

Abstract
Sirtuins are NAD+-dependent protein deacylases/ADP-ribosyltransferases that have emerged as candidate targets for new therapeutics to treat metabolic disorders and other diseases, including cancer. The sirtuin SIRT5 resides primarily in the mitochondrial matrix, and catalyzes the removal of negatively charged lysine acyl modifications; succinyl, malonyl, and glutaryl groups. Evidence has now accumulated to document the roles of SIRT5 as a significant regulator of cellular homeostasis, in a context- and cell-type specific manner, as has been observed previously for other sirtuin family members. SIRT5 regulates protein substrates involved in glycolysis, TCA cycle, fatty acid oxidation, electron transport chain, ketone body formation, nitrogenous waste management, and ROS detoxification, among other processes. SIRT5 plays pivotal roles in cardiac physiology and stress responses, and is involved in the regulation of numerous aspects of myocardial energy metabolism. SIRT5 is implicated in neoplasia, as both a tumor promoter and suppressor in a context-specific manner, and may serve a protective function in the setting of neurodegenerative disorders. Here, we review the current understanding of functional impacts of SIRT5 on its metabolic targets, and its molecular functions in both normal and pathological conditions. Finally, we will discuss the potential utility of SIRT5 as a drug target and also summarize the current status, progress, and challenges in developing small molecule compounds to modulate SIRT5 activity with high potency and specificity.
Keywords: succinylation, glutarylation, malonylation, mitochondria, inhibitors
Introduction
Protein post-translational modifications (PTMs) – covalent chemical modifications of amino acid side chains or termini of proteins – influence protein structure and function, and thus play critical roles in regulating nearly every aspect of cellular biology (Audagnotto and Dal Peraro 2017; Sabari et al. 2017). Lysine acylation comprises a diverse family of evolutionarily conserved, reversible PTMs, which are derived from acyl-coenzyme A (CoA) thioesters (Wagner and Hirschey 2014; Hirschey and Zhao 2015). The prototypical PTM of this class is lysine acetylation (Kac), functions of which have been described extensively in the context of nuclear histones and many non-nuclear proteins, including mitochondrial proteins (Kumar and Lombard 2017a; Sabari et al. 2017). Recent advances in proteomics have revealed the existence of a large number of additional lysine acyl modifications, including succinylation, malonylation, glutarylation, crotonylation, beta-hydroxyisobutyrylation, 3-hydroxy 3-methylglutaryl, and fatty acylation, among many others (Sabari et al. 2017).
Identifying the acyltransferases catalyzing the addition of these acyl groups remains a subject of active investigation. In this regard, a recent study by Wang et al. found that the α-ketoglutarate dehydrogenase (α-KGDH) complex interacts with the lysine acetyltransferase 2A (KAT2A, aka GCN5) and functions as a histone H3 succinyltransferase (Wang Y et al. 2017). The α-KGDH complex, which catalyzes the conversion of α-ketoglutarate (α-KG) to succinyl-CoA, translocates to the nucleus and binds together with KAT2A in the promoter regions of genes. This allows KAT2A to access locally generated succinyl-CoA and mediate histone succinylation, in spite of the relatively low concentration of succinyl-CoA in the nucleus overall (Wang Y et al. 2017). Another recent study by Kurmi et al. reported that in addition to its carnitine palmitoyl transferase activity, carnitine palmitoyl transferase 1A (CPT1A) also possesses succinyltransferase activity (Kurmi et al. 2018). Using a SILAC based quantitative succinylation proteomics approach, Kurmi and colleagues identified 171 lysine sites on 101 proteins as potential targets of CPT1A in 293T cells; notably, ~50% of these targets were cytosolic proteins. Strikingly, enolase 1 contained highest number of succinylation sites in CPT1A expressing 293T cells. CPT1A mediated succinylation reduced the activity of enolase (Kurmi et al. 2018).
However, accumulating evidence strongly suggests that many of these lysine acyl modifications occur via a spontaneous, non-enzymatic mechanism, involving reaction of acyl-CoA species with lysine ε–amino groups, especially in the mitochondrial matrix (Wagner and Hirschey 2014). The presence of the thioester functional group renders acyl-CoAs inherently highly reactive biomolecules. Moreover, the alkaline pH in mitochondrial matrix provides a favorable chemical environment for non-enzymatic conjugation to occur. A recent study from the Hirschey laboratory showed that negatively charged dicarboxyl CoA thioesters with four- and five-carbon acyl backbones, e.g. succinyl-CoA and glutaryl-CoA, can undergo an intramolecular reaction to form high-energy cyclic anhydride intermediates, which in turn robustly react with free lysine ε–amino groups (Kumar and Lombard 2017a; Wagner et al. 2017). Acetyl-CoA and other acyl-CoA species are unable to move freely across the inner mitochondrial membrane. In mitochondria, acetyl-CoA condenses with oxaloacetate to form citrate, which is exported to the cytosol (Pietrocola et al. 2015). Citrate can freely diffuse through nuclear pore complexes (Paine et al. 1975). In the cytosol as well as in the nucleus, citrate is cleaved by ATP-citrate lyase into acetyl-CoA and oxaloacetate (Wellen et al. 2009; Chypre et al. 2012). Citrate-derived acetyl-CoA is then used to drive acetylation of non-mitochondrial proteins, including nuclear histones (Wellen et al. 2009).
The spontaneous accumulation of these lysine acyl modifications may deleteriously impact many biochemical processes such as glycolysis, the tricarboxylic acid (TCA) cycle, and fatty acid oxidation (FAO) among many others, resulting in the disruption of cellular metabolic homeostasis (Wagner and Hirschey 2014). Strikingly, the stoichiometry of Kac has been suggested to be very low under normal conditions in wild-type cells and tissues (<1%) (Lombard et al. 2015; Weinert et al. 2015), implying the existence of efficient mechanisms to remove Kac as well other lysine acyl modifications. Sirtuins (see below) function as homeostatic regulators in part by catalyzing removal of these PTMs. In turn, these lysine acyl PTMs have, in many cases, been shown to play important roles in regulating aspects of the biology of the modified proteins.
Sirtuins are a class of nicotinamide adenine dinucleotide (NAD+)-dependent protein deacylases and/or ADP ribosyltransferases, which share homology with the yeast silent information regulator 2 (Sir2) protein (Kumar and Lombard 2015). Based on their NAD+ dependence, sirtuins were initially classified as class III histone deacetylases (North and Verdin 2004; Imai SI and Guarente 2016). Owing to their ability to remove a wide array of acyl modifications from cellular proteins, sirtuins are now known to regulate diverse biological processes: DNA repair, gene expression, cell survival, metabolism, aging, and many others (Chalkiadaki and Guarente 2015). In mammals, the sirtuin family comprises seven members (SIRT1–7), which possess conserved NAD+-binding and catalytic domains, while their flanking N- and C-termini are distinct from one another, and contribute to differences in subcellular localization, enzymatic activity, and substrate specificity among sirtuin proteins (Kumar and Lombard 2017b). SIRT1 and SIRT2 are predominantly found in nucleus and cytoplasm respectively, and both proteins shuttle between these compartments (Michishita et al. 2005; Vaquero et al. 2006; Tanno et al. 2007). SIRT3, SIRT4, and SIRT5 primarily localize to mitochondrial matrix (Onyango et al. 2002; Schwer et al. 2002; Michishita et al. 2005; Haigis et al. 2006; Ahuja et al. 2007; Cooper and Spelbrink 2008; Nakagawa et al. 2009), whereas SIRT6 and SIRT7 are primarily nuclear and nucleolar proteins respectively (Michishita et al. 2005; Ford et al. 2006; Mostoslavsky et al. 2006). The sirtuin-catalyzed deacylation reaction consumes NAD+ as a co-substrate, and generates 2’-O-acyl-ADP-ribose and the sirtuin feedback inhibitor nicotinamide (NAM), resulting in the release of a deacylated substrate (Imai S and Guarente 2010). SIRT1, SIRT2, and SIRT3 possess robust deacetylase and long chain deacylase activities (Michishita et al. 2005; Lombard et al. 2007; Feldman et al. 2013; Rauh et al. 2013; Bao et al. 2014; Teng et al. 2015; Kumar and Lombard 2017b). SIRT4 exhibits ADP-ribosyltransferase (Haigis et al. 2006), deacylase (Anderson et al. 2017) as well as substrate-specific deacetylase (Michishita et al. 2005) and lipoamidase activities (Mathias et al. 2014). SIRT6 possesses deacetylase (Michishita et al. 2008; Kaidi et al. 2010; Tasselli et al. 2017), ADP-ribosyltransferase (Liszt et al. 2005; Mao et al. 2011; Van Meter et al. 2014) and long-chain deacylase (Feldman et al. 2013; Wang WW et al. 2016) activities, and SIRT7 mediates deacetylation (Barber et al. 2012; Chen S et al. 2013; Ryu et al. 2014; Chen S et al. 2016), histone desuccinylation (Li L et al. 2016), and long-chain deacylation reactions (Tong et al. 2017).
Among the mitochondrial sirtuins, SIRT5 displays unique affinity for negatively charged acyl lysine modifications, and performs protein desuccinylation, demalonylation, and deglutarylation reactions (Du J et al. 2011; Peng et al. 2011; Park et al. 2013; Rardin et al. 2013; Tan et al. 2014; Nishida et al. 2015). Until recently, SIRT5 did not appear to represent a particularly attractive target for investigation, since it possesses extremely weak deacetylase activity (North and Verdin 2004; Du J et al. 2011), and moreover no obvious phenotypes or marked metabolic abnormalities were observed in SIRT5-null mice under basal conditions (Lombard et al. 2007; Nakagawa et al. 2009; Yu et al. 2013). However, over the past several years, aided tremendously by the advent of advanced high-resolution proteomics approaches, the existence of a large number of novel PTMs have been revealed (Sabari et al. 2017) and SIRT5 was identified as the principle regulator of three of these modifications (Hershberger, Martin, et al. 2017; Kumar and Lombard 2017b). These discoveries have led to the recent identification of a plethora of SIRT5 targets and physiologic functions.
Here, we comprehensively discuss the current state of knowledge regarding SIRT5, its cellular targets, and its functional roles in mitochondrial metabolic pathways and other cellular processes, in context of both normal and disease conditions. Finally, we highlight recent advances in the generation of potent and selective SIRT5 inhibitors, as valuable research tools, and even as potential eventual therapeutic agents.
Overview of SIRT5 expression, distribution and physiological relevance
Phylogenetically, SIRT5 is distinct from other mammalian sirtuins, and belongs to the so-called class III sirtuin family (Frye 2000), a family that includes mostly prokaryotic sirtuins. The human SIRT5 gene encodes two main SIRT5 isoforms, SIRT5iso1 and SIRT5iso2 comprising 310 amino acids and 299 amino acids respectively, which differ slightly from one other at their C-termini (Mahlknecht et al. 2006; Matsushita et al. 2011) (Fig. 1A). In addition, two other human SIRT5 isoforms (SIRT5iso3 and SIRT5iso4) have been reported in NCBI database (Coordinators 2018); however, no current data are available regarding their expression, localization or functional properties. SIRT5iso3 is identical to SIRT5iso1 except that it lacks an internal sequence of 18 amino acids, while in SIRT5iso4 the initial 108 amino acids of SIRT5iso1 are missing, including the mitochondrial localization sequence. Otherwise, SIRT5iso4 completely aligns with amino acids 109–310 of SIRT5iso1. The mouse Sirt5 gene encodes a single protein of 310 amino acids (Voelter-Mahlknecht and Mahlknecht 2013), corresponding to human SIRT5 isoform 1. SIRT5 displays broad tissue distribution, with highest levels in brain, heart, liver, kidney, muscle, and testis (Michishita et al. 2005; Nakagawa et al. 2009). SIRT5 expression is reported to be regulated by two major cellular regulators of metabolism, peroxisome proliferator-activated receptor coactivator-1α (PGC-1α), and AMP-activated protein kinase (AMPK) (Buler et al. 2014). PGC-1α overexpression or food withdrawal increased SIRT5 mRNA and protein levels in mouse primary hepatocytes, whereas overexpression or metformin-mediated activation of AMPK inhibited SIRT5 expression (Buler et al. 2014).
Figure 1: Isoforms and biochemical activities of SIRT5.

(A) The four different SIRT5 isoforms. The N-terminus, containing a 36 amino acid (aa) canonical mitochondrial localization signal (MLS) (violet) and an additional stretch of 72 aa (yellow), is present in isoforms 1, 2 and 3, but absent in isoform 4. Eighteen aa (Blue) from aa 189–206 of isoform 1 is absent in isoform 3. The C-terminus (black; aa 286–310 of isoform 1) is identical in isoforms 1, 3 and 4, while the C-terminal portion of isoform 2 (green; aa 286–299 of isoform 2) differs in both sequence and length from other isoforms. The remainder of the sequence, common to all isoforms, is shown in red. (B) SIRT5-catalyzed deacylation (desuccinylation/demalonylation/deglutarylation) reactions consume nicotinamide adenine dinucleotide (NAD+) as a co-substrate, and generate the sirtuin feedback inhibitor nicotinamide (NAM), 2’-O-acyl (succinyl/malonyl/glutaryl)-ADP-ribose, and a deacylated substrate. A color version of the figure is available online.
Intriguing new data suggest that SIRT5 polymorphisms may impact human lifespan. The presence of a single-nucleotide polymorphism (SNP) (rs9382222) in a conserved region of the SIRT5 promoter correlates with reduced SIRT5 mRNA expression levels in the anterior cingulate cortex (ACC) region of the brain in individuals with the CC genotype relative to individuals with the CT genotype (Glorioso et al. 2011). The CC genotype is associated with an “older” ACC, as assessed by expression of age regulated transcripts (Glorioso et al. 2011). A subsequent study by TenNapel et al. analyzed the relationship of SIRT5 SNPs with human lifespan, and reported that SNP rs2841505 is associated with slightly reduced lifespan in cohort members with a GG genotype compared to those with other genotypes (TenNapel et al. 2014). Another SIRT5 SNP, rs4712047, displayed gender-specific impact on the lifespan; females with a GG genotype exhibited an increased lifespan compared to those with a GA or a AA genotype, whereas males with a GG genotype displayed decreased lifespan compared to males with the other genotypes (TenNapel et al. 2014). Recently, Donlon et al. reported the association of SIRT5 SNP rs2253217 with human lifespan; cohort members with TT genotype live longer than those with TC or CC genotype (Donlon et al. 2017). These provocative studies need to be replicated in other, genetically distinct populations, and in larger groups, and the functional significance of these polymorphisms on SIRT5 expression more firmly established.
SIRT5 is predominantly a mitochondrial matrix protein (Dryden et al. 2003; Michishita et al. 2005; Schlicker et al. 2008; Nakagawa et al. 2009). However, several groups have reported that a significant portion of SIRT5 localizes to the cytosol, peroxisomes, and nucleus as well (Geng et al. 2011; Matsushita et al. 2011; Park et al. 2013; Chen XF et al. 2018). Consistently, in addition to mitochondrial proteins, a large number of cytosolic and nuclear proteins exhibit increased succinylation, malonylation, and glutarylation upon SIRT5 deletion (Park et al. 2013; Rardin et al. 2013; Tan et al. 2014; Nishida et al. 2015; Sadhukhan et al. 2016; Hershberger, Abraham, et al. 2017). Despite the broad expression and unique activity profile of SIRT5, which is non-redundant with other mitochondrial sirtuins, Sirt5 knockout (KO) mice display no strong phenotypes or major metabolic abnormalities, and germline ablation of Sirt5 is well tolerated in mice under basal, unstressed conditions (Lombard et al. 2007; Nakagawa et al. 2009; Yu et al. 2013). Notably, Sirt5 KO mice on the C57BL/6 background are born at a sub-Mendelian ratio, an effect not observed in Sirt5 KO 129/J background animals (Lombard et al. 2007; Yu et al. 2013). The mechanistic basis for this phenotype has not been determined.
Since the elucidation of SIRT5’s biochemical activities, significant progress has been made in deciphering physiological functions of SIRT5, both in the contexts of normal cell biology and under pathological circumstances. SIRT5 is now known to play significant roles in maintaining metabolic and cellular homeostasis by regulating a variety of processes, including glucose oxidation, ketone body formation, FAO, ammonia detoxification, and ROS management, topics we return to in greater depth subsequently. Furthermore, SIRT5 has emerged as a critical player in maintaining cardiac health and neuronal viability upon stress, and also functions as tumor promotor or tumor suppressor, in a context specific manner.
SIRT5 biochemical activities and its target PTMs
Although SIRT5 was initially characterized as deacetylase (Frye 2000), it possesses very weak or undetectable deacetylase activity (North and Verdin 2004; Lombard et al. 2007; Du J et al. 2011; Peng et al. 2011). Among the newly discovered PTMs, lysine succinylation (Ksucc), malonylation (Kmal), and glutarylation (Kglu), derived from succinyl-CoA, malonyl-CoA, and glutaryl-CoA respectively, have emerged as functionally important modifications, but most likely have distinct functions from Kac in regulating metabolism and other cellular processes (Park et al. 2013; Rardin et al. 2013; Tan et al. 2014; Nishida et al. 2015; Sadhukhan et al. 2016; Hershberger, Abraham, et al. 2017). Notably, the presence of succinyl, malonyl, or glutaryl moiety confers upon a modified lysine residue a negative charge at physiological pH (Hirschey and Zhao 2015). SIRT5 preferentially catalyzes the removal of these negatively charged acidic modifications, thereby functioning as the dominant cellular desuccinylase, demalonylase, and deglutarylase (Fig. 1B). (Du J et al. 2011; Peng et al. 2011; Park et al. 2013; Rardin et al. 2013; Tan et al. 2014; Nishida et al. 2015). As discussed in subsequent sections, several groups have reported that SIRT5 can functionally deacetylate several specific substrates. Whether SIRT5 actually possesses substrate-specific deacetylase activity, or alternatively whether these results represent artifacts of SIRT5 overexpression and/or cross-reactivity of anti-PTM antibodies, remains an unresolved issue in the field. Compared to other sirtuins, SIRT5 possess a larger acyl binding pocket (Peng et al. 2011), able to accommodate these acyl modifications, which are bulkier than an acetyl group. Furthermore, the presence of alanine (Ala86), arginine (Arg105), and tyrosine (Tyr102) residues in the catalytic pocket of SIRT5 appears to be responsible for its specificity for negatively charged acyl groups (Du J et al. 2011). Notably, SIRT7 has been reported to desuccinylate histones (Li L et al. 2016), however SIRT7 does not localize to mitochondria, strongly suggesting that SIRT5 activity is non-redundant with other sirtuins in mitochondria, and likely elsewhere in the cell as well. Tan et al. showed that SIRT5 is selective only for 3–5 carbon chains acidic acyl modifications, and displays no detectable activity against either an acetyl modification, a neutral 2 carbon group, or an adipoyl, a 6-carbon acidic modification (Tan et al. 2014). Sirt5 ablation in mice causes a dramatic increase of Ksucc, Kmal, and Kglu levels, globally across multiple tissues and embryonic fibroblasts, while it has very little impact on Kac levels (Lombard et al. 2007; Du J et al. 2011; Peng et al. 2011; Park et al. 2013; Rardin et al. 2013; Tan et al. 2014; Nishida et al. 2015; Sadhukhan et al. 2016; Hershberger, Abraham, et al. 2017).
Using high resolution quantitative mass spectrometry approaches, several independent proteomic screens have been performed to identify SIRT5 targets with Ksucc, Kglu, and/or Kmal PTMs. In this regard, Park et al. carried out proteomics analysis of Ksucc peptides in Sirt5 KO mouse liver and mouse embryonic fibroblasts (MEFs), and identified a total of 2,565 succinylation sites on 779 proteins (Park et al. 2013). More than 90% of quantifiable sites showed hypersuccinylation in Sirt5 KO MEFs, strongly supporting the role of SIRT5 as a major regulator of Ksucc in mammals. KEGG pathway analysis revealed that Ksucc has potential impacts on enzymes participating in processes such as amino acid degradation, the TCA cycle, and fatty acid metabolism (Park et al. 2013). Similarly, Rardin et al. identified a total of 1190 unique Ksucc sites in mouse liver mitochondria, among which 386 sites on 140 proteins displayed hypersuccinylation upon SIRT5 loss (Rardin et al. 2013). Reactome analysis revealed that most of these proteins are involved in metabolic pathways, including β-oxidation and ketogenesis (Rardin et al. 2013). Recent succinyl-proteome analysis identified a number of functionally important SIRT5 targets in murine hearts, suggesting a potential role for SIRT5 mediated desuccinylation in maintaining cardiac energy metabolism (Boylston et al. 2015; Sadhukhan et al. 2016; Hershberger, Abraham, et al. 2017). We discuss these studies in more depth subsequently.
In addition to Ksucc, proteomic screening has also been performed to identify SIRT5 target Kglu and Kmal sites. Tan et al. identified 683 Kglu sites on 191 proteins, which are enriched in liver from Sirt5 KO mice (Tan et al. 2014). Remarkably, about 67% of identified Kglu sites overlapped with Ksucc sites, indicating the existence of some proteins potentially regulated by more than one PTM; e.g. Carbamoyl-phosphate synthase 1 (CPS1), previously known to be acetylated and succinylated, was also found glutarylated and regulated by SIRT5 (Tan et al. 2014). Colak and colleagues reported the existence of 4,016 Kmal sites on 1,395 proteins in Sirt5 KO mouse liver; 274 of identified malonylated proteins were exclusively present in mitochondria (Colak et al. 2015). Similarly, depletion of malonyl-CoA decarboxylase (MCD), which catalyzes the conversion of malonyl-CoA into acetyl-CoA and CO2, resulted in >2-fold increase in the levels of 461 Kmal sites in human fibroblasts. In addition, 1452 Kmal sites were reported only in MCD KO human fibroblasts (Colak et al. 2015). Integration of GO and KEGG analyses of Kmal targets, identified in Sirt5 KO mouse liver and MCD KO human fibroblasts, found that proteins involved in fatty acid metabolism were preferentially heavily malonylated. Consistently, human fibroblasts with increased lysine malonylation displayed impaired mitochondrial function and FAO (Colak et al. 2015). Nishida et al. identified 1,137 Kmal sites on 430 proteins in mouse liver (Nishida et al. 2015). 183 of the identified Kmal sites on 120 proteins display significant enrichment in Sirt5 KOs. Ingenuity Pathway Analysis (IPA) identified glycolysis as the top SIRT5-regulated pathway. Consistently, glycolytic flux was reduced in Sirt5 KO primary hepatocytes compared to WT controls (Nishida et al. 2015).
Despite the identification of hundreds of SIRT5 substrates in these studies, including numerous metabolic enzymes, our understanding of the biological significance and regulation of Ksucc, Kmal, and Kglu is still in its infancy. However, as we discuss in upcoming sections, these PTMs are emerging as important players in regulating metabolism and cellular physiology.
Roles of SIRT5 in maintaining metabolic homeostasis
SIRT5 regulates glycolysis, the TCA cycle and the electron transport chain
A number of studies suggest that SIRT5 plays roles in regulating and coordinating activities of multiple enzymes involved in glycolysis, the TCA cycle and the electron transport chain (ETC) (Fig. 2). Nishida et al. showed that SIRT5 demalonylates glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and other glycolytic enzymes and thus promotes glycolytic flux (Nishida et al. 2015). GAPDH catalyzes conversion of glyceraldehyde-3-phosphate to D-1,3-bisphosphoglycerate, and physically interacts with SIRT5 (Marcon et al. 2015). Substitution of lysine residue 184 in GAPDH with glutamic acid, a malonyl-lysine mimic, inhibited its enzymatic activity (Nishida et al. 2015), suggesting that SIRT5 promotes the activity of GAPDH and perhaps other glycolytic enzymes via demalonylation. Consequently, hepatocytes from Sirt5 KO mice display reduced glycolytic flux (Nishida et al. 2015).
Figure 2: Metabolic regulation by SIRT5.
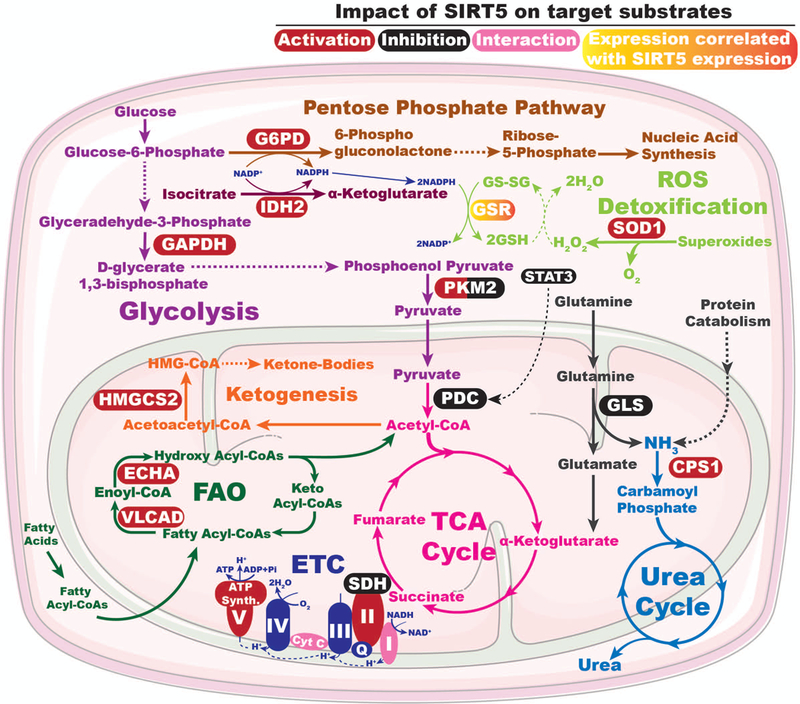
SIRT5 plays roles in maintaining metabolic homeostasis by regulating the activities and/or expression levels of enzymes involved in multiple metabolic pathways. See text for details. G6PD: Glucose-6-phosphate dehydrogenase, IDH2: Isocitrate dehydrogenase 2, GAPDH: Glyceraldehyde phosphate dehydrogenase, PKM2: pyruvate kinase muscle isozyme 2, GSR: Glutathione reductase, GSH: Reduced glutathione, GS-SG: oxidized glutathione, STAT3: Signal transduction and activator of transcription 3 , SOD1: Cu/Zn superoxide dismutase, ROS: Reduced oxygen species, PDC: Pyruvate dehydrogenase complex, TCA: Tricarboxylic acid, ETC: Electron transport chain, Cyt C: Cytochrome C, I-IV: Complex I-IV, Q: Quinolone, SDH/complex II: Succinate dehydrogenase, FAO: Fatty acid oxidation, ECHA: Enoyl-coenzyme A hydratase, VLCAD: Very long chain acyl-CoA dehydrogenase, HMGCS2: 3-hydroxy-3-methylglutaryl CoA synthase 2, GLS: Glutaminase, CPS1: Carbamoyl phosphate synthetase 1. A color version of the figure is available online.
Wang et al. identified pyruvate kinase M2 (PKM2) as another glycolytic enzyme whose activity is regulated by SIRT5-mediated desuccinylation (Wang F et al. 2017). PKM2 catalyzes the last step of glycolysis, the conversion of phosphoenolpyruvate to pyruvate. SIRT5 desuccinylates PKM2 at lysine 311 to increase pyruvate kinase activity (Wang F et al. 2017), further suggesting that SIRT5 promotes glycolytic flux. Paradoxically however, loss of Sirt5 in LPS-activated macrophages redirects cellular metabolism in favor of glycolysis (Wang F et al. 2017). LPS-activated macrophages undergo a metabolic switch similar to the Warburg effect in tumor cells, suggesting context-specific roles of SIRT5 in regulating glycolytic flux. The Warburg effect refers to a common feature of many cancer cells, which show high levels of glycolysis even in the presence of sufficient oxygen to support mitochondrial respiration (Liberti and Locasale 2016). The Warburg effect has been proposed to provide a metabolic framework allowing for rapid biosynthesis of macromolecules to support growth, proliferation, and survival of cancer cells (Liberti and Locasale 2016). In contrast, Xiangyun and colleagues reported that SIRT5 desuccinylates PKM2 at lysine 498 to inhibit its activity in tumor cells (Xiangyun et al. 2017).
Pyruvate dehydrogenase complex (PDC) catalyzes the oxidation of pyruvate into acetyl-CoA, which subsequently enters the TCA cycle. SIRT5 has been shown to repress the activity of PDC by direct desuccinylation (Park et al. 2013), and by deacetylating signal transduction and activator of transcription 3 (STAT3) (Xu YS et al. 2016). The acetylated form of STAT3 translocates to mitochondria, where it associates with, and activates, PDC, resulting in accelerated conversion of pyruvate to acetyl-CoA, which correlates with elevated mitochondrial membrane potential and increased ATP synthesis. SIRT5-mediated deacetylation of STAT3 inhibits its mitochondrial translocation, which in turn suppresses pyruvate metabolism (Xu YS et al. 2016). Consequently, SIRT5 depletion results in elevated PDC activity that leads to increased pyruvate-dependent cellular respiration (Park et al. 2013). Paradoxically however, significantly reduced malate/pyruvate-driven respiration was observed in SIRT5-deficient HEK293 cells (Zhang Y et al. 2017). Likewise, overexpression of SIRT5 enhanced ATP synthesis and oxygen consumption in HepG2 cells (Buler et al. 2014), again highlighting the context specificity of SIRT5 function.
SIRT5 desuccinylates and activates isocitrate dehydrogenase 2 (IDH2) (Zhou et al. 2016), an enzyme that catalyzes the NADP+-dependent oxidative decarboxylation of isocitrate to α-KG, generating NADPH and CO2 (Fujii et al. 2016). An IDH2 isoform; IDH3, catalyzes a similar reaction in the TCA cycle, converting NAD+ to NADH and could conceivably be activated by SIRT5. In contrast, SIRT5-catalyzed desuccinylation inhibits the activity of another TCA cycle enzyme complex, succinate dehydrogenase (SDH) (Park et al. 2013). SDH, aka complex II of the ETC, catalyzes oxidation of succinate to fumarate with concomitant conversion of ubiquinone to ubiquinol. Consequently, SIRT5-depletion leads to an increase in SDH activity, associated with enhanced succinate dependent cellular respiration (Park et al. 2013). Recently, Zhang et al. have shown that SIRT5 electrostatically binds to cardiolipin and desuccinylates inner mitochondrial membrane proteins, including multiple subunits of all four ETC complexes and ATP synthase, to promote respiratory chain function (Zhang Y et al. 2017). As a result, Sirt5 KO liver homogenates display impaired enzymatic activities of Complex II and ATP synthase, and reduced Complex II-driven respiration (Zhang Y et al. 2017). The discrepancies in SIRT5-dependent phenotypes with regard to Complex II activity and respiration observed between these two studies may be attributed in part to distinct approaches for measuring Complex II activity and mitochondrial respiration. SIRT5 also interacts with complex I subunit NDUFA4 (Marcon et al. 2015) and cytochrome C (Schlicker et al. 2008), however, the functional relevance of these interactions has not been elucidated.
Roles of SIRT5 in fatty acid β-oxidation
β-oxidation is a four-step process of fatty acid breakdown into acetyl-CoA, which takes place in mitochondria (Houten and Wanders 2010). The first step involves the dehydrogenation of fatty acyl-CoAs of varying lengths by the members of acyl-CoA dehydrogenase (ACAD) enzyme family, converting them into enoyl-CoA. Enoyl-CoA species then sequentially undergo hydration, oxidation and thiolysis by mitochondrial trifunctional (MTF) complex, resulting in the release of acetyl group as acetyl-CoA (Houten and Wanders 2010). Three ACAD family members -- very long-chain acyl-CoA dehydrogenase (VLCAD), long-chain acyl-CoA dehydrogenase (LCAD), and medium-chain acyl-CoA dehydrogenase (MCAD), which show preferences for fatty acid substrates of varying chain length -- and the trifunctional enzyme subunits a and b (aka ECHA and ECHB) are all hypersuccinylated at multiple sites in Sirt5 KO mice liver tissue (Rardin et al. 2013). SIRT5 ablation results in modestly impaired β-oxidation and accumulation of medium- and long-chain acylcarnitines in liver and muscles of Sirt5 KO mice (Rardin et al. 2013). VLCAD works in close association with carnitine palmitoyltransferase-2 and MTF complex, all of which localize to the inner mitochondrial membrane through electrostatic interaction with cardiolipin (Fould et al. 2010; Kashfi et al. 2011; Zhang Y et al. 2015). Notably, SIRT5, in cooperation with SIRT3, deacylates VLCAD, thereby promoting its mitochondrial membrane localization and activity (Zhang Y et al. 2015). SIRT5-mediated desuccinylation and SIRT3 catalyzed deacetylation of K299 stabilizes association of the essential flavin adenine dinucleotide (FAD) cofactor in the active site of VLCAD to upregulates VLCAD activity (Zhang Y et al. 2015). Furthermore, deacetylation of K507 by SIRT3 and desuccinylation of K482, K492, and K507 by SIRT5 in a domain near the C-terminus of VLCAD promote cardiolipin binding to VLCAD. Consequently, VLCAD from Sirt3- and Sirt5 KO mice liver exhibits reduced binding to cardiolipin (Zhang Y et al. 2015). These results suggest that SIRT5 cooperates with SIRT3 to facilitate FAO, by promoting the activity and mitochondrial membrane localization of VLCAD. Recently, SIRT5 has been shown to desuccinylate and activate ECHA in mice myocardium (Sadhukhan et al. 2016). As a result, SIRT5-deficient hearts exhibit impaired fatty acid metabolism and reduced ATP production during energy-demanding conditions such as fasting and exercise (Sadhukhan et al. 2016), further highlighting the role of SIRT5 in promoting FAO (Fig. 2).
SIRT5 promotes ketone body production
SIRT5 also plays a role in regulating ketone body formation by desuccinylating and modulating the activity of 3-hydroxy-3-methylglutaryl CoA synthase 2 (HMGCS2) (Fig. 2), and potentially other enzymes involved in ketogenesis (Rardin et al. 2013). Under fasting conditions, ketone bodies serve as an important source of energy for tissues such as heart, skeletal muscle, and brain. Loss of SIRT5 results in hypersuccinylation and reduced activity of HMGCS2 (Rardin et al. 2013), an enzyme that catalyzes the initial step in the conversion of acetyl-CoA into ketone bodies. Consequently, Sirt5 KO mice display reduced β-hydroxybutyrate levels during fasting (Rardin et al. 2013).
SIRT5 promotes ROS detoxification
As a consequence of oxidative metabolism, mitochondria produce the majority of cellular reactive oxygen species (ROS) (Zorov et al. 2014). While low levels of ROS can serve as redox messengers (Holmstrom and Finkel 2014), excessive ROS levels are damaging to cellular macromolecules and can promote cell death via the intrinsic apoptotic pathway (Sohal and Weindruch 1996; Circu and Aw 2010). To mitigate the deleterious effects of ROS, cells have evolved numerous antioxidant defense systems. For instance, superoxide dismutases catalyze the conversion of superoxide into oxygen and hydrogen peroxide, which is later converted to water by catalase or glutathione peroxidase (Kuciel and Mazurkiewicz 2004). In this context, SIRT5 binds to, desuccinylates, and activates Cu/Zn superoxide dismutase (SOD1). Consequently, SOD1-mediated ROS detoxification is significantly increased when SOD1 is co-overexpressed with SIRT5 (Lin ZF et al. 2013). In order to catalyze conversion of peroxides to water, glutathione peroxidases use glutathione in its reduced form, GSH (Kuciel and Mazurkiewicz 2004). NADPH, a major intracellular reductant, plays a key role in regenerating GSH from the oxidized form, GSSG (Balendiran et al. 2004). In this regard, SIRT5 desuccinylates and deglutarylates IDH2 and glucose-6-phosphate dehydrogenase (G6PD), respectively, to activate these proteins (Zhou et al. 2016). Since both IDH2 and G6PD are major NADPH-producing enzymes, SIRT5 plays a key role in promoting NADPH production and attenuating cellular ROS levels. Furthermore, SIRT5-deficient non-small cell lung cancer cells exhibit reduced expression of glutathione reductase (GSR) (Lu et al. 2014), which catalyzes the regeneration of GSH from GSSG (Balendiran et al. 2004). As a consequence, SIRT5 knockdown (KD) or KO cells display reduced levels of NADPH and GSH, resulting in an impaired ability to scavenge ROS and increased sensitivity to oxidative stress (Zhou et al. 2016). Moreover, SIRT5 desuccinylates and inhibits PKM2, diverting glucose-derived carbon into the pentose phosphate pathway and thus generating reducing potential for antioxidant responses (Xiangyun et al. 2017). In a recent study Liang et al. reported that SIRT5 overexpression reduces the levels of ROS and protects neuroblastoma cells against hydrogen peroxide-induced toxicity (Liang et al. 2017). Similarly, SIRT5-depleted rat cardiomyocytes are more sensitive to oxidative stress-induced death, while SIRT5 overexpression is protective in this context (Liu B et al. 2013). Moreover, it has been suggested that SIRT5 may directly deacetylate the Forkhead protein Foxo3A to promote its nuclear localization (Fig. 3), which in turn promotes expression of genes involved in antioxidant defense (Wang Y et al. 2015). Collectively, these studies indicate that SIRT5 plays multiple roles in coordinating cellular anti-oxidant defense mechanisms (Figs. 2 & 3).
Figure 3: Regulation of gene expression by SIRT5.

SIRT5 expression positively correlates with expression of NRF2, a transcription factor that promotes expression of genes involved in xenobiotic metabolism and redox homeostasis. SIRT5 is also reported to deacetylate and enhance nuclear localization of the transcription factor FOXO3A, which promotes the expression of genes involved in antioxidant defense. NRF2: Nuclear factor erythroid-2-related factor 2, FOXO3A: Fork head box O3A. A color version of the figure is available online.
SIRT5 contributes to nitrogenous waste management
Nitrogenous wastes such as ammonia and uric acid, produced during protein and nucleic acid catabolism, can exert detrimental effects in cells if they accumulate beyond the cell’s ability to detoxify and/or export these molecules. For detoxification and disposal, ammonia is converted to urea by a process termed the urea or ornithine cycle, which primarily takes place in liver (Haussinger 1990; Meijer et al. 1990). The initial committed step of the urea cycle, conversion of ammonia into carbamoyl phosphate, is catalyzed by the enzyme carbamoyl phosphate synthetase (CPS1) (Haussinger 1990; Meijer et al. 1990), which is deacylated and activated by SIRT5 (Fig. 2) (Nakagawa et al. 2009; Du J et al. 2011; Tan et al. 2014). Sirt5 KO mice exhibit reduced CPS1 activity, and show modestly elevated levels of blood ammonia during physiological conditions of high amino acid catabolism, such as fasting or calorie restriction (CR) (Nakagawa et al. 2009; Du J et al. 2011). Conversely, SIRT5 overexpression in mice leads to increased hepatic CPS1 activity (Ogura et al. 2010). This observation, along with the increased expression of Sirt5 mRNA in the liver of mice observed during CR (Ogura et al. 2010), suggests that SIRT5 might play a role in the metabolic adaptation to CR, via regulation of CPS1 activity and the urea cycle. In this regard, reduced expression and activity of SIRT5 have been suggested to induce the accumulation of follicular fluid ammonia in granulosa and cumulus cells of women with reduced ovarian reserve or advanced maternal age, potentially via decreased CPS1 activity (Pacella-Ince et al. 2014). In non-hepatic cells, SIRT5 suppresses ammonia production by desuccinylating and inhibiting the activity of glutaminase (Fig. 2) (Polletta et al. 2015), an enzyme that catalyzes the conversion of glutamine to glutamate, producing ammonia as a by-product. In the livers of SIRT5-overexpressing transgenic mice, SIRT5 deacetylates and activates urate oxidase, an enzyme catalyzing the conversion of urate to allantoin, the last step of purine catabolism in most mammals, but without a functional homolog in humans (Alvarez-Lario and Macarron-Vicente 2010; Nakamura et al. 2012).
SIRT5 maintains cardiac homeostasis under stress
SIRT5 is now known to regulate a variety of physiologic processes, and to represent a significant guardian of metabolic and cellular homeostasis. In this context, recent work has revealed roles of SIRT5 in maintaining cardiac homeostasis, especially under stress conditions (Fig. 4). Both human and murine hearts express SIRT5 at higher levels relative to other tissues (Michishita et al. 2005; Nakagawa et al. 2009). In this context, succinyl-CoA, the precursor of lysine succinylation, is the most abundant acyl-CoA species in cardiac tissue (Sadhukhan et al. 2016). As a consequence, Sirt5 KO hearts display predominant enrichment of lysine succinylated proteins (Boylston et al. 2015; Sadhukhan et al. 2016; Hershberger, Abraham, et al. 2017). Boylston et al. compared the lysine succinylomes of mitochondria isolated from WT and Sirt5 KO hearts, and identified 46 uniquely succinylated proteins and 9 hypersuccinylated proteins in Sirt5 KO heart mitochondria (Boylston et al. 2015). Using pathway enrichment analysis, the authors found that many of these proteins participate in processes such as oxidative phosphorylation, FAO, ketogenesis, branched chain amino acid catabolism, and the TCA cycle (Boylston et al. 2015). In order to assess the potential impact of SIRT5 on ischemia-reperfusion (IR) injury, a model for myocardial infarction in humans, Boylston et al. subjected Sirt5 null and WT mice to IR injury using a Langendorff perfusion model. They found that SIRT5-deficent hearts display increased susceptibility to IR injury and impaired recovery after the IR insult (Boylston et al. 2015). Consistent with previous reports (Park et al. 2013), SIRT5 deficiency was associated with increased lysine succinylation of succinate dehydrogenase (SDH) in heart (Boylston et al. 2015). SIRT5 mediated desuccinylation inhibits SDH activity (Park et al. 2013), and inhibition of SDH activity using dimethyl malonate (a precursor of the SDH inhibitor malonate) has been reported to be protective in an in vivo model of cardiac IR injury (Chouchani et al. 2014). Inhibition of SDH with dimethyl malonate reduced superoxide production and rescued Sirt5 KO hearts from their increased susceptibility to IR injury (Boylston et al. 2015), indicating the importance of the role of SIRT5-mediated regulation of SDH in the heart.
Figure 4: Roles of SIRT5 in health and pathophysiological conditions.
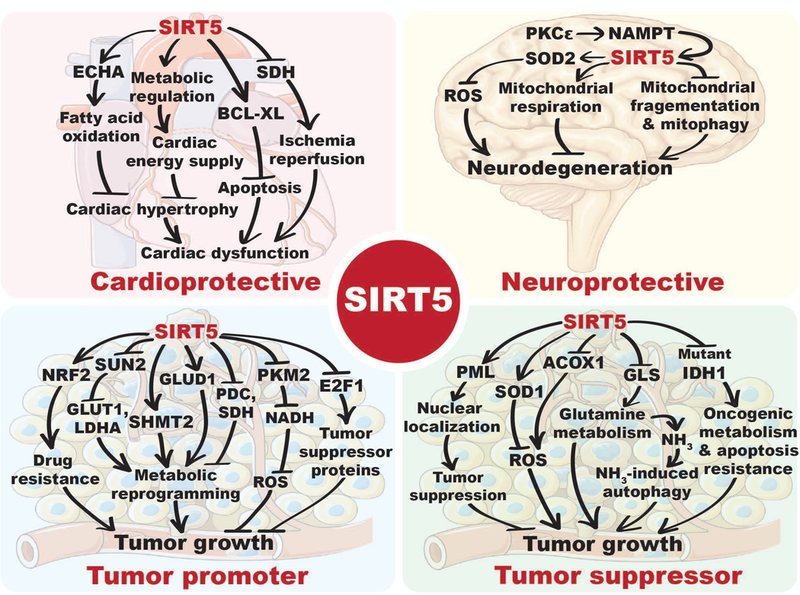
SIRT5 regulates the activities, localization and/or expression of multiple substrates involved in diverse cellular processes, including metabolism, apoptosis or redox homeostasis, which are crucial for maintaining cardiac health, neuronal viability and regulating cancer biology. ECHA: Enoyl-coenzyme A hydratase, SDH: Succinate dehydrogenase, BCL-XL: B cell lymphoma-XL, PKCε: Protein kinase C epsilon, NAMPT: Nicotinamide phosphoribosyl transferase, SOD2: Manganese superoxide dismutase, ROS: Reduced oxygen species, NRF2: Nuclear factor erythroid-2-related factor 2, SUN2: SUN domain-containing protein 2, GLUT1: Glucose transporter 1, LDHA: Lactate dehydrogenase A, SHMT2: Mitochondrial serine hydroxymethyltransferase, PDC: Pyruvate dehydrogenase complex, SOD1: Cu/Zn superoxide dismutase, PKM2: Pyruvate kinase muscle isozyme 2, NADH: Reduced nicotinamide adenine dinucleotide, E2F1: E2F transcription factor 1, PML: Pro-myelocytic leukemia protein, GLS: Glutaminase, IDH1: Isocitrate dehydrogenase 1, ACOX1: acyl-CoA oxidase 1, GLUD1: glutamate dehydrogenase 1. A color version of the figure is available online.
In a recent study, Sadhukhan et al. identified 124 succinylated proteins as potential targets of SIRT5 in heart; more than 75% of these proteins were mitochondrial (Sadhukhan et al. 2016). Pathway enrichment analysis revealed that a majority of these proteins are involved in branched-chain amino acids metabolism, the TCA cycle, fatty acid metabolism, oxidative phosphorylation, propanoate metabolism, pyruvate metabolism, and ATP synthesis (Sadhukhan et al. 2016). Among potential SIRT5 targets, Sadhukhan and colleagues focused on ECHA (Enoyl-CoA Hydratase Alpha), aka HADHA (Hydroxyacyl-CoA Dehydrogenase Alpha), a subunit of mitochondrial trifunctional protein required for FAO, to possess the most numerous succinylation sites (28 succinylated lysine residues), the majority of which (26 out of 28) were identified only in Sirt5 KO heart. Consistently, ECHA was among the 9 hypersuccinylated mitochondrial proteins in Sirt5 KO hearts identified by Boylston and colleagues (Boylston et al. 2015). SIRT5-mediated desuccinylation activates ECHA (Sadhukhan et al. 2016) and therefore, SIRT5 deficiency is associated with impaired myocardial fatty acid metabolism and reduced ATP production in aged hearts during energetically demanding conditions, such as fasting and exercise (Sadhukhan et al. 2016). Sirt5 KOs exhibit both an impaired shortening fraction and ejection fraction and develop cardiac hypertrophy with age (Sadhukhan et al. 2016).
In a recent study, Herschberger et al. evaluated the role of SIRT5 in cardiac stress responses, using an established model of pressure overload-induced hypertrophy induced by traverse aortic constriction (TAC). Sirt5 ablation led to development of severe cardiac dysfunction in response to TAC, and was associated with increased mortality (Hershberger, Abraham, et al. 2017). The authors identified 766 unique proteins that displayed at least a 2-fold increase in succinylation in Sirt5 KOs. IPA revealed that SIRT5 targets are primarily involved in metabolic pathways such as oxidative phosphorylation, FAO, ketogenesis, branched chain amino acid catabolism, TCA cycle, and glutaryl-CoA degradation (Hershberger, Abraham, et al. 2017).
Interestingly, oxidative phosphorylation, fatty acid metabolism/oxidation, branched-chain amino acid metabolism, and the TCA cycle are common metabolic pathways identified in three independent profiling studies of the cardiac succinylome, strongly suggesting that SIRT5-mediated desuccinylation plays important roles in regulating cardiac metabolism during metabolic stress. Consistently, the SIRT5 protein is upregulated in the rat cardiomyocytes during intermittent hypoxia (Zhu et al. 2012). Furthermore, SIRT5 has been shown to physically interact with the anti-apoptotic protein Bcl-XL to protect cardiomyocytes from oxidative stress induced apoptosis (Liu B et al. 2013). Taken together, these findings highlight important roles for SIRT5 in cardioprotection under stress conditions, potentially via regulation of metabolism.
Importantly, all studies to date cataloging phenotypes of SIRT5 deficiency in the heart have used global Sirt5 KO animals, lacking SIRT5 in every cell from the time of conception onwards. The formal possibility remains that SIRT5 exerts some of its cardioprotective functions in a non-cardiomyocyte-autonomous manner, for example through roles in inflammatory cells, vascular endothelium, cardiac fibroblasts, and/or other cell types. In this regard, the mitochondrial sirtuin SIRT3 inhibits fibrosis in the heart and other tissues via functions in fibroblasts (Sundaresan et al. 2015). Only analysis of mouse strains wherein Sirt5 has been deleted in specific cell populations will address this important topic.
Janus-faced roles of SIRT5 in Cancer
Diverse roles for mammalian sirtuins relevant for cancer biology have been described, including maintenance of genomic stability, regulation of metabolic reprogramming, and modification of the tumor microenvironment (Chalkiadaki and Guarente 2015; Bringman-Rodenbarger et al. 2017). Given their diversity of function, it is perhaps not surprising that sirtuins can act as tumor suppressors in some cases and tumor promoters in others. In contrast to certain other sirtuins, especially SIRT1, roles for SIRT5 in cancer are only beginning to be elucidated. The chromosomal region; 6p23, encompassing the SIRT5 locus is highly unstable, and frequently altered across a variety of cancer types (Takeshita et al. 2004; Mahlknecht et al. 2006; Cancer Genome Atlas Research 2011; Bringman-Rodenbarger et al. 2017). Furthermore, altered levels of SIRT5 mRNA expression have been reported in different cancers relative to their normal counterparts. In some cancer types – e.g. non-small cell lung cancer (NSCLC), breast tumors, Waldenstrom’s macroglobulinemia, hepatocellular carcinoma and colorectal cancer – SIRT5 mRNA levels are elevated (Sun et al. 2011; Lu et al. 2014; Igci et al. 2016; Chang et al. 2018; Wang YQ et al. 2018), while in others – endometrial carcinoma, head and neck squamous cell carcinoma and hepatocellular carcinoma – SIRT5 expression were significantly reduced (Lai et al. 2013; Bartosch et al. 2016; Chen XF et al. 2018). These findings suggest that, like other sirtuins, SIRT5 could potentially play context-specific roles in human cancer (Fig. 4).
SIRT5 as tumor promoter
A study by Lu et al. highlighted a potential role for SIRT5 in facilitating lung cancer growth and drug resistance by promoting expression of NRF2 (Fig. 3) and its downstream targets (Lu et al. 2014). NRF2 is a transcription factor that promotes expression of a large suite of genes involved in oxidative and xenobiotic defense (Jaiswal 2004). SIRT5 has been found to be overexpressed in advanced NSCLC, and high SIRT5 expression correlated with poor prognosis (Lu et al. 2014). SIRT5 KD suppressed the proliferation of NSCLC cell lines, and reduced expression of NRF2 and its downstream targets were observed in SIRT5 KD cells, resulting in their increased susceptibility to genotoxic drugs (Lu et al. 2014). Another study elucidating roles of SIRT5 in lung cancer showed that SIRT5 negatively regulates the expression of SUN2, a key component of the LINC (linker of nucleoskeleton and cytoskeleton) complex (Lv et al. 2015). SUN2 expression is reduced in lung cancer, and serves as a predictor of overall poor survival. Conversely, SUN2 overexpression inhibits growth and migration of lung cancer cells and sensitizes them to cisplatin-induced apoptosis (Lv et al. 2015). SUN2 seems to play its tumor suppressor role by downregulating expression of GLUT1 and LDHA to inhibit Warburg metabolism. Consistent with the observations of Lu et al., this study reported the association of high SIRT5 expression with poor patient survival, and further highlighted a potential role of SIRT5 in promoting Warburg-type metabolism by repressing SUN2 expression (Lv et al. 2015). Altered activities of SIRT5 targets, PDC and SDH (Park et al. 2013), have been implicated in neoplasia and cancer cell metabolic reprogramming (Stacpoole 2017; Zhao et al. 2017), further supporting the notion of SIRT5 as a candidate regulator of metabolism in cancer cells.
In a recent study, Yang et al. uncovered a role of SIRT5 in folate metabolism to promote tumor cell growth (Yang X et al. 2017). In this pathway, mitochondrial serine hydroxymethyltransferase (SHMT2) catalyzes the reversible, concurrent conversion of serine to glycine and tetrahydrofolate (THF) to 5,10-methylenetetrahydrofolate (5,10-CH2-THF), an essential intermediate for purine biosynthesis (Ducker and Rabinowitz 2017). SIRT5 interacts with, and desuccinylates SHMT2 at lysine 280, resulting in its enzymatic activation (Yang X et al. 2017). Conversely, SIRT5 ablation results in SHMT2 hypersuccinylation and attenuated activity, associated with impaired growth of U2OS cells. U2OS and HCT116 cells expressing a succinylation mimic mutant (K280E) of SHMT2 exhibited strikingly reduced proliferation and tumor growth in vitro and in vivo (Yang X et al. 2017). Notably, folate metabolism is a target of several approved chemotherapy drugs (Hagner and Joerger 2010). These results suggest that SIRT5 inhibitors might prove useful to target this pathway.
Another recent study revealed that SIRT5 binds to and desuccinylates pyruvate kinase M2 (PKM2) at lysine 498, resulting in inhibition of its pyruvate kinase activity (Xiangyun et al. 2017). PKM2 exists either as fully functional tetrameric pyruvate kinase, or as dimer that exhibit very low pyruvate kinase activity, but localizes to nucleus and possesses protein kinase activity (Gao X et al. 2012; Yang W et al. 2012; Jiang et al. 2014; Wang F et al. 2017). As pyruvate kinase, in the last step of glycolysis, PKM2 catalyzes the transfer of phosphate from phosphoenolpyruvate (PEP) to ADP, resulting in formation of pyruvate and ATP. Notably, glycolytic intermediates serve as precursors for synthesis of macromolecules necessary to support tumor cell growth and proliferation (Pavlova and Thompson 2016). Thus, inhibition of PKM2 activity leads to accumulation of glycolytic intermediates to facilitate tumor growth. In this context, SIRT5 deficiency, or treatment with the promiscuous SIRT5 inhibitor suramin, induces increased PKM2 activity that leads to reduced proliferation of A549 cells, consistent with the notion that PKM2 hypersuccinylation inhibits tumor cell proliferation (Xiangyun et al. 2017). Indeed, substitution of endogenous PKM2 with a succinylation mimetic mutant K498E decreases cellular NADPH production, sensitizes them to oxidative stress, and represses A549 proliferation and tumor-forming ability (Xiangyun et al. 2017).
However, in contrast to this study, another group showed that SIRT5 desuccinylates PKM2 at lysine 311 to increase its activity, whereas succinylation of lysine 498 had no effect on pyruvate kinase activity (Wang F et al. 2017). As noted, increased PKM2 succinylation at lysine 311 causes a reduction in its pyruvate kinase activity, while promoting protein kinase activity (Wang F et al. 2017). Interestingly, increased reactive oxygen species (ROS) levels promotes SIRT5 interaction with PKM2, reducing both succinylation and activity of PKM2, indicating that ROS induce SIRT5 to curtail further ROS production via a feedback mechanism (Bringman-Rodenbarger et al. 2017; Xiangyun et al. 2017). Furthermore, SIRT5 has been reported to be overexpressed in hepatocellular carcinoma (HCC), where high SIRT5 expression correlates with overall poor survival (Chang et al. 2018). SIRT5-depleted HCC cells display reduced proliferation and invasion in vitro. SIRT5 KD induces the expression of transcription factor E2F1 (Chang et al. 2018), which exhibits tumor suppressor activity in HCC (Zhan et al. 2014), strongly suggesting that SIRT5 promotes HCC via inhibition of E2F1 expression.
Recently, Wang et al. showed that SIRT5 expression levels are upregulated in colorectal cancer (CRC) tissues and cell lines, and increased expression of SIRT5 is associated with poor prognosis in CRC (Wang YQ et al. 2018). SIRT5 KD CRC cell lines display reduced proliferation. Overexpression of WT SIRT5, but not a H158Y catalytic mutant, promotes tumorigenesis of HCT116 cells in a xenograft model, suggesting that SIRT5 catalytic activity is required for the oncogenic role of SIRT5 in this cancer type. In this context, SIRT5 plays a role in regulating glutamine metabolism in CRC cells by deglutarylation and activation of glutamate dehydrogenase 1 (GLUD1) (Wang YQ et al. 2018), an enzyme that catalyzes conversion of glutamate to α-KG. Consequently, SIRT5 ablated cells exhibit reduced α-KG generation, resulting in reduced anaplerotic entry of glutamine into the TCA cycle, which in turn suppresses the malignant phenotypes of CRC cells (Wang YQ et al. 2018).
Recent work links SIRT5 to therapeutic resistance in CRC. SIRT5-expressing WT KRAS CRC cells exhibit remarkable resistance to chemotherapies, or targeted agents such as the EGFR inhibitor cetuximab, and serve as major contributor to recurrence in CRC patients (Du Z et al. 2018). Indeed, high expression of SIRT5 is associated with a shorter time to post therapy recurrence and overall poor survival in WT KRAS CRC patients. Consistent with prior results (Park et al. 2013), Du and colleagues showed that SIRT5 demalonylates and inactivates SDH subunit A (SDHA), resulting in the accumulation of succinate, which in turn binds with and activates thioredoxin reductase 2 (TrxR2) (Du Z et al. 2018). Since TrxR2 is a ROS scavenger protein, its activation imbalances redox homeostasis, a critical factor in the development of chemotherapeutic resistance. Furthermore, inactivation of SDHA causes an increased succinate/α-KG ratio in SIRT5 expressing cells, resulting in inhibition of α-KG dependent dioxygenases (Du Z et al. 2018). α-KG-dependent dioxygenases are involved in DNA and histone demethylation and thus their inhibition results in epigenetic dysregulation, which in turn promotes tumorigenesis and resistance to targeted therapy (Xu W et al. 2011; Du Z et al. 2018).
Taken together, these studies strongly implicate SIRT5 as an oncogene in a variety of cancer types.
SIRT5 as tumor suppressor
As with other sirtuins, roles for SIRT5 in cancer appear to be highly context-specific, and some reports now highlight tumor suppressor functions of SIRT5. SIRT5 desuccinylates and activates SOD1, resulting in reduced ROS levels (Lin ZF et al. 2013). Suppression of SOD1 activity via succinylation seems to play an important role in lung tumor cell growth, since cells expressing a desuccinylation mimetic of SOD1 exhibited reduced proliferation (Lin ZF et al. 2013), implying a tumor suppressor function of SIRT5. Guan and colleagues reported that both SIRT1 and SIRT5 mediate H2O2-induced deacetylation of the tumor suppressor promyelocytic leukemia protein (PML), which is essential for its nuclear localization to exert tumor suppressor function (Guan et al. 2014). Consequently, SIRT1 KD HeLa cells exhibit fewer H2O2-induced PML-nuclear bodies and resist H2O2-induced cell death. Ectopic expression of wild-type SIRT5 restored H2O2-induced cell death in SIRT1 KD HeLa cells (Guan et al. 2014).
Mutant IDH1 or IDH2 neomorphs possess the ability to convert α-KG to R-2-hydroxyglutarate (R-2HG) (Dang et al. 2009; Fujii et al. 2016). These IDH mutant alleles are frequently identified in glioma, acute myeloid leukemia, and other tumor types such as chondrosarcoma (Clark et al. 2016). R-2HG is an α-KG analog and inhibits α-KG-dependent dioxygenases (Xu W et al. 2011). Recently, Li et al. reported that R-2HG promotes cancer metabolism and apoptotic resistance by inhibiting SDH, resulting in succinyl-CoA accumulation and mitochondrial protein hypersuccinylation (Li F et al. 2015). Glioma samples and cells harboring an IDH1 R132H mutation exhibit increased protein succinylation, leading to mitochondrial dysfunction and accumulation of the anti-apoptotic protein BCL-2, conferring apoptosis resistance (Li F et al. 2015). Strikingly, ectopic SIRT5 expression reversed the metabolic defects and apoptosis resistance in IDH1 mutant glioma cells and impaired their growth in vitro and in vivo (Li F et al. 2015), implying a tumor-suppressor function of SIRT5.
Poletta et al. have shown that SIRT5 binds to, desuccinylates and inhibits glutaminase (Polletta et al. 2015), an enzyme catalyzing hydrolysis of glutamine to glutamate and ammonia. SIRT5 overexpression decreases ammonia production, and reduced ammonia-induced autophagy and mitophagy are observed in SIRT5-ovrexpressing human breast cancer MDA-MB-231 cells and C2C12 cells. Conversely, silencing or inhibition of SIRT5 results in increased ammonia production and ammonia-induced autophagy (Polletta et al. 2015). Ammonia-induced autophagy can play a protective role in tumor cells, rendering them able to survive chemotherapy or other environmental stresses such as hypoxia or nutrient starvation. Furthermore, many cancer cells require exogenous glutamine to support survival and proliferation in vitro, since glutamine serves as an anaplerotic substrate to replenish the TCA cycle via α-KG, a product of glutamine catabolism (Cluntun et al. 2017). Therefore, SIRT5 activation may impair the ability of tumor cells to metabolize glutamine, reducing ATP production and sensitizing them to chemotherapy and other stresses.
Recently, Chen et al. reported the existence of a peroxisomal fraction of SIRT5 that desuccinylates and inhibits acyl-CoA oxidase 1 (ACOX1) (Chen XF et al. 2018). In peroxisomes, ACOX1 is a rate-limiting enzyme in FAO and is a major contributor of H2O2 production (Zeng and Li 2004). Aberrant activation of ACOX1 leads to disruption of FAO and redox homeostasis in hepatic tissues, resulting in chronic liver disease and hepatocellular carcinoma (HCC). Active ACOX1 exists as a dimer; SIRT5-mediated desuccinylation prevents its dimerization, resulting in reduced activity of ACOX1 in cultured cells and mouse liver tissues (Chen XF et al. 2018). Consequently, SIRT5 depletion elevates H2O2 production and oxidative DNA damage, which is attenuated by ACOX1 KD (Chen XF et al. 2018). Moreover, reduced expression of SIRT5 in HCC patient tissues is associated with increased succinylation and activity of ACOX1, suggesting that downregulation of SIRT5 expression leads to increased oxidative DNA damage in HCC patients (Chen XF et al. 2018).
Role of SIRT5 in the pathogenesis of neurodegenerative disorders
Mitochondrial dysfunction and oxidative stress have been implicated in the pathogenesis of many neurodegenerative diseases such as Parkinson’s disease (Gao J et al. 2017). Mitochondrial functions including energy production, oxidative phosphorylation, redox homeostasis, calcium buffering, and apoptotic signaling pathways are essential for neuronal viability (Gao J et al. 2017). As a quality control mechanism for maintaining their functionality under metabolic or other stressed conditions, mitochondria undergo fusion and fission, resulting in mitochondrial elongation and fragmentation, respectively (Meyer et al. 2017). In this regard, Guedouari et al. have shown that SIRT5 induces mitochondrial elongation under certain stress conditions such as starvation (Guedouari et al. 2017). Sirt5 depletion results in increased mitochondrial fragmentation and increased mitophagy during starvation, indicating SIRT5 protects mitochondria from fragmentation and degradation under these conditions (Guedouari et al. 2017). Additionally, as previously noted, SIRT5 plays significant roles in inhibiting mitochondrial ROS levels. Given the crucial roles of mitochondria in neuronal viability, SIRT5 seems well-positioned to suppress the onset and/or pace of neurodegenerative disease (Fig. 4).
Parkinson’s disease (PD) is characterized by progressive loss of nigrostriatal dopaminergic neurons and a motor deficit (Beitz 2014). 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a small molecule that induces selective loss of nigrostriatal dopaminergic neurons in mice, is widely used as a PD model (Przedborski et al. 2001). MPTP is specifically taken up by astrocytes and serotonergic neurons, and converted by monoamine oxidase-B into its active form, 1-methyl-4-phenylpyridinium (MPP+). MPP+ is then secreted into extracellular space and selectively taken via the dopamine transporter of dopaminergic neurons, where it suppresses mitochondrial respiration by inhibiting complex I, causing reduced ATP production and increased ROS levels, activating cell death pathways (Williams and Ramsden 2005). Strikingly, SIRT5 deficiency aggravates MPTP-induced nigrostriatal dopaminergic degeneration in mice (Liu L et al. 2015). Furthermore, MPTP treatment induces increased SIRT5 expression in brain, whereas following MPTP treatment, Sirt5 ablated mice brain striata display reduced levels of the mitochondrial antioxidant enzyme manganese superoxide dismutase (SOD2) (Liu L et al. 2015). These findings suggest that SIRT5 alleviates MPTP-induced nigrostriatal dopaminergic degeneration by suppressing mitochondrial-derived ROS levels. Mechanisms of SOD2 downregulation in this context have not been elucidated.
Epilepsy is a common neurological disorder characterized by recurrent spontaneous seizures (Pitkanen and Lukasiuk 2011). Kainate (KA), an analog of glutamate, exerts neuro-excitotoxic and epileptogenic effects by acting on glutamate receptors (Wang Q et al. 2005). The KA-induced seizure rodent model can be used to study human temporal lobe epilepsy (Ben-Ari and Cossart 2000). A recent study by Li and Liu revealed neuroprotective roles of SIRT5 in response to KA-induced epileptic seizure (Li F and Liu 2016). KA exposure leads to increased SIRT5 expression in the hippocampus. Conversely, SIRT5 loss dramatically promotes hippocampal neuronal loss and degeneration in KA-exposed mice, and also provokes more reactive astrogliosis in the hippocampus (Li F and Liu 2016). As a consequence, Sirt5 deletion results in severe response to epileptic seizures, and Sirt5 KO mice display strikingly increased mortality after KA treatment (Li F and Liu 2016). Notably, the protective effects of SIRT5 in KA-induced epilepsy seems to be independent of its role in ROS management (Li F and Liu 2016).
Cerebral ischemia induces dysfunction in neuronal mitochondria and cell death (Lin HW et al. 2011). Protein kinase C epsilon (PKCε), a serine/threonine kinase implicated in mitochondrial protection, is an important signaling molecule involved in neuroprotection against ischemic insult (Raval et al. 2007; Dave et al. 2008; Dave et al. 2009; Della-Morte et al. 2011). PKCε enhances expression of nicotinamide phosphoribosyl transferase (NAMPT) (Morris-Blanco et al. 2014), the enzyme catalyzing the rate limiting step of the NAD+ salvage pathway. NAMPT deficiency has been reported to exacerbate neurodegeneration following cerebral ischemia (Zhang W et al. 2010; Wang P et al. 2012). In this context, a recent study by Morris-Blanco et al. showed that increased NAMPT expression is necessary to confer PKCε mediated neuroprotection (Morris-Blanco et al. 2016). Moreover, PKCε activation results in increased expression, and desuccinylase activity of SIRT5 in isolated rat cortical mitochondria, in a NAMPT-dependent manner. Strikingly, cortical mitochondria isolated from Sirt5 KO mice display significantly reduced oxygen consumption, and PKCε fails to prevent cortical degeneration in Sirt5 KO mice following ischemic injury (Morris-Blanco et al. 2016), strongly suggesting that SIRT5 is involved in regulating mitochondrial bioenergetics and PKCε-mediated neuroprotection against cerebral ischemia. Indeed, a subsequent study by Koronowski and colleagues revealed alterations in metabolic pathways including purine metabolism, nitrogen metabolism and malate-aspartate shuttle among several others in Sirt5 KO cortex (Koronowski et al. 2018). Notably, PKCε activation causes an alteration in purine metabolic pathway in WT but not in Sirt5 KO cortex, indicating that SIRT5 plays a role in PKCε-mediated ischemic tolerance by regulating purine metabolism (Koronowski et al. 2018).
Targeting SIRT5 via small molecules
Given its numerous molecular functions in the context of many different pathologies, a great deal of interest exists in targeting SIRT5 therapeutically, either through activation or inhibition by small molecules, depending on context. In addition, SIRT5-specific modulators could provide valuable tools in deciphering in vivo functions and regulation of SIRT5. Although many SIRT5 modulating compounds have been described, most suffer from poor potency and/or low selectivity, limiting their in vivo utility (Table 1). For instance, the prototypical SIRT5 inhibitor suramin inhibits SIRT5 at low micromolar levels by blocking both its substrate and NAD+ binding sites, but also inhibits other sirtuins with comparable potencies (Schuetz et al. 2007; Trapp et al. 2007; Suenkel et al. 2013). Similarly, resveratrol has been shown to inhibit desuccinylase -- and promote deacetylase -- activity of SIRT5, in a substrate-specific manner. However, resveratrol is a well-known activator of SIRT1 and also exhibits SIRT3 inhibitory activity (Howitz et al. 2003; Milne et al. 2007; Gertz et al. 2012; Lakshminarasimhan et al. 2013). A product of the sirtuin reaction, nicotinamide (NAM), inhibits SIRT5 desuccinylase activity with an IC50 value of 21μM, without affecting SIRT5 deacetylase activity, even at a concentration of ~100μM (Fischer et al. 2012). The presence of the Arg105 residue in SIRT5 catalytic pocket is thought to be responsible for this differential effect of NAM on SIRT5 activities (Fischer et al. 2012). Similarly, the indole GW5074 has been shown to inhibit SIRT5 desuccinylase activity (IC50=19.5μM), but it has weaker effects on SIRT5-catalyzed deacetylation (Suenkel et al. 2013). However, NAM is a pan sirtuin inhibitor (Gertz and Steegborn 2016; Madsen et al. 2016; Pannek et al. 2017) and GW5074 is a potent kinase and SIRT2 inhibitor (Trapp et al. 2006), making them unsuitable for in vivo studies focused on SIRT5. In addition to NAM, sirtinol, cambinol, and thiobarbiturate-containing compounds have also been shown to inhibit SIRT5 (Maurer et al. 2012). Unfortunately, these compounds also lack SIRT5 specificity, and inhibit other sirtuins at comparable potencies (Grozinger et al. 2001; Mai et al. 2005; Heltweg et al. 2006; Maurer et al. 2012). By contrast, Polletta et al. chemically synthesized a small molecule MC3482, which inhibited SIRT5 selectively vis-a-vis SIRT1 and SIRT3 (Polletta et al. 2015). Treatment of the human breast cancer cell line MDA-MB-231 and mouse C2C12 myoblasts with 50μM MC3482 resulted in ~42% inhibition of SIRT5 desuccinylase activity, without significantly affecting SIRT1 activity at similar concentration. Only 8% inhibition of SIRT3 activity was observed in the presence of 50μM MC3482 (Polletta et al. 2015). Recently, through screening using a microdroplet-based approach, we identified several approved drugs as SIRT5 inhibitors, with IC50 values in the low micromolar range (Guetschow et al. 2016). In this screening, the polyphenol anthralin was identified as most potent SIRT5 inhibitor (IC50= 0.1μM), but further investigation will be required to evaluate selectivity and mechanism of action (Guetschow et al. 2016).
Table 1:
Small molecule SIRT5 modulators and their impact on sirtuin activities.
| Compound | Impact on SIRT5 | Impact on other sirtuins |
Reference |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Desuccinylation | Deglutarylation | Demalonylation | Deacetylation | SIRT1 | SIRT2 | SIRT3 | SIRT4 | SIRT6 | SIRT7 | ||
| Suramin | Inhibition (IC50=~50μM) |
Unknown | Unknown | Inhibition (IC50=14.2–22μM) |
Inhibition (IC50= 297nM) |
Inhibition (IC50= 1150nM) |
Unknown | Unknown | Unknown | Unknown | (Schuetz et al. 2007; Trapp et al. 2007; Suenkel et al. 2013) |
| Resveratrol | Inhibition (~25% at 0.2mM) |
Unknown | Unknown | Activation (2.5 fold at 0.2mM) |
Activation (2–8 fold, EC50= 50–100μM) |
Activation (1.5 fold at >300μM) |
Activation (1.5 fold at >300μM) Inhibition (10–50% at 0.2mM) |
Unknown | Unknown | Unknown | (Howitz et al. 2003; Milne et al. 2007; Gertz et al. 2012) |
| Nicotinamide (NAM) | Inhibition (IC50=21–28μM) |
Inhibition (IC50=99+30μM) |
Unknown | Inhibition (IC50=0.7–1.6mM) |
Inhibition (IC50= 68–180μM) |
Inhibition (IC50= 35–44μM) |
Inhibition (IC50= 53–76μM) |
Inhibition (IC50 = 13±2μM) |
Inhibition (IC50= 120–150μM) |
Inhibition (IC50= Un-known) |
(Fischer et al. 2012; Madsen et al. 2016; Pannek et al. 2017) |
| GW5074 | Inhibition (IC50=19.5 ± 7.3μM) |
Unknown | Unknown | Inhibition (IC50= 97.8 ± 18.6μM) |
Unknown | >60% Inhibition at
12.5μM |
Unknown | Unknown | Unknown | Unknown | (Trapp et al. 2006; Suenkel et al. 2013) |
| Sirtinol | Inhibition (IC50=48.9 ± 6.3μM) |
Unknown | Unknown | No effect up to 100μM | Inhibition (IC50= 131μM) |
Inhibition (IC50= 38μM) |
24% inhibition at 50μM |
Unknown | Unknown | Unknown | (Grozinger et al. 2001; Mai et al. 2005; Maurer et al. 2012; Suenkel et al. 2013) |
| Cambinol | Inhibition (IC50=42.5 ± 1.5μM) |
Unknown | Unknown | 42% inhibition at 300μM |
Inhibition (IC50= 56μM) |
Inhibition (IC50= 59μM) |
No effect up to 300μM | Unknown | Unknown | Unknown | (Heltweg et al. 2006; Maurer et al. 2012) |
| Thiobarbiturate-2 |
Inhibition (IC50=2.3 ± 0.2μM) |
Unknown | Unknown | Unknown | Inhibition (IC50= 5.3±0.7μM) |
Inhibition (IC50= 9.7±1.6μM) |
41% inhibition at 50μM |
Unknown | Unknown | Unknown | (Maurer et al. 2012) |
| MC3482 | Inhibition (~42% inhibition at 50μM) |
Unknown | Unknown | No effect at 50μM as per Western Blot analysis | No effect at 50μM | Unknown | 8% inhibition at 50μM |
Unknown | Unknown | Unknown | (Polletta et al. 2015) |
| Anthralin | Inhibition (IC50=0.1μM) |
Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | (Guetschow et al. 2016) |
| H3K9TSu | Inhibition (IC50=5μM) |
Unknown | Unknown | Unknown | No effect up to 100μM | No effect up to 100μM | No effect up to 100μM | Unknown | Unknown | Unknown | (He et al. 2012) |
| 3-methyl-3-phenyl-succinyl-CPS1 | Inhibition (Ki=4.3±0.32μM) |
Unknown | Unknown | Unknown | <1% inhibition at 50μM | 4.23±2.16% inhibition at
50μM |
<1% inhibition at 50μM | Unknown | Unknown | Unknown | (Roessler et al. 2014) |
| Nε-carboxyethyl-thiocarbamoyl-lysine | Inhibition (IC50=5.0±1.9μM) |
Unknown | Unknown | Unknown | Inhibition (IC50= >100μM) |
Unknown | Unknown | Unknown | Inhibition (IC50= ~2.4mM) |
Unknown | (Zang et al. 2015) |
| Compound-5 | Inhibition (IC50=7.5±4.0μM) |
Unknown | Unknown | Unknown | Inhibition (IC50= >1000μM) |
Inhibition (IC50= >1000μM) |
Inhibition (IC50= >200μM) |
Unknown | Inhibition (IC50= >200μM) |
Unknown | (Liu J et al. 2016) |
| Compound-19 | Inhibition (IC50=9.26±2.09μM) |
Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | (Liu S et al. 2017) |
| Compound-37 | Inhibition (IC50=5.59±0.75μM) |
Unknown | Unknown | Unknown | Unknown |
No effect up to 600μM | Unknown | Unknown | No effect up to 600μM | Unknown | (Liu S et al. 2017) |
| Compound-10 | Unknown | Inhibition (IC50=0.83±0.01μM) |
Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | (Rajabi et al. 2017) |
| Compound-15 | Unknown | Inhibition (IC50=0.45±0.05μM) |
Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | (Rajabi et al. 2017) |
| Compound-29 | Inhibition (IC50=0.15±0.02μM) |
Inhibition (IC50=0.37±0.1μM) |
Unknown | Unknown | No effect at 10μM | No effect at 10μM | No effect at 10μM | Unknown | No effect at 10μM | Unknown | (Rajabi et al. 2017) |
| Compound-48 | Inhibition (IC50=0.42±0.01μM) |
Inhibition (IC50=0.26±0.01μM) |
Unknown | Unknown | No effect at 10μM | No effect at 10μM | No effect at 10μM | Unknown | No effect at 10μM | Unknown | (Rajabi et al. 2017) |
| Compound-49 | Inhibition (IC50=0.17±0.01μM) |
Inhibition (IC50=0.11±0.02μM) |
Unknown | Unknown | No effect at 10μM | No effect at 10μM | No effect at 10μM | Unknown | No effect at 10μM | Unknown | (Rajabi et al. 2017) |
| Compound-50 | Inhibition (IC50=0.31±0.01μM) |
Inhibition (IC50=0.23±0.05μM) |
Unknown | Unknown | No effect at 10μM | No effect at 10μM | No effect at 10μM | Unknown | No effect at 10μM | Unknown | (Rajabi et al. 2017) |
| Compound-33.2 | Inhibition (IC50=40nM) |
Inhibition (IC50=30.3±3.5nM) |
Unknown | Unknown | No effect at 50μM | No effect at 50μM | No effect at 50μM | Unknown | No effect at 50μM | Unknown | (Kalbas et al. 2018) |
| Compound-35.2 | Unknown | Inhibition (IC50=15.4±9.5nM, Ki=7.0nM) |
Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | (Kalbas et al. 2018) |
| Compound-39.2 | Unknown | Inhibition (IC50=19.1±14.1nM, Ki=13.6+3.8nM) |
Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | (Kalbas et al. 2018) |
Efforts are now underway in many laboratories to develop more potent and selective SIRT5 modulators. The substrate binding cleft and NAD+ binding pocket are attractive target sites for developing competitive inhibitors to inhibit sirtuin enzymatic activity. However, due to the high sequence conservation of the NAD+ binding site among different sirtuins, blocking NAD+ binding is likely not an effective strategy for development of SIRT5-selective inhibitors. In contrast, SIRT5-specific residues (Tyr102, Arg105, and Ala86) in the substrate binding pocket of SIRT5 are responsible for its unique ability to target acidic acyl-lysine modifications, and thus targeting the substrate-binding pocket appears to represent a more promising approach to develop SIRT5 selective inhibitors. Indeed, thioacetyl peptides have been shown to inhibit sirtuin deacetylase activity by forming stable covalent intermediates, preventing substrate binding and processing. These structural elements have been termed ‘catalytic mechanism-based sirtuin inhibitory warheads’ (Fatkins et al. 2006; Smith and Denu 2007; Hawse et al. 2008; Jiang et al. 2017). He et al. synthesized a histone H3 lysine 9 thiosuccinyl peptide (H3K9TSu), which exhibited inhibition of SIRT5 activity (IC50=5μM) without affecting SIRT1–3 activities up to 100μM (He et al. 2012). In order to evaluate the effect of lysine acyl modifications on SIRT5 activity, Roessler et al. screened different acyl variants of the lysine side chain of a CPS1-based peptide inhibitor, and identified 3-methyl-3-phenyl-succinyl-CPS1 as most potent (Ki = 4.3±0.32 μM) and highly selective inhibitor of SIRT5 (Roessler et al. 2014). Recently Zang et al. identified Nε-carboxyethyl-thiocarbamoyl-lysine as a potent thiourea-based SIRT5 inhibitory warhead (IC50 = 5.0 ± 1.9μM), which shows selectivity versus SIRT1 and SIRT6 (IC50 values of >100 μM and ~2400 μM respectively) (Zang et al. 2015). Notably, thiourea-based inhibitory warheads appear to circumvent cytotoxicity issues resulting from the use of thioamide harboring Nε-thioacyl-lysines type warheads, suggesting that thiourea-based inhibitors may represent safer approach for targeting sirtuins therapeutically (Jiang et al. 2017). However, due to the fact that linear peptides are generally associated with limited bio-stability and poor membrane permeability, they are not considered as ideal tools for research and therapeutic purposes.
Peptide macrocyclization, i.e. converting linear peptide to cyclic peptide, is an established approach to enhance bio-stability and cell membrane permeabilization of linear peptides. Depending on its functional groups, a peptide can be cyclized in four different ways: head-to-tail (C-terminus to N-terminus), head-to-side chain, side chain-to-tail or side chain-to-side chain (White and Yudin 2011). Zheng and co-workers developed a side chain to side chain cyclized pentapeptide (compound-5) (Liu J et al. 2016), which harbored a central Nε-carboxyethyl-thiocarbamoyl-lysine (Zang et al. 2015), and central five residue fragment of an H3K9TSu peptide (He et al. 2012). Compound-5 was more stable relative to its linear counterpart against proteolysis, and behaved as fairly strong and selective inhibitor of the SIRT5-catalyzed deacylation reaction, with an IC50 value of 7.5 ± 4.0μM, versus IC50 values of between 200 and 1000μM for SIRT1/2/3/6 (Liu J et al. 2016). In a recent study using virtual screening approach targeting the Tyr102 and Arg105 residues of SIRT5, followed by biochemical testing, Liu et al. identified compound-19 (containing (E)-2-cyano-N-phenyl-3-(5-phenylfuran-2-yl) acrylamide as core scaffold), which inhibited SIRT5 with an IC50 value of 9.26 ± 2.09μM (Liu S et al. 2017). To identify still more potent SIRT5 inhibitors, the authors generated a series of (E)-2-cyano-N-phenyl-3-(5-phenylfuran-2-yl) acrylamide derivatives, and found that compound-37 (bearing 2-fluorobenzonitrile as amide-N substituent) exhibited the most potent inhibition of SIRT5, with an IC50 value of 5.59 ± 0.75μM (Liu S et al. 2017). Further biochemical analysis revealed that compound-37 acts via competitive inhibition and displays considerable selectivity for SIRT5 over SIRT2 and SIRT6 (Liu S et al. 2017).
Recently, Rajabi et al. evaluated more than 70 compounds in a structure-activity relationship (SAR) study and identified SIRT5 selective inhibitors that exhibited sub-micromolar potency via a slow, tight-binding mechanism (Rajabi et al. 2017). The authors first screened various C-terminal variants of Nε-thioglutaryllysine, which have carboxybenzyl (Cbz) protected α–amino group. Based on this initial screen, Compound-10 and Compound-15 were identified as most potent thioamide based inhibitors, with IC50 values of 0.83 ± 0.01μM and 0.45 ± 0.05μM respectively. Of interest, a thiourea analogue of Compound-10, Compound-29 proved to be more potent, which exhibited IC50 value of 0.37 ± 0.1μM. Finally, the authors generated sulfonamide analogues of Cbz group and introduced steric bulk at C-terminus in context of Compound-29, to prepare Compounds-48, 49 and 50, which exhibited IC50 values against SIRT5 of 0.26 ± 0.01μM, 0.11 ± 0.02μM, and 0.23 ± 0.05μM respectively. Remarkably, Compounds-29, 48, 49 and 50 displayed excellent selectivity for SIRT5 over SIRT1–3 and 6 (Rajabi et al. 2017).
Recently, Kalbas et al. synthesized and screened 3-(arylthio)succinylated- and 3-(benzylthio)succinylated peptide derivatives, yielding the most potent SIRT5 inhibitors developed to date (Kalbas et al. 2018). Among the 3-(arylthio)succinyl scaffold-harboring inhibitors, naphthylthio derivative 33.2 exhibited the most efficient SIRT5 inhibition, with an IC50 value of 30.3+3.5nM. Moreover, it displayed high selectivity for SIRT5, with no effect on the activities of SIRT1, SIRT2, SIRT3 and SIRT6, observed even at concentrations up to 50μM. Strikingly, the 3-(benzylthio)succinyl scaffold-bearing naphthylmethylthio derivative 35.2 exhibited improved inhibition of SIRT5, with an IC50 value of 15.4+9.5nM and calculated Ki value of 7nM (Kalbas et al. 2018). Inspired by the high SIRT5 selectivity of 33.2, Kalbas and colleagues further developed an affinity probe 39.2 for human SIRT5, by replacing the benzoyl moiety in 33 by a spacer molecule and biotinyl residue, which enable immobilization on streptavidin coated surfaces. 39.2 displayed improved SIRT5 inhibition over 33.2 with an IC50 value of 19.1+14.1nM and Ki value of 13.6+3.8nM, and pulled down SIRT5 with remarkable selectively over SIRT2 and SIRT3 (Kalbas et al. 2018).
Collectively, these studies have generated potent and selective SIRT5 inhibitors; however, none of these agents have been evaluated for their in vivo effects in either cells or whole animals. Additionally, the number of SIRT5 selective compounds with high efficacy are rather limited. Therefore, high throughput screening approaches and further medicinal chemistry-based optimization campaigns are warranted to identify more potent and selective compounds. Besides fluorescence-based assays, a number of other high-throughput screening (HTS) assay systems have been developed to aid such efforts (Li Y et al. 2015; Roessler et al. 2015; Guetschow et al. 2016).
Concluding remarks
With recent advances in proteomics, a variety of novel lysine acyl modifications have been discovered, which has led to the recognition that sirtuins function as lysine deacylases, rather than strictly as deacetylases. SIRT5 has now been identified as the major cellular activity responsible for desuccinylation, demalonylation, and deglutarylation in mammalian cells, and numerous proteomics studies have elucidated a landscape of hundreds of SIRT5 substrates. SIRT5 plays roles in maintaining metabolic and cellular homeostasis in a cell- and context-specific manner, by regulating the activities of substrates involved in metabolic processes including glycolysis, TCA cycle, FAO, the ETC, ketone body formation, nitrogenous waste management, and ROS detoxification. SIRT5 plays particularly important functions in cardiac heath, especially in the context of aging or stress. Roles for SIRT5 in the context of other diseases, including cancer and neurodegenerative disorders, are beginning to be described. There is little doubt that we have merely scratched the surface in understanding the biological functions of this sirtuin. This effort will be aided by development of potent and selective small molecule SIRT5 modulators, efforts towards which are well underway in many laboratories.
Acknowledgements
The authors would like to thank members of Lombard laboratory for helpful discussions. Some portions of Figures 2, 3, and 4 were obtained and modified from Servier Medical Art from Servier (http://www.servier.com/Powerpoint-image-bank).
Footnotes
Declaration of Interest
The Lombard laboratory is supported by awards from NIH (R01GM101171, 2R01HL114858, and R21AG053561), the Glenn Foundation for Medical Research, the Melanoma Research Alliance, the Harrington Discovery Institute, AACR, and the University of Michigan Comprehensive Cancer Center. The authors declare no competing financial interests.
References
- Ahuja N, Schwer B, Carobbio S, Waltregny D, North BJ, Castronovo V, Maechler P, Verdin E. 2007. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J Biol Chem. 282(46):33583–33592. [DOI] [PubMed] [Google Scholar]
- Alvarez-Lario B, Macarron-Vicente J. 2010. Uric acid and evolution. Rheumatology (Oxford). 49(11):2010–2015. [DOI] [PubMed] [Google Scholar]
- Anderson KA, Huynh FK, Fisher-Wellman K, Stuart JD, Peterson BS, Douros JD, Wagner GR, Thompson JW, Madsen AS, Green MF et al. 2017. SIRT4 Is a Lysine Deacylase that Controls Leucine Metabolism and Insulin Secretion. Cell Metab. 25(4):838–855 e815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audagnotto M, Dal Peraro M. 2017. Protein post-translational modifications: In silico prediction tools and molecular modeling. Comput Struct Biotechnol J. 15:307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balendiran GK, Dabur R, Fraser D. 2004. The role of glutathione in cancer. Cell Biochem Funct. 22(6):343–352. [DOI] [PubMed] [Google Scholar]
- Bao X, Wang Y, Li X, Li XM, Liu Z, Yang T, Wong CF, Zhang J, Hao Q, Li XD. 2014. Identification of ‘erasers’ for lysine crotonylated histone marks using a chemical proteomics approach. Elife. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber MF, Michishita-Kioi E, Xi Y, Tasselli L, Kioi M, Moqtaderi Z, Tennen RI, Paredes S, Young NL, Chen K et al. 2012. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature. 487(7405):114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartosch C, Monteiro-Reis S, Almeida-Rios D, Vieira R, Castro A, Moutinho M, Rodrigues M, Graca I, Lopes JM, Jeronimo C. 2016. Assessing sirtuin expression in endometrial carcinoma and non-neoplastic endometrium. Oncotarget. 7(2):1144–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beitz JM. 2014. Parkinson’s disease: a review. Front Biosci (Schol Ed). 6:65–74. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cossart R. 2000. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 23(11):580–587. [DOI] [PubMed] [Google Scholar]
- Boylston JA, Sun J, Chen Y, Gucek M, Sack MN, Murphy E. 2015. Characterization of the cardiac succinylome and its role in ischemia-reperfusion injury. J Mol Cell Cardiol. 88:73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringman-Rodenbarger LR, Guo AH, Lyssiotis CA, Lombard DB. 2017. Emerging roles for SIRT5 in metabolism and cancer. Antioxid Redox Signal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buler M, Aatsinki SM, Izzi V, Uusimaa J, Hakkola J. 2014. SIRT5 is under the control of PGC-1alpha and AMPK and is involved in regulation of mitochondrial energy metabolism. FASEB J. 28(7):3225–3237. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research N. 2011. Integrated genomic analyses of ovarian carcinoma. Nature. 474(7353):609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkiadaki A, Guarente L. 2015. The multifaceted functions of sirtuins in cancer. Nat Rev Cancer. 15(10):608–624. [DOI] [PubMed] [Google Scholar]
- Chang L, Xi L, Liu Y, Liu R, Wu Z, Jian Z. 2018. SIRT5 promotes cell proliferation and invasion in hepatocellular carcinoma by targeting E2F1. Mol Med Rep. 17(1):342–349. [DOI] [PubMed] [Google Scholar]
- Chen S, Blank MF, Iyer A, Huang B, Wang L, Grummt I, Voit R. 2016. SIRT7-dependent deacetylation of the U3–55k protein controls pre-rRNA processing. Nat Commun. 7:10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Seiler J, Santiago-Reichelt M, Felbel K, Grummt I, Voit R. 2013. Repression of RNA polymerase I upon stress is caused by inhibition of RNA-dependent deacetylation of PAF53 by SIRT7. Mol Cell. 52(3):303–313. [DOI] [PubMed] [Google Scholar]
- Chen XF, Tian MX, Sun RQ, Zhang ML, Zhou LS, Jin L, Chen LL, Zhou WJ, Duan KL, Chen YJ et al. 2018. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC et al. 2014. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 515(7527):431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chypre M, Zaidi N, Smans K. 2012. ATP-citrate lyase: a mini-review. Biochem Biophys Res Commun. 422(1):1–4. [DOI] [PubMed] [Google Scholar]
- Circu ML, Aw TY. 2010. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 48(6):749–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark O, Yen K, Mellinghoff IK. 2016. Molecular Pathways: Isocitrate Dehydrogenase Mutations in Cancer. Clin Cancer Res. 22(8):1837–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cluntun AA, Lukey MJ, Cerione RA, Locasale JW. 2017. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer. 3(3):169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak G, Pougovkina O, Dai L, Tan M, Te Brinke H, Huang H, Cheng Z, Park J, Wan X, Liu X et al. 2015. Proteomic and Biochemical Studies of Lysine Malonylation Suggest Its Malonic Aciduria-associated Regulatory Role in Mitochondrial Function and Fatty Acid Oxidation. Mol Cell Proteomics. 14(11):3056–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper HM, Spelbrink JN. 2008. The human SIRT3 protein deacetylase is exclusively mitochondrial. Biochem J. 411(2):279–285. [DOI] [PubMed] [Google Scholar]
- Coordinators NR. 2018. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 46(D1):D8–D13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC et al. 2009. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 462(7274):739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave KR, Anthony Defazio R, Raval AP, Dashkin O, Saul I, Iceman KE, Perez-Pinzon MA, Drew KL. 2009. Protein kinase C epsilon activation delays neuronal depolarization during cardiac arrest in the euthermic arctic ground squirrel. J Neurochem. 110(4):1170–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave KR, DeFazio RA, Raval AP, Torraco A, Saul I, Barrientos A, Perez-Pinzon MA. 2008. Ischemic preconditioning targets the respiration of synaptic mitochondria via protein kinase C epsilon. J Neurosci. 28(16):4172–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della-Morte D, Raval AP, Dave KR, Lin HW, Perez-Pinzon MA. 2011. Post-ischemic activation of protein kinase C epsilon protects the hippocampus from cerebral ischemic injury via alterations in cerebral blood flow. Neurosci Lett. 487(2):158–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlon TA, Morris BJ, Chen R, Masaki KH, Allsopp RC, Willcox DC, Tiirikainen M, Willcox BJ. 2017. Analysis of Polymorphisms in 58 Potential Candidate Genes for Association with Human Longevity. J Gerontol A Biol Sci Med Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryden SC, Nahhas FA, Nowak JE, Goustin AS, Tainsky MA. 2003. Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol Cell Biol. 23(9):3173–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH et al. 2011. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 334(6057):806–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Liu X, Chen T, Gao W, Wu Z, Hu Z, Wei D, Gao C, Li Q. 2018. Targeting a Sirt5-Positive Subpopulation Overcomes Multidrug Resistance in Wild-Type Kras Colorectal Carcinomas. Cell Rep. 22(10):2677–2689. [DOI] [PubMed] [Google Scholar]
- Ducker GS, Rabinowitz JD. 2017. One-Carbon Metabolism in Health and Disease. Cell Metab. 25(1):27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatkins DG, Monnot AD, Zheng W. 2006. Nepsilon-thioacetyl-lysine: a multi-facet functional probe for enzymatic protein lysine Nepsilon-deacetylation. Bioorg Med Chem Lett. 16(14):3651–3656. [DOI] [PubMed] [Google Scholar]
- Feldman JL, Baeza J, Denu JM. 2013. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J Biol Chem. 288(43):31350–31356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer F, Gertz M, Suenkel B, Lakshminarasimhan M, Schutkowski M, Steegborn C. 2012. Sirt5 deacylation activities show differential sensitivities to nicotinamide inhibition. PLoS One. 7(9):e45098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford E, Voit R, Liszt G, Magin C, Grummt I, Guarente L. 2006. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 20(9):1075–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fould B, Garlatti V, Neumann E, Fenel D, Gaboriaud C, Arlaud GJ. 2010. Structural and functional characterization of the recombinant human mitochondrial trifunctional protein. Biochemistry. 49(39):8608–8617. [DOI] [PubMed] [Google Scholar]
- Frye RA. 2000. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun. 273(2):793–798. [DOI] [PubMed] [Google Scholar]
- Fujii T, Khawaja MR, DiNardo CD, Atkins JT, Janku F. 2016. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med. 21(117):373–380. [PubMed] [Google Scholar]
- Gao J, Wang L, Liu J, Xie F, Su B, Wang X. 2017. Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases. Antioxidants (Basel). 6(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Wang H, Yang JJ, Liu X, Liu ZR. 2012. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell. 45(5):598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng YQ, Li TT, Liu XY, Li ZH, Fu YC. 2011. SIRT1 and SIRT5 activity expression and behavioral responses to calorie restriction. J Cell Biochem. 112(12):3755–3761. [DOI] [PubMed] [Google Scholar]
- Gertz M, Nguyen GT, Fischer F, Suenkel B, Schlicker C, Franzel B, Tomaschewski J, Aladini F, Becker C, Wolters D et al. 2012. A molecular mechanism for direct sirtuin activation by resveratrol. PLoS One. 7(11):e49761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz M, Steegborn C. 2016. Using mitochondrial sirtuins as drug targets: disease implications and available compounds. Cell Mol Life Sci. 73(15):2871–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glorioso C, Oh S, Douillard GG, Sibille E. 2011. Brain molecular aging, promotion of neurological disease and modulation by sirtuin 5 longevity gene polymorphism. Neurobiol Dis. 41(2):279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grozinger CM, Chao ED, Blackwell HE, Moazed D, Schreiber SL. 2001. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J Biol Chem. 276(42):38837–38843. [DOI] [PubMed] [Google Scholar]
- Guan D, Lim JH, Peng L, Liu Y, Lam M, Seto E, Kao HY. 2014. Deacetylation of the tumor suppressor protein PML regulates hydrogen peroxide-induced cell death. Cell Death Dis. 5:e1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedouari H, Daigle T, Scorrano L, Hebert-Chatelain E. 2017. Sirtuin 5 protects mitochondria from fragmentation and degradation during starvation. Biochim Biophys Acta. 1864(1):169–176. [DOI] [PubMed] [Google Scholar]
- Guetschow ED, Kumar S, Lombard DB, Kennedy RT. 2016. Identification of sirtuin 5 inhibitors by ultrafast microchip electrophoresis using nanoliter volume samples. Anal Bioanal Chem. 408(3):721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagner N, Joerger M. 2010. Cancer chemotherapy: targeting folic acid synthesis. Cancer Manag Res. 2:293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G et al. 2006. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 126(5):941–954. [DOI] [PubMed] [Google Scholar]
- Haussinger D 1990. Nitrogen metabolism in liver: structural and functional organization and physiological relevance. Biochem J. 267(2):281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawse WF, Hoff KG, Fatkins DG, Daines A, Zubkova OV, Schramm VL, Zheng W, Wolberger C. 2008. Structural insights into intermediate steps in the Sir2 deacetylation reaction. Structure. 16(9):1368–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Du J, Lin H. 2012. Thiosuccinyl peptides as Sirt5-specific inhibitors. J Am Chem Soc. 134(4):1922–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heltweg B, Gatbonton T, Schuler AD, Posakony J, Li H, Goehle S, Kollipara R, Depinho RA, Gu Y, Simon JA et al. 2006. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 66(8):4368–4377. [DOI] [PubMed] [Google Scholar]
- Hershberger KA, Abraham DM, Martin AS, Mao L, Liu J, Gu H, Locasale JW, Hirschey MD. 2017. Sirtuin 5 is required for mouse survival in response to cardiac pressure overload. J Biol Chem. 292(48):19767–19781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger KA, Martin AS, Hirschey MD. 2017. Role of NAD(+) and mitochondrial sirtuins in cardiac and renal diseases. Nat Rev Nephrol. 13(4):213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Zhao Y. 2015. Metabolic Regulation by Lysine Malonylation, Succinylation, and Glutarylation. Mol Cell Proteomics. 14(9):2308–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmstrom KM, Finkel T. 2014. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 15(6):411–421. [DOI] [PubMed] [Google Scholar]
- Houten SM, Wanders RJ. 2010. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis. 33(5):469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL et al. 2003. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 425(6954):191–196. [DOI] [PubMed] [Google Scholar]
- Igci M, Kalender ME, Borazan E, Bozgeyik I, Bayraktar R, Bozgeyik E, Camci C, Arslan A. 2016. High-throughput screening of Sirtuin family of genes in breast cancer. Gene. 586(1):123–128. [DOI] [PubMed] [Google Scholar]
- Imai S, Guarente L. 2010. Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends Pharmacol Sci. 31(5):212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai SI, Guarente L. 2016. It takes two to tango: NAD(+) and sirtuins in aging/longevity control. NPJ Aging Mech Dis. 2:16017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal AK. 2004. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med. 36(10):1199–1207. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Li X, Yang W, Hawke DH, Zheng Y, Xia Y, Aldape K, Wei C, Guo F, Chen Y et al. 2014. PKM2 regulates chromosome segregation and mitosis progression of tumor cells. Mol Cell. 53(1):75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Liu J, Chen D, Yan L, Zheng W. 2017. Sirtuin Inhibition: Strategies, Inhibitors, and Therapeutic Potential. Trends Pharmacol Sci. 38(5):459–472. [DOI] [PubMed] [Google Scholar]
- Kaidi A, Weinert BT, Choudhary C, Jackson SP. 2010. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science. 329(5997):1348–1353. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kalbas D, Liebscher S, Nowak T, Meleshin M, Pannek M, Popp C, Alhalabi Z, Bordusa F, Sippl W, Steegborn C et al. 2018. Potent and selective inhibitors of human Sirtuin 5. J Med Chem. [DOI] [PubMed] [Google Scholar]
- Kashfi K, Mynatt RL, Park EA, Cook GA. 2011. Membrane microenvironment regulation of carnitine palmitoyltranferases I and II. Biochem Soc Trans. 39(3):833–837. [DOI] [PubMed] [Google Scholar]
- Koronowski KB, Khoury N, Morris-Blanco KC, Stradecki-Cohan HM, Garrett TJ, Perez-Pinzon MA. 2018. Metabolomics Based Identification of SIRT5 and Protein Kinase C Epsilon Regulated Pathways in Brain. Front Neurosci. 12:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuciel R, Mazurkiewicz A. 2004. Formation and detoxification of reactive oxygen species. Biochem Mol Biol Educ. 32(3):183–186. [DOI] [PubMed] [Google Scholar]
- Kumar S, Lombard DB. 2015. Mitochondrial sirtuins and their relationships with metabolic disease and cancer. Antioxid Redox Signal. 22(12):1060–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Lombard DB. 2017a. Cycling around Lysine Modifications. Trends Biochem Sci. 42(7):501–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Lombard DB. 2017b. For Certain, SIRT4 Activities! Trends Biochem Sci. 42(7):499–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurmi K, Hitosugi S, Wiese EK, Boakye-Agyeman F, Gonsalves WI, Lou Z, Karnitz LM, Goetz MP, Hitosugi T. 2018. Carnitine Palmitoyltransferase 1A Has a Lysine Succinyltransferase Activity. Cell Rep. 22(6):1365–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CC, Lin PM, Lin SF, Hsu CH, Lin HC, Hu ML, Hsu CM, Yang MY. 2013. Altered expression of SIRT gene family in head and neck squamous cell carcinoma. Tumour Biol. 34(3):1847–1854. [DOI] [PubMed] [Google Scholar]
- Lakshminarasimhan M, Rauh D, Schutkowski M, Steegborn C. 2013. Sirt1 activation by resveratrol is substrate sequence-selective. Aging (Albany NY). 5(3):151–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, He X, Ye D, Lin Y, Yu H, Yao C, Huang L, Zhang J, Wang F, Xu S et al. 2015. NADP(+)-IDH Mutations Promote Hypersuccinylation that Impairs Mitochondria Respiration and Induces Apoptosis Resistance. Mol Cell. 60(4):661–675. [DOI] [PubMed] [Google Scholar]
- Li F, Liu L. 2016. SIRT5 Deficiency Enhances Susceptibility to Kainate-Induced Seizures and Exacerbates Hippocampal Neurodegeneration not through Mitochondrial Antioxidant Enzyme SOD2. Front Cell Neurosci. 10:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Shi L, Yang S, Yan R, Zhang D, Yang J, He L, Li W, Yi X, Sun L et al. 2016. SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat Commun. 7:12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Huang W, You L, Xie T, He B. 2015. A FRET-based assay for screening SIRT5 specific modulators. Bioorg Med Chem Lett. 25(8):1671–1674. [DOI] [PubMed] [Google Scholar]
- Liang F, Wang X, Ow SH, Chen W, Ong WC. 2017. Sirtuin 5 is Anti-apoptotic and Anti-oxidative in Cultured SH-EP Neuroblastoma Cells. Neurotox Res. 31(1):63–76. [DOI] [PubMed] [Google Scholar]
- Liberti MV, Locasale JW. 2016. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci. 41(3):211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HW, Thompson JW, Morris KC, Perez-Pinzon MA. 2011. Signal transducers and activators of transcription: STATs-mediated mitochondrial neuroprotection. Antioxid Redox Signal. 14(10):1853–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin ZF, Xu HB, Wang JY, Lin Q, Ruan Z, Liu FB, Jin W, Huang HH, Chen X. 2013. SIRT5 desuccinylates and activates SOD1 to eliminate ROS. Biochem Biophys Res Commun. 441(1):191–195. [DOI] [PubMed] [Google Scholar]
- Liszt G, Ford E, Kurtev M, Guarente L. 2005. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J Biol Chem. 280(22):21313–21320. [DOI] [PubMed] [Google Scholar]
- Liu B, Che W, Zheng C, Liu W, Wen J, Fu H, Tang K, Zhang J, Xu Y. 2013. SIRT5: a safeguard against oxidative stress-induced apoptosis in cardiomyocytes. Cell Physiol Biochem. 32(4):1050–1059. [DOI] [PubMed] [Google Scholar]
- Liu J, Huang Y, Zheng W. 2016. A Selective Cyclic Peptidic Human SIRT5 Inhibitor. Molecules. 21(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Peritore C, Ginsberg J, Shih J, Arun S, Donmez G. 2015. Protective role of SIRT5 against motor deficit and dopaminergic degeneration in MPTP-induced mice model of Parkinson’s disease. Behav Brain Res. 281:215–221. [DOI] [PubMed] [Google Scholar]
- Liu S, Ji S, Yu ZJ, Wang HL, Cheng X, Li WJ, Jing L, Yu Y, Chen Q, Yang LL et al. 2017. Structure-based discovery of new selective small-molecule sirtuin 5 inhibitors. Chem Biol Drug Des. [DOI] [PubMed] [Google Scholar]
- Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A et al. 2007. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 27(24):8807–8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard DB, Dash BP, Kumar S. 2015. Acetyl-ed question in mitochondrial biology? EMBO J. 34(21):2597–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Zuo Y, Feng Y, Zhang M. 2014. SIRT5 facilitates cancer cell growth and drug resistance in non-small cell lung cancer. Tumour Biol. 35(11):10699–10705. [DOI] [PubMed] [Google Scholar]
- Lv XB, Liu L, Cheng C, Yu B, Xiong L, Hu K, Tang J, Zeng L, Sang Y. 2015. SUN2 exerts tumor suppressor functions by suppressing the Warburg effect in lung cancer. Sci Rep. 5:17940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen AS, Andersen C, Daoud M, Anderson KA, Laursen JS, Chakladar S, Huynh FK, Colaco AR, Backos DS, Fristrup P et al. 2016. Investigating the Sensitivity of NAD+-dependent Sirtuin Deacylation Activities to NADH. J Biol Chem. 291(13):7128–7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlknecht U, Ho AD, Letzel S, Voelter-Mahlknecht S. 2006. Assignment of the NAD-dependent deacetylase sirtuin 5 gene (SIRT5) to human chromosome band 6p23 by in situ hybridization. Cytogenet Genome Res. 112(3–4):208–212. [DOI] [PubMed] [Google Scholar]
- Mai A, Massa S, Lavu S, Pezzi R, Simeoni S, Ragno R, Mariotti FR, Chiani F, Camilloni G, Sinclair DA. 2005. Design, synthesis, and biological evaluation of sirtinol analogues as class III histone/protein deacetylase (Sirtuin) inhibitors. J Med Chem. 48(24):7789–7795. [DOI] [PubMed] [Google Scholar]
- Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, Seluanov A, Gorbunova V. 2011. SIRT6 promotes DNA repair under stress by activating PARP1. Science. 332(6036):1443–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcon E, Jain H, Bhattacharya A, Guo H, Phanse S, Pu S, Byram G, Collins BC, Dowdell E, Fenner M et al. 2015. Assessment of a method to characterize antibody selectivity and specificity for use in immunoprecipitation. Nat Methods. 12(8):725–731. [DOI] [PubMed] [Google Scholar]
- Mathias RA, Greco TM, Oberstein A, Budayeva HG, Chakrabarti R, Rowland EA, Kang Y, Shenk T, Cristea IM. 2014. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell. 159(7):1615–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita N, Yonashiro R, Ogata Y, Sugiura A, Nagashima S, Fukuda T, Inatome R, Yanagi S. 2011. Distinct regulation of mitochondrial localization and stability of two human Sirt5 isoforms. Genes Cells. 16(2):190–202. [DOI] [PubMed] [Google Scholar]
- Maurer B, Rumpf T, Scharfe M, Stolfa DA, Schmitt ML, He W, Verdin E, Sippl W, Jung M. 2012. Inhibitors of the NAD(+)-Dependent Protein Desuccinylase and Demalonylase Sirt5. ACS Med Chem Lett. 3(12):1050–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer AJ, Lamers WH, Chamuleau RA. 1990. Nitrogen metabolism and ornithine cycle function. Physiol Rev. 70(3):701–748. [DOI] [PubMed] [Google Scholar]
- Meyer JN, Leuthner TC, Luz AL. 2017. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology. 391:42–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL, Barrett JC et al. 2008. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 452(7186):492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. 2005. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. 16(10):4623–4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB et al. 2007. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 450(7170):712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris-Blanco KC, Cohan CH, Neumann JT, Sick TJ, Perez-Pinzon MA. 2014. Protein kinase C epsilon regulates mitochondrial pools of Nampt and NAD following resveratrol and ischemic preconditioning in the rat cortex. J Cereb Blood Flow Metab. 34(6):1024–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris-Blanco KC, Dave KR, Saul I, Koronowski KB, Stradecki HM, Perez-Pinzon MA. 2016. Protein Kinase C Epsilon Promotes Cerebral Ischemic Tolerance Via Modulation of Mitochondrial Sirt5. Sci Rep. 6:29790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM et al. 2006. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 124(2):315–329. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Lomb DJ, Haigis MC, Guarente L. 2009. SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell. 137(3):560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Ogura M, Ogura K, Tanaka D, Inagaki N. 2012. SIRT5 deacetylates and activates urate oxidase in liver mitochondria of mice. FEBS Lett. 586(23):4076–4081. [DOI] [PubMed] [Google Scholar]
- Nishida Y, Rardin MJ, Carrico C, He W, Sahu AK, Gut P, Najjar R, Fitch M, Hellerstein M, Gibson BW et al. 2015. SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Mol Cell. 59(2):321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North BJ, Verdin E. 2004. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 5(5):224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura M, Nakamura Y, Tanaka D, Zhuang X, Fujita Y, Obara A, Hamasaki A, Hosokawa M, Inagaki N. 2010. Overexpression of SIRT5 confirms its involvement in deacetylation and activation of carbamoyl phosphate synthetase 1. Biochem Biophys Res Commun. 393(1):73–78. [DOI] [PubMed] [Google Scholar]
- Onyango P, Celic I, McCaffery JM, Boeke JD, Feinberg AP. 2002. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc Natl Acad Sci U S A. 99(21):13653–13658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacella-Ince L, Zander-Fox DL, Lane M. 2014. Mitochondrial SIRT5 is present in follicular cells and is altered by reduced ovarian reserve and advanced maternal age. Reprod Fertil Dev. 26(8):1072–1083. [DOI] [PubMed] [Google Scholar]
- Paine PL, Moore LC, Horowitz SB. 1975. Nuclear envelope permeability. Nature. 254(5496):109–114. [DOI] [PubMed] [Google Scholar]
- Pannek M, Simic Z, Fuszard M, Meleshin M, Rotili D, Mai A, Schutkowski M, Steegborn C. 2017. Crystal structures of the mitochondrial deacylase Sirtuin 4 reveal isoform-specific acyl recognition and regulation features. Nat Commun. 8(1):1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, Xie Z, Zhang Y, Zwaans BM, Skinner ME et al. 2013. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell. 50(6):919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, Thompson CB. 2016. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 23(1):27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C, Lu Z, Xie Z, Cheng Z, Chen Y, Tan M, Luo H, Zhang Y, He W, Yang K et al. 2011. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol Cell Proteomics. 10(12):M111 012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. 2015. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 21(6):805–821. [DOI] [PubMed] [Google Scholar]
- Pitkanen A, Lukasiuk K. 2011. Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. 10(2):173–186. [DOI] [PubMed] [Google Scholar]
- Polletta L, Vernucci E, Carnevale I, Arcangeli T, Rotili D, Palmerio S, Steegborn C, Nowak T, Schutkowski M, Pellegrini L et al. 2015. SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy. 11(2):253–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V, Naini AB, Jakowec M, Petzinger G, Miller R, Akram M. 2001. The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): a technical review of its utility and safety. J Neurochem. 76(5):1265–1274. [DOI] [PubMed] [Google Scholar]
- Rajabi N, Auth M, Troelsen KR, Pannek M, Bhatt DP, Fontenas M, Hirschey MD, Steegborn C, Madsen AS, Olsen CA. 2017. Mechanism-Based Inhibitors of the Human Sirtuin 5 Deacylase: Structure-Activity Relationship, Biostructural, and Kinetic Insight. Angew Chem Int Ed Engl. 56(47):14836–14841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, Guo A, Gut P, Sahu AK, Li B et al. 2013. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 18(6):920–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauh D, Fischer F, Gertz M, Lakshminarasimhan M, Bergbrede T, Aladini F, Kambach C, Becker CF, Zerweck J, Schutkowski M et al. 2013. An acetylome peptide microarray reveals specificities and deacetylation substrates for all human sirtuin isoforms. Nat Commun. 4:2327. [DOI] [PubMed] [Google Scholar]
- Raval AP, Dave KR, DeFazio RA, Perez-Pinzon MA. 2007. epsilonPKC phosphorylates the mitochondrial K(+) (ATP) channel during induction of ischemic preconditioning in the rat hippocampus. Brain Res. 1184:345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler C, Nowak T, Pannek M, Gertz M, Nguyen GT, Scharfe M, Born I, Sippl W, Steegborn C, Schutkowski M. 2014. Chemical probing of the human sirtuin 5 active site reveals its substrate acyl specificity and peptide-based inhibitors. Angew Chem Int Ed Engl. 53(40):10728–10732. [DOI] [PubMed] [Google Scholar]
- Roessler C, Tuting C, Meleshin M, Steegborn C, Schutkowski M. 2015. A Novel Continuous Assay for the Deacylase Sirtuin 5 and Other Deacetylases. J Med Chem. 58(18):7217–7223. [DOI] [PubMed] [Google Scholar]
- Ryu D, Jo YS, Lo Sasso G, Stein S, Zhang H, Perino A, Lee JU, Zeviani M, Romand R, Hottiger MO et al. 2014. A SIRT7-dependent acetylation switch of GABPbeta1 controls mitochondrial function. Cell Metab. 20(5):856–869. [DOI] [PubMed] [Google Scholar]
- Sabari BR, Zhang D, Allis CD, Zhao Y. 2017. Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol. 18(2):90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadhukhan S, Liu X, Ryu D, Nelson OD, Stupinski JA, Li Z, Chen W, Zhang S, Weiss RS, Locasale JW et al. 2016. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc Natl Acad Sci U S A. 113(16):4320–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CF, Steegborn C. 2008. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J Mol Biol. 382(3):790–801. [DOI] [PubMed] [Google Scholar]
- Schuetz A, Min J, Antoshenko T, Wang CL, Allali-Hassani A, Dong A, Loppnau P, Vedadi M, Bochkarev A, Sternglanz R et al. 2007. Structural basis of inhibition of the human NAD+-dependent deacetylase SIRT5 by suramin. Structure. 15(3):377–389. [DOI] [PubMed] [Google Scholar]
- Schwer B, North BJ, Frye RA, Ott M, Verdin E. 2002. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol. 158(4):647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BC, Denu JM. 2007. Mechanism-based inhibition of Sir2 deacetylases by thioacetyl-lysine peptide. Biochemistry. 46(50):14478–14486. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Weindruch R. 1996. Oxidative stress, caloric restriction, and aging. Science. 273(5271):59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacpoole PW. 2017. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. J Natl Cancer Inst. 109(11). [DOI] [PubMed] [Google Scholar]
- Suenkel B, Fischer F, Steegborn C. 2013. Inhibition of the human deacylase Sirtuin 5 by the indole GW5074. Bioorg Med Chem Lett. 23(1):143–146. [DOI] [PubMed] [Google Scholar]
- Sun JY, Xu L, Tseng H, Ciccarelli B, Fulciniti M, Hunter ZR, Maghsoudi K, Hatjiharissi E, Zhou Y, Yang G et al. 2011. Histone deacetylase inhibitors demonstrate significant preclinical activity as single agents, and in combination with bortezomib in Waldenstrom’s macroglobulinemia. Clin Lymphoma Myeloma Leuk. 11(1):152–156. [DOI] [PubMed] [Google Scholar]
- Sundaresan NR, Bindu S, Pillai VB, Samant S, Pan Y, Huang JY, Gupta M, Nagalingam RS, Wolfgeher D, Verdin E et al. 2015. SIRT3 Blocks Aging-Associated Tissue Fibrosis in Mice by Deacetylating and Activating Glycogen Synthase Kinase 3beta. Mol Cell Biol. 36(5):678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita A, Naito K, Shinjo K, Sahara N, Matsui H, Ohnishi K, Beppu H, Ohtsubo K, Horii T, Maekawa M et al. 2004. Deletion 6p23 and add(11)(p15) leading to NUP98 translocation in a case of therapy-related atypical chronic myelocytic leukemia transforming to acute myelocytic leukemia. Cancer Genet Cytogenet. 152(1):56–60. [DOI] [PubMed] [Google Scholar]
- Tan M, Peng C, Anderson KA, Chhoy P, Xie Z, Dai L, Park J, Chen Y, Huang H, Zhang Y et al. 2014. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 19(4):605–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanno M, Sakamoto J, Miura T, Shimamoto K, Horio Y. 2007. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J Biol Chem. 282(9):6823–6832. [DOI] [PubMed] [Google Scholar]
- Tasselli L, Zheng W, Chua KF. 2017. SIRT6: Novel Mechanisms and Links to Aging and Disease. Trends Endocrinol Metab. 28(3):168–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng YB, Jing H, Aramsangtienchai P, He B, Khan S, Hu J, Lin H, Hao Q. 2015. Efficient demyristoylase activity of SIRT2 revealed by kinetic and structural studies. Sci Rep. 5:8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TenNapel MJ, Lynch CF, Burns TL, Wallace R, Smith BJ, Button A, Domann FE. 2014. SIRT6 minor allele genotype is associated with >5-year decrease in lifespan in an aged cohort. PLoS One. 9(12):e115616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Z, Wang M, Wang Y, Kim DD, Grenier JK, Cao J, Sadhukhan S, Hao Q, Lin H. 2017. SIRT7 Is an RNA-Activated Protein Lysine Deacylase. ACS Chem Biol. 12(1):300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp J, Jochum A, Meier R, Saunders L, Marshall B, Kunick C, Verdin E, Goekjian P, Sippl W, Jung M. 2006. Adenosine mimetics as inhibitors of NAD+-dependent histone deacetylases, from kinase to sirtuin inhibition. J Med Chem. 49(25):7307–7316. [DOI] [PubMed] [Google Scholar]
- Trapp J, Meier R, Hongwiset D, Kassack MU, Sippl W, Jung M. 2007. Structure-activity studies on suramin analogues as inhibitors of NAD+-dependent histone deacetylases (sirtuins). ChemMedChem. 2(10):1419–1431. [DOI] [PubMed] [Google Scholar]
- Van Meter M, Kashyap M, Rezazadeh S, Geneva AJ, Morello TD, Seluanov A, Gorbunova V. 2014. SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat Commun. 5:5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquero A, Scher MB, Lee DH, Sutton A, Cheng HL, Alt FW, Serrano L, Sternglanz R, Reinberg D. 2006. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 20(10):1256–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelter-Mahlknecht S, Mahlknecht U. 2013. Cloning, chromosomal characterization and FISH mapping of the NAD(+)-dependent histone deacetylase gene sirtuin 5 in the mouse. Int J Oncol. 43(1):237–245. [DOI] [PubMed] [Google Scholar]
- Wagner GR, Bhatt DP, O’Connell TM, Thompson JW, Dubois LG, Backos DS, Yang H, Mitchell GA, Ilkayeva OR, Stevens RD et al. 2017. A Class of Reactive Acyl-CoA Species Reveals the Non-enzymatic Origins of Protein Acylation. Cell Metab. 25(4):823–837 e828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GR, Hirschey MD. 2014. Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol Cell. 54(1):5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Wang K, Xu W, Zhao S, Ye D, Wang Y, Xu Y, Zhou L, Chu Y, Zhang C et al. 2017. SIRT5 Desuccinylates and Activates Pyruvate Kinase M2 to Block Macrophage IL-1beta Production and to Prevent DSS-Induced Colitis in Mice. Cell Rep. 19(11):2331–2344. [DOI] [PubMed] [Google Scholar]
- Wang P, Guan YF, Du H, Zhai QW, Su DF, Miao CY. 2012. Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy. 8(1):77–87. [DOI] [PubMed] [Google Scholar]
- Wang Q, Yu S, Simonyi A, Sun GY, Sun AY. 2005. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol Neurobiol. 31(1–3):3–16. [DOI] [PubMed] [Google Scholar]
- Wang WW, Zeng Y, Wu B, Deiters A, Liu WR. 2016. A Chemical Biology Approach to Reveal Sirt6-targeted Histone H3 Sites in Nucleosomes. ACS Chem Biol. 11(7):1973–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Guo YR, Liu K, Yin Z, Liu R, Xia Y, Tan L, Yang P, Lee JH, Li XJ et al. 2017. KAT2A coupled with the alpha-KGDH complex acts as a histone H3 succinyltransferase. Nature. 552(7684):273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhu Y, Xing S, Ma P, Lin D. 2015. SIRT5 prevents cigarette smoke extract-induced apoptosis in lung epithelial cells via deacetylation of FOXO3. Cell Stress Chaperones. 20(5):805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YQ, Wang HL, Xu J, Tan J, Fu LN, Wang JL, Zou TH, Sun DF, Gao QY, Chen YX et al. 2018. Sirtuin5 contributes to colorectal carcinogenesis by enhancing glutaminolysis in a deglutarylation-dependent manner. Nat Commun. 9(1):545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert BT, Moustafa T, Iesmantavicius V, Zechner R, Choudhary C. 2015. Analysis of acetylation stoichiometry suggests that SIRT3 repairs nonenzymatic acetylation lesions. EMBO J. 34(21):2620–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. 2009. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 324(5930):1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CJ, Yudin AK. 2011. Contemporary strategies for peptide macrocyclization. Nat Chem. 3(7):509–524. [DOI] [PubMed] [Google Scholar]
- Williams AC, Ramsden DB. 2005. Autotoxicity, methylation and a road to the prevention of Parkinson’s disease. J Clin Neurosci. 12(1):6–11. [DOI] [PubMed] [Google Scholar]
- Xiangyun Y, Xiaomin N, Linping G, Yunhua X, Ziming L, Yongfeng Y, Zhiwei C, Shun L. 2017. Desuccinylation of pyruvate kinase M2 by SIRT5 contributes to antioxidant response and tumor growth. Oncotarget. 8(4):6984–6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT et al. 2011. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 19(1):17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YS, Liang JJ, Wang Y, Zhao XJ, Xu L, Xu YY, Zou QC, Zhang JM, Tu CE, Cui YG et al. 2016. STAT3 Undergoes Acetylation-dependent Mitochondrial Translocation to Regulate Pyruvate Metabolism. Sci Rep. 6:39517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK, Lu Z. 2012. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 150(4):685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Wang Z, Li X, Liu B, Liu M, Liu L, Chen S, Ren M, Wang Y, Yu M et al. 2017. SHMT2 desuccinylation by SIRT5 drives cancer cell proliferation. Cancer Res. [DOI] [PubMed] [Google Scholar]
- Yu J, Sadhukhan S, Noriega LG, Moullan N, He B, Weiss RS, Lin H, Schoonjans K, Auwerx J. 2013. Metabolic characterization of a Sirt5 deficient mouse model. Sci Rep. 3:2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang W, Hao Y, Wang Z, Zheng W. 2015. Novel thiourea-based sirtuin inhibitory warheads. Bioorg Med Chem Lett. 25(16):3319–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J, Li D. 2004. Expression and purification of his-tagged rat peroxisomal acyl-CoA oxidase I wild-type and E421 mutant proteins. Protein Expr Purif. 38(1):153–160. [DOI] [PubMed] [Google Scholar]
- Zhan L, Huang C, Meng XM, Song Y, Wu XQ, Miu CG, Zhan XS, Li J. 2014. Promising roles of mammalian E2Fs in hepatocellular carcinoma. Cell Signal. 26(5):1075–1081. [DOI] [PubMed] [Google Scholar]
- Zhang W, Xie Y, Wang T, Bi J, Li H, Zhang LQ, Ye SQ, Ding S. 2010. Neuronal protective role of PBEF in a mouse model of cerebral ischemia. J Cereb Blood Flow Metab. 30(12):1962–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Bharathi SS, Rardin MJ, Lu J, Maringer KV, Sims-Lucas S, Prochownik EV, Gibson BW, Goetzman ES. 2017. Lysine desuccinylase SIRT5 binds to cardiolipin and regulates the electron transport chain. J Biol Chem. 292(24):10239–10249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Bharathi SS, Rardin MJ, Uppala R, Verdin E, Gibson BW, Goetzman ES. 2015. SIRT3 and SIRT5 regulate the enzyme activity and cardiolipin binding of very long-chain acyl-CoA dehydrogenase. PLoS One. 10(3):e0122297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, Mu X, You Q. 2017. Succinate: An initiator in tumorigenesis and progression. Oncotarget. 8(32):53819–53828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Wang F, Sun R, Chen X, Zhang M, Xu Q, Wang Y, Wang S, Xiong Y, Guan KL et al. 2016. SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. EMBO Rep. 17(6):811–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu WZ, Wu XF, Zhang Y, Zhou ZN. 2012. Proteomic analysis of mitochondrial proteins in cardiomyocytes from rats subjected to intermittent hypoxia. Eur J Appl Physiol. 112(3):1037–1046. [DOI] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Sollott SJ. 2014. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 94(3):909–950. [DOI] [PMC free article] [PubMed] [Google Scholar]