude of evoked desynchronized events without baseline spontaneous activity, we used the following equation: [(postLED amp × postLED frq) − (preLED amp × preLED frq)] / (postLED frq − preLED frq) where amp is amplitude and frq is frequency. Calculated LEv-Sr2+ mEPSC amplitudes between the two recording conditions (with and without TTX) showed no significant changes (NR = 21.02 ± 2.8 pA, NRTTX = 16.44 ± 1.4 pA, unpaired Student’s t test p = 0.2080; mean DF = 18.61 ± 2.4 pA, DFTTX = 20.8 ± 2.3 pA, unpaired t test p = 0.5200). Therefore, the data were pooled for Figure 3.
E/I ratio
Cortical L4 principal neurons were patched in voltage-clamp configuration with internal solution consisting of (in mM): 130 Cs-gluconate, 8 KCl, 1 EGTA, 10 HEPES, 4 ATP, and 5 QX-314, pH 7.4, 285–295 mOsm. Only recordings with Rs < 25 MΩ and Ri > 150 MΩ were included in the analysis. Monosynaptic EPSCs and disynaptic inhibitory postsynaptic currents (IPSCs) were recorded from each cell at the reversal potential for inhibitory currents and excitatory currents, respectively. After compensation for the junction potential, EGABA was −52 mV and Eglu was 0 mV. Thalamic terminals were activated with light flashes (LED 455-nm, 5-ms duration) delivered at several intensities until responses maximized, but did not evoke polysynaptic events. E/I ratios were calculated at each stimulation intensity for each cell. We noticed a tendency for E/I values to increase along with stimulation power until it reached a plateau at higher intensities (Figure S3). Therefore, we averaged the first three values of E/I ratio at intensities that produced the plateau for each cell and report this as the E/I ratio.
Biocytin labeling
Biocytin was added to the internal solution during most experiments to allow confirmation of neuronal location and morphology. Cortical slices were fixed in 4% paraformaldehyde overnight at 4°C, rinsed 10 minutes in 0.1 M phosphate buffer (PB) at room temperature and permeabilized in 2% Triton X-100 for one hour. Slices were then incubated in avidin-Texas red conjugate (Fisher) diluted 1:2000 in 1% Triton X-100 overnight at 4°C. Slices were washed twice in 0.1 M PB and mounted on glass slides with Prolong Gold antifade (Invitrogen). Images were taken on a laser scanning confocal microscope (Zeiss LSM 700) to confirm location of recorded cells post hoc.
Monocular deprivation
Mice were deeply anesthetized with isoflurane gas (3%) in an induction chamber. After the disappearance of toe pinch response, animals were transferred to a stereotaxic apparatus where oxygen supply was supplemented with isoflurane (1%–2%). The upper and lower margins of one eyelid were trimmed and sutured (PSD II; Ethicon) shut. The eye contralateral to the hemisphere being imaged was always sutured. Animals of the same sex were housed together (2–3 mice/cage) and disqualified if sutures opened.
Optical imaging of intrinsic signals
Mice were placed in a stereotaxic apparatus under constant supply of an oxygen/isoflurane mixture (0.7%–1.5% isoflurane), supplemented by a single injection of chlorprothixene (0.2 μg/g, ip). Body temperature was maintained at 37°C and heart rate was monitored throughout the experiment. The skull over V1 on the left hemisphere (contralateral to lid suture) was exposed and washed with hydrogen peroxide. Low melting point agarose (3%) and a glass coverslip were placed over the exposed area and allowed to solidify. V1 responses were recorded using the method previously developed by Kalatsky and Stryker (2003) and optimized for ocular dominance (OD) measurements (Cang et al., 2005). Optical images of cortical intrinsic signals were acquired using a Dalsa CCD camera (Dalsa, Waterloo, Canada) controlled by custom software. The surface vasculature and intrinsic signals were visualized with illumination wavelengths of 555-nm and 610-nm, respectively. After focusing on prominent vasculature marks, the camera was focused 600-μm below the surface. A high refresh rate monitor (ViewSonic) was placed 25-cm in front of the animal for stimulus presentation. The visual stimulus presented was restricted to the binocular visual field (5° to +15° azimuth) and consisted of a horizontal bar (x = 5°, y = 0°, width = 20) continuously presented for 5 minutes in upward (90°) and downward (270°) directions to each eye separately. The cortical response at the stimulus frequency was extracted by Fourier analysis and used to calculate the ocular dominance index (ODI). Two maps were averaged per eye for each animal to compute the ODI following the formula: (C−I)/(C+I) where C and I are the response magnitudes of each pixel to visual stimulation to the contralateral (C) and ipsilateral (I) eye respectively. The binocular area was selected as a region of interest (ROI) in the ODI map and the ODI values within this region were averaged.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analysis was performed using Prism 7.0 (GraphPad Software; RRID: SCR_002798) and data are presented as mean ± SEM. Sample number for each experiment can be found in the figure legends. Unpaired two-tailed Student’s t test was used to compare recordings between NR and DF groups. Paired two-tailed t tests were used to compare ODI and eye-specific cortical activation responses measured from the same animals before and after MD. In all cases, p <0.05 was considered statistically significant.
Supplementary Material
Highlights.
Thalamocortical (TC) plasticity is limited in adults
A short duration of deafening specifically recovers TC-LTP in adult V1
Deafening increases NMDAR function and E/I balance at TC synapses
Deafening accelerates ocular dominance plasticity in adult V1
ACKNOWLEDGMENTS
This work was supported by NIH grant R01-EY014882 (H.-K.L.) and the David M. Rubenstein Fund for Hearing Research (A.M.L. and H.-K.L.). The authors would like to thank Varun Chokshi and Dr. Jessica Whitt for help on TC-LTP experiments and Drs. Kristina Nielsen and Alfredo Kirkwood for helpful discussions on this project and the manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.08.072.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Barkat TR, Polley DB, and Hensch TK (2011). A critical period for auditory thalamocortical connectivity. Nat. Neurosci 14, 1189–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth AL, and Malenka RC (2001). NMDAR EPSC kinetics do not regulate the critical period for LTP at thalamocortical synapses. Nat. Neurosci 4, 235–236. [DOI] [PubMed] [Google Scholar]
- Cang J, Kalatsky VA, Lowel S, and Stryker MP (2005). Optical imaging of the intrinsic signal as a measure of cortical plasticity in the mouse. Vis. Neurosci 22, 685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittajallu R, and Isaac JT (2010). Emergence of cortical inhibition by coordinated sensory-driven plasticity at distinct synaptic loci. Nat. Neurosci 13, 1240–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S, Jeong JH, Ko S, Yu X, Kim YH, Isaac JTR, and Koretsky AP (2017). Peripheral sensory deprivation restores critical-period-like plasticity to adult somatosensory thalamocortical inputs. Cell Rep 19, 2707–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crair MC, and Malenka RC (1995). A critical period for long-term potentiation at thalamocortical synapses. Nature 375, 325–328. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Lewis TJ, and Connors BW (2007). Synaptic basis for intense thalamocortical activation of feedforward inhibitory cells in neocortex. Nat. Neurosci 10, 462–468. [DOI] [PubMed] [Google Scholar]
- Daw MI, Ashby MC, and Isaac JT (2007). Coordinated developmental recruitment of latent fast spiking interneurons in layer IV barrel cortex. Nat. Neurosci 10, 453–61. [DOI] [PubMed] [Google Scholar]
- Dehghani N, Peyrache A, Telenczuk B, Le Van Quyen M, Halgren E, Cash SS, Hatsopoulos NG, and Destexhe A (2016). Dynamic balance of excitation and inhibition in human and monkey neocortex. Sci. Rep 6, 23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai NS, Cudmore RH, Nelson SB, and Turrigiano GG (2002). Critical periods for experience-dependent synaptic scaling in visual cortex. Nat. Neurosci 5, 783–789. [DOI] [PubMed] [Google Scholar]
- Dudek SM, and Friedlander MJ (1996). Developmental down-regulation of LTD in cortical layer IV and its independence of modulation by inhibition. Neuron 16, 1097–1106. [DOI] [PubMed] [Google Scholar]
- Frenkel MY, and Bear MF (2004). How monocular deprivation shifts ocular dominance in visual cortex of young mice. Neuron 44, 917–923. [DOI] [PubMed] [Google Scholar]
- Froemke RC (2015). Plasticity of cortical excitatory-inhibitory balance.Annu. Rev. Neurosci 38, 195–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froemke RC, Merzenich MM, and Schreiner CE (2007). Asynaptic memory trace for cortical receptive field plasticity. Nature 450, 425–429. [DOI] [PubMed] [Google Scholar]
- Gabernet L, Jadhav SP, Feldman DE, Carandini M, and Scanziani M (2005). Somatosensory integration controlled by dynamic thalamocortical feed-forward inhibition. Neuron 48, 315–327. [DOI] [PubMed] [Google Scholar]
- Gil Z, Connors BW, and Amitai Y (1999). Efficacy of thalamocortical and intracortical synaptic connections: quanta, innervation, and reliability. Neuron 23, 385–397. [DOI] [PubMed] [Google Scholar]
- Goel A, and Lee HK (2007). Persistence of experience-induced homeostatic synaptic plasticity through adulthood in superficial layers of mouse visual cortex. J. Neurosci 27, 6692–6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JA, and Stryker MP (1996). Experience-dependent plasticity of binocular responses in the primary visual cortex of the mouse. J. Neurosci 16, 3274–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, and Cang J (2016). Binocular matching of thalamocortical and intracortical circuits in the mouse visual cortex. eLife 5, e22032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Iwasaki S, Mizuta K, Arai M, and Mineta H (2007). Pattern of cochlear damage caused by short-term kanamycin application using the round window microcatheter method. Acta Otolaryngol 127, 116–121. [DOI] [PubMed] [Google Scholar]
- He HY, Hodos W, and Quinlan EM (2006). Visual deprivation reactivates rapid ocular dominance plasticity in adult visual cortex. J. Neurosci 26, 2951–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He HY, Ray B, Dennis K, and Quinlan EM (2007). Experience-dependent recovery of vision following chronic deprivation amblyopia. Nat. Neurosci 10, 1134–1136. [DOI] [PubMed] [Google Scholar]
- House DR, Elstrott J, Koh E, Chung J, and Feldman DE (2011). Parallel regulation of feedforward inhibition and excitation during whisker map plasticity. Neuron 72, 819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B, Huang ZJ, Morales B, and Kirkwood A (2005). Maturation of GABAergic transmission and the timing of plasticity in visual cortex. Brain Res. Brain Res. Rev 50, 126–133. [DOI] [PubMed] [Google Scholar]
- Jiang B, Treviño M, and Kirkwood A (2007). Sequential development of long-term potentiation and depression in different layers of the mouse visual cortex. J. Neurosci 27, 9648–9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalatsky VA, and Stryker MP (2003). New paradigm for optical imaging: temporally encoded maps of intrinsic signal. Neuron 38, 529–545. [DOI] [PubMed] [Google Scholar]
- Kirkwood A, Lee HK, and Bear MF (1995). Co-regulation of long-term potentiation and experience-dependent synaptic plasticity in visual cortex by age and experience. Nature 375, 328–331. [DOI] [PubMed] [Google Scholar]
- Kloc M, and Maffei A (2014). Target-specific properties of thalamocortical synapses onto layer 4 of mouse primary visual cortex. J. Neurosci 34, 15455–15465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, and Whitt JL (2015). Cross-modal synaptic plasticity in adult primary sensory cortices. Curr. Opin. Neurobiol 35, 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainardi M, Landi S, Gianfranceschi L, Baldini S, De Pasquale R, Berardi N, Maffei L, and Caleo M (2010). Environmental enrichment potentiates thalamocortical transmission and plasticity in the adult rat visual cortex. J. Neurosci. Res 88, 3048–3059. [DOI] [PubMed] [Google Scholar]
- McGuire B, Fiorillo B, Ryugo DK, and Lauer AM (2015). Auditory nerve synapses persist in ventral cochlear nucleus long after loss of acoustic input in mice with early-onset progressive hearing loss. Brain Res 1605, 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montey KL, and Quinlan EM (2011). Recovery from chronic monocular deprivation following reactivation of thalamocortical plasticity by dark exposure. Nat. Commun 2, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, and Seeburg PH (1994). Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540. [DOI] [PubMed] [Google Scholar]
- Petrus E, Isaiah A, Jones AP, Li D, Wang H, Lee HK, and Kanold PO (2014). Crossmodal induction of thalamocortical potentiation leads to enhanced information processing in the auditory cortex. Neuron 81, 664–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrus E, Rodriguez G, Patterson R, Connor B, Kanold PO, and Lee HK (2015). Vision loss shifts the balance of feedforward and intracortical circuits in opposite directions in mouse primary auditory and visual cortices. J. Neurosci 35, 8790–8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham TA, Graham SJ, Suzuki S, Barco A, Kandel ER, Gordon B, and Lickey ME (2004). A semi-persistent adult ocular dominance plasticity in visual cortex is stabilized by activated CREB. Learn. Mem 11, 738–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpot BD, Sekhar AK, Shouval HZ, and Bear MF (2001). Visual experience and deprivation bidirectionally modify the composition and function of NMDA receptors in visual cortex. Neuron 29, 157–169. [DOI] [PubMed] [Google Scholar]
- Philpot BD, Espinosa JS, and Bear MF (2003). Evidence for altered NMDA receptor function as a basis for metaplasticity in visual cortex. J. Neurosci 23, 5583–5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan EM, Olstein DH, and Bear MF (1999). Bidirectional, experience-dependent regulation of N-methyl-D-aspartate receptor subunit composition in the rat visual cortex during postnatal development. Proc. Natl. Acad. Sci. USA 96, 12876–12880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranson A, Cheetham CE, Fox K, and Sengpiel F (2012). Homeostatic plasticity mechanisms are required for juvenile, but not adult, ocular dominance plasticity. Proc. Natl. Acad. Sci. USA 109, 1311–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaugh G, and Vicini S (1999). Distinct synaptic and extrasynaptic NMDA receptors in developing cerebellar granule neurons. J. Neurosci. 19, 10603–10610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, and Stryker MP (2008). Distinctive features of adult ocular dominance plasticity. J. Neurosci 28, 10278–10286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NB, Frenkel MY, Philpot BD, Nakazawa K, Tonegawa S, and Bear MF (2003). NMDA receptor-dependent ocular dominance plasticity in adult visual cortex. Neuron 38, 977–985. [DOI] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, and Jan LY (1994). Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368, 144–147. [DOI] [PubMed] [Google Scholar]
- Watt AJ, van Rossum MC, MacLeod KM, Nelson SB, and Turrigiano GG (2000).Activity coregulates quantal AMPA and NMDA currents at neocortical synapses. Neuron 26, 659–670. [DOI] [PubMed] [Google Scholar]
- Xue M, Atallah BV, and Scanziani M (2014). Equalizing excitation-inhibition ratios across visual cortical neurons. Nature 511, 596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Chung S, Chen DY, Wang S, Dodd SJ, Walters JR, Isaac JT, and Koretsky AP (2012). Thalamocortical inputs show post-critical-period plasticity. Neuron 74, 731–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Jiao YY, and Sun QQ (2011). Developmental maturation of excitation and inhibition balance in principal neurons across four layers of somatosensory cortex. Neuroscience 174, 10–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.