

Abstract
In many biomolecular interactions, changes in the assembly states and structural conformations of participants can act as a complementary reporter of binding to functional and thermodynamic assays. This structural information is captured by a number of structural biology and biophysical techniques that are viable either as primary screens in small-scale applications or as secondary screens to complement higher throughput methods. In particular, small-angle X-ray scattering (SAXS) reports the average distance distribution between all atoms after orientational averaging. Such information is important when for example investigating conformational changes involved in inhibitory and regulatory mechanisms where binding events do not necessarily cause functional changes. Thus, we summarise here the current and prospective capabilities of SAXS-based screening in the context of other methods that yield structural information. Broad guidelines are also provided to assist readers in preparing screening protocols that are tailored to available X-ray sources.
Keywords: Small-angle scattering, Screening, Structural biology
Introduction
The identification of an optimal ligand-receptor combination by screening can be performed by a broad array of methods, in which one searches the chemical space of biomolecular interactions to discover candidates for further investigation. Screening occurs most frequently in pharmacological lead discovery (Ciulli 2013; Renaud et al. 2016) where a macromolecule relevant to disease is assayed with an existing lead or fragment library containing between molecules, seeking candidates that may best be modified to interfere with the receptor’s pathogenic function while maintaining clinical viability (Payne et al. 2007; Arkin et al. 2014; Erlanson et al. 2016). Applications in fundamental research also exist, such as in proteomics to screen for potential binding partners of a newly isolated protein, cellular targeting, or in nucleic acid interactions. Since the total chemical space is astronomical relative to existing capability, it is necessary to optimise both throughput and library content. Here, literature applies the term high-throughput screening to methodological advances that significantly improve performance via parallelisation, detection speed, and other modifications.
The drawbacks of high-throughput variants often includes a concomitant sacrifice in measurement precision or total information obtained. Thus, secondary screening techniques are included in screening strategies to provide verification of initial leads and further elucidate their observed interactions. For example, in drug discovery contexts a highly parallelisable thermal-shift assay may be paired with dual polarization interferometry to probe both binding kinetics as well as potential structural changes (Grøftehauge et al. 2015). Whereas when investigating protein–nucleic-acid interactions, the initial sequence patterns from an RNA-binding assay may be further filtered by small-angle X-ray scattering (SAXS) to optimise for stoichiometries ideal for structural characterisation (Chen et al. 2018). The judicious use of complementary approaches during screening increases the quality of preliminary information available for decision making.
Since a majority of high-throughput assays work via the detection of thermodynamics or functional effects exerted by bound ligands, secondary screening methods should provide a source of independent information. Here, various structural biology and biophysical methods can yield the requisite data on the nature of conformational or configurational changes triggered by binding, if any (Fig. 1). Notably, their current role as secondary screens is usually dictated by increased sample requirements or protocol complexity relative to other alternatives. The type and amount of structural information depends on the experiment: For example, X-ray crystallography and nuclear magnetic resonance (NMR) methods provide rich, atomistic details on ligand placement and contacts necessary to conduct rational drug design. In contrast, fluorescence labelling and surface plasmon resonance (SPR) provide rapid, coarse structural characterisation ideal for larger-scale applications. Thus, a high-level understanding of respective techniques will assist in choosing an appropriate method for the biological system under investigation (Table 1).
Fig. 1.
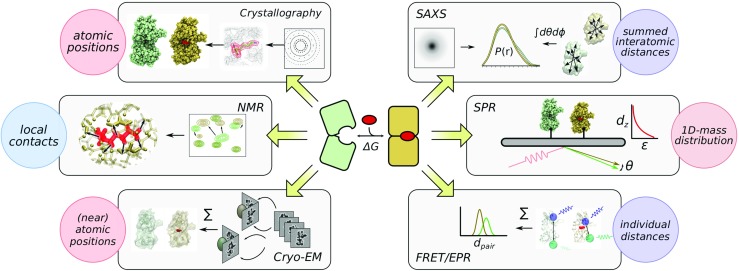
A simplified schematic depicting structural biology and biophysical techniques that can be deployed in screening applications. Figure prepared with Inkscape-0.92, VMD-1.9.3, and Gimp-2.8
Table 1.
Overview of structure-based screening techniques with two thermodynamics assays for comparison
| Name | Information attained | Advantages | Technique-specific requirements |
|---|---|---|---|
| X-ray crystallography | 3D spatial electron density | Extensive information useful for | Reliable crystallisation conditions with atomic |
| further refinement | resolution | ||
| Cryo-EM | 3D spatial electron density | As crystallography without requiring | Requires extensive, target-specific protocol |
| crystallisation conditions | optimisation | ||
| SAXS | 1D pair-distance | Solution measurement in native | Purifiable to monodisperse or oligodisperse |
| distribution of electron density | environment, applicable to | conditions, sufficient conformational changes | |
| protein disorder | |||
| receptor-based | local contacts | Extensive information useful for | Sufficiently small size and M solution |
| NMR | further refinement | concentrations of labelled species | |
| ligand-based | limited local contacts | parallelisable measurements for | Fast binding and unbinding kinetics, library |
| NMR | fragment libraries | optimisation to avoid artefacts from non-specific | |
| interactions and aggregation | |||
| SPR | limited 1D mass distribution | fast measurement and sample | Functionally-neutral tethering site or reliable |
| reuse | adsorption mechanism | ||
| Fluorescence labelling | pair-distance distribution of | parallelisable measurement and | Functionally-neutral labelling site |
| two sites | potential for in-vivo assays | ||
| Isothermal | thermodynamics | High accuracy thermodynamics | M solution concentrations of receptor and ligand |
| calorimetry | measurement with relative stoichiometry | ||
| data | |||
| Thermal-shift | thermodynamics | Simple experiment with massive | none |
| assay | parallelisability |
This review aims to report the increasing and potential usage of solution X-ray scattering to conduct small-scale screening experiments, and provide arguments as to its particular role in the gamut of structural-based techniques. As a dedicated solution technique (Putnam et al. 2007; Hura et al. 2009; Grant et al. 2011; Svergun et al. 2013), SAXS is capable of characterising solution stoichiometries (Ando et al. 2012; Tuukkanen and Svergun 2014; Cordeiro et al. 2017; Chen et al. 2018), complex configuration (Yang et al. 2010; Vestergaard and Sayers 2014) and even transient structural changes (Cammarata et al. 2008; Cardia et al. 2008; Kantrowitz 2012; Neutze and Moffat 2012). Current synchrotron setups offer source intensities that permit precise measurements sufficient to conduct investigation into disordered protein conformations (Tompa 2012; Kikhney and Svergun 2015; Cordeiro et al. 2017) as well as weak interactions (Tuukkanen and Svergun 2014) relevant to current drug targets. These capabilities provide a unique niche for SAXS-based screening. Below, we provide executive summaries of several current and potential alternatives to further contextualise the application of scattering techniques. An overview of scattering theory and practice follows, leading into general advice for tailoring screening protocols to the broad range of X-ray sources.
Summaries of four structural biology and biophysical methods
Nuclear magnetic resonance
NMR spectroscopy is an established approach within drug discovery (van Dongen et al. 2002; Meyer and Peters 2003; Harner et al. 2013; Dias and Ciulli 2014; Erlanson et al. 2016) and molecular interactions (Williamson 2013; Collins et al. 2015; Liu et al. 2016) due to its provision of detailed, spatial information on binding interfaces and in some cases ligand orientations (Skjærven et al. 2013). Here, the seminal publication by (Shuker et al. 1996) demonstrated the characterisation of structure-activity relationships based on observed chemical-shift perturbations (Williamson 2013) on the receptor as caused by bound ligand. A range of variant procedures have since been developed based on ligand signals to circumvent size-limitations on NMR signal quality, while losing the ability of directly map the binding site. These include saturation transfer difference (Mayer and Meyer 1999; Wagstaff et al. 2013, STD,), WaterLOGSY (Dalvit et al. 2000; Dalvit et al. 2001; Huang et al. 2017), and INPHARMA (Sánchez-Pedregal et al. 2005; Skjærven et al. 2013), all of which function by the transfer of magnetization from an arbitrary-sized receptor to the smaller ligand. Thus, evidence of binding is demonstrated by presence of magnetization on ligands in the mixture. Alternative formulations based on enhanced relaxation also exist in the form of target-immobilised NMR screening (Vanwetswinkel et al. 2005) and paramagnetic labelling schemes (Jahnke 2002).
Although a range of NMR methods are available, applicability can be restricted due to inherit size limitations of this method (Felli and Brutscher 2009; Lee et al. 2014). Receptor-based direct mapping of binding interfaces using chemical shift perturbations are only feasible with signals-to-noise ratios, which still allow for detection of signals after addition of ligands. This method is routinely applicable for systems up to 40 kDa of size provided they have globular shape and sufficient solubility and stability. In contrast, ligand-based experiments can be measured irrespective of receptor size (Cala et al. 2014). However, binding and unbinding kinetics must be faster than experiment timescales for saturation transfer techniques to work, making this method amenable for detection of ligands binding in a range between and M. Both categories see use in fragment-based drug discovery (Tsao et al. 2006; Friberg et al. 2013; Murray et al. 2010; Prati et al. 2015), and analogous techniques can also be applied to investigate protein-protein interactions (Barile and Pellecchia 2014).
X-ray crystallography
X-ray crystallography offers the possibility of directly yielding the atomic coordinates of bound ligands within the receptor active site, ideal for downstream rational and in-silico drug design. This makes the technique very attractive for lead discovery as long as the target can be reliably crystallised at sufficient resolution both with and without ligand, which may be challenging due to potential conformational changes (Hassell et al. 2007). Where that condition is satisfied, parallised protocols with ligand-cocktail mixtures permit the techniques to be deployed as a primary screen (Davies and Tickle 2011; Patel et al. 2014). The excess saturation conditions and additional stabilisation of bound complex by crystal contacts also make possible the detection of leads with very weak affinities, which may be difficult to discover using other methods (Schiebel et al. 2016).
On the other hand, the lengthy process involved in establishing crystallization conditions for novel proteins imposes a significant limitation on applicability of crystallographic methods, if these have not been found in previous available studies. This biases their application towards known and well-studied systems (Zheng et al. 2015), particularly in membrane-bound scenarios such as G-protein coupled receptors (Ranganathan et al. 2017). Given that transporters and membrane-bound receptors are critical to major cellular processes and thus highly attractive targets, crystallographic screening remains an important source of structural information. While solubilisation in membrane-mimics such as nanodiscs (Ritchie et al. 2009; Bayburt and Sligar 2010) can bring other techniques into play (Berthaud et al. 2012; Skar-Gislinge et al. 2015; Puthenveetil et al. 2017, e.g. NMR and SAXS,), the intensive process involved has so far preferenced its utilisation in ligand screening towards receptor-immobilised platforms (Das et al. 2009; Congreve et al. 2011; Wilcox et al. 2015).
Cryo-electron microscopy
The recent advent and increasing feasibility of cryo-EM maps at near-atomic and atomic resolutions have inspired discussions on their potential drug discovery applications (Merk et al. 2016; Renaud et al. 2018). The breakthroughs in both resolution required for direct atomic placement (< 3Å) and minimum resolvable particle sizes are regarded as key advances for the technique to become viable as a means to directly visualise bound complexes, particularly for systems that are not known to crystallise. Similar to crystallography, the discovery of such 3D-structure will create starting points for rational drug design not hitherto possible. Although the current throughput is not yet viable for high-throughput screening, significant efforts are being carried out on automation and sample optimisation similar to that once required to establish crystallographic screening protocols.
Surface plasmon resonance
Another common biophysical approach to fragment-based screening is SPR spectroscopy (Navratilova and Hopkins 2010; Chavanieu and Pugnière 2016; Zeng et al. 2017), where structural and mass changes of biomolecules tethered closely to a metallic substrate alters the local refractive index and thus the total internal reflection behaviour of light beams on the opposing side. Light microscopy for detection brings advantages of millisecond time resolution and thus an ability to directly observe binding kinetics (Salamon et al. 1997b; Daghestani and Day 2010). The fixation process enables sample reuse across multiple titrations, which is a key advantage in large-scale applications (Piliarik et al. 2005; Maynard et al. 2009). On the other hand, a suitable anchoring site on the receptor must be found that does not perturb its native function or stability significantly. Notably, membrane-embedded targets can either be anchored by tethering the entire membrane above the substrate (Salamon et al. 1997a; Patching 2014) or attaching nanodisc-solubilised proteins to functionalised surfaces (Das et al. 2009; Congreve et al. 2011). In all, SPR seems to be a persuasive option for intensive screening scenarios where the detection of structural change itself is sufficient for the purposes of candidate selection.
Small-angle X-ray scattering
Basic primer
With the major structure-based and biophysical screening methods introduced above, we devote the remaining space to critical niches that small-angle scattering fulfils. For a thorough coverage of basic principles of scattering, we recommend other excellent reviews both on the theory (Putnam et al. 2007; Rambo and Tainer 2013; Kikhney and Svergun 2015) and practical aspects (Skou et al. 2014; Gabel 2015; Trewhella 2016; Bernadó et al. 2017) of X-ray (SAXS) and neutron (SANS) scattering. A minimal introduction of scattering is given here to establish an elementary understanding of the information obtained via scattering experiments.
The exposure of a biomolecular solution to an incoming, collimated source results in the elastic scattering of photons by an angle (Fig. 2a) from all solute and solvent atoms (among other phenomena), with an intensity that decays rapidly as a function of increasing angle. The momentum transfer is generally adopted to remove dependence of this scattering on wavelength in the final analysis. To obtain the contribution of solvated macromolecules, scattering intensities of the sample solution and an equivalent buffer solution containing all components except the molecule of interest are measured then subtracted. The difference pattern due to the macromolecule alone represents the intra- and intermolecular interference patterns summed over all solutions orientations and conformations. Under dilute concentrations where intermolecular effects are insignificant, an obtained by SAXS/SANS is directly proportional to sample concentration and contains information on the conformational average distances between scattering atoms.
Fig. 2.

A proposed method to conduct SAXS-based screening (see main text for details). Prepared mixtures between receptors and ligands are submitted to SAXS measurements at beamlines with automated liquid handling capabilities (a). Scattering profiles of individual components and respective buffers are used to fit the mixture scattering profiles (b), where the goodness-of-fit χlin is taken as a proxy for complex formation (c) and subsequent affinity calculations. Direct comparison metrics between scattering profiles at equal ratios can be used to visualise ligand-dependence (d), as well as compute a titration similarity metric for classification of binding responses (e). Figure prepared with Inkscape-0.92, Xmgrace-5.1.23, and matplotlib-2.2.2. Permission to use image of the Arinax sample-handling robot kindly provided by Ralf Siebrecht
The key distance information provided by small-angle scattering methods in dilute conditions can be summarised in form of a weighted pair-distance distributions between all scattering centers (Putnam et al. 2007, §2.3.3). is related to by indirect Fourier transform, with the weights determined by the product of the net scattering contrast at each scattering site, evaluated as the difference between solute and buffer scattering densities. For X-ray sources, scattering is determined by the electron densities and thus contributions to is dominated by non-hydrogen nuclei. Whereas, neutron scattering yields an analogous that is instead dominated by the location and ratio of protons versus almost all other nuclei, due to the proton’s negative scattering cross-section. Both SAXS and SANS patterns can in principle be precisely measured, which yield sufficient accuracy to detect even small perturbations to the conformational ensemble (Rambo and Tainer 2013; Kikhney and Svergun 2015).
Applications in structural biology
It is this sensitivity to global distances that makes SAXS and SANS valuable contributors to integrative structural biology. The missing high-resolution information from scattering can be readily furnished via other sources including crystallography (Putnam et al. 2007; Grant et al. 2011) and NMR (Grishaev et al. 2005; Madl et al. 2011; Carlomagno 2014; Hennig and Sattler 2014; Rossi et al. 2015). Such combination enables the isolation of complex structures where SAXS can be used as a guide in rigid-body docking (Petoukhov and Svergun 2005; Jiménez-García et al. 2015; Xia et al. 2015; Schneidman-Duhovny et al. 2016; Schindler et al. 2016), elastic perturbations (Gorba et al. 2008; Zheng and Tekpinar 2011), atomistic simulations (Björling et al. 2015; Chen and Hub 2015a; Kimanius et al. 2015), or trivially as post hoc removal of incompatible structural models (Karaca and Bonvin 2013; Hennig et al. 2013). Moreover, mixtures of monodisperse species can be decomposed (Schneidman-Duhovny et al. 2016; Bernadó et al. 2007) when either the relative populations or the component scattering are already known. Here the decomposition of a five-state ensemble between the trimeric Proliferating Cell Nuclear Antigen plus its disordered binding partner p15PAF (Cordeiro et al. 2017), is a prime example where structural information, population states, and binding affinity can be extracted from a complex oligodisperse system. Additionally, SAXS is often applied to characterise the extent of molecular flexibility in systems ranging from folded proteins (Makowski et al. 2011), linker-connected domains (Yang et al. 2010; RóŻycki et al. 2011; Receveur-Bréchot and Durand 2012), to complete disorder (Bernadó and Svergun 2011; Rambo and Tainer 2011; Kikhney and Svergun 2015). Amongst the vast existing structural biology applications of SAXS, we believe that many such cases could potentially be adapted in screening studies. A selection is presented in Table 2 to illustrate the types of structural information that scattering can provide in the context of SAXS-based titrations.
Table 2.
Examples of SAXS-based titrations or screening
| System | Source | Type | Description and notes |
|---|---|---|---|
| Protein conformational change | |||
| Leucine/Isoleucine/Valine | Olah et al. (1993) | titration | contraction of bi-lobe domain upon ligand binding. |
| binding protein | Early example using lab-based source. | ||
| Transient photoexcitation | Cammarata et al. (2008) | time-resolved SAXS | laser-triggered detachment of carbon monoxide |
| of haemoglobin | enables solution characterisation of the relaxed | ||
| state(s). | |||
| Aspartate transcarbamoylase | Cardia et al. (2008), | time-resolved SAXS | self-regulation of enzyme by substrates and |
| Kantrowitz (2012) | regulators | ||
| Protein domain assembly | |||
| B12-dependent | Ando et al. (2012) | titration | association of two domains in a mixture of 1:1 and |
| methyltransferase | 2:1 stoichiometry | ||
| Ribonucleotide reductase | Ando et al. (2016) | titration | subunit in dimer-hexamer equilibrium, and |
| subunit dimer | |||
| Antibody self-association | Fukuda et al. (2017); Tian et al. (2014) | screening | technical studies on antibody stability as a function |
| of concentration, pH, buffers and viscosity, | |||
| important in clinical applications | |||
| Linker-connected domain configurations | |||
| Src kinase | Jamros et al. (2010); Yang et al. (2010) | two-point measurement | Assembly state of regulatory domains in the |
| presence of SH3 signal peptide | |||
| Sxl-lethal RNA-binding | Chen et al. (2018) | titration | Equilibrium between apo-protein, 1:1 protein- |
| protein | RNA complex, and 2:2 protein-RNA complex. | ||
| Disordered protein interactions | |||
| Proliferating Cell | Cordeiro et al. (2017) | titration and population | association of trimer-of-dimer PCNA with |
| Nuclear Antigen, p15PAF | modelling | disordered p15 in 3:0–3:3 stoichiometries | |
| α-synuclein | Herranz-Trillo et al. (2017) | time-resolved SAXS and | decomposition of species during amyloid fibril |
| population modelling | genesis | ||
| Micellular compositions | |||
| various detergents used in | Lipfert et al. (2007), | titration | Shift in micellular size distributions and |
| membrane protein solubilisation | Oliver et al. (2013) | associated shape. | |
| Nucleic-acid conformations | |||
| dT30 with MgCl2 | Meisburger et al. (2013) | titration | Alteration of polymer properties due to |
| coordinating ions. | |||
In contrast to SAXS, SANS sees particular uses in situations involving more than two binding partners where the scattering difference between hydrogen and deuterium can be leveraged in contrast variation (Jacrot 1976; Heller 2010; Gabel 2015) studies, initially used to estimate the composition and spatial distribution of large multi-component proteins, nucleoproteins, and viruses by varying H2O/D2O ratios (Cusack et al. 1985; Mangel et al. 1990; Svergun et al. 1994). Further distinct measurements can be conducted by selective deuteration, achievable at the sub-component level and more recently the level of individual domains in a multi-domain protein via segmental labelling (Freiburger et al. 2015; Sonntag et al. 2017). The acquisition of such extensive scattering datasets together with NMR restraints (Madl et al. 2011; Carlomagno 2014; Hennig and Sattler 2014) provided a critical breakthrough in efforts to characterise flexible protein–nucleic acid assemblies (Lapinaite et al. 2013; Hennig et al. 2013; Hennig et al. 2014). While the potential to provide extensive characterisation makes neutron scattering an attractive cousin to X-rays, the lower intensity of neutron sources greatly limit the throughput of SANS measurements. Until significant developments can be made on the level of detection and workflow optimisation such as on microfluidic lab-chip devices (Lopez et al. 2015; Adamo et al. 2017; Pham et al. 2017)), we expect SAXS to remain the primary avenue for high-throughput scattering.
SAXS-based screening
To summarise the advantages of SAXS-based screening, we briefly describe several examples in the literature starting with our recent work on the binding profile of Sex-lethal protein (Sxl) and followed by other applications and methodologies.
Classification of protein-ligand interactions
As a proof-of-principle for SAXS-based screening of protein-ligand interactions, the selectivity profile of Sxl was investigated in a screen of thirty-five oligonucleotides assuming existing biochemical knowledge of its preference versus polyuridine with guanidine substitutions (Chen et al. 2018). The overall procedure is illustrated in Fig. 2. Initial selection of candidates are based on sparse sequence space coverage, scanning bipartite and tripartite uridine/cytidine compositions, oligonucleotide length, and guanidine-substitution position (not shown). Twelve eight-point titrations per 96-well plate are conducted to maintain compatibility with existing beamline automation robots, with additional measurements of nucleotide scattering to enable subtraction of unbound ligand contributions.
At concentrations where interparticle scattering are insignificant, the measured SAXS profile of a mixture between unbound proteins, ligands, and their complexes can be expressed by a linear sum of components. Figure 2b represents the general case where ligand scattering significantly deviates from buffer scattering and the respective receptor and ligand buffers differ. We note that this is reducible to standard buffer subtraction procedures when ligand scattering is not differentiable from buffer scattering and buffer conditions are shared between protein and ligand. By fitting the scattering profiles of individual components to the final mixture scattering profile, we derive a residual quantity that represents departure from the linear sum (Fig. 2c):
| 1 |
χlin is linearly proportional to the amount of formed complex and magnitude of structural perturbations. Under conditions that permit ligand-saturation, the bound complex curve can be unambiguously derived and associated with a saturated . The effective affinity can be computed from the titration curve. In the current scenario where RNA-excess conditions trigger sample oligomerisation, was computed instead by assuming a shared binding mechanism across all RNA-titrations. This enables simultaneous fitting across multiple titrations to derive respective affinities and shared saturated .
The buffer-subtracted curves can also be directly compared using a number of curve similarity metrics (Fig. 2d, c.f. Hura et al. 2013; Franke et al. 2015). Hierarchical clustering can be further performed on the metrics that preserving linear distance such as symmetrised pairwise-χ to more clearly summarise binding mechanisms. When aggregated across all titration points, the overall “distance” between titrations of two ligand a and b is useful for classifying binding interactions without assuming any structural knowledge:
| 2 |
| 3 |
When clustering was performed on the Sxl-RNA interactions screen, we uncovered a range of solution stoichiometries consisting of 1:1, 2:2, and mixtures thereof, corresponding to low and high apparent resulting from combined analysis. Results were further confirmed by EOM modelling using the published crystallographic conformation (Handa et al. 1999), Notably, this stoichiometry switching was not evident in accompanying isothermal calorimetry and NMR measurements. Thus SAXScreen provides useful structural information on optimising species purity for downstream structural biology work, and we expect it to be useful in other applications as a complement to thermodynamics-based assays.
Screening for concentration and environmental effects
One direct application of screening is to investigate in detail the dependence of the structural ensemble upon environmental conditions such as temperature, buffer composition and pH tolerance. Several studies included in Table 2 have conducted small-scale screens in this manner such as Tian et al. (2014) and Fukuda et al. (2017), who examine optimal conditions for the formulation and delivery of antibody therapeutics. The particular sensitivity of SAXS towards inter-particle scattering is leveraged to identify conditions that avoid unwanted oligomerisation and associated viscosity. Here, the sample requirements for scattering measurements is significantly smaller than that required for then-current rheological methods. Similarly, labs such as Lipfert et al. (2007) and Oliver et al. (2013) leverage SAXS to probe the solution morphology of detergent micelles commonly used in membrane protein solubilisation. Although individual micelles might be highly diverse in shape, essential parameters can be captured in ellipsoidal models that describe the average size, shape, and approximate packing as a function of total concentration. Numerous other applications such as protein disorder, protein-detergent complexation, and solvation layer densities showcase the ability of scattering methods to detect small magnitude perturbations as long as they are sufficient to influence the entire distance distribution (Møller et al. 2013; Cordeiro et al. 2017; Kim and Gabel 2015; Koutsioubas et al. 2013; Chen and Hub 2015b; Henriques et al. 2018). These perturbations can in principle also be screened to study environmental influences on molecular structure.
Microfluidic chip screening platform
Although our own work utilises broadly available 96-well plates setups for screening, it is well worth mentioning the ongoing improvements in microfluidic chip platforms as a means to conduct screening. For purposes of illustration, we restrict ourselves here to two sister reports by Teychené lab in the understanding that multiple labs have also contributed towards this sub-field (Lafleur et al. 2011; Møller et al. 2013; Watkin et al. 2017; Schwemmer et al. 2016; Pham et al. 2017), with alternative on-chip mixing schemes (Lee et al. 2011; You et al. 2015).Pham et al. (2017) reports the fabrication and initial testing of a four-channel mixing device intended for screening crystallisation conditions, whereas Rodríguez-Ruiz et al. (2017) reports its immediate implications for on-chip structural characterisation. The authors’ implementation of chip-based SAXS screening relies upon the creation of stable water-in-oil droplets by joining three aqueous channels into a primary channel containing immiscible fluorous oil. Precise regulation of relative rates between the aqueous channels determine final concentrations in the droplet, and thus enabling coverage of protein concentration and crystallisation conditions, or equivalently titration points in a protein-ligand-buffer setup. Droplet size is itself regulated by relative oil-water velocities and synchronised with the X-ray beam to minimise damage and interface scattering artefacts. This results in the measurement of one to three SAXS measurements per droplet, each equivalent to individual exposure frames of a capillary-based setup. The authors use lysozyme to demonstrate the dependence of inter-particle interactions on salt concentration, as well as a standard concentration series.
The chief advantages of microfluidic platforms include sample-efficiency, consistency, and immediate measurement. For a theoretical application to ligand screening, the estimated consumption of 2 μl per titration point1 is an order of magnitude smaller than minimum volumes in the well-plate setup. On-chip generation of droplets followed by UV-characterisation prevents stochastic sample handling errors that can occur while preparing plates. The main challenges in adapting microfluidics for screening consists of a higher sample concentration required to produce an equivalent angular q-resolution and throughput, and rapid exchange of ligand identity between titrations.
10–100 nm ultrastructural organisation
It should also be noted that the applicable length-scales of SAXS reaches above the single molecule interactions covered here: a simple adjustment of sample-detector distances enables investigations of cellular structures, meso-scale phenomena, and beyond. We highlight here the application of SAXS-based screening by von Gundlach et al. (2016) towards tracking the effect of antibiotics on ultrastructural changes in E. coli cells, by measuring scattering between nm− 1, corresponding to length scales of nm.
The authors demonstrate that alterations in physical characteristics such as membrane integrity and nucleoid distribution can be indirectly detected and used to classify mechanisms of action by novel agents, showing by example the unique scattering changes of polymerase inhibitors and membrane disruptors versus protein synthesis inhibitors. The principle-component analysis used here is another valid ab initio method that does not invoke structural assumptions, and can be utilised alongside hierarchical clustering to distinguish between ligand responses. The observed differences in SAXS were also shown to correlate with visual changes of TEM images. Independently, the authors also make efforts at mapping the time-dependence of structural alterations with decreasing cell viability. This work exemplifies the potential for SAXS to offer real-time study of cellular and sub-cellular ultrastructure.
General considerations for screening
Due to the relative uniqueness of synchrotron platforms in terms of beamline characteristics, sample automation and other aspects, it is not feasible to include in this review a comprehensive formulation of a SAXS-based screening protocol. These hardware and software characteristics influence the optimal sample volumes, concentrations and expected throughput. As such, we recommend initial contact with beamline staff and research labs for assistance in tailoring experimental proposals to the specific advantages of respective beamlines. A number of notable items will be discussed below to cover broadly applicable aspects.
Available screening platforms
In terms of screening capabilities, most synchrotron beamlines around the world possess automated sample measurement and analysis capabilities. The authors have recently tested the BioSAXS sample changer (Round et al. 2015) that is currently implemented at ESRF and PETRA-III, which is also available at Diamond Light Source. These utilise 96-well plates as the means of sample preparation followed by robotic loading and measurements. We include a representative list of equivalent automation setups below for other beamlines: SIBYLS (Classen et al. 2013), SOLEIL (David and Pérez 2009), Stanford SRL (Martel et al. 2012), Australian Synchrotron (Kirby et al. 2013), CHESS (Acerbo et al. 2015), and the Shanghai SRF (Li et al. 2016). At the time of writing, the biological SAXS beamlines at MAX-IV Sweden, SESAME, and Taiwan Photon Source remain in various stages of preparation, for which we also expect feasibility of screening applications.
Although the above list suggests that screening can be carried out at numerous beamlines, there currently exists no detailed performance comparisons specific to biomolecular titrations. We note here that lab-based sources may serve as a readily available and more reproducible platform to conduct screening. While the lower intensities extend required measurement times by 102 103-fold and thus impose relatively strict limits on protein stability, measurement protocols are in principle identical to synchrotron sources.
Sample consumption
For screens conducted using small-angle X-ray scattering, a balance must be struck between information gained versus total throughput. The precision and total information gained from a single titration is proportional to sample concentration and volume, number of titration points, and measurement time. Since synchrotron sites currently recommend protein sample parameters of volumes 20l and concentrations of 0.210.0 mg mL− 1, a single ten-point titration theoretically consumes 100 μg of materials. The destructive nature of measurement implies that sample consumption scales directly with the size of the screen—thus, for large-scale efforts and low-yield targets, it may be more cost-effective to adopt alternatives that re-use valuable binding partners.
Further improvements in these consumption rates are largely contingent upon hardware upgrades. Minimum sample volumes for standard capillary-flow measurements are formulated based on sample cell dimensions as dictated by the incoming beam geometry. For example, the beam geometry provided by G1 at MacCHESS (Acerbo et al. 2015) consists of a m cross section with an optimised path length of 2 mm, resulting in an instantaneous exposure volume of 0.125 μl. The sample is flowed through this volume to minimise radiation damage during measurement, resulting in a final recommendation of 15 or 30 μl. Similarly, the geometry of P12 at PETRA (120 × 200 × 1700μm) results in a recommendation of 20 μl. Sample-handling robotics may also implement dead volumes to prevent collisions, which further increase the practical minimum volume. In these contexts, a broader adoption of microfluidic platforms as discussed above is likely to significantly improve future sample utilisation by an order of magnitude.
Throughput
Our current experience with the capillary-based measurements at ESRF Grenoble and PETRA-III Hamburg indicate a performance of 45 h per plate in practice (Chen et al. 2018). The majority of time is spent conducting necessary capillary cleaning and transport using the Arinax sample handler, noting that recommended exposure times (110 seconds) permit a theoretical floor of 315 min per plate. This latter value is stated to be achievable according to SIBYLS’s setup2 for solution-based SAXS. In powder-diffraction applications, the Australian Synchrotron3 proposes a benchmark of 5 min per plate. Thus, significant increases can be achieved with more specialised setups that minimise downtime between sample exposures. Given an optimised logistics profile, practical throughput can be increased to 104 measurements per day at current beamline intensities.
Sensitivity and titration coverage
A final limitation in throughput is the choice of titration points that ideally covers conditions spanning between apo- and saturation. In the context of a simple two-state, receptor-ligand binding where one partner possesses buffer-like scattering (featureless over the q-range relevant to SAXS,) the receptor concentrations used in the measurement and the precision at which the bound fraction can be evaluated determines the strongest distinguishable (Fig. 3). For instance, a5% uncertainty on the bound fraction implies that all ligands possessing stronger than 10− 2 of receptor concentration are indistinguishable. Here, beamline-specific reference data can be utilised to estimate uncertainty values. In contrast, detection of weak s is a matter of including relevant titration points potentially followed by subtraction of detectable unbound ligand scattering. Thus, we suggest a higher concentration of titration points around 1:1-ratios for improved coverage of strong-affinity ligands.
Fig. 3.

Family of titration curves at constant receptor concentration [R], for a simple two-state interaction. Colors indicate expected bound-fractions for a range of KD values relative to [R]: 10− 3 (maroon), 10− 2 (red), 10− 1 (orange), 100 (green), 101 (turquoise), 102 (blue), and 103 (violet). Figure prepared with Xmgrace-5.1.23
Although the base method does not assume any knowledge about molecular structure, external knowledge can significantly improve the accuracy of estimated bound fractions. For instance, available atomistic structures can be utilised to generate predicted scattering curves for the apo- and holo- states (Knight and Hub 2015; Schneidman-Duhovny et al. 2016; Franke et al. 2017), which can be used to directly model the relevant populations. We also note that Sedlak et al. (2017) has proposed a quantitative error model to recapitulate the measurement process. When used together with predicted scattering curves, this can assist with determining optimal concentrations at which to perform titrations.
Summary
Small-angle scattering has a unique niche amongst the structural biological screening techniques in that it supplies information on solution conformations at low material costs per measurement, albeit using a destructive measurement. Since the common high-throughput assays detect binding thermodynamics or biological function, the structural information can be used to independently confirm affinity predictions and binding mechanisms of initial hits. These range from distinguishing agonists versus inhibitors to optimising binding partners for downstream structural biology. However, serial measurements limit the current SAXS throughput to 103 titrations per day with current improvements focussed upon reducing the downtime due to sample handling and cell cleaning. This restricts SAXS-based approaches currently to smaller-scale investigations and screening with curated libraries.
At the time of writing, there remains numerous avenues to further investigate SAXS-based screening such as feasibility on lab-based sources, parallelisation and protocol reproducibility across synchrotron sites. This review has collated the essential information necessary to conduct small-scale screening, with which we hope to popularise the utilisation of existing beamline automation capabilities and encourage further development in throughput. We note that data curation support remains relatively underdeveloped compared to structural modelling applications—the current formatting of SAXS repositories SASBDB (Valentini et al. 2015) and BIOISIS do not yet support composite entries for reporting titrations. Nevertheless, we recommend contribution of summary titration data and the adoption of recent publishing standards (Trewhella et al. 2017). These actions will ease the development of comparable protocols between different setups. As the ongoing efforts to improve throughput and efficiency bear fruit, we hope to see an increasing role of small-angle scattering within the arsenal of structural biology screening methods.
Author Contributions
P.C. and J.H. co-wrote the article.
Funding information
P.C. was supported by the EIPOD postdoctoral programme cofunded by the European Molecular Biology Laboratory (EMBL) and the Marie Curie Actions Cofund grant MSCACOFUND-FP no. 664726. J.H. gratefully acknowledges the EMBL and the German Research Council (Deutsche Forschungsgemeinschaft, DFG) for support via an Emmy-Noether Fellowship and the Priority Programme SPP1935.
Compliance with Ethical Standards
Conflict of interest
P.C. declares that he has no conflict of interest. J.H. declares that he has no conflict of interest.
Footnotes
Based on observed droplet volume of order 20 nl and droplets required to produce the reported average using 100 ms exposure time at BM29, ESRF Grenoble.
http://www.synchrotron.org.au/images/AOF2017/10-SAXS-Charlotte-Conn.pdf, slide 47 of 63.
Supplementary Material
None.
Contributor Information
Po-chia Chen, Email: pchen@embl.de.
Janosch Hennig, Email: janosch.hennig@embl.de.
References
- Acerbo AS, Cook MJ, Gillilan RE. Upgrade of MacCHESS facility for x-ray scattering of biological macromolecules in solution. J Synchrotron Radiat. 2015;22(1):180–186. doi: 10.1107/S1600577514020360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamo M, Poulos AS, Miller RM, Lopez CG, Martel A, Porcar L, Cabral JaT. Rapid contrast matching by microfluidic SANS. Lab Chip. 2017;17(9):1559–1569. doi: 10.1039/c7lc00179g. [DOI] [PubMed] [Google Scholar]
- Ando N, Kung Y, Can M, Bender G, Ragsdale SW, Drennan CL. Transient B12-dependent methyltransferase complexes revealed by small-angle x-ray scattering. J Am Chem Soc. 2012;134(43):17945–17954. doi: 10.1021/ja3055782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando N, Li H, Brignole EJ, Thompson S, McLaughlin MI, Page JE, Asturias FJ, Stubbe J, Drennan CL. Allosteric inhibition of human ribonucleotide reductase by dATP entails the stabilization of a Hexamer. Biochemistry. 2016;55(2):373–381. doi: 10.1021/acs.biochem.5b01207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin MR, Tang Y, Wells JA. Small-molecule inhibitors of protein–protein interactions: progressing toward the reality. Chem Biol. 2014;21(9):1102–1114. doi: 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barile E, Pellecchia M. NMR-based approaches for the identification and optimization of inhibitors of protein–protein interactions. Chem Rev. 2014;114(9):4749–4763. doi: 10.1021/cr500043b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayburt TH, Sligar SG. Membrane protein assembly into Nanodiscs. FEBS Lett. 2010;584(9):1721–1727. doi: 10.1016/j.febslet.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernadó P, Svergun DI. Structural analysis of intrinsically disordered proteins by small-angle x-ray scattering. Mol BioSyst. 2011;8(1):151–167. doi: 10.1039/c1mb05275f. [DOI] [PubMed] [Google Scholar]
- Bernadó P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural characterization of flexible proteins using small-angle x-ray scattering. J Am Chem Soc. 2007;129(17):5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- Bernadó Pau, Shimizu Nobutaka, Zaccai Giuseppe, Kamikubo Hironari, Sugiyama Masaaki. Solution scattering approaches to dynamical ordering in biomolecular systems. Biochimica et Biophysica Acta (BBA) - General Subjects. 2018;1862(2):253–274. doi: 10.1016/j.bbagen.2017.10.015. [DOI] [PubMed] [Google Scholar]
- Berthaud A, Manzi J, Pérez J, Mangenot S. Modeling detergent organization around Aquaporin-0 using small-angle x-ray scattering. J Am Chem Soc. 2012;134(24):10080–10088. doi: 10.1021/ja301667n. [DOI] [PubMed] [Google Scholar]
- Björling Alexander, Niebling Stephan, Marcellini Moreno, van der Spoel David, Westenhoff Sebastian. Deciphering Solution Scattering Data with Experimentally Guided Molecular Dynamics Simulations. Journal of Chemical Theory and Computation. 2015;11(2):780–787. doi: 10.1021/ct5009735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cala O, Guillière F, Krimm I. NMR-based analysis of protein–ligand interactions. Anal Bioanal Chem. 2014;406(4):943–956. doi: 10.1007/s00216-013-6931-0. [DOI] [PubMed] [Google Scholar]
- Cammarata M, Levantino M, Schotte F, Anfinrud PA, Ewald F, Choi J, Cupane A, Wulff M, Ihee H. Tracking the structural dynamics of proteins in solution using time-resolved wide-angle X-ray scattering. Nat Methods. 2008;5(10):881–886. doi: 10.1038/nmeth.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardia JP, Eldo J, Xia J, O’Day EM, Tsuruta H, Gryncel KR, Kantrowitz ER. Use of L-asparagine and N-phosphonacetyl-L-asparagine to investigate the linkage of catalysis and homotropic cooperativity in E. coli aspartate transcarbomoylase. Proteins. 2008;71(3):1088–1096. doi: 10.1002/prot.21760. [DOI] [PubMed] [Google Scholar]
- Carlomagno T. Present and future of NMR for RNA–protein complexes: a perspective of integrated structural biology. J Magn Reson. 2014;241:126–136. doi: 10.1016/j.jmr.2013.10.007. [DOI] [PubMed] [Google Scholar]
- Chavanieu A, Pugnière M. Developments in SPR fragment screening. Expert Opin Drug Discovery. 2016;11(5):489–499. doi: 10.1517/17460441.2016.1160888. [DOI] [PubMed] [Google Scholar]
- Chen Po-chia, Hub Jochen S. Interpretation of Solution X-Ray Scattering by Explicit-Solvent Molecular Dynamics. Biophysical Journal. 2015;108(10):2573–2584. doi: 10.1016/j.bpj.2015.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Po-chia, Hub Jochen S. Structural Properties of Protein–Detergent Complexes from SAXS and MD Simulations. The Journal of Physical Chemistry Letters. 2015;6(24):5116–5121. doi: 10.1021/acs.jpclett.5b02399. [DOI] [PubMed] [Google Scholar]
- Chen Pc, Masiewicz P, Rybin V, Svergun D, Hennig J. A general small-angle x-ray scattering-based screening protocol validated for protein–RNA interactions. ACS Comb Sci. 2018;20(4):197–202. doi: 10.1021/acscombsci.8b00007. [DOI] [PubMed] [Google Scholar]
- Ciulli Alessio. Protein-Ligand Interactions. Totowa, NJ: Humana Press; 2013. Biophysical Screening for the Discovery of Small-Molecule Ligands; pp. 357–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Classen S, Hura GL, Holton JM, Rambo RP, Rodic I, McGuire PJ, Dyer K, Hammel M, Meigs G, Frankel KA, Tainer JA. Implementation and performance of SIBYLS: a dual endstation small-angle x-ray scattering and macromolecular crystallography beamline at the advanced light source. J Appl Crystallogr. 2013;46(Pt 1):1–1. doi: 10.1107/S0021889812048698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins KM, Oregioni A, Robertson LE, Kelly G, Ramos A. Protein–RNA specificity by high-throughput principal component analysis of NMR spectra. Nucl Acids Res. 2015;43(6):e41–e41. doi: 10.1093/nar/gku1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congreve Miles, Rich Rebecca L., Myszka David G., Figaroa Francis, Siegal Gregg, Marshall Fiona H. Methods in Enzymology. 2011. Fragment Screening of Stabilized G-Protein-Coupled Receptors Using Biophysical Methods; pp. 115–136. [DOI] [PubMed] [Google Scholar]
- Cordeiro TN, Chen PC, De Biasio A, Sibille N, Blanco FJ, Hub JS, Crehuet R, Bernadó P. Disentangling polydispersity in the PCNA-p15PAF complex, a disordered, transient and multivalent macromolecular assembly. Nucl Acids Res. 2017;45(3):1501–1515. doi: 10.1093/nar/gkw1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro Tiago N., Herranz-Trillo Fátima, Urbanek Annika, Estaña Alejandro, Cortés Juan, Sibille Nathalie, Bernadó Pau. Biological Small Angle Scattering: Techniques, Strategies and Tips. Singapore: Springer Singapore; 2017. Structural Characterization of Highly Flexible Proteins by Small-Angle Scattering; pp. 107–129. [DOI] [PubMed] [Google Scholar]
- Cusack S, Ruigrok RWH, Krygsman PCJ, Mellemam JE. Structure and composition of influenza virus: a small-angle neutron scattering study. J Mol Biol. 1985;186(3):565–582. doi: 10.1016/0022-2836(85)90131-7. [DOI] [PubMed] [Google Scholar]
- Daghestani HN, Day BW. Theory and applications of surface plasmon resonance, resonant mirror, resonant waveguide grating, and dual polarization interferometry biosensors. Sensors. 2010;10(11):9630–9646. doi: 10.3390/s101109630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalvit C, Pevarello P, Tatò M, Veronesi M, Vulpetti A, Sundström M. Identification of compounds with binding affinity to proteins via magnetization transfer from bulk water*. J Biomol NMR. 2000;18(1):65–68. doi: 10.1023/a:1008354229396. [DOI] [PubMed] [Google Scholar]
- Dalvit C, Fogliatto G, Stewart A, Veronesi M, Stockman B. WaterLOGSY as a method for primary NMR screening: practical aspects and range of applicability. J Biomol NMR. 2001;21(4):349–359. doi: 10.1023/a:1013302231549. [DOI] [PubMed] [Google Scholar]
- Das A, Zhao J, Schatz GC, Sligar SG, Van Duyne RP. Screening of type I and II drug binding to human cytochrome P450-3A4 in Nanodiscs by localized surface plasmon resonance spectroscopy. Anal Chem. 2009;81(10):3754–3759. doi: 10.1021/ac802612z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Pérez J. Combined sampler robot and high-performance liquid chromatography: a fully automated system for biological small-angle x-ray scattering experiments at the synchrotron SOLEIL SWING beamline. J Appl Crystallogr. 2009;42(5):892–900. [Google Scholar]
- Davies Thomas G., Tickle Ian J. Topics in Current Chemistry. Berlin, Heidelberg: Springer Berlin Heidelberg; 2011. Fragment Screening Using X-Ray Crystallography; pp. 33–59. [DOI] [PubMed] [Google Scholar]
- Dias DM, Ciulli A. NMR approaches in structure-based lead discovery: recent developments and new frontiers for targeting multi-protein complexes. Prog Biophys Mol Biol. 2014;116(2-3):101–112. doi: 10.1016/j.pbiomolbio.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dongen Maria, Weigelt Johan, Uppenberg Jonas, Schultz Johan, Wikström Mats. Structure-based screening and design in drug discovery. Drug Discovery Today. 2002;7(8):471–478. doi: 10.1016/s1359-6446(02)02233-x. [DOI] [PubMed] [Google Scholar]
- Erlanson DA, Fesik SW, Hubbard RE, Jahnke W, Jhoti H. Twenty years on: the impact of fragments on drug discovery. Nat Rev Drug Discovery. 2016;15(9):605. doi: 10.1038/nrd.2016.109. [DOI] [PubMed] [Google Scholar]
- Felli IC, Brutscher B. Recent advances in solution NMR: fast methods and heteronuclear direct detection. ChemPhysChem. 2009;10(9-10):1356–1368. doi: 10.1002/cphc.200900133. [DOI] [PubMed] [Google Scholar]
- Franke D, Jeffries CM, Svergun DI. Correlation map, a goodness-of-fit test for one-dimensional x-ray scattering spectra. Nat Methods. 2015;12(5):419–422. doi: 10.1038/nmeth.3358. [DOI] [PubMed] [Google Scholar]
- Franke D, Petoukhov MV, Konarev PV, Panjkovich A, Tuukkanen A, Mertens HDT, Kikhney AG, Hajizadeh NR, Franklin JM, Jeffries CM, Svergun DI. ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J Appl Crystallogr. 2017;50(4):1212–1225. doi: 10.1107/S1600576717007786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiburger L, Sonntag M, Hennig J, Li J, Zou P, Sattler M. Efficient segmental isotope labeling of multi-domain proteins using Sortase A. J Biomol NMR. 2015;63(1):1–8. doi: 10.1007/s10858-015-9981-0. [DOI] [PubMed] [Google Scholar]
- Friberg A, Vigil D, Zhao B, Daniels RN, Burke JP, Garcia-Barrantes PM, Camper D, Chauder BA, Lee T, Olejniczak ET, Fesik SW. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J Med Chem. 2013;56(1):15–30. doi: 10.1021/jm301448p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Watanabe A, Hayasaka A, Muraoka M, Hori Y, Yamazaki T, Imaeda Y, Koga A. Small-scale screening method for low-viscosity antibody solutions using small-angle x-ray scattering. Eur J Pharm Biopharm. 2017;112:132–137. doi: 10.1016/j.ejpb.2016.11.027. [DOI] [PubMed] [Google Scholar]
- Gabel Frank. Methods in Enzymology. 2015. Small-Angle Neutron Scattering for Structural Biology of Protein–RNA Complexes; pp. 391–415. [DOI] [PubMed] [Google Scholar]
- Gorba C, Miyashita O, Tama F. Normal-mode flexible fitting of high-resolution structure of biological molecules toward one-dimensional low-resolution data. Biophys J. 2008;94(5):1589–1599. doi: 10.1529/biophysj.107.122218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant TD, Luft JR, Wolfley JR, Tsuruta H, Martel A, Montelione GT, Snell EH. Small angle x-ray scattering as a complementary tool for high-throughput structural studies. Biopolymers. 2011;95(8):517–530. doi: 10.1002/bip.21630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishaev A, Wu J, Trewhella J, Bax A. Refinement of multidomain protein structures by combination of solution small-angle x-ray scattering and NMR data. J Am Chem Soc. 2005;127(47):16621–16628. doi: 10.1021/ja054342m. [DOI] [PubMed] [Google Scholar]
- Grøftehauge MK, Hajizadeh NR, Swann MJ, Pohl E. Protein–ligand interactions investigated by thermal shift assays (TSA) and dual polarization interferometry (DPI) Acta Crystallogr D. 2015;71(1):36–44. doi: 10.1107/S1399004714016617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gundlach AR, Garamus VM, Gorniak T, Davies HA, Reischl M, Mikut R, Hilpert K, Rosenhahn A. Small angle x-ray scattering as a high-throughput method to classify antimicrobial modes of action. Biochim Biophys Acta, Biomembr. 2016;1858(5):918–925. doi: 10.1016/j.bbamem.2015.12.022. [DOI] [PubMed] [Google Scholar]
- Handa N, Nureki O, Kurimoto K, Kim I, Sakamoto H, Shimura Y, Muto Y, Yokoyama S. Structural basis for recognition of the tra mRNA precursor by the Sex-lethal protein. Nature. 1999;398(6728):579–585. doi: 10.1038/19242. [DOI] [PubMed] [Google Scholar]
- Harner MJ, Frank AO, Fesik SW. Fragment-based drug discovery using NMR spectroscopy. J Biomol NMR. 2013;56(2):65–75. doi: 10.1007/s10858-013-9740-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassell AM, An G, Bledsoe RK, Bynum JM, Carter HL, Deng SJJ, Gampe RT, Grisard TE, Madauss KP, Nolte RT, Rocque WJ, Wang L, Weaver KL, Williams SP, Wisely GB, Xu R, Shewchuk LM. Crystallization of protein ligand complexes. Acta Crystallogr D Biol Crystallogr. 2007;63(1):72–79. doi: 10.1107/S0907444906047020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller WT. Small-angle neutron scattering and contrast variation: A powerful combination for studying biological structures. Acta Crystallogr D. 2010;66(11):1213–1217. doi: 10.1107/S0907444910017658. [DOI] [PubMed] [Google Scholar]
- Hennig J, Sattler M. The dynamic duo: Combining NMR and small angle scattering in structural biology. Prot Sci. 2014;23(6):669–682. doi: 10.1002/pro.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig J, Wang I, Sonntag M, Gabel F, Sattler M. Combining NMR and small angle x-ray and neutron scattering in the structural analysis of a ternary protein-RNA complex. J Biomol NMR. 2013;56(1):17–30. doi: 10.1007/s10858-013-9719-9. [DOI] [PubMed] [Google Scholar]
- Hennig J, Militti C, Popowicz GM, Wang I, Sonntag M, Geerlof A, Gabel F, Gebauer F, Sattler M. Structural basis for the assembly of the Sxl-Unr translation regulatory complex. Nature. 2014;515(7526):287–290. doi: 10.1038/nature13693. [DOI] [PubMed] [Google Scholar]
- Henriques João, Arleth Lise, Lindorff-Larsen Kresten, Skepö Marie. On the Calculation of SAXS Profiles of Folded and Intrinsically Disordered Proteins from Computer Simulations. Journal of Molecular Biology. 2018;430(16):2521–2539. doi: 10.1016/j.jmb.2018.03.002. [DOI] [PubMed] [Google Scholar]
- Herranz-Trillo Fátima, Groenning Minna, van Maarschalkerweerd Andreas, Tauler Romà, Vestergaard Bente, Bernadó Pau. Structural Analysis of Multi-component Amyloid Systems by Chemometric SAXS Data Decomposition. Structure. 2017;25(1):5–15. doi: 10.1016/j.str.2016.10.013. [DOI] [PubMed] [Google Scholar]
- Huang R, Bonnichon A, Claridge TDW, Leung IKH. Protein-ligand binding affinity determination by the waterLOGSY method: An optimised approach considering ligand rebinding. Sci Rep. 2017;7:43727. doi: 10.1038/srep43727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hura GL, Menon AL, Hammel M, Rambo RP, Poole FLII, Tsutakawa SE, Jenney FEJr, Classen S, Frankel KA, Hopkins RC, Yang SJ, Scott JW, Dillard BD, Adams MWW, Tainer JA. Robust, high-throughput solution structural analyses by small angle x-ray scattering (SAXS) Nat Methods. 2009;6(8):606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hura GL, Budworth H, Dyer KN, Rambo RP, Hammel M, McMurray CT, Tainer JA. Comprehensive objective maps of macromolecular conformations by quantitative SAXS analysis. Nat Methods. 2013;10(6):453–454. doi: 10.1038/nmeth.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacrot B. The study of biological structures by neutron scattering from solution. Rep Prog Phys. 1976;39(10):911. [Google Scholar]
- Jahnke W. Spin labels as a tool to identify and characterize protein ligand interactions by NMR spectroscopy. ChemBioChem. 2002;3(2-3):167–173. doi: 10.1002/1439-7633(20020301)3:2/3<167::AID-CBIC167>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Jamros MA, Oliveira LC, Whitford PC, Onuchic JN, Adams JA, Blumenthal DK, Jennings PA. Proteins at work: a combined small angle x-ray scattering and theoretical determination of the multiple structures involved on the protein kinase functional landscape. J Biol Chem. 2010;285(46):36121–36128. doi: 10.1074/jbc.M110.116947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-García B, Pons C, Svergun DI, Bernadó P, Fernández-Recio J. PyDockSAXS: protein protein complex structure by SAXS and computational docking. Nucl Acids Res. 2015;43(W1):W356–W361. doi: 10.1093/nar/gkv368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantrowitz ER. Allostery and cooperativity in Escherichia coli aspartate transcarbamoylase. Arch Biochem Biophys. 2012;519(2):81–90. doi: 10.1016/j.abb.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaca E, Bonvin AMJJ. On the usefulness of ion-mobility mass spectrometry and SAXS data in scoring docking decoys. Acta Crystallogr D. 2013;69(5):683–694. doi: 10.1107/S0907444913007063. [DOI] [PubMed] [Google Scholar]
- Kikhney AG, Svergun DI. A practical guide to small angle x-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015;589(19, Part A):2570–2577. doi: 10.1016/j.febslet.2015.08.027. [DOI] [PubMed] [Google Scholar]
- Kim HS, Gabel F. Uniqueness of models from small-angle scattering data: the impact of a hydration shell and complementary NMR restraints. Acta Crystallogr D. 2015;71(1):57–66. doi: 10.1107/S1399004714013923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimanius D, Pettersson I, Schluckebier G, Lindahl E, Andersson M. SAXS-guided metadynamics. J Chem Theory Comput. 2015;11(7):3491–3498. doi: 10.1021/acs.jctc.5b00299. [DOI] [PubMed] [Google Scholar]
- Kirby NM, Mudie ST, Hawley AM, Cookson DJ, Mertens HDT, Cowieson N, Samardzic-Boban V. A low-background-intensity focusing small-angle x-ray scattering undulator beamline. J Appl Crystallogr. 2013;46(6):1670–1680. [Google Scholar]
- Knight CJ, Hub JS. WAXSiS: a web server for the calculation of SAXS/WAXS curves based on explicit-solvent molecular dynamics. Nucl Acids Res. 2015;43(W1):W225–W230. doi: 10.1093/nar/gkv309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsioubas A, Berthaud A, Mangenot S, Pérez J. Ab initio and all-atom modeling of detergent organization around aquaporin-0 based on SAXS data. J Phys Chem B. 2013;117(43):13588–13594. doi: 10.1021/jp407688x. [DOI] [PubMed] [Google Scholar]
- Lafleur JP, Snakenborg D, Nielsen SS, Møller M, Toft KN, Menzel A, Jacobsen JK, Vestergaard B, Arleth L, Kutter JP. Automated microfluidic sample-preparation platform for high-throughput structural investigation of proteins by small-angle x-ray scattering. J Appl Crystallogr. 2011;44(5):1090–1099. [Google Scholar]
- Lapinaite A, Simon B, Skjaerven L, Rakwalska-Bange M, Gabel F, Carlomagno T. The structure of the box C/D enzyme reveals regulation of RNA methylation. Nature. 2013;502(7472):519–523. doi: 10.1038/nature12581. [DOI] [PubMed] [Google Scholar]
- Lee CY, Chang CL, Wang YN, Fu LM. Microfluidic mixing: a review. Int J Mol Sci. 2011;12(5):3263–3287. doi: 10.3390/ijms12053263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Okuno Y, Cavagnero S. Sensitivity enhancement in solution NMR: Emerging ideas and new frontiers. J Magn Reson. 2014;241:18–31. doi: 10.1016/j.jmr.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Li X, Wang Y, Liu G, Zhou P, Wu H, Hong C, Bian F, Zhang R. The new NCPSS BL19u2 beamline at the SSRF for small-angle x-ray scattering from biological macromolecules in solution. J Appl Crystallogr. 2016;49(5):1428–1432. doi: 10.1107/S160057671601195X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipfert J, Columbus L, Chu VB, Lesley SA, Doniach S. Size and shape of detergent micelles determined by small-angle x-ray scattering. J Phys Chem B. 2007;111(43):12427–12438. doi: 10.1021/jp073016l. [DOI] [PubMed] [Google Scholar]
- Liu Z, Gong Z, Dong X, Tang C. Transient protein protein interactions visualized by solution NMR. Biochim Biophys Acta Proteins Proteomics. 2016;1864(1):115–122. doi: 10.1016/j.bbapap.2015.04.009. [DOI] [PubMed] [Google Scholar]
- Lopez CG, Watanabe T, Martel A, Porcar L, Cabral JaT. Microfluidic-SANS: flow processing of complex fluids. Sci Rep. 2015;5:7727. doi: 10.1038/srep07727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madl T, Gabel F, Sattler M. NMR and small-angle scattering-based structural analysis of protein complexes in solution. J Struct Biol. 2011;173(3):472–482. doi: 10.1016/j.jsb.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Makowski L, Gore D, Mandava S, Minh D, Park S, Rodi DJ, Fischetti RF. X-ray solution scattering studies of the structural diversity intrinsic to protein ensembles. Biopolymers. 2011;95(8):531–542. doi: 10.1002/bip.21631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangel WF, Lin BH, Ramakrishnan V. Characterization of an extremely large, ligand-induced conformational change in plasminogen. Science. 1990;248(4951):69–73. doi: 10.1126/science.2108500. [DOI] [PubMed] [Google Scholar]
- Martel A, Liu P, Weiss TM, Niebuhr M, Tsuruta H. An integrated high-throughput data acquisition system for biological solution x-ray scattering studies. J Synchrotron Radiat. 2012;19(3):431–434. doi: 10.1107/S0909049512008072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer M, Meyer B. Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew Chem Int Ed. 1999;38(12):1784–1788. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1784::AID-ANIE1784>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Maynard JA, Lindquist NC, Sutherland JN, Lesuffleur A, Warrington AE, Rodriguez M, Oh SH. Surface plasmon resonance for high-throughput ligand screening of membrane-bound proteins. Biotechnol J. 2009;4(11):1542–1558. doi: 10.1002/biot.200900195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisburger SP, Sutton JL, Chen H, Pabit SA, Kirmizialtin S, Elber R, Pollack L. Polyelectrolyte properties of single stranded DNA measured using SAXS and single-molecule FRET: Beyond the wormlike chain model. Biopolymers. 2013;99(12):1032–1045. doi: 10.1002/bip.22265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merk A, Bartesaghi A, Banerjee S, Falconieri V, Rao P, Davis M, Pragani R, Boxer M, Earl LA, Milne JL, Subramaniam S. Breaking cryo-EM resolution barriers to facilitate drug discovery. Cell. 2016;165(7):1698–1707. doi: 10.1016/j.cell.2016.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer B, Peters T. NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew Chem Int Ed. 2003;42(8):864–890. doi: 10.1002/anie.200390233. [DOI] [PubMed] [Google Scholar]
- Møller M, Nielsen SS, Ramachandran S, Li Y, Tria G, Streicher W, Petoukhov MV, Cerione RA, Gillilan RE, Vestergaard B. Small angle x-ray scattering studies of mitochondrial glutaminase c reveal extended flexible regions, and link oligomeric state with enzyme activity. PLOS ONE. 2013;8(9):e74783. doi: 10.1371/journal.pone.0074783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CW, Carr MG, Callaghan O, Chessari G, Congreve M, Cowan S, Coyle JE, Downham R, Figueroa E, Frederickson M, Graham B, McMenamin R, O’Brien MA, Patel S, Phillips TR, Williams G, Woodhead AJ, Woolford AJA. Fragment-based drug discovery applied to hsp90. Discovery of two lead series with high ligand efficiency. J Med Chem. 2010;53(16):5942–5955. doi: 10.1021/jm100059d. [DOI] [PubMed] [Google Scholar]
- Navratilova I, Hopkins AL. Fragment screening by surface plasmon resonance. ACS Med Chem Lett. 2010;1(1):44–48. doi: 10.1021/ml900002k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neutze R, Moffat K. Time-resolved structural studies at synchrotrons and x-ray free electron lasers: Opportunities and challenges. Curr Opin Struct Biol. 2012;22(5):651–659. doi: 10.1016/j.sbi.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah GA, Trakhanov S, Trewhella J, Quiocho FA. Leucine/isoleucine/valine-binding protein contracts upon binding of ligand. J Biol Chem. 1993;268(22):16241–16247. [PubMed] [Google Scholar]
- Oliver RC, Lipfert J, Fox DA, Lo RH, Doniach S, Columbus L. Dependence of micelle size and shape on detergent alkyl chain length and head group. PLoS ONE. 2013;8(5):e62488. doi: 10.1371/journal.pone.0062488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patching SG. Surface plasmon resonance spectroscopy for characterisation of membrane protein ligand interactions and its potential for drug discovery. Biochim Biophys Acta Biomembr. 2014;1838(1, Part A):43–55. doi: 10.1016/j.bbamem.2013.04.028. [DOI] [PubMed] [Google Scholar]
- Patel D, Bauman JD, Arnold E. Advantages of crystallographic fragment screening: Functional and mechanistic insights from a powerful platform for efficient drug discovery. Prog Biophys Mol Biol. 2014;116(2):92–100. doi: 10.1016/j.pbiomolbio.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat Rev Drug Discovery. 2007;6(1):29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- Petoukhov MV, Svergun DI. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys J. 2005;89(2):1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham N, Radajewski D, Round A, Brennich M, Pernot P, Biscans B, Bonneté F, Teychené S. Coupling high throughput microfluidics and small-angle x-ray scattering to study protein crystallization from solution. Anal Chem. 2017;89(4):2282–2287. doi: 10.1021/acs.analchem.6b03492. [DOI] [PubMed] [Google Scholar]
- Piliarik M, Vaisocherová H, Homola J. A new surface plasmon resonance sensor for high-throughput screening applications. Biosens Bioelectron. 2005;20(10):2104–2110. doi: 10.1016/j.bios.2004.09.025. [DOI] [PubMed] [Google Scholar]
- Prati F, De Simone A, Armirotti A, Summa M, Pizzirani D, Scarpelli R, Bertozzi SM, Perez DI, Andrisano V, Perez-Castillo A, Monti B, Massenzio F, Polito L, Racchi M, Sabatino P, Bottegoni G, Martinez A, Cavalli A, Bolognesi ML. 3,4-Dihydro-1,3,5-triazin-2(1H)-ones as the first dual BACE-1/GSK-3β fragment hits against Alzheimer’s disease. ACS Chem Neurosci. 2015;6(10):1665–1682. doi: 10.1021/acschemneuro.5b00121. [DOI] [PubMed] [Google Scholar]
- Puthenveetil R, Nguyen K, Vinogradova O. Nanodiscs and solution NMR: Preparation, application and challenges. Nanotechnol Rev. 2017;6(1):111–126. doi: 10.1515/ntrev-2016-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: Defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40(3):191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the porod-Debye law. Biopolymers. 2011;95(8):559–571. doi: 10.1002/bip.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA. Super-resolution in solution x-ray scattering and its applications to structural systems biology. Ann Rev Biophys. 2013;42(1):415–441. doi: 10.1146/annurev-biophys-083012-130301. [DOI] [PubMed] [Google Scholar]
- Ranganathan A, Heine P, Rudling A, Plückthun A, Kummer L, Carlsson J. Ligand discovery for a peptide-binding GPCR by structure-based screening of fragment- and lead-like chemical libraries. ACS Chem Biol. 2017;12(3):735–745. doi: 10.1021/acschembio.6b00646. [DOI] [PubMed] [Google Scholar]
- Receveur-Bréchot V, Durand D. How random are intrinsically disordered proteins? A small angle scattering perspective. Curr Protein Pept Sci. 2012;13(1):55–75. doi: 10.2174/138920312799277901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renaud JP, Cw Chung, Danielson UH, Egner U, Hennig M, Hubbard RE, Nar H. Biophysics in drug discovery: Impact, challenges and opportunities. Nat Rev Drug Discovery. 2016;15(10):679–698. doi: 10.1038/nrd.2016.123. [DOI] [PubMed] [Google Scholar]
- Renaud Jean-Paul, Chari Ashwin, Ciferri Claudio, Liu Wen-ti, Rémigy Hervé-William, Stark Holger, Wiesmann Christian. Cryo-EM in drug discovery: achievements, limitations and prospects. Nature Reviews Drug Discovery. 2018;17(7):471–492. doi: 10.1038/nrd.2018.77. [DOI] [PubMed] [Google Scholar]
- Ritchie T.K., Grinkova Y.V., Bayburt T.H., Denisov I.G., Zolnerciks J.K., Atkins W.M., Sligar S.G. Methods in Enzymology. 2009. Reconstitution of Membrane Proteins in Phospholipid Bilayer Nanodiscs; pp. 211–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Ruiź I, Radajewski D, Charton S, Phamvan N, Brennich M, Pernot P, Bonneté F, Teychené S, Rodriguez-Ruiź I, Radajewski D, Charton S, Phamvan N, Brennich M, Pernot P, Bonneté F, Teychené S. Innovative high-throughput SAXS methodologies based on photonic lab-on-a-chip sensors: Application to macromolecular studies. Sensors. 2017;17(6):1266. doi: 10.3390/s17061266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi P, Shi L, Liu G, Barbieri CM, Lee HW, Grant TD, Luft JR, Xiao R, Acton TB, Snell EH, Montelione GT, Baker D, Lange OF, Sgourakis NG. A hybrid NMR/SAXS-based approach for discriminating oligomeric protein interfaces using Rosetta. Proteins. 2015;83(2):309–317. doi: 10.1002/prot.24719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round A, Felisaz F, Fodinger L, Gobbo A, Huet J, Villard C, Blanchet CE, Pernot P, McSweeney S, Roessle M, Svergun DI, Cipriani F. BioSAXS sample changer: A robotic sample changer for rapid and reliable high-throughput X-ray solution scattering experiments. Acta Crystallogr D. 2015;71(1):67–75. doi: 10.1107/S1399004714026959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RóŻycki B, Kim YC, Hummer G. SAXS ensemble refinement of ESCRT-III CHMP3 conformational transitions. Structure. 2011;19(1):109–116. doi: 10.1016/j.str.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamon Zdzislaw, Macleod H.Angus, Tollin Gordon. Surface plasmon resonance spectroscopy as a tool for investigating the biochemical and biophysical properties of membrane protein systems. I: Theoretical principles. Biochimica et Biophysica Acta (BBA) - Reviews on Biomembranes. 1997;1331(2):117–129. doi: 10.1016/s0304-4157(97)00004-x. [DOI] [PubMed] [Google Scholar]
- Salamon Zdzislaw, Macleod H.Angus, Tollin Gordon. Surface plasmon resonance spectroscopy as a tool for investigating the biochemical and biophysical properties of membrane protein systems. II: Applications to biological systems. Biochimica et Biophysica Acta (BBA) - Reviews on Biomembranes. 1997;1331(2):131–152. doi: 10.1016/s0304-4157(97)00003-8. [DOI] [PubMed] [Google Scholar]
- Sánchez-Pedregal VM, Reese M, Meiler J, Blommers MJJ, Griesinger C, Carlomagno T. The INPHARMA method: Protein-mediated interligand NOEs for pharmacophore mapping. Angew Chem Int Ed. 2005;44(27):4172–4175. doi: 10.1002/anie.200500503. [DOI] [PubMed] [Google Scholar]
- Schiebel J, Radeva N, Krimmer SG, Wang X, Stieler M, Ehrmann FR, Fu K, Metz A, Huschmann FU, Weiss MS, Mueller U, Heine A, Klebe G. Six biophysical screening methods miss a large proportion of crystallographically discovered fragment hits: A case study. ACS Chem Biol. 2016;11(6):1693–1701. doi: 10.1021/acschembio.5b01034. [DOI] [PubMed] [Google Scholar]
- Schindler Christina E.M., de Vries Sjoerd J., Sasse Alexander, Zacharias Martin. SAXS Data Alone can Generate High-Quality Models of Protein-Protein Complexes. Structure. 2016;24(8):1387–1397. doi: 10.1016/j.str.2016.06.007. [DOI] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Hammel M, Tainer JA, Sali A. FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucl Acids Res. 2016;44(W1):W424–W429. doi: 10.1093/nar/gkw389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwemmer F, E Blanchet C, Spilotros A, Kosse D, Zehnle S, T Mertens HD, A Graewert M, Rössle M, Paust N, I Svergun D, von Stetten F, Zengerle R, Mark D. LabDisk for SAXS: A centrifugal microfluidic sample preparation platform for small-angle X-ray scattering. Lab Chip. 2016;16(7):1161–1170. doi: 10.1039/c5lc01580d. [DOI] [PubMed] [Google Scholar]
- Sedlak SM, Bruetzel LK, Lipfert J. Quantitative evaluation of statistical errors in small-angle X-ray scattering measurements. J Appl Crystallogr. 2017;50(2):621–630. doi: 10.1107/S1600576717003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-Affinity ligands for proteins: SAR by NMR. Science. 1996;274(5292):1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- Skar-Gislinge N, Kynde SaR, Denisov IG, Ye X, Lenov I, Sligar SG, Arleth L. Small-angle scattering determination of the shape and localization of human cytochrome p450 embedded in a phospholipid nanodisc environment. Acta Crystallogr D Biol Crystallogr. 2015;71(12):2412–2421. doi: 10.1107/S1399004715018702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skjærven L, Codutti L, Angelini A, Grimaldi M, Latek D, Monecke P, Dreyer MK, Carlomagno T. Accounting for conformational variability in protein–ligand docking with NMR-guided rescoring. J Am Chem Soc. 2013;135(15):5819–5827. doi: 10.1021/ja4007468. [DOI] [PubMed] [Google Scholar]
- Skou S, Gillilan RE, Ando N. Synchrotron-based small-angle X-ray scattering of proteins in solution. Nat Protocols. 2014;9(7):1727–1739. doi: 10.1038/nprot.2014.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonntag M, Jagtap PKA, Simon B, Appavou MS, Geerlof A, Stehle R, Gabel F, Hennig J, Sattler M. Segmental, domain-Selective perdeuteration and small-Angle neutron scattering for structural analysis of multi-Domain proteins. Angew Chem Int Ed. 2017;56(32):9322–9325. doi: 10.1002/anie.201702904. [DOI] [PubMed] [Google Scholar]
- Svergun DI, Koch MHJ, Pedersen JS, Serdyuk IN. Structural model of the 50 S subunit of escherichia coli ribosomes from solution scattering: II. Neutron scattering study. J Mol Biol. 1994;240(1):78–86. doi: 10.1006/jmbi.1994.1419. [DOI] [PubMed] [Google Scholar]
- Svergun DI, Koch MHJ, Timmins PA, May RP (2013) Small angle X-Ray and neutron scattering from solutions of biological macromolecules. Oxford University Press. 10.1093/acprof:oso/9780199639533.001.0001
- Tian X, Langkilde AE, Thorolfsson M, Rasmussen HB, Vestergaard B. Small-angle X-ray scattering screening complements conventional biophysical analysis: Comparative structural and biophysical analysis of monoclonal antibodies IgG1, IgG2, and IgG4. J Pharmaceut Sci. 2014;103(6):1701–1710. doi: 10.1002/jps.23964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P. Intrinsically disordered proteins: A 10-year recap. Trends Biochem Sci. 2012;37(12):509–516. doi: 10.1016/j.tibs.2012.08.004. [DOI] [PubMed] [Google Scholar]
- Trewhella J. Small-angle scattering and 3D structure interpretation. Curr Opin Struct Biol. 2016;40:1–7. doi: 10.1016/j.sbi.2016.05.003. [DOI] [PubMed] [Google Scholar]
- Trewhella J, Duff AP, Durand D, Gabel F, Guss JM, Hendrickson WA, Hura GL, Jacques DA, Kirby NM, Kwan AH, Pérez J, Pollack L, Ryan TM, Sali A, Schneidman-Duhovny D, Schwede T, Svergun DI, Sugiyama M, Tainer JA, Vachette P, Westbrook J, Whitten AE. 2017 publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution: An update. Acta Crystallogr D. 2017;73(9):710–728. doi: 10.1107/S2059798317011597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao DHH, Sutherland AG, Jennings LD, Li Y, Rush TS, Alvarez JC, Ding W, Dushin EG, Dushin RG, Haney SA, Kenny CH, Karl Malakian A, Nilakantan R, Mosyak L. Discovery of novel inhibitors of the zipa/FtsZ complex by NMR fragment screening coupled with structure-based design. Bioorg Med Chem. 2006;14(23):7953–7961. doi: 10.1016/j.bmc.2006.07.050. [DOI] [PubMed] [Google Scholar]
- Tuukkanen AT, Svergun DI. Weak protein ligand interactions studied by small-angle X-ray scattering. FEBS J. 2014;281(8):1974–1987. doi: 10.1111/febs.12772. [DOI] [PubMed] [Google Scholar]
- Valentini E, Kikhney AG, Previtali G, Jeffries CM, Svergun DI. SASBDB, a repository for biological small-angle scattering data. Nucl Acids Res. 2015;43(D1):D357–D363. doi: 10.1093/nar/gku1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanwetswinkel Sophie, Heetebrij Robert J., van Duynhoven John, Hollander Johan G., Filippov Dmitri V., Hajduk Philip J., Siegal Gregg. TINS, Target Immobilized NMR Screening: An Efficient and Sensitive Method for Ligand Discovery. Chemistry & Biology. 2005;12(2):207–216. doi: 10.1016/j.chembiol.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Vestergaard B, Sayers Z. Investigating increasingly complex macromolecular systems with small-angle X-ray scattering. IUCrJ. 2014;1(6):523–529. doi: 10.1107/S2052252514020843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagstaff JL, Taylor SL, Howard MJ. Recent developments and applications of saturation transfer difference nuclear magnetic resonance (STD NMR) spectroscopy. Mol BioSyst. 2013;9(4):571–577. doi: 10.1039/c2mb25395j. [DOI] [PubMed] [Google Scholar]
- Watkin Serena A.J., Ryan Timothy M., Miller Antonia G., Nock Volker M., Pearce F. Grant, Dobson Renwick C.J. X-ray Scattering. 2017. Microfluidics for Small-Angle X-ray Scattering. [Google Scholar]
- Wilcox KC, Marunde MR, Das A, Velasco PT, Kuhns BD, Marty MT, Jiang H, Luan CH, Sligar SG, Klein WL. Nanoscale synaptic membrane mimetic allows unbiased high throughput screen that targets binding sites for Alzheimer’s-Associated Aβ Oligomers. PLoS ONE. 2015;10(4):e0125263. doi: 10.1371/journal.pone.0125263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson MP. Using chemical shift perturbation to characterise ligand binding. Prog Nucl Magn Reson Spectrosc. 2013;73:1–16. doi: 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Xia B, Mamonov A, Leysen S, Allen KN, Strelkov SV, Paschalidis IC, Vajda S, Kozakov D. Accounting for observed small angle X-ray scattering profile in the protein protein docking server cluspro. J Comput Chem. 2015;36(20):1568–1572. doi: 10.1002/jcc.23952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Blachowicz L, Makowski L, Roux B. Multidomain Assembled States of hck tyrosine kinase in solution. Proc Natl Acad Sci USA. 2010;107(36):15757–15762. doi: 10.1073/pnas.1004569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You JB, Kang K, Tran TT, Park H, Hwang WR, Kim JM, Im SG. PDMS-based turbulent microfluidic mixer. Lab Chip. 2015;15(7):1727–1735. doi: 10.1039/c5lc00070j. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Hu R, Wang L, Gu D, He J, Wu SY, Ho HP, Li X, Qu J, Gao BZ, Shao Y. Recent advances in surface plasmon resonance imaging: Detection speed, sensitivity, and portability. Nanophotonics. 2017;6(5):1017–1030. [Google Scholar]
- Zheng H, Handing KB, Zimmerman MD, Shabalin IG, Almo SC, Minor W. X-ray crystallography over the past decade for novel drug discovery where are we heading next? Expert Opin Drug Discovery. 2015;10(9):975–989. doi: 10.1517/17460441.2015.1061991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Tekpinar M. Accurate flexible fitting of high-Resolution protein structures to small-Angle x-Ray Scattering Data Using a coarse-Grained model with Implicit Hydration Shell. Biophys J. 2011;101(12):2981–2991. doi: 10.1016/j.bpj.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]