

Abstract
Within the last decade, it has become clear that DNA replication and transcription are routinely in conflict with each other in growing cells. Much of the seminal work on this topic has been carried out in bacteria, specifically, Escherichia coli and Bacillus subtilis; therefore, studies of conflicts in these species deserve special attention. Collectively, the recent findings on conflicts have fundamentally changed the way we think about DNA replication in vivo. Furthermore, new insights on this topic have revealed that the conflicts between replication and transcription significantly influence many key parameters of cellular function, including genome organization, mutagenesis, and evolution of stress response and virulence genes. In this review, we discuss the consequences of replication-transcription conflicts on the life of bacteria and describe some key strategies cells use to resolve them. We put special emphasis on two critical aspects of these encounters: (a) the consequences of conflicts on replisome stability and dynamics, and (b) the resulting increase in spontaneous mutagenesis.
Keywords: DNA replication, transcription, replication-transcription conflicts
1. INTRODUCTION
Efficient and accurate replication of chromosomal DNA is an essential process in all life-forms. DNA replication in most bacteria starts at a single origin and proceeds bidirectionally until it reaches a defined terminal region. Recent advances on this topic have revealed that the process of replication is far more complicated than previously thought. The DNA is not naked: The chromosome is covered with various DNA-binding proteins, including RNA polymerases (RNAPs). We now know that the presence of RNAPs on the chromosome affects DNA replication in a significant manner. The replication machinery (the replisome) is, in essence, on an obstacle course, and the transcription machinery is the main obstacle on its path. The lack of temporal and spatial separation of these two processes generates inevitable conflicts between them, with profound consequences not only on replication, but also on mutagenesis and evolution.
Depending on which strand serves as the template for RNAP, the transcription machinery can either move codirectionally (leading-strand genes) or head-on (lagging-strand genes) relative to the movement of the replication machinery (Figure 1). The existence of genes in the two orientations leads to two types of conflicts, both of which have negative consequences, but to different degrees. Codirectional collisions occur pervasively at highly transcribed regions or at the sites of backtracked RNAPs, thereby stalling the replisome and leading to reassembly of the replication machinery outside of the origin, also known as replication restart (11, 35, 40). Although they can compromise genome integrity by exacerbating collisions, backtracked RNAPs coordinate many critical processes, including the rate of transcription elongation, and coupling of transcription and translation (44). However, head-on collisions have much more severe consequences, even at regions with low levels of transcription. A single RNAP moving head-on can dislodge the replisome in vitro (26, 50). In vivo, in a head-on collision, the replisome disassembles, the DNA breaks, and local mutation rates more than double (7, 14, 24, 28, 38, 39, 41, 48, 55, 65). Therefore, despite the common assumption in the field, the consequences of head-on conflicts are not necessarily linked to transcription levels. Rather, the orientation of a gene is the true determinant of conflict severity, although as many experiments have demonstrated, increased transcription in either orientation undoubtedly worsens the conflict.
Figure 1.
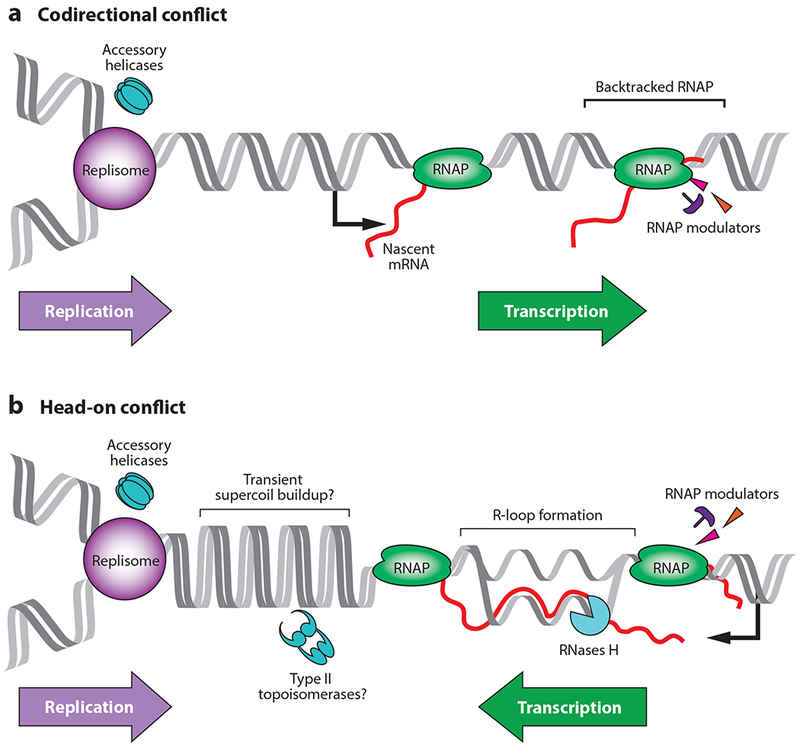
Replication-transcription conflicts occur in two different orientations. Codirectional conflicts (a) occur when a gene is encoded on the leading strand. Head-on conflicts (b) occur when a gene is encoded on the lagging strand. Accessory helicases and RNA polymerase (RNAP) modulators aid in the resolution of both types of conflicts. Accessory helicases (PcrA, UvrD, Rep, DinG) likely arrive at conflict regions together with the replication fork. In contrast, RNAP modulators (DksA, GreA/B) are likely present at the transcription unit through their interactions with RNAP. The modulators regulate the movement and activity of RNAP when necessary (for instance, during backtracking), helping resolve conflicts. Head-on conflicts increase the frequency and/or stability of R-loops, leading to replisome stalling, mutations, and reduced gene expression. Models of conflict-mediated topological disturbance predict that positive supercoiling builds up transiently between the replication and transcription machineries at head-on gene regions, as depicted. This could drive R-loop formation. Accessory helicases, RNases H, and possibly type II topoisomerases resolve head-on conflicts. How type II topoisomerases may be recruited to conflict regions is unclear. Figure adapted from Reference 24.
2. CONFLICT REDUCTION AND PREVENTION
2.1. Orientation Bias
Initial genomic organization analyses of bacterial chromosomes have shown that there are almost always more genes encoded on the leading strand versus the lagging strand, especially when it comes to the most highly transcribed and essential genes (53, 54, 72). It is assumed that this gene orientation bias is a result of the migration of genes to the leading strand over evolutionary time. However, this assumption was recently tested using GC skew analysis, a robust method capable of identifying inversions in genomes. The evolutionary inversion record of individual genes showed that a significant percentage of head-on genes were originally codirectional. This pattern is consistent across divergent bacterial species (32). Nevertheless, the co-orientation bias of highly transcribed and essential genes reduces the number of severe head-on conflicts, replisome-stalling and restart events, and DNA breaks, increasing replication efficiency and preserving genomic stability. However, this avoidance mechanism does not seem to apply to a large number of core genes—some of which are key to surviving environmental changes, are associated with virulence (in the case of pathogens), or both (24, 48). These genes are highly induced in replicating cells, which generates a cacophony of head-on conflicts (4, 42, 43, 58). These recent findings challenge previous assumptions surrounding genome organization. It seems that evolutionary pressures prevent a completely co-oriented genome and/or drive the maintenance of (and inversion to) the head-on orientation for certain genes.
2.2. Temporal Regulation
Discreet separation of transcription and replication would completely solve the issue of conflicts. However, it is difficult to imagine temporal separation of replication and transcription in bacteria, as these processes occur simultaneously. It is noteworthy that although the two processes occur concurrently, given the conditional expression pattern of head-on genes, there seems to be at least one type of temporal regulatory mechanism that reduces the frequency and severity of conflicts (4, 42, 43, 58). In other words, many head-on genes are expressed conditionally, such as during stress response. This is reminiscent of eukaryotic cell cycle regulation of gene expression, where replication and some transcription can occur separately. However, there is still transcription during S phase in most eukaryotic cells, which generates replication-transcription conflicts (19, 20). Therefore, in all known cases, although temporal regulation can reduce conflict frequency, complete avoidance of collisions through such mechanisms does not occur.
So, neither genome orientation bias or temporal regulation can circumvent the problem of conflicts. Is it possible to completely avoid conflicts? If so, why haven’t cells evolved such mechanisms? Perhaps resolution of conflicts by various mechanisms (discussed below) can sufficiently support cellular proliferation. Alternatively, as we proposed previously, there may be some evolutionary advantage to conditionally inducing head-on conflicts at some genes (see Section 6 and the opinion piece Reference 34).
3. CONSEQUENCES FOR DNA REPLICATION
3.1. Replisome Stalling
Many different types of obstacles, including DNA lesions, secondary DNA structures, and DNA-binding proteins, can stall the replisome. However, recent findings suggest that the most common cause of replisome stalling in vivo is RNAP (28, 35, 65). The majority of endogenous replication fork stalling detected by various methods is caused by transcription (28, 35). However, regardless of the cause, stalled replication forks, in general, are a major problem for growing cells and must be dissolved and restarted in order for replication to be completed and the chromosomes to be segregated into daughter cells.
The ability for transcription to hinder replication in living cells was first observed by placing an origin of replication on either side of a single ribosomal RNA gene operon in Escherichia coli. In this experiment, French (14) provided the first piece of evidence that transcription slows down replication in vivo. Her experiments also showed, for the first time, that a head-on conflict is worse than a codirectional conflict. In this study, the replisome proceeded at a relatively normal rate when faced with a codirectionally oriented operon. However, when the collision occurred in the head-on orientation, its progression was significantly delayed. In vitro experiments performed by Lui & Alberts (26) using a reconstituted phage replication system produced similar results with regard to gene orientation and replication stalling. Later, in vivo experiments using plasmid-based systems in E. coli found that replisome stalling is increased when the collision occurs in the head-on orientation (40, 41). Consistent with these findings, in vitro experiments using a reconstituted E. coli replisome stalls more severely in the face of a head-on RNAP compared to a codirectional RNAP (49, 50). Inverting the ribosomal gene operons in E. coli to the head-on orientation only leads to significant stalling events in the absence of accessory helicases in vivo; nevertheless, this effect is orientation dependent (2). Interestingly, Bacillus subtilis appears to be much more sensitive to head-on conflicts: Inverting ribosomal operons in B. subtilis has severe consequences for replication even in the presence of conflict resolution factors such as accessory helicases (60, 65). Recent work in B. subtilis showed that this phenomenon is not specific to rRNA genes. Even a single coding gene can severely stall the replisome when it is highly expressed in the head-on, but not codirectional, orientation (24, 31, 39). This particular experiment measuring replication stalling at a single coding gene in the two orientations has not been performed in E. coli.
It is worth noting that codirectional collisions are not without consequence. On a plasmid-based system, it was shown that the replisome stalls at transcriptional terminators (40). Interestingly, the consequences of codirectional conflicts become more severe than those of head-on conflicts if the replication fork encounters backtracked RNAPs (11). Using chromatin immunoprecipitation (ChIP) in B. subtilis, it was shown that, even in wild-type cells, the replicative helicase DnaC (a proxy for replisome stalling), as well as replication restart and helicase loader proteins DnaD and DnaB, was enriched at the codirectionally oriented rRNA operons, indicating that replisome stalling is significant at regions of high transcription, even though they are codirectionally expressed relative to replication (35). The ChIP-based detection of replication stalling at the rRNA genes was later confirmed by direct analysis of replication intermediates using 2D gel electrophoresis, which detects replication fork intermediates directly (31). Additionally, when essential conflict resolution factors were depleted, an increased number of stalling events at even more codirectionally oriented genes (coding genes, distinct from rRNA loci) were discovered, indicating that in fact codirectionality does not solve the problem of conflicts and occurs pervasively during growth in rich media (31). This finding suggested that, in living cells, the replisome alone cannot effectively displace RNAPs, which contrasts with some of the prior in vitro findings that suggested codirectional encounters between the replisome and RNAP are benign (26, 49). It is important to note, however, that in the in vitro studies, the number of RNAPs oriented codirectionally was roughly 1/50 of that found on an rRNA operon or other highly transcribed ribosomal protein genes in vivo (6; see also supplementary discussion in Reference 35). Therefore, the discrepancies between the in vitro and in vivo studies were likely due to the difference in the number of RNAPs ahead of the fork in the reconstituted system.
3.2. Replisome Disassembly
Much of what is known about the consequences of conflicts on replication has been determined through both in vitro and population-based, in vivo studies. Genetic and molecular biology studies detecting replication restart proteins at conflict regions implied that at least some components of the replisome disassemble in a conflict (30, 35, 38). However, direct evidence for replisome disassembly due to conflicts in vivo did not exist until recently. Furthermore, the majority of in vivo studies on this topic took population-based approaches, in cells at various stages of replication. Therefore, it was unclear how frequently the replisome disassembles during a single cell cycle. In vivo single-molecule fluorescence microscopy (SMFM) became an extraordinarily useful tool to answer questions about the dynamics of conflicts in single cells. Recent work characterized the stoichiometry and lifetimes of replication proteins in growing cells (in both B. subtilis and E. coli) (28). These studies found that, remarkably, (a) a majority of cells have only one active replisome a significant proportion of the time, and (b) the replisome complexes are extremely short lived and stay intact for only several minutes rather than the expected 40 minutes during a cell cycle. The replication time is likely longer on agarose pads used to grow cells under the microscope, where the doubling time of cells is roughly 180 minutes. Therefore, although the SMFM studies estimated that the replisome disassembles at least five times per cell cycle—still a striking number, much higher than expected based on prior predictions—this is likely an underestimate due to the longer doubling time of cells under the microscope, which was used to obtain these numbers (28). These findings overall are significant given the expectation that there should be two replication forks per cell (and thus, two replisome complexes) and given that replication was thought to be largely a continuous process in vivo. Excitingly, the SMFM studies explained why only one replisome was detected for much of the cell cycle: Mangiameli et al. (28) found that shutting off transcription by brief rifampicin treatment recovers the second replication fork in the majority of cells and significantly lengthens the lifetime of replisome complexes. These results are consistent with conflicts causing pervasive replisome disassembly every cell cycle and the rate of DNA replication in living cells being largely a function of how frequently (and the degree of severity) by which the replisome encounters RNAPs. Consistent with the conflict model, mutations in rpoB, the beta subunit of RNAP, which rescue phenotypes associated with conflicts in genetic studies (1, 7, 63), also recovered the second fork in the majority of the cells.
3.3. Replication Restart
DNA replication initiates through DnaA activity at the origin, oriC, and proceeds bidirectionally to the terminus. In vitro studies of replisome components led to the model that DNA replication could proceed from origin to terminus without interruption due to the stability of these purified, reconstituted complexes. However, genetic studies had identified the Pri proteins long ago, and through a large body of work, demonstrated that Pri proteins are essential in bacteria, they recognize stalled replication fork structures, and they reassemble the replisome outside of oriC (37). Presumably, if the replisome synthesized DNA largely uninterrupted from oriC to the terminus, replication restart mechanisms would not be essential. The results of SMFM studies showing the high frequency of transcription impeding replication and disassembling the replisome in vivo finally provide at least one explanation for the essentiality of restart proteins. Mangiameli et al. (28) showed that PriA in B. subtilis and DnaC in E. coli are critical for fork recovery after transcription-induced disassembly, demonstrating that replication restart proteins are essential conflict resolution factors, and are critical for completion of replication every single cell cycle.
3.4. Fork Reversal
It has been proposed that head-on conflicts lead to replication fork reversal through reannealing of the nascent DNA strands to each other at stalled forks. Early work showed that phenotypes associated with loss of Holliday junction resolvase RuvABC and double-stranded break–processing complex RecBCD can be rescued by mutations in the β and β’ subunits of RNAP (30, 63). In subsequent work, it was shown in E. coli that inversion of rRNA genes increases the need for the double-stranded end processing enzyme RecBC, but not the homologous repair enzyme RecA in rich media (7). These results suggested that there are double-stranded ends generated at sites of head-on conflicts which are not necessarily recombination substrates. Given that RecA, which is required for homologous recombination, is not required for surviving head-on rRNA genes, the simplest explanation for this observation is that there are reversed forks at these regions being processed by RecBC and/or Ruv proteins (Figure 2). If so, these reversed forks are processed by double-stranded end resection, leading to the eventual reassembly of the replisome.
Figure 2.
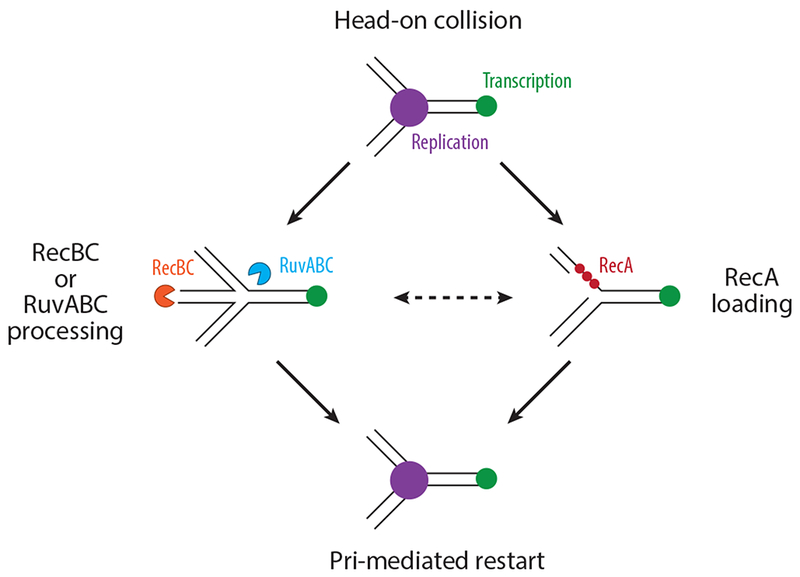
Models of potential fork-processing pathways downstream of a head-on collision. A head-on collision between replication (purple) and transcription (green) can lead to a reversed replication fork (left pathway), which is processed by RecBC (orange) and/or Ruv proteins (blue). These collisions can also result in RecA (red) loading (right pathway). RecBC processing may result in RecA loading, or RecA loading could result in initiation of replication fork reversal (dashed arrow). In both cases, replication restart occurs through Pri protein–mediated replisome reassembly.
Interestingly, data from studies in B. subtilis led to a slightly different model for how replication forks are processed and restarted at a head-on conflict region (38). It is important to first note that in contrast to the E. coli studies that used untranslated rRNA genes, the B. subtilis studies used coding genes, so the systems were different: different organisms and different conflict models. In any case, the two studies did lead to slightly different conclusions. Million-Weaver et al. (38) assessed the viability of strains containing the engineered conflicts in the absence of different DNA repair and recombination factors. These studies concluded that RecA, its loaders (RecO and AddAB), and the Holliday resolvase RecU are required for loading of replication restart proteins at the site of the head-on conflict as well as the subsequent efficiency of survival. These results suggested that DNA breaks and/or single-stranded gaps that need RecA processing are generated at head-on-conflict regions in B. subtilis. In E. coli, rRNA-induced head-on conflicts did not require RecA for viability, in contrast with findings from B. subtilis. The B. subtilis results, however, cannot rule out the possibility that the conflict is inducing RecA-dependent replication fork reversal (with subsequent processing by RecU and/or AddAB), which has been observed in vitro (52) and in eukaryotes upon replication stress (70). It remains to be determined if head-on transcription in B. subtilis leads to RecA-dependent replication fork reversal or whether the forks are processed differently in the only two organisms examined to date.
4. NUCLEIC ACID BARRIERS
4.1. R-Loops
Why are the consequences of head-on conflicts more severe than those of codirectional conflicts? Are collisions between replication and transcription driven by protein-protein encounters, or does the nature of the local DNA structure exacerbate conflicts? Recent work with both prokaryotic and eukaryotic organisms has demonstrated that head-on conflict severity is largely a result of R-loop formation (2, 18, 24, 68, 71). R-loops are nucleic acid structures where a nascent mRNA anneals to the coding DNA strand outside the transcription bubble, leaving the displaced noncoding DNA single stranded (62). Similar to head-on replication-transcription conflicts, R-loops can stall the replisome and lead to genomic instability (16, 68). R-loop formation was therefore predicted to be one of the major underlying reasons for the detrimental consequences of head-on conflicts. Two studies using inverted rRNA operons provided indirect evidence for the involvement of R-loops in conflicts (2, 71). Both studies showed that modulating levels of RNase HI (the enzyme that processes R-loops in E. coli) affected the survival of cells with inverted rRNA operons. But it was not until recently that R-loop formation was directly measured in bacteria (24). An antibody specific for RNA:DNA hybrids was used to measure R-loops at both endogenous and engineered conflict regions in B. subtilis. In wild-type cells, R-loops were more abundant at head-on conflict regions compared to codirectional conflict regions. Furthermore, loss of RNase HIII (the enzyme that processes R-loops in B. subtilis) increased R-loop abundance and completely stalled the replisome at the conflict regions, but only when the genes were oriented head-on to replication (Figure 1). It is presumed that R-loop formation at head-on genes is problematic for replication forks because the replicative helicase in prokaryotes moves 5’ to 3’ on the lagging strand and is not capable of unwinding RNA:DNA hybrids, which would form on the lagging strand in head-on conflicts (56). Interestingly, however, this model is likely incorrect given that the eukaryotic replicative helicase functions on the leading strand, yet the replisome is still impeded by R-loops resulting from head-on conflicts (18). Therefore, it seems that the R-loops are a significant problem for DNA replication progression regardless of which strand the helicase traverses. Despite these recent advances however, many important questions with regard to the role of R-loops and their formation remains. For example, what about transcription in the head-on orientation promotes R-loop formation, and why is it that the replisome cannot get past a stable R-loop if it is head-on, as was shown recently (18, 23, 24)? Are the R-loops simply more stable at a head-on conflict region, or do they form more frequently at these loci compared to codirectional conflict regions? Another question relates to DNA topology: Does the supercoiling status of the DNA at the conflict region play a role in conflict severity, and in particular R-loop formation? Below, we discuss some aspects of why supercoiling is potentially an interesting, yet largely unexplored, factor in conflicts and perhaps R-loop formation at conflict regions.
4.2. Supercoiling
Both transcription and replication require unwinding of the DNA duplex; thus, they generate positive supercoiling ahead (27). This would predict positive supercoil buildup at head-on conflict regions specifically, which would potentially stall and/or reverse replication forks in vivo. Only two studies have tested this model. In one study, 2D gel electrophoresis analysis of E. coli plasmids showed more DNA knots accumulated when RNAPs met replication head-on (47). These data are consistent with the model that the accumulation of positive supercoils at the head-on conflict region could have been distributed behind the fork to the newly synthesized DNA, subsequently entangling it. In another study, again analyzing plasmids in E. coli, replisome stalling was observed only within the transcription unit (41). This result contrasts with the model that positive supercoiling at head-on conflict regions is the cause of stalling. However, it should be noted that this study utilized a small transcription unit (only 400 bp) located on a small ColE1 plasmid. Because the amount of supercoiling is a function of the length of DNA unwound by either replication or transcription, the short length of the examined gene may have masked potential effects of supercoiling on replication stalling at head-on conflict regions. It will be important for future studies to analyze genes of different lengths in order to deepen our understanding of how supercoiling may disrupt DNA replication at head-on conflict regions. Furthermore, both aforementioned studies were carried out in wild-type E. coli cells where supercoiling was being actively resolved by topoisomerases. Experimental detection of (potentially transient) supercoils at conflict regions may require brief inactivation of topoisomerases that presumably resolve topological problems in wild-type cells. If positive supercoils accumulate at head-on conflict regions, then the consequences of head-on transcription may extend to neighboring genes, far beyond the conflict point—a phenomenon with interesting and potentially important implications.
If what is speculated about transient positive-supercoil accumulation at head-on conflict regions is true, then DNA gyrase, a type II topoisomerase that resolves positive supercoiling, may be enriched at head-on genes. Interestingly, DNA gyrase activity, coupled with transcription has been shown to promote R-loops in vitro (9, 29). Thus, the two types of nucleic acid structures discussed here may be intimately linked and, together, be responsible for many of the detrimental outcomes of head-on conflicts, as has been discussed recently (23). Given the catastrophic effects of R-loop formation at head-on conflict regions, it will be important for future studies to tease apart the connection between head-on conflicts, supercoiling, and R-loops.
5. CONFLICT RESOLUTION
5.1. Helicases: PcrA, Rep, UvrD, DinG
Cells possess a variety ofmechanisms to facilitate replication through transcription units. Accessory helicases can remove RNAPs and other protein complexes that would otherwise hinder DNA replication (17). In E. coli, cells are sensitive to inversion of rRNA operons when lacking the accessory helicases DinG and either Rep or UvrD (2). This is consistent with the idea that these types of accessory helicases can remove RNAPs from DNA, which has been demonstrated in vitro (26, 49). The B. subtilis essential accessory helicase PcrA plays a similar role in conflict resolution. Partial depletion of PcrA sensitizes cells to severe conflicts of both orientations (31). Given both the in vitro and in vivo evidence, it seems that the major role of accessory helicases is to remove stalled RNAP complexes from the DNA template to facilitate replication progression beyond the conflict point (Figure 1). However, recent work has suggested that DinG in E. coli and the Pif1 helicase in yeast are also involved in R-loop removal (2, 3). Therefore, although it is clear that accessory helicases are important for conflict resolution, whether their primary role in vivo is to directly remove RNAPs or to unwind R-loops has yet to be determined.
5.2. R-Loop Processing Enzymes: RNase HIII/RNase HI
As discussed above, R-loop formation at head-on genes leads to increased stalling of the replisome, increased mutagenesis, and decreased gene expression (24). RNase H enzymes, which degrade the RNA component of an RNA:DNA hybrid, found at Okazaki fragments and R-loops, are therefore critical conflict resolution factors (2, 18, 24, 46, 71) (Figure 1). In prokaryotes, RNase HI and RNase HIII enzymes degrade long RNA:DNA hybrids (45). Interestingly, RNase HIII and high co-orientation bias in the genomes are highly correlated (24). This implies that R-loop formation at head-on genes are particularly problematic for organisms with a high strand bias. Alternatively, this correlation may imply that organisms with a low co-orientation bias employ mechanisms other than RNase H enzymes (such as helicases) that can resolve R-loops at head-on genes. For example, in E. coli (where about 55% of genes are codirectional), RNase HI overexpression rescued growth defects that were apparent only in cells lacking the accessory helicase DinG, suggesting that RNase HI activity is not required to remove R-loops in wild-type cells (2). Additionally, other R-loop dependent defects in E. coli are apparent in cells lacking topoisomerase I or the essential replication initiation factor DnaA (8, 10, 64). In B. subtilis (where about 75% of genes are codirectional), RNase HIII becomes essential if there is high transcription in the head-on orientation (24). Furthermore, in wild-type cells, the increased mutation rate of head-on genes can be abolished through overexpression of RNase HIII. This suggests that the R-loop formation at head-on gene regions in B. subtilis is significant, even in wild-type cells, and highlights the fundamental importance of studying conflicts in different organisms. For example, RNase H is essential in Mycobacterium tuberculosis (67). Could this essentiality be linked to conflicts?
Although RNase H enzymes are conserved from bacteria to humans, B. subtilis cells lacking RNase HIII are viable in the laboratory, which suggests that Okazaki fragment processing is accomplished through redundant mechanisms (15). If the majority of genes are oriented codi-rectionally, then what is the physiologic role of RNase HIII in wild-type cells? Head-on stress response genes are not expressed to wild-type levels in the absence of RNase HIII, and this results in survival defects when the cells are exposed to stress (24). This is consistent with observations in E. coli, where R-loop resolution was found to be important for expression of stress response genes (5). Importantly, the inability to respond to stress is not limited to exogenous stresses introduced in laboratory culture. It was shown that Listeria monocytogenes, which is closely related to B. subtilis, is impaired for intracellular survival without RNase HIII (24). In L. monocytogenes some critical virulence genes (such as prfA) are oriented head-on. Presumably, some or all of these head-on virulence genes have impaired expression in cells lacking RNase HIII. This suggests that conflict resolution is also needed for intracellular pathogens to survive host defenses. Taken together, it seems RNase H enzymes are critical conflict-resolution factors, in particular when bacteria experience changes in environmental conditions.
5.3. RNAP Modulators: Gre, DksA, Rho, NusG
Factors that modulate RNAP movement can significantly alter the outcomes of a replication-transcription conflict (1, 7). E. coli DksA acts as both a transcription initiation and an elongation factor. Conflict is exacerbated in dksA mutants when they are starved for amino acids (61). Under these conditions, replication is blocked, which results in increased DNA damage. This is probably due to the ability of DksA to rescue backtracked RNAPs. Under amino acid starvation, translational pausing is increased, which uncouples translation and transcription. Without DksA, backtracked RNAPs would pile up. If not resolved, backtracked RNAPs act as a barrier to replisome progression, as previously proposed (11).
The transcription elongation factors GreA/B bind to the secondary channel of RNAP and promote its ability to cleave nascent transcripts. This activity aids in rescuing stalled RNAP complexes. In vitro, GreB helps removal of stalled elongation complexes in the face of a head-on replisome (49). Under amino acid starvation conditions, GreA can suppress the loss of DksA and promote replisome progression (61). This is consistent with studies using plasmid systems in E. coli, demonstrating that Gre proteins prevent genome instability by rescuing backtracked RNAPs (11). However, new work showing that Gre factors help favor transcriptional fidelity over recombinational repair may warrant revisiting current models of how RNAP modulators alleviate conflicts (59). Inhibition of recombination at conflict regions by the Gre proteins might prove to be deleterious given how important recombination is for restarting stalled replication forks at conflict regions (36, 38).
Termination of transcription in bacteria is accomplished by either mRNA secondary structure or the trans-acting factors Rho and NusG. Presumably, if transcription is not efficiently terminated, RNAPs could pile up, creating a barrier to replication. Rho and NusG act together to terminate transcription of specific subsets of genes. Recent work has demonstrated that Rho-dependent transcription termination can alleviate some of the inhibition of replication by transcription, thus preventing genomic instability (11, 66).
6. THE ROLE OF CONFLICTS IN MUTAGENESIS AND EVOLUTION
6.1. Spontaneous Mutagenesis
Arguably, one ofthe most important outcomes ofreplication-transcription conflicts is the increase in mutagenesis. In E. coli and B. subtilis, when a gene is in the head-on orientation it has an increased mutation rate compared to when it is co-oriented with replication (12, 39, 48, 55, 60). The significance of this phenomenon was first appreciated in 2013, when the rates of nonsynonymous and convergent mutations were measured for genes in the two orientations in B. subtilis (48). Importantly, both the increased mutagenesis and markers of adaptive evolution were also found to be prevalent in head-on genes of many divergent bacterial pathogens (32). In all species analyzed, a significantly higher rate of nonsynonymous mutations was found for head-on genes compared to codirectional genes. The original study in B. subtilis demonstrated that gene orientation–dependent differences in mutagenesis are transcription dependent (46). To date, similar results have been observed in several other studies, by three different groups, using a total of six reporter genes (hisC, metB, leuC, lacZ, thyP3, and rpoB) placed under various promoters, and examined at least at three different loci on the chromosome (12, 39, 55, 60). Each of these studies isolated the effect of gene orientation on the rate of spontaneous mutagenesis, resulting in a general consensus: Transcription increases mutation rates for genes of both orientations, but more so (roughly 2.5-fold) for the head-on orientation.
6.2. The Mechanism Behind Increased Mutagenesis of Head-On Genes
Several studies have investigated the molecular mechanisms of conflict-induced head-on gene mutagenesis. It was demonstrated that in B. subtilis, the differential rate of mutagenesis in the two orientations depends on the error-prone polymerase PolY1, which apparently functions in the same pathway as the transcription-coupled repair factor Mfd (39). PolY1 and Mfd (and the repair protein UvrA) were required for the increased mutagenesis of head-on genes as measured by three different reporters, hisC, metB, and leuC, which were each placed at two different chromosomal loci (39). These studies showed clearly that the loss of PolY1 or Mfd results in the same mutation rate for both the codirectional and head-on alleles (which is still higher than the rate observed in the absence of transcription). Although PolYl and Mfd are classically considered to be stationary-phase mutagenesis factors, fluctuation analyses indicated that the orientation- and transcription-dependent mutations measured in these studies arose during growth. This was a critical point, as the results of the fluctuation analyses clearly demonstrated that conflict-induced mutagenesis occurs during active replication and is distinct from stress-induced mutagenesis and stationary phase mutagenesis, which occur in the absence of replication (13). Recent work has demonstrated that the increase in mutagenesis of head-on genes also depends on R-loops, which we now know play critical roles in conflict severity (24). These findings hint at a connection between R-loops and transcription-coupled repair, which was previously suggested (69).
The effect of PolY1 and transcription on mutagenesis of head-on genes was further supported by a study in 2015 using comparative genomics analyses of retained base substitutions for all core genes (95% or more conservation) within sequenced genomes of B. subtilis (39). This study reported that the well-known mutational signature of TLS polymerases (T→C and A→G) are more prevalent and more strongly correlated with gene length in head-on genes compared to codirectional genes (39). One weakness of this analysis was that the converse C→T and G→A substitutions could not be separated from the former set. Therefore, future evolutionary analyses using ancestral genomes are needed to pinpoint the exact directionality of conflict-induced base substitutions. Determining the exact nature of the base substitutions generated by conflicts will provide further insight into both the molecular mechanisms causing mutagenesis at conflict regions and the downstream consequences of conflict-induced mutagenesis.
One recent study by Sankar and colleagues (55) examined the effect of transcription and orientation on the inactivating mutations that arose in a forward genetic reporter using a phage gene, thyP3. They confirmed previous results: Sankar et al. found that the head-on thyP3 reporter gene accumulated single base pair substitutions at a higher rate than codirectional thyP3 within the open reading frame. However, they also found that a single promoter base substitution (A→C) was overrepresented in the thyP3 reporter system, but this particular substitution was apparently independent of PolY1 and caused by a deamination event at the promoter (55).
An alternative, sequence context–based mechanism for the higher mutation rates of head-on genes was later proposed by Schroeder et al. In their study, the authors found that 5′-CCG-3′, 5′-GCG-3′, and 5′-CAC-3′ triplets have the highest transition rates in mutation accumulation (MA) line experiments (MA line experiments are further described below). Schroeder et al. (57) pointed out that these triplets are overrepresented in head-on genes. This led them to propose that the previously observed increase in retained base substitutions in head-on genes within B. subtilis genomes was due to the overabundance of these triplet sequences (57). However, this model was largely disproven by a follow-up study. Neither triplet sequences nor sequence context in general contributed to the increased rates of nonsynonymous mutations in head-on genes (33). This was not a surprising finding given that these particular triplet sequences were not present (or abundant) in the reporters used in prior studies that determined that the head-on orientation increases mutation rates.
6.3. Determining the Nature of Mutations Induced by Conflicts
To identify the evolutionary consequences of conflict-induced mutagenesis, it is imperative to determine the nature of mutations caused by these encounters. Although reporter genes can be a great tool to study mutation rates, using a single gene to describe the mutational footprint of conflicts, especially in the presence of selection, may generate misleading results. Reverse genetic reporters, where only a limited number of base pair changes can return the gene back to functionality (hisC, metB, etc.) or confer rifampicin resistance (rpoB), should not be used as a tool to describe the mutational footprint of conflicts. Forward genetic reporters generally allow for the detection of many more types of spontaneous mutations and therefore may be a better tool for determining mutational footprints, but even these types of reporters must be used with caution. For example, Sankar and colleagues (55) utilized the thyP3 gene to determine conflict-induced mutational footprints. Expression of thyP3 is toxic in the presence of trimethoprim, and therefore any mutation that inactivates its expression can be identified by sequencing the viable colonies after selection. They predominantly found promoter mutations in their experiments, which they attributed to conflicts (55). These types of inactivating mutations, however, may be overrepresented in this system because of the toxic nature of the thyP3 reporter. Base pair substitutions are less likely than indels or promoter substitutions to inactivate any given gene. Therefore, fewer of these types of mutations will be detected in forward-genetic assays that demand inactivation of toxic genes, such as thyP3. Therefore, the thyP3-based conclusion that conflicts predominantly generate promoter mutations should only be considered with these caveats in mind. All reporter systems therefore have fundamental constraints that need to be carefully considered if one is investigating mutational footprints, especially given that sequence context is well known to affect mutagenesis as well (22). So how can one accurately determine the nature of mutations caused by conflicts? We discuss better approaches below.
6.4. Genome-Wide Mutation Rate Experiments
Genome-wide measurements of mutagenesis are critical to studying the effect of gene orientation and conflicts on mutagenesis in an unbiased manner. Unfortunately, experimental determination of mutation rates is currently quite challenging because mutation rates across the genome are extremely low in wild-type cells. MA lines offer one genome-wide approach for experimentally measuring mutation rates. Typical MA line experiments include serial passage of parallel clonal populations for thousands of generations. These populations accumulate mutations across the genome, unless they are lethal. Coupling this method with whole-genome sequencing of each clonal line allows for the measurement of mutation rates in a relatively unbiased manner. However, MA line experiments have three critical drawbacks: (a) They do not control for transcription level or gene orientation, i.e., they do not measure the mutation rates of the same gene in either orientation when transcribed (or repressed) to the same level; (b) in the context of our discussion here, given that MA line experiments seek to minimize stress, head-on genes are generally repressed during these experiments and therefore do not experience conflicts; and (c) to detect enough mutations, the majority of these experiments must be performed in mutant backgrounds (such as a mutant of the mismatch repair protein MutS), where the cells are flooded with mutations of only a single source. As such, the nature of MA line experiments (where cells are flooded with fork errors because they are mismatch-repair defective) prevents detection of the true footprint of endogenous mutations in wild-type cells, especially the mutations that are derived from sources independent of the replication fork. Importantly, in the context of conflicts, MA lines do not allow for a robust analysis of gene orientation effects on mutagenesis, and the mutation rates of head-on genes are likely to be underestimated (25, 42, 43, 57). This hypothesis may explain the inconsistencies between reporter-based experiments, where a clear effect of orientation was isolated and identified, and MA line experiments, where such effects are either subtle or nonexistent (57).
Determining both mutation rates and the nature of the mutations caused by conflicts requires new approaches. Recently improved next-generation sequencing (NGS) increases mutation detection by expanding the number of bases subject to change. Such methods are useful in part because they analyze the full range of sequences, transcription levels, and other genomic architectural features endemic to naturally occurring genomes. However, standard NGS data sets contain one base detection error for every 103–104 bases, making it impossible to detect mutations as they arise within a population—at a rate of roughly 1 in 1010 nucleotides per generation, depending on the species in question. The various limitations inherent to the current cadre of methods (including the reporter-based, MA line, and forward genetic assays discussed above) may be overcome, at least in part, with use of new techniques. In particular, techniques that reduce the effective error rate of NGS could facilitate the detection of the ultrarare mutations that spontaneously arise within mixed populations. Such techniques will allow for a significantly more robust analysis of head-on versus codirectional reporter genes because they will identify the mutational spectra in and around a given reporter gene, irrespective of the phenotypic effects of those mutations (no selection bias). This is a superior method compared to reverse genetic analysis performed with hisC952, metB5, and leuC427, which identified rate but not mutational spectra, and the forward genetic analysis of thyP3, which only identified mutations that inactivate the gene. Two such methods, dubbed duplex sequencing and maximum-depth sequencing, were recently developed by the Loeb (51) and Nudler (21) labs, respectively. The recently published maximum depth-technique impressively determined a substitution rate of 10−5 per nucleotide prior to fixation. Hence, a more accurate and unbiased measurement of both mutation rate and spectra can be determined using these methods.
6.5. The Impact of Conflict-Induced Mutations on Evolution of Stress Response and Virulence
Experimental work demonstrated that head-on genes have a higher mutation rate than codirectional genes, and that this effect is independent of gene sequence and chromosomal location. Analysis of B. subtilis core genes showed that head-on genes have a higher rate of retained non-synonymous mutations, suggesting they have a higher spontaneous mutation rate (48). This was confirmed experimentally through careful mutation rate measurements in wild-type cells using various reporter genes (24, 39, 48). These observations strongly suggest that conflicts can affect bacterial evolution by accelerating the mutagenesis of chromosomally encoded head-on genes. If so, it would be important to consider the functions of head-on genes. Are they a random assortment of genes, or is there a pattern to their function? As discussed above, few head-on genes are expressed during growth in rich media, but many are upregulated during exposure to stress or during pathogenesis (4, 42, 43, 58). This repression/induction pattern should prevent conflicts and decrease mutation rates during normal growth. Conversely, during stress (e.g., during host invasion), the transcription of relevant head-on genes should be derepressed, leading to conflicts and an increase in their mutation rates. Hence, replication-transcription conflicts provide a simple and elegant mechanism by which cells can fine-tune their own evolution, ratcheting up or down the mutation rate of highly localized regions of the DNA like a rheostat. Therefore, it makes sense that genes involved in stress response—those that might need to change rapidly due to emerging existential threats to the cell—are major targets. The presence of key stress response and virulence genes in the head-on orientation implies that the evolution of stress tolerance and pathogenesis was potentially driven at least in part by conflicts. Further research in this area could yield exciting new insights.
ACKNOWLEDGMENTS
The authors would like to thank Christopher Merrikh and Maureen Thomason, as well as the anonymous reviewers, for critical discussions and review of this manuscript. K.S.L. is supported by an NIH Training Grant (AI055396), and H.M. is supported by the NIH New Innovator award (DP2GM110773).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Baharoglu Z, Lestini R, Duigou S, Michel B. 2010. RNA polymerase mutations that facilitate replication progression in the rep uvrD recF mutant lacking two accessory replicative helicases. Mol. Microbiol 77(2):324–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boubakri H, de Septenville AL, Viguera E, Michel B. 2010. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J. 29(1):145–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boulé J-B,Zakian VA. 2007. The yeast Pif1p DNA helicase preferentially unwinds RNA DNA substrates. Nucleic Acids Res. 35(17):5809–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Camejo A, Buchrieser C, Couvé E, Carvalho F, Reis O, et al. 2009. In vivo transcriptional profiling of Listeria monocytogenes and mutagenesis identify new virulence factors involved in infection. PLOS Pathog. 5(5):e1000449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng B, Rui S, Ji C, Gong VW, Van Dyk TK, et al. 2003. RNase H overproduction allows the expression of stress-induced genes in the absence of topoisomerase I. FEMS Microbiol. Lett 221(2):237–42 [DOI] [PubMed] [Google Scholar]
- 6.Condon C, French S, Squires C, Squires CL. 1993. Depletion of functional ribosomal RNA operons in Escherichia coli causes increased expression of the remaining intact copies. EMBOJ. 12(11):4305–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Septenville AL, Duigou S, Boubakri H, Michel B. 2012. Replication fork reversal after replication-transcription collision. PLOS Genet. 8(4):e1002622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dimude JU, Stockum A, Midgley-Smith SL, Upton AL, Foster HA, et al. 2015. The consequences of replicating in the wrong orientation: bacterial chromosome duplication without an active replication origin. mBio 6(6):e01294–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drolet M, Bi X, Liu LF. 1994. Hypernegative supercoiling of the DNA template during transcription elongation in vitro. J. Biol. Chem 269(3):2068–74 [PubMed] [Google Scholar]
- 10.Drolet M, Phoenix P, Menzel R, Massé E, Liu LF, Crouch RJ. 1995. Overexpression of RNase H partially complements the growth defect of an Escherichia coli AtopA mutant: R-loop formation is a major problem in the absence of DNA topoisomerase I. PNAS 92(8):3526–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dutta D, Shatalin K, Epshtein V, Gottesman ME, Nudler E. 2011. Linking RNA polymerase backtracking to genome instability in E. coli. Cell 146(4):533–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fijalkowska IJ, Jonczyk P, Tkaczyk MM, Bialoskorska M, Schaaper RM. 1998. Unequal fidelity of leading strand and lagging strand DNA replication on the Escherichia coli chromosome. PNAS 95(17):10020–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foster PL. 2006. Methods for determining spontaneous mutation rates. Methods Enzymol. 409:195–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.French S 1992. Consequences of replication fork movement through transcription units in vivo. Science 258(5086):1362–65 [DOI] [PubMed] [Google Scholar]
- 15.Fukushima S, Itaya M, Kato H, Ogasawara N, Yoshikawa H. 2007. Reassessment of the in vivo functions of DNA polymerase I and RNase H in bacterial cell growth. J. Bacteriol 189(23):8575–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gan W, Guan Z, Liu J, Gui T, Shen K, et al. 2011. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 25(19):2041–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guy CP, Atkinson J, Gupta MK, Mahdi AA, Gwynn EJ, et al. 2009. Rep provides a second motor at the replisome to promote duplication of protein-bound DNA. Mol. Cell 36(4):654–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamperl S, Bocek MJ, Saldivar JC, Swigut T, Cimprich KA. 2017. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell 170(4):774–86.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamperl S, Cimprich KA. 2016. Conflict resolution in the genome: how transcription and replication makeitwork. Cell 167(6):1455–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Helmrich A, Ballarino M, Tora L.2011. Collisions betweenreplication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 44(6):966–77 [DOI] [PubMed] [Google Scholar]
- 21.Jee J, Rasouly A, Shamovsky I, Akivis Y, Steinman SR, et al. 2016. Rates and mechanisms of bacterial mutagenesis from maximum-depth sequencing. Nature 534(7609):693–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koch RE. 1971. The influence of neighboring base pairs upon base-pair substitution mutation rates. PNAS 68(4):773–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuzminov A 2018. When DNA topology turns deadly—RNA polymerases dig in their R-loops to stand their ground: new positive and negative (super)twists in the replication–transcription conflict. Trends Genet. 34:111–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lang KS, Hall AN, Merrikh CN, Ragheb M, Tabakh H, et al. 2017. Replication-transcription conflicts generate R-loops that orchestrate bacterial stress survival and pathogenesis. Cell 170(4):787–99.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee H, Popodi E, Tang H, Foster PL. 2012. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. PNAS 109(41):E2774–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu B, Alberts BM. 1995. Head-on collision between a DNA replication apparatus and RNA polymerase transcription complex. Science 267(5201):1131–37 [DOI] [PubMed] [Google Scholar]
- 27.Liu LF, Wang JC. 1987. Supercoiling of the DNA template during transcription. PNAS 84(20):7024–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mangiameli SM, Merrikh CN, Wiggins PA, Merrikh H. 2017. Transcription leads to pervasive replisome instability in bacteria. eLife 6:e19848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masse E, Drolet M. 1999. Relaxation oftranscription-induced negative supercoiling is an essential function of Escherichia coli DNA topoisomerase I. J. Biol. Chem 274(23):16654–58 [DOI] [PubMed] [Google Scholar]
- 30.McGlynn P, Lloyd RG. 2000. Modulation of RNA polymerase by (p)ppGpp reveals a RecG-dependent mechanism for replication fork progression. Cell 101(1):35–45 [DOI] [PubMed] [Google Scholar]
- 31.Merrikh CN, Brewer BJ, Merrikh H. 2015. The B. subtilis accessory helicase PcrA facilitates DNA replication through transcription units. PLOS Genet. 11(6):e1005289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merrikh CN, Merrikh H. 2018. Gene inversion increases evolvability in bacteria. bioRxiv 293571. 10.1101/293571 [DOI]
- 33.Merrikh CN, Weiss E, Merrikh H. 2016. The accelerated evolution of lagging strand genes is independent of sequence context. Genome Biol. Evol 8(12):3696–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Merrikh H 2017. Spatial and temporal control of evolution through replication-transcription conflicts. Trends Microbiol. 25:515–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Merrikh H, Machón C, Grainger WH, Grossman AD, Soultanas P. 2011. Co-directional replication-transcription conflicts lead to replication restart. Nature. 470(7335):554–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Michel B, Grompone G, Florès M-J, Bidnenko V. 2004. Multiple pathways process stalled replication forks. PNAS 101(35):12783–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michel B, Sandler SJ. 2017. Replication restart in bacteria. J. Bacteriol 199(13):e00102–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Million-Weaver S, Samadpour AN, Merrikh H. 2015. Replication restart after replication-transcription conflicts requires RecAin Bacillus subtilis. J. Bacteriol 197(14):2374–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Million-Weaver S, Samadpour AN, Moreno-Habel DA, Nugent P, Brittnacher MJ, et al. 2015. An underlying mechanism for the increased mutagenesis of lagging-strand genes in Bacillus subtilis. PNAS 112(10):E1096–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mirkin EV, Castro Roa D, Nudler E, Mirkin SM. 2006. Transcription regulatory elements are punctuation marks for DNA replication. PNAS 103(19):7276–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mirkin EV, Mirkin SM. 2005. Mechanisms of transcription-replication collisions in bacteria. Mol. Cell. Biol 25(3):888–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mostertz J, Scharf C, Hecker M, Homuth G. 2004. Transcriptome and proteome analysis of Bacillus subtilis gene expression in response to superoxide and peroxide stress. Microbiology 150(2):497–512 [DOI] [PubMed] [Google Scholar]
- 43.Nicolas P, Mader U, Dervyn E, Rochat T, Leduc A, et al. 2012. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science 335(6072):1103–6 [DOI] [PubMed] [Google Scholar]
- 44.Nudler E 2012. RNA polymerase backtracking in gene regulation and genome instability. Cell 149(7):1438–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohtani N, Haruki M, Morikawa M, Crouch RJ, Itaya M, Kanaya S. 1999. Identification of the genes encoding Mn2+-dependent RNase HII and Mg2+-dependent RNase HIII from Bacillus subtilis: classification of RNases H into three families. Biochemistry 38:605–18 [DOI] [PubMed] [Google Scholar]
- 46.Ohtani N, Haruki M, Morikawa M, Kanaya S. 1999. Molecular diversities of RNases H. J. Biosci. Bioeng 88(1):12–19 [DOI] [PubMed] [Google Scholar]
- 47.Olavarrieta L, Hernández P, Krimer DB, Schvartzman JB. 2002. DNA knotting caused by head-on collision of transcription and replication. J. Mol. Biol 322(1):1–6 [DOI] [PubMed] [Google Scholar]
- 48.Paul S, Million-Weaver S, Chattopadhyay S, Sokurenko E, Merrikh H. 2013. Accelerated gene evolution through replication-transcription conflicts. Nature 495(7442):512–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pomerantz RT, O’Donnell M. 2008. The replisome uses mRNA as a primer after colliding with RNA polymerase. Nature 456(7223):762–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pomerantz RT, O’Donnell M. 2010. Direct restart of a replication fork stalled by a head-on RNA polymerase. Science 327(5965):590–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reid-Bayliss KS, Loeb LA. 2017. Accurate RNA consensus sequencing for high-fidelity detection of transcriptional mutagenesis-induced epimutations. PNAS 114(35):9415–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robu ME, Inman RB, Cox MM. 2001. RecA protein promotes the regression of stalled replication forks in vitro. PNAS 98(15):8211–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rocha EPC, Danchin A. 2003. Essentiality, not expressiveness, drives gene-strand bias in bacteria. Nat. Genet 34(4):377–78 [DOI] [PubMed] [Google Scholar]
- 54.Rocha EPC, Danchin A. 2003. Gene essentiality determines chromosome organisation in bacteria. Nucleic Acids Res. 31(22):6570–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sankar TS, Wastuwidyaningtyas BD, Dong Y, Lewis SA, Wang JD. 2016. The nature of mutations induced by replication-transcription collisions. Nature 535(7610):178–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santamaría D, de la Cueva G, Martínez-Robles ML, Krimer DB, Hernandez P, Schvartzman JB. 1998. DnaB helicase is unable to dissociate RNA-DNAhybrids: its implication in the polar pausing of replication forks at ColE1 origins. J. Biol. Chem 273(50):33386–96 [DOI] [PubMed] [Google Scholar]
- 57.Schroeder JW, Hirst WG, Szewczyk GA, Simmons LA. 2016. The effect of local sequence context on mutational bias of genes encoded on the leading and lagging strands. Curr. Biol 26(5):692–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scortti M, Monzó HJ, Lacharme-Lora L, Lewis DA, Vázquez-Boland JA. 2007. The PrfA virulence regulon. Microbes Infect. 9(10):1196–207 [DOI] [PubMed] [Google Scholar]
- 59.Sivaramakrishnan P, Sepúlveda LA, Halliday JA, Liu J, Núñez MAB, et al. 2017. The transcription fidelity factor GreA impedes DNA break repair. Nature 550(7675):214–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Srivatsan A, Tehranchi A, MacAlpine DM, Wang JD. 2010. Co-orientation of replication and transcription preserves genome integrity. PLOS Genet. 6(1):e1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tehranchi AK, Blankschien MD, Zhang Y, Halliday JA, Srivatsan A, et al. 2010. The transcription factor DksA prevents conflicts between DNA replication and transcription machinery. Cell 141(4):595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thomas M, White RL, Davis RW. 1976. Hybridization of RNA to double-stranded DNA: formation of R-loops. PNAS 73(7):2294–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Trautinger BW, Lloyd RG. 2002. Modulation of DNA repair by mutations flanking the DNA channel through RNA polymerase. EMBOJ. 21(24):6944–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Usongo V, Nolent F, Sanscartier P, Tanguay C, Broccoli S, et al. 2008. Depletion of RNase HI activity in Escherichia coli lacking DNA topoisomerase I leads to defects in DNA supercoiling and segregation. Mol. Microbiol 69(4):968–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang JD, Berkmen MB, Grossman AD. 2007. Genome-wide coorientation of replication and transcription reduces adverse effects on replication in Bacillus subtilis. PNAS 104(13):5608–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Washburn RS, Gottesman ME. 2011. Transcription termination maintains chromosome integrity. PNAS 108(2):792–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Watkins HA, Baker EN. 2010. Structural and functional characterization of an RNase HI domain from the bifunctional protein Rv2228c from Mycobacterium tuberculosis. J. Bacteriol 192(11):2878–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wellinger RE, Prado F, Aguilera A. 2006. Replication fork progression is impaired by transcription in hyperrecombinant yeast cells lacking a functional THO complex. Mol. Cell. Biol 26(8):3327–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wimberly H, Shee C, Thornton PC, Sivaramakrishnan P, Rosenberg SM, Hastings PJ. 2013. R-loops and nicks initiate DNA breakage and genome instability in non-growing Escherichia coli. Nat. Commun 4:2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, et al. 2015. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol 208(5):563–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang Y, Mooney RA, Grass JA, Sivaramakrishnan P, Herman C, et al. 2014. DksA guards elongating RNA polymerase against ribosome-stalling-induced arrest. Mol. Cell 53(5):766–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zheng W-X, Luo C-S, Deng Y-Y, Guo F-B, Koonin EV, et al. 2015. Essentiality drives the orientation bias of bacterial genes in a continuous manner. Sci. Rep 5:16431. [DOI] [PMC free article] [PubMed] [Google Scholar]