

Abstract
Previous work established a coumarin scaffold as a starting point for inhibition of Mycobacterium tuberculosis (Mtb) FadD32 enzymatic activity. After further profiling of the coumarin inhibitor 4 revealed chemical instability, we discovered that a quinoline ring circumvented this instability and had the advantage of offering additional substitution vectors to further optimize. Ensuing SAR studies gave rise to quinoline-2-carboxamides with potent anti-tubercular activity. Further optimization of ADME/PK properties culminated in 21b that exhibited compelling in vivo efficacy in a mouse model of Mtb infection.
Keywords: Mycobacterium tuberculosis, FadD32 inhibitor, structure-activity relationship, quinoline-2-carboxamide, in vivo efficacy
Graphical Abstract
To create your abstract, type over the instructions in the template box below. Fonts or abstract dimensions should not be changed or altered.
Despite global efforts to improve diagnosis and make current therapy more readily available, tuberculosis (TB) remains one of the deadliest diseases in the world. In 2016, an estimated 10.4 million new cases were reported, and nearly 1.7 million people died from the disease.1 The standard treatment regimen for active, drug-sensitive TB disease includes a six-month course of four antimicrobial drugs (isoniazid, rifampin, ethambutol, and pyrazinamide). However, the emergence of multi-drug resistant (MDR) and extensively drug resistant (XDR) TB has made control of this disease much more challenging. As a result, discovery of new agents with novel mechanisms of action that can overcome existing resistance is of critical importance to global health.
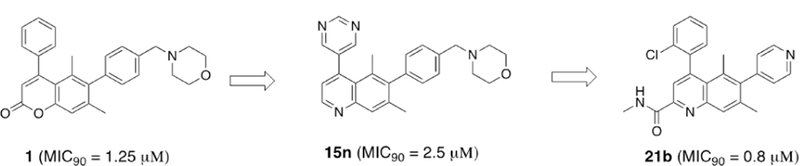
We previously identified substituted coumarin inhibitors that kill Mycobacterium tuberculosis by inhibiting fatty acid degradation protein D32 (FadD32), an enzyme required for mycolic acid biosynthesis.2,3 Mycolic acid biosynthesis is one of the few well-validated pathways in anti-tubercular drug development, as a key enzyme in this pathway (InhA) is targeted by isoniazid (INH). Coumarin compounds such as 1–3 demonstrated good-to-moderate whole cell activity (MIC90 = 0.24 – 5.6 μM) and provided a starting point to validate FadD32 as a viable target for the treatment of TB (Fig. 1). 4,5
Fig. 1.
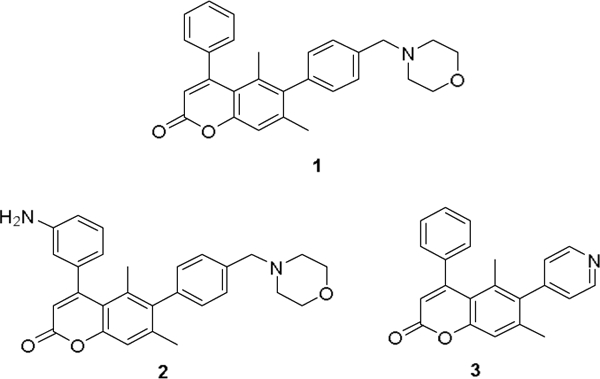
Coumarins previously identified as potent anti-TB agents
Despite encouraging in vivo activity of the initial coumarin hit, we recognized that potential chemical reactivity of the lactone ring could lead to formation of covalent adducts that could introduce unwanted toxicity and preclude further development. Indeed, as part of our initial coumarin SAR efforts, we observed that electron- poor ring systems such as 4 were not stable towards organic bases such as morpholine, affording equimolar levels of ring-opened product 5 after prolonged time at ambient temperature (Scheme 1). Similar hydrolysis has been observed in other known coumarin compounds.6
Scheme 1.
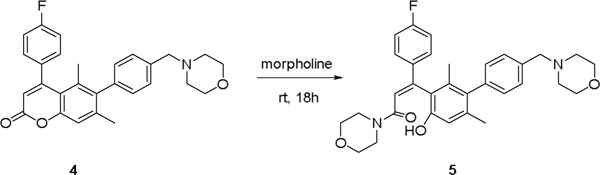
Coumarin core undergoes ring opening under basic conditions
To eliminate this source of hydrolytic instability, we initiated an effort to explore heterocyclic replacements of the coumarin ring. Several 5/6- and 6/6-bicyclic scaffolds including benzofuran, 2-quinolone, and quinoline were investigated, of which the latter was found to be the most potent (Fig. 2). Based on the encouraging activity as well as the additional substitution possibilities afforded by the quinoline ring system, we selected this chemically-stable heterocycle for further investigation.
Fig. 2.
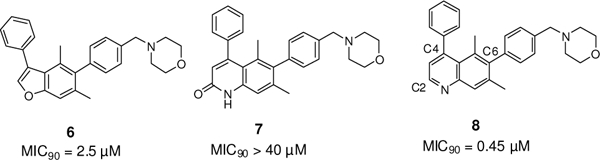
Heterocyclic replacements for the unstable coumarin core
The quinoline core of 11 was synthesized via Conrad-Limpach cyclization of the substituted aniline 9.7,8,9 Ester hydrolysis followed by decarboxylation afforded 4-hydroxyquinoline 12, which served as a key intermediate for late-stage diversification of two distinct vectors off the quinoline ring. Suzuki coupling of 12 with boronate 13 permitted exploration of carbon-linked substituents. Functionalization at the C4 position was enabled via conversion of the hydroxyl to the triflate, with a range of C4 variants including 8 and 15a-o accessible by this synthetic strategy (Scheme 2).
Scheme 2.
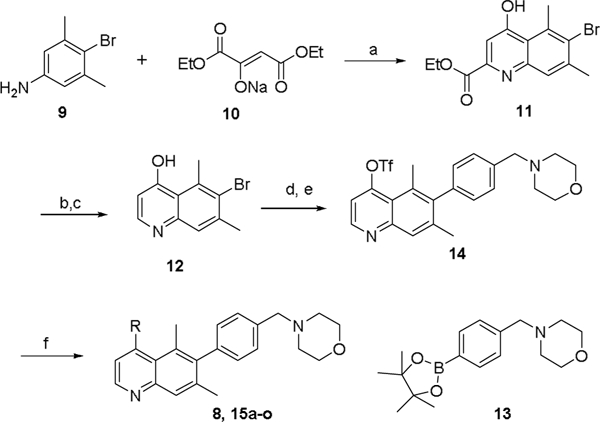
Synthesis of C2 non-substituted quinoline analogs. Reagents and conditions: a) TsOH.H2O, toluene, reflux then Ph2O, 250 oC, 83%; b) NaOH, H2O/THF, 90%; c) Ph2O, 250 oC, 75%; d) XPhos-Pd-G2, 13, Na2CO3, dioxane/H2O, 50%; e) Tf2O, Et3N, CH2Cl2, 66%; f) Pd(PPh3)4, R-B(OH)2, Et3N, dioxane, 100 oC, 8–93%.
This efficient synthetic route also allowed the preparation of 2- substituted quinoline derivatives (Scheme 3). First, conversion of the hydroxyl group in 11 into triflate 16 followed by Suzuki coupling with 2-chlorophenylboronic acid afforded the bromoquinoline 17. A second Suzuki reaction with 4-pyridyl boronic acid at the more-hindered C6 position afforded the key intermediate 18. Reduction with LiAlH4 afforded primary alcohol 19, while treatment with MeMgBr afforded tertiary alcohol 20. Amide-ester exchange with different primary amines led to the formation of amides 21. Alternatively, amides 23 were prepared by standard amide coupling with acid 22.
Scheme 3.
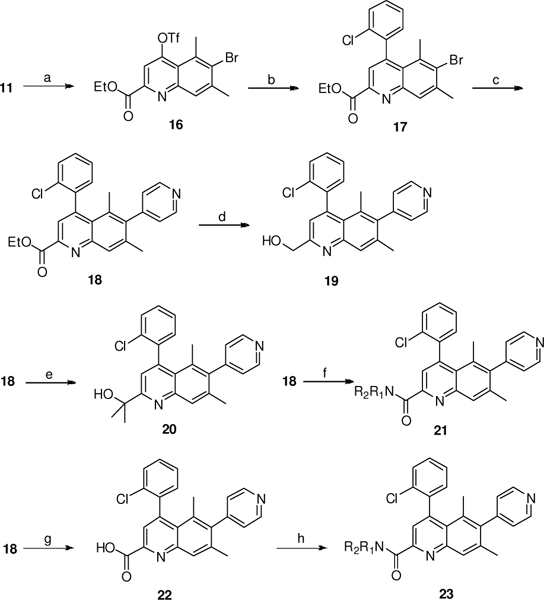
Synthesis of C2 substituted quinoline analogs 18–23. Reagents and conditions: a) Tf2O, Et3N, CH2Q2; b) o-ClC6H4B(OH)2, Pd(PPh3)4, Et3N, dioxane, 100 oC, 60%, 2 steps; c) XPhos-Pd-G2, 4-pyridyl boronic acid, Na2CO3, dioxane/H2O, 60 oC, 66%; d) LiAlH4, THF, 52%; e) MeMgBr, THF, −20 oC, 35%; f) R1R2NH, MeOH, 10–70%; g) NaOH, THF/H2O, 70%; h) R1R2NH, EDCI, HOBt, iPr2EtN, DMF, 11–45%
The synthetic routes above enabled the systematic exploration of several directional vectors emanating from the quinoline core. At the C4 position, the chemically-stable quinoline ring now permitted exploration of electron-poor substituents like halogen- substituted aryl rings. Slight improvements in MIC were indeed observed (MIC90 of 15a = 0.3 μM), however these potency gains were offset by poor kinetic aqueous solubility. We were able to improve solubility of phenyl-substituted quinolines by introduction of ortho-substituted analogs with reduced planarity such as 15d, however these analogs were less potent (MIC90 = 2.5 μm).
Given the low solubility of phenyl-substituted quinolines, we then turned our attention to heterocyclic C4 substitution patterns. Several analogs (15j, 15m, 15n) showed improved aqueous solubility (pH 7.4 solubility > 150 μM), albeit with reduced whole-cell activity.
Having identified a more soluble quinoline FadD32 inhibitor with micromolar inhibition of Mtb growth (15n), we initiated ADME profiling to prioritize additional properties for optimization (Table 2). Compound 15n showed moderate levels of binding to proteins from mouse and human plasma, which suggests appreciable levels of free compound would be circulating in the plasma. Unfortunately, the metabolic stability of 15n was unacceptably low; this rapid clearance in vitro was corroborated by an in vivo pharmacokinetic study in mice (plasma clearance = 65.1 mL/min/kg following 1 mg/kg intravenous dose).
Table 2.
In vitro ADME parameters of 15n
| Mouse | Human | ||
|---|---|---|---|
| Plasma protein binding (%) a | 92.8 | 95.2 | |
| Microsomal stability (% remaining at 60 min) a | <5 | 11.6 | |
All experiments were performed at least twice in duplicate, and data reported are mean values.
Due to the high clearance of 15n, our optimization efforts next focused on replacing metabolically-labile substituents with groups less prone to oxidation. One opportunity from our earlier SAR efforts was to install an electron-poor chlorophenyl group at the C2 position, as we recognized the potential for improved potency. We also elected to replace the benzyl morpholine C6 group with heteroaromatic groups like pyridine which lack C-H bonds adjacent to oxygen and nitrogen. Compound 15p (MIC90 = 5 μM, see Table 3) therefore served as our template for further optimization.
Table 3.
C2 SAR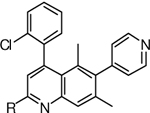
| Compd | R |
Mtb H37Rv MIC90 (μM) |
Solubility10 (μM) |
|---|---|---|---|
| 15p | H | 5 | 21 |
| 18 | CO2Et | 10 | 9 |
| 19 | CH2OH | 3.75 | 32 |
| 20 | CMe2OH | >40 | 38 |
| 21a | CONH2 | 1.25 | 8 |
| 21b | CONHMe | 0.8 | 15 |
| 21c | CONHiPr | 2.5 | 4 |
| 21d | CONHCH2CH2F | 1.25 | 5 |
| 21e | CONHCH2CH2OH | 0.3 | 25 |
| 21f | CONHCH2CH2NMe2 | 0.3 | 263 |
| 22 | CO2H | 80 | 226 |
| 23a | CONMe2 | 20 | 157 |
| 23b |  |
5 | 41 |
| 23c | 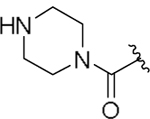 |
10 | n.d. |
| 23d | 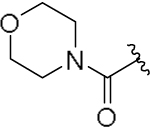 |
15 | 145 |
A key advantage of the quinoline scaffold is the opportunity to functionalize the 2-position, a growth vector not accessible in the original coumarin scaffold. The C2 ester intermediate 11 provides ready access to a series of quinoline-2-carboxamides. Gratifyingly, installation of a methyl amide at C2 (compound 21b) resulted in a 6-fold potency improvement compared with no substitution at this quinoline ring position. Tertiary amides (23a-d) were less active than primary and secondary amides (21a-f), suggesting the importance of a free N-H for anti-tubercular activity. Among various secondary amides, several hydrophilic functional groups including basic amines (e.g., 21f) were tolerated, providing a useful handle for modulating overall physicochemical and off-target properties of quinoline FadD32 inhibitors.
To confirm the primary biochemical target of these modified quinoline derivatives, we tested their growth inhibition against mutant strains of Mtb that were resistant to inhibition by coumarin FadD32 inhibitors (Table 4). Resistance mutations to coumarin- based inhibitors had been previously mapped to E120 and F291 in the fadD32 gene. Since the discovery of these resistance mutations, a crystal structure of FadD32 bound to a substrate-based inhibitor has been solved.11,12 F291 is located at the entrance of the putative pantetheine-binding tunnel of FadD32, at the interface of its N-and C-terminal domains. Given previous biochemical work establishing the inhibition of acyl group transfer (and not acyl adenylation) as the likely mode of action of coumarin FadD32 inhibitors, our current model situates the inhibitor at the entrance to the phosphopantetheine tunnel used by PKS13 ACP to block approach to the active site or prevent the transition to the thiolation conformation. † Despite the additional substitution patterns introduced on the quinoline inhibitors described above, compounds 8, 15m, 15n, and 21b all show markedly less activity versus the E120 and F291 mutants, compared to wild-type, suggesting that they are still targeting FadD32.
Table 4.
Comparison of MIC90s of selected quinoline compounds against wild type and mutant FadD32
| Compd | H37Rv MIC90 (μM) |
FadD32 mutant MIC90 (μM) |
|---|---|---|
| 8 | 0.45 | 15.6 |
| 15m | 2.5 | 100 |
| 15n | 2.5 | 100 |
| 21b | 0.8 | 6.25 |
The observed cross-resistance of FadD32 mutants to coumarin and quinoline inhibitors provided confidence in the biological target and impetus to re-affirm in vivo. Of the most potent compounds with sub-micromolar MICs (21b, 21e, and 21f), 21b was chosen for in vivo investigation in a mouse model of Mtb infection based on its cross-species microsomal stability profile as well as its minimal cytotoxicity (Table 5). In an 8-day acute model of tuberculosis infection (1 × 105 CFU of H37Rv by intratracheal infection), 21b showed a nearly 4-log CFU reduction in a dose- dependent manner with no tolerability issue observed up to the highest dose of 50 mg/kg (Fig. 3). The calculated ED99 of 7.8 mg/kg (95% CI: 6.9–8.7 mg/kg) provides a reasonable starting point for human dose prediction.
Table 5.
Comparison of in vitro ADME and toxicity parameters of selected quinoline compounds
| Compd |
Mtb H37Rv MIC90 (μM) |
mLM stability (%R at 60 min) a |
hLM stability (%R at 60 min) a |
Cytotoxicity HepG2 IC50 (uM) a |
|---|---|---|---|---|
| 21b | 0.8 | 19.1 | 19.4 | 60 |
| 21e | 0.3 | 26.2 | 8.0 | 100 |
| 21f | 0.3 | 24.0 | 3.2 | 16 |
All experiments were performed at least twice in duplicate, and data reported are mean values.
Fig. 3.
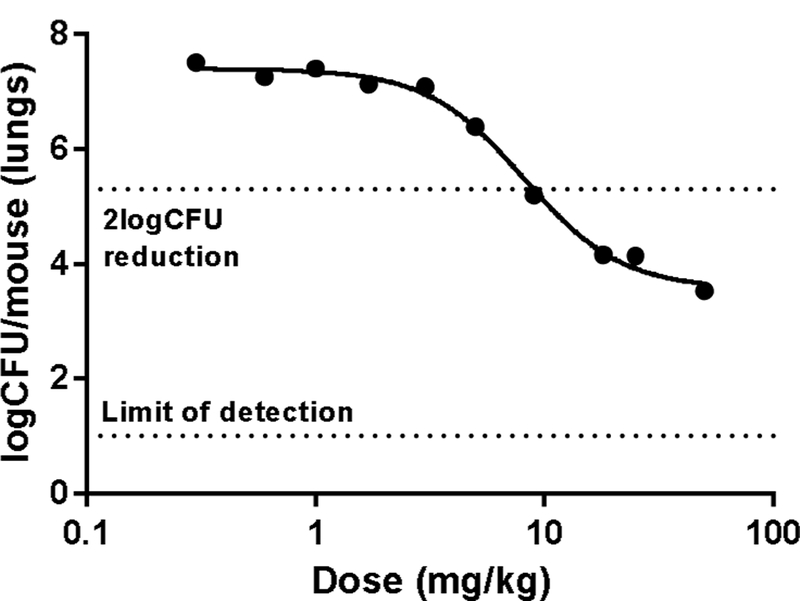
Lung CFU reduction in an acute murine model of TB infection by once-daily oral administration of compound 21b.13 Each dot represents data from 1 mouse. Data were fitted to a sigmoidal curve to estimate ED99 (dose that shows 2 log CFUs reduction compared to untreated mice).
In conclusion, we have discovered a series of quinoline compounds which address a chemical liability inherent to the previous coumarin hit and provide additional handles for medicinal chemistry optimization. From these quinoline-based FadD32 inhibitors, we were able to achieve sub-micromolar inhibition of Mtb growth with molecules bearing sufficient solubility and exposure for in vivo study in mice. Additional efforts to biophysically characterize the direct binding of quinoline inhibitors to the FadD32 protein and their progression to advanced candidates will be reported in due course.
Supplementary Material
Table 1.
C4 SAR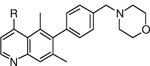
| Compd | R |
Mtb H37Rv MIC90 (μM) |
Solubility10 (μM) |
|---|---|---|---|
| 15a | 2-ClPh | 0.3 | n.d. |
| 15b | 3-ClPh | 1.5 | 1 |
| 15c | 4-ClPh | >80 | n.d. |
| 15d | 2-CNPh | 2.5 | 24 |
| 15e | 2-Et2NCH2Ph | 2.5 | 59 |
| 15f | 2-HO2CPh | 40 | 222 |
| 15g | 2-H2NC(O)Ph | 20 | n.d. |
| 15h | 2-Cl-3-MeOPh | 10 | 2 |
| 15i | 2,6-diMe-Ph | 5 | n.d. |
| 15j | 3-pyridyl | 5 | 209 |
| 15k | 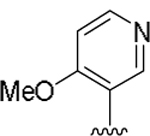 |
2.5 | 169 |
| 15l | 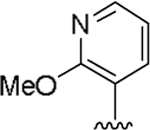 |
2.5 | 1 |
| 15m | 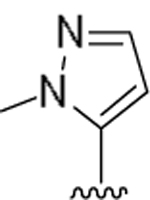 |
2.5 | 243 |
| 15n | 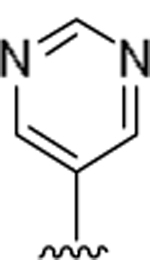 |
2.5 | >230 |
Highlights.
New quinoline scaffolds show improved chemical stability; handle for further optimization
Subsequent SAR identified quinoline-2- acrboxamides with potent antitubercular activity
Hit-to-lead efforts yielded a potent FadD32 inhibitor with notable in vivo efficacy in mouse model of TB infection
Acknowledgments
The authors would like to thank Patrick O’ Hearn and Stephen Johnston and Patrick Harran of the Center for the Development of Therapeutics (CDoT) at Broad Institute for the ADME work, and William Reiley at TICRO BioServices for the in vivo efficacy studies. This work was supported by the Broad Institute Tuberculosis donor group and the Pershing Square Foundation and NIH R01 AI132300.
Abbreviations
- MIC90
minimum inhibitory concentration for 90%
- Mtb
Mycobacterium tuberculosis
- CFU
colony-forming unit
- ED99
effective dose for 99% CFU count reduction
- mLM
mouse liver microsome
- hLM
human liver microsome
- 95% CI
95% confidence interval
Footnotes
Our efforts to confirm the proposed binding mode of quinoline-based FadD32 inhibitors will be reported elsewhere.
References
- 1.WHO Global tuberculosis report 2017. http://www.who.int/tb/publications/global_report/en/; 2018 Accessed 15 April 2018.
- 2.Stanley SA; Kawate T; Iwase N; Shimizu M; Clarworthy AE; Kazyanskaya E; Sacchettini JC; Ioerger TR; Siddiqi NA; Minami S; Aquadro JA; Grant SS; Rubin EJ; Hung DT Diarylcoumarins inhibit mycolic acid biosynthesis and kill mycobacterium tuberculosis by targeting FadD32, Proc. Natl. Acad. Sci. U.S.A.2013; 110: 11565–11570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawate T; Iwase N; Shimizu M; Stanley SA; Wellington S; Kazyanskaya E; Hung DT Synthesis and structure-activity relationships of phenyl-substituted coumarins with anti-tubercular activity that target FadD32, Bioorg. Med. Chem. Lett. 2013; 23: 6052–6059. [DOI] [PubMed] [Google Scholar]
- 4.Zhang W; Lun S; Wang SH; Jiang XW;Yang F; Tang J; Manson AL; Earl AM; Gunosewoyo H; Bishai WR;Yu LF Identification of novel coumestan derivatives as polyketide synthase 13 inhibitors against mycobacterium tuberculosis. J. Med. Chem. 2018; 61(3): 791–803. [DOI] [PubMed] [Google Scholar]
- 5.Guillet V; Galandrin S; Maveyraud L; Ladevèze S; Mariaule V,; Von C; Eynard, N; Daffe, M; Marrakchi H,; Mourey L Insight into Structure-Function Relationships and Inhibition of the Fatty Acyl-AMP Ligase (FadD32) orthologs from mycobacteria. J. Biol. Chem. 2016; 291(15): 7973–7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maresca A; Temperini C; Vu H; Pham NB; Poulsen S-A; Scozzafava A; Quinn RJ; Supuran CT. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors, J. Am. Chem. Soc. 2009; 131: 3057–3062. [DOI] [PubMed] [Google Scholar]
- 7.Gould RG; Jacobs WA The synthesis of certain substituted quinolines and 5,6-benzoquinolines. J. Am Chem. Soc. 1939; 61: 2890–2895. [Google Scholar]
- 8.Hoglund LPJ; Silver S; Endstrom MT; Salo H; Tauber A; Kyyronen H-K; Saarenketo P; Hoffren A-M; Kokko K; Pohjanoksa K; Sallinen J; Savola J-M; Wurster S; Kallatsa OA Structure-activity relationship of quinoline derivatives as potent and selective a2c-Adrenoceptor antagonists. J. Med. Chem. 2006; 49: 6351–6363. [DOI] [PubMed] [Google Scholar]
- 9.Brouet JC; Gu S; Peet NP, Williams JD, A survey of solvents for the conrad-Limpach synthesis of 4- hydroxyquinolones Synth. Commun. 2009; 39(9):5193–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kestranek A; Chervenak A; Longenberger J; Placko S Chemiluminescent Nitrogen Detection (CLND) to Measure Kinetic Aqueous Solubility. Curr. Protoc. Chem. Biol. 2013, 5, 269–280. [DOI] [PubMed] [Google Scholar]
- 11.Kuhn ML; Alexander E; Minasov G; Page HJ; Warwrzak Z; Shuvalova L; Flores KJ; Wilson DJ; Shi. C; Aldrich CC; Anderson WF. Structure of the essential Mtb FadD32 enzyme: A promising drug target for treating tuberculosis ACS Infect. Dis. 2016; 2(8): 579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li W; Gu S; Fleming J; Bi L Crystal structure of FadD32, an enzyme essential for mycolic acid biosynthesis in mycobacteria. Sci. Rep 2015. December 2;5:15493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.All animal studies were ethically reviewed and carried out in accordance with European Directive 2010/63/EEC and the GSK Policy on the Care, Welfare and Treatment of Animals.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.