

Abstract
We have developed a widely applicable nucleophilic (radio)fluoro-click reaction of ynamides with readily available and easy handling KF(18F). The reactions exhibited high functional group tolerance and needed only ambient atmosphere. Most importantly, this is the first 18F addition protocol to C-C unsaturated bonds with extraordinarily high radiochemical yields.
Keywords: alkali fluoride, hydrogen bonding network, fluorination, radio-fluorination, HFIP
Due to fluorine’s unique properties such as a small size and a metabolically resistant C-F bond, the selective substitution of hydrogen by fluorine constitutes a key strategy in drug discovery and material research.[1],[2] More specifically, positron emission tomography (PET) based on the radioactive fluorine isotope (18F) has become increasingly important in diagnosis and drug discovery.[3] But common fluorination reagents, including nucleophilic fluorination reagents (e.g., HF-based reagents, DAST) and electrophilic fluorination reagents (e.g., NFSI and Selectfluor) are expensive, corrosive or toxic, and corresponding fluorination processes often have very poor atom-economy. Among fluorination reagents, alkali metal fluorides (MF), such as KF, are inexpensive and easy to handle. Especially for radioactive fluorine isotope introduction, the primary source of 18F is the alkali metal salt of 18F-. For the use of MF as fluorination reagent,[4] especially in the introduction of 18F to organic molecules,[5] the most common method is the nucleophilic displacement of alkyl or aryl halides, pseudo halides, ammonium or iodonium salts (Scheme 1a). In recent years, there has been great progress in transition metal catalyzed (radio)fluorinations using MF (Scheme 1b).[6], [7] However, these metal catalyzed processes need more sophisticated conditions and are usually limited to the synthesis of aryl fluorides. Clearly, there is a need and a market for expanding the use of alkali metal fluorides to other types of fluorination reactions, such as addition reactions. We believe that the introduction of readily available MF (18F) to an alkyne, under simple conditions and with great efficiency, could pave the way to applications in medicine and other fields in a manner not too dissimilar to the click reaction (copper-catalyzed reaction of an organo azide with an alkyne).[8] Thus, we have named this transformation a fluoro-click reaction.
Scheme 1.
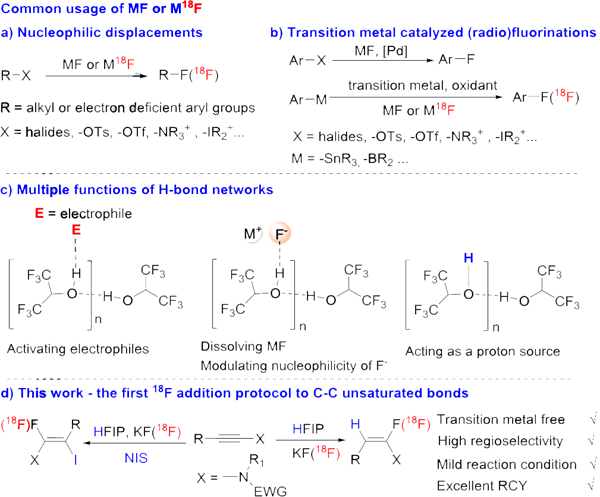
Nucleophilic (radio)fluoro-click reaction using alkali fluorides.
We propose that a hydrogen-bonding network can activate alkali metal fluorides such as KF. Due to σ-cooperativity or non-additivity (hydrogen bond energy of a chain of H-bonds can be greater than the total energies of the individual links),[9] strong hydrogen bonding donors such as HFIP (hexafluoro-2-propanol) could form a H-bond network or aggregation (Scheme 1c).[10] This hydrogen-bonding network is a better H-bond donor than a single HFIP, activating the electrophile via strong hydrogen bonding interaction (Scheme 1c). Indeed, hydrogen bonding donor solvents like HFIP have been shown to provide significant rate enhancements in many reactions,[10a-c, 10f] and kinetic data suggest that hydrogen-bonding solvent aggregates play an important role.[10d]
Another problem of MF salts is their poor solubility in most organic solvents. Since fluoride itself is a good hydrogen bonding acceptor, a H-bond network could complex with MF and make it highly soluble. Indeed, alkali fluorides such as KF have good solubility in HFIP at room temperature. Also, it has been reported that F- is a better nucleophile than other halides (Br-/I-) in hydrogen-bonding donor solvents like t-BuOH, and t-BuOH due to their positive effect on the ‘effective fluoride nucleophilicity’.[11] Because HFIP is a stronger H-bond donor than those alcohols, we expect it will have an even stronger effect in the modulation of the nucleophilicity of fluoride. Lastly, a H-bond network generated from HFIP could also act as a proton source in hydrofluorination (Scheme 1c), as shown by Doyle and coworkers in their copper-catalyzed H-F insertion into α-diazocarbonyl compounds using HFIP as proton source.[12] We are now glad to report the first 18F addition protocol to an alkyne via a nucleophilic (radio)fluoro-click reaction, enabled by a hydrogen bonding cluster and using the readily available KF(18F) (Scheme 1d).
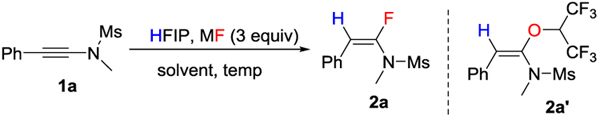
We used the hydrofluorination of ynamide 1a as our model reaction (Table 1). Ynamides are readily available compounds that have found wide applications in organic synthesis.[13] Although the hydrofluorination of ynamides have been reported,[14] these methods are based on hydrogen fluoride or silver fluoride as fluorination reagents, all of which are not environmentally friendly. More importantly, the introduction of 18F is difficult using these methods. Initially, we chose HFIP as our hydrogen bonding activator and proton source. Screening of various alkali metal fluorides indicated that metal fluorides with bulkier counterions gave better results (Table 1, entries 1-4, efficiency LiF < NaF < KF ~ CsF). Because KF and CsF gave similarly good results, and KF is more inexpensive, we chose KF as our fluorination reagent. Also, a higher temperature promoted the formation of HFIP addition by-product 2a’ (Table 1, entry 5). We investigated the effect of solvents (Table 1, entries 5-7). Reducing the amount of HFIP (Table 1, entry 6) or using the weaker hydrogen-bonding donor solvent trifluoroethanol (TFE) diminished the reactivity (Table 1, entry 7). The amount of HFIP addition by-product 2a’ was effectively reduced by increasing the number of equivalents of KF (Table 1, entry 8).
Table 1.
Screening of reaction conditions.[a]
 |
| Entry | MF | Temp / °C | Solvent | Yield (%) 2a:2a’:1a |
|---|---|---|---|---|
| 1 | LiF | rt | HFIP | 3/4/93 |
| 2 | NaF | rt | HFIP | 6/4/90 |
| 3 | KF | rt | HFIP | 64/31/4 |
| 4 | CsF | rt | HFIP | 62/33/5 |
| 5 | KF | 70 | HFIP | 32/59/9 |
| 6 | KF | 70 | HFIP/DCE (1:1) | 33/34/32 |
| 7 | KF | rt | TFE | 2/0/98 |
| 8 | KFc | rt | HFIP | 84/14/1 |
Reaction conditions: 1a (0.2 mmol), MF (0.6 mmol), solvent (0.4 mL), 8 h.
Determined by GC-MS analysis.
6 equiv of KF was used.
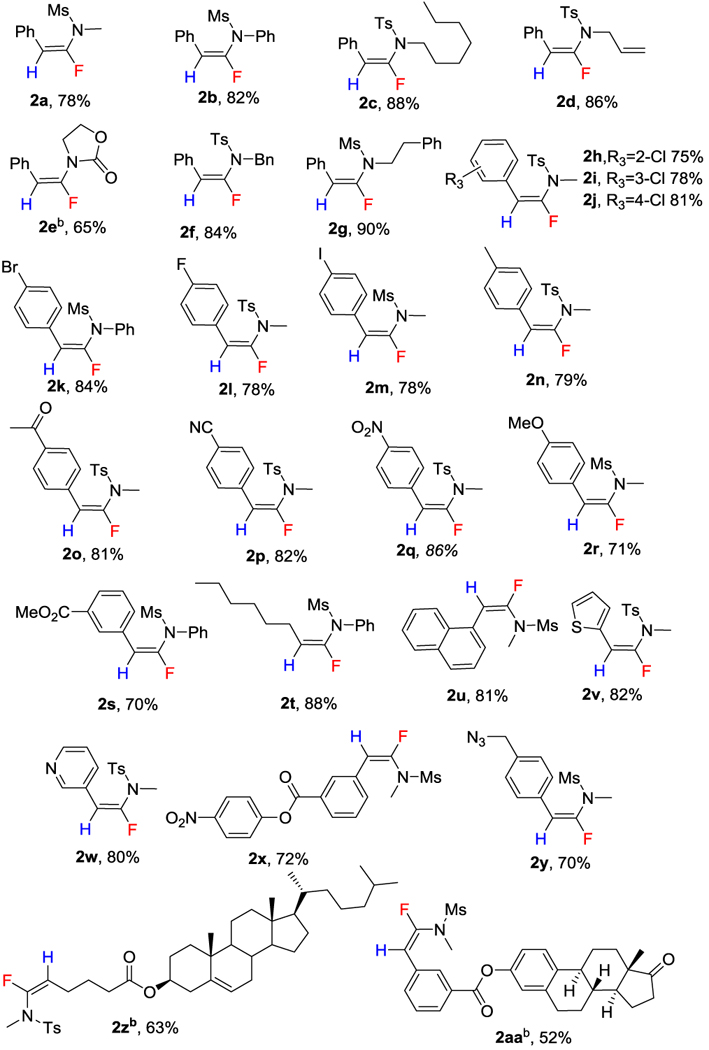
Having found the optimized conditions, we then explored the scope and functional group tolerance of our hydrofluorination protocol (Table 2). First, we evaluated the effect of R2 substitution in ynamides 1 (Table 2, 2a-2g). The structure of R2 (alkyl groups, aryl groups and allyl group, benzyl group, heterocyclic groups) played only a minor role good yields of 2 were obtained regardless (Table 2, 2a-2g). We evaluated the effect of R1 substitution (Table 2, 2h-2u). When R1 was a benzylic group, substitution patterns (ortho, meta, para) and the presence of electron donating groups or electron withdrawing groups on R1 exerted little influence on the efficiency of the reaction (Table 2, 2h-2s). This reaction also tolerated a wide variety of other R1 variations: simple alkyl group (Table 2, 2t), fused aromatic (Table 2, 2u), or even heteroaromatics, such as thiophene and pyridine (Table 2 , 2v-2w). To demonstrate the applicability of our method, we synthesized ynamides tethered to activated ester and azido functionalities (Table 2, 2x-2y) or complex natural products (Table 2, 2z, 2aa); in all these cases our reaction worked very well.
Table 2.
Scope for the synthe sis of α-fluoroenamides 3.a
 |
|---|
 |
Reaction conditions: 1 (0.2 mmol), KF (1.2 mmol), HFIP (0.5 mL), rt. All yields are isolated yields.
Run at 60 °C.
We were quite pleased to find that our protocol could be used in the radio fluorination of ynamides 1 (Table 3). In all cases, we got excellent radiochemical yields (RCY). Various functional groups, including halogen, ester, nitrile, and nitro did not affect the efficiency of the reaction, and heterocycles such as thiophene and pyridine were well tolerated. To demonstrate the applicability of our method in biological applications, we prepared ynamide 2x, an organoazide (Table 3, [F18]2aa) and a biologically active compound (Table 3, [F18]2z) with great efficiency. In principle, an activated ester could be tethered to biomolecules such as peptides or proteins easily in radiochemistry via formation of amide linkage.[15] And the azide linker could be easily attached to bioactive compounds via click-chemistry.[16] It should be noted that our method does not require the use of expensive polycyclic multidentate cation ligands such as K222, which has been commonly used in other radiofluorination protocols to increase the nucleophilicity of 18F-.
Table 3.
Radio fluorination of ynamides 1.a
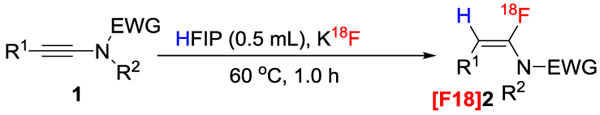 |
|---|
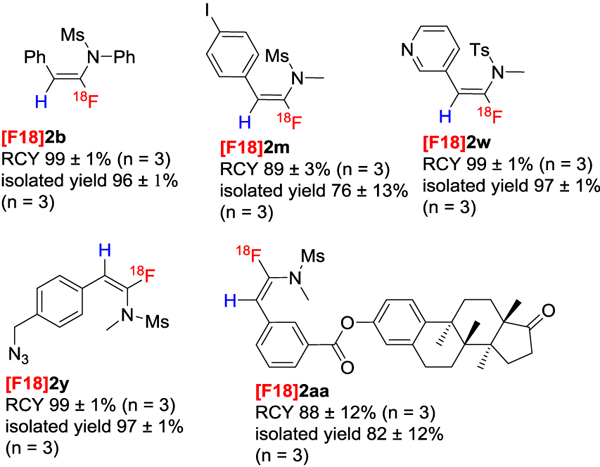 |
Reaction conditions: 1 (0.015 mmol) in HFIP (100 μl) was mixed with 18F (~ 0.3 mCi, in 50 μL HFIP), 60 oC, 1 h. Reactions were performed 3 times; radiochemical yields were determined by radio-HPLC and are given in the form mean ± standard deviation.
Our system was also extended to iodofluorinations[14a] (Table 4). When the iodination reagent NIS was incorporated to our fluorination system (KF/HFIP), we obtained the iodofluorination product 3 (Table 4a). The yields were only moderate, possibly due to the relatively instability of the resulting vinyl-iodides. To our delight, the corresponding radio-iodofluorination was very efficient: close to 80% RCY yield was obtained (Table 4b).
Table 4.
 |
 |
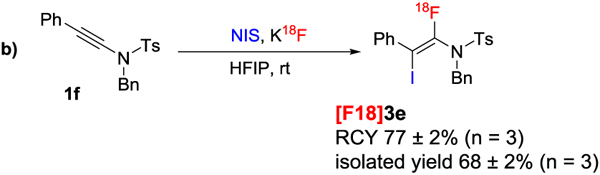 |
Reaction conditions: 1 (0.2 mmol), KF (1.2 mmol), NIS (0.24 mmol) HFIP (0.4 mL), DCM (0.1 mL), rt, 8 h.
Reaction conditions: 1 (0.015 mmol),NIS (2.7mg) in HFIP (100 μl) was mixed with 18F (~ 0.3 mCi, in 50 μl HFIP), 60°C, 1 h. Reactions were performed 3 times; radiochemical yields were determined by radio-HPLC and are given in the form mean ± standard deviation.
We also investigated the serum stability of our synthesized 18F-tracer in fetal bovine serum (incubated at 37 oC for 2 h, see section 7 of SI). Our results, based on three radiotracers (2b, 2w, 2y) showed that they were stable for up to 2 h in serum.
Our methodology is scalable (Scheme 2a) and the reaction products could be further utilized in transition metal catalyzed cross-coupling reactions. As shown in Scheme 2b and 2c, the Suzuki coupling of 2m with phenyl boronic acid gave moderate yield of the coupling product 4a, and the Sonogashira coupling of 2m only furnished the aromatic substitution product 4b.
Scheme 2.
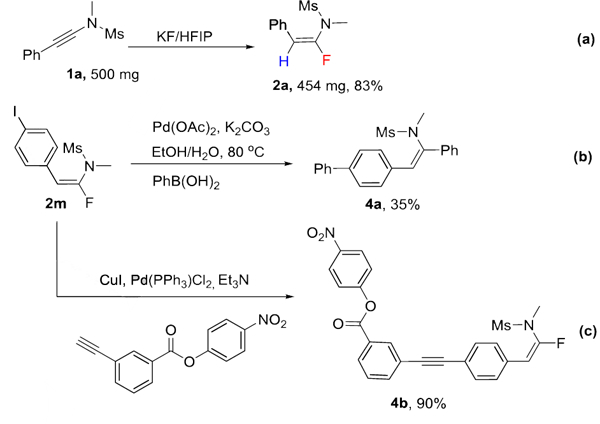
Gram scale reaction and further synthetic manipulations.
The proposed mechanism is shown in Scheme 3. The hydrogen-bonding network generated from HFIP facilitates the rate-determining proton transfer step, which produces the key intermediate--keteniminium A.[17] Keteniminium A possesses a linear geometry,[17] with its upper face being sterically hindered by the R1 group, thus favoring the nucleophile (fluoride) syn approach (formation of the syn-addition product) (Scheme 3., top).[18] On the other hand, in the presence of NIS, an iododium B forms instead because NIS is a strong electrophile; ring-opening of B by fluoride yields the anti-addition product 3 (Scheme 3., bottom).
Scheme 3.

Proposed mechanism.
In summary, we have developed a widely applicable synthesis of α-fluoroenamides using KF(18F). The reactions exhibited high functional groups tolerance and needed only ambient atmosphere. Most importantly, this is the first 18F addition protocol to C-C unsaturated bonds with extraordinary high radiochemical yields. Other (radio)fluorination systems based on KF/HFIP system are currently being investigated in our laboratories.
Supplementary Material
Acknowledgements
We are grateful to the National Science Foundation of China (NSFC-21672035) and China Recruitment Program of Global Experts for financial support. G.B.H. is grateful to the National Institutes of Health for financial support (1R01GM121660-01). X.Z thanks the China Scholarship Council for financial support. X.Z. is also grateful to Prof. Xiangming Zhu (University College Dublin) for his support and encouragement.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Xiaojun Zeng, College of Chemistry, Chemical Engineering and Biotechnology Donghua University, Shanghai 201620, China bo.xu@dhu.edu.cn.
Dr Junling Li, Department of Diagnostic Radiology, University of Louisville, Louisville, KY 40292 USA..
Dr Chin K. Ng, Department of Diagnostic Radiology, University of Louisville, Louisville, KY 40292 USA..
Gerald B. Hammond, Department of Chemistry, University of Louisville, Louisville, KY 40292 USA., gb.hammond@louisville.edu
Dr Bo Xu, College of Chemistry, Chemical Engineering and Biotechnology Donghua University, Shanghai 201620, China bo.xu@dhu.edu.cn.
References
- [1].a) Chambers RD, Fluorine in organic chemistry, Blackwell Publishing Ltd./CRC Press, Boca Raton, Fl, 2004; [Google Scholar]; b) Hiyama T, Organofluorine compounds, chemistry and applications, Springer-Verlag, Berlin, 2000; [Google Scholar]; c) Kirsch P, Modern fluoroorganic chemistry, Wiley-VCH, Weinheim, 2004; [Google Scholar]; d) Muller K, Faeh C, Diederich F, Science 2007, 317, 1881–1886; [DOI] [PubMed] [Google Scholar]; e) Schlosser M, Angew. Chem. Int. Ed. 1998, 37, 1496; [DOI] [PubMed] [Google Scholar]; f) Soloshonok VA, Fluorine-containing synthons, ACS symposium series 911, Oxford University Press,Washington, D.C, 2005; [Google Scholar]; g) Uneyama K, Organofluorine Chemistry, Blackwell publishing, Oxford, 2006. [Google Scholar]
- [2].Zeng X, Liu S, Shi Z, Liu G, Xu B, Angew. Chem. Int. Ed. 2016, 55, 10032–10036. [DOI] [PubMed] [Google Scholar]
- [3].Cole EL, Stewart MN, Littich R, Hoareau R, Scott PJH, Curr. Top. Med. Chem. 2014, 14, 875–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Champagne PA, Desroches J, Hamel J-D, Vandamme M, Paquin J-F, Chem. Rev. 2015, 115, 9073–9174. [DOI] [PubMed] [Google Scholar]
- [5].a) van der Born D, Pees A, Poot AJ, Orru RVA, Windhorst AD, Vugts DJ, Chem. Soc. Rev. 2017, 46, 4709–4773; [DOI] [PubMed] [Google Scholar]; b) Krishnan HS, Ma L, Vasdev N, Liang SH, Chem. Eur. J, n/a–n/a; [Google Scholar]; c) Clark J, O’Hagan D, J. Fluorine Chem. 2017; [Google Scholar]; d) Qingzhi Z, Sergio DA, Ian NF, Lutz FS, Matteo Z, David OH, Chemistry - A European Journal 2016; [Google Scholar]; e) Thompson S, Onega M, Ashworth S, Fleming IN, Passchier J, Hagan DO, Chem. Commun. 2015; [DOI] [PubMed] [Google Scholar]; f) Li J, Gray BD, Pak KY, Ng CK, J. Labelled Compd. Radiopharm. 2012, 55, 149–154; [Google Scholar]; g) Sadovski O, Hicks JW, Parkes J, Raymond R, Nobrega J, Houle S, Cipriano M, Fowler CJ, Vasdev N, Wilson AA, Biorg. Med. Chem. 2013, 21, 4351–4357; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Tredwell M, Gouverneur V, Angew. Chem. Int. Ed. 2012, 51, 11426–11437; [DOI] [PubMed] [Google Scholar]; i) Gengyang Y, Feng W, Nickeisha AS, Lu W, Benjamin HR, Neil V, Pingping T, Steven HL, Chem. Commun. 2016; [Google Scholar]; j) Mohammad BH, Sanjay T, Yong-Sok L, Cheryl LM, Shuiyu L, Victor WP, J. Org. Chem. 2015. [Google Scholar]
- [6].Watson DA, Su M, Teverovskiy G, Zhang Y, Garcia-Fortanet J, Kinzel T, Buchwald SL, Science 2009, 325, 1661–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Taylor NJ, Emer E, Preshlock S, Schedler M, Tredwell M, Verhoog S, Mercier J, Genicot C, Gouverneur V, J. Am. Chem. Soc. 2017, 139, 8267–8276; [DOI] [PubMed] [Google Scholar]; b) Brooks AF, Topczewski JJ, Ichiishi N, Sanford MS, Scott PJH, Chem. Sci. 2014, 5, 4545–4553; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Makaravage KJ, Brooks AF, Mossine AV, Sanford MS, Scott PJH, Org. Lett. 2016, 18, 5440–5443; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Mossine AV, Brooks AF, Makaravage KJ, Miller JM, Ichiishi N, Sanford MS, Scott PJH, Org. Lett. 2015, 17, 5780–5783; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Preshlock S, Calderwood S, Verhoog S, Tredwell M, Huiban M, Hienzsch A, Gruber S, Wilson TC, Taylor NJ, Cailly T, Schedler M, Collier TL, Passchier J, Smits R, Mollitor J, Hoepping A, Mueller M, Genicot C, Mercier J, Gouverneur V, Chem. Commun. 2016, 52, 8361–8364; [DOI] [PubMed] [Google Scholar]; f) Tredwell M, Preshlock SM, Taylor NJ, Gruber S, Huiban M, Passchier J, Mercier J, Génicot C, Gouverneur V, Angew. Chem. Int. Ed. 2014, 53, 7751–7755; [DOI] [PubMed] [Google Scholar]; g) Shi H, Braun A, Wang L, Liang SH, Vasdev N, Ritter T, Angew. Chem. Int. Ed. 2016, 55, 10786–10790; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Neumann CN, Hooker JM, Ritter T, Nature 2016, 534, 369–373; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Matthew SM, Stephen T, Allen FB, Shane WK, Peter JHS, Melanie SS, Org. Lett. 2017. [Google Scholar]
- [8].a) Thirumurugan P, Matosiuk D, Jozwiak K, Chem. Rev. 2013, 113, 4905–4979; [DOI] [PubMed] [Google Scholar]; b) Tiwari VK, Mishra BB, Mishra KB, Mishra N, Singh AS, Chen X, Chem. Rev. 2016, 116, 3086–3240; [DOI] [PubMed] [Google Scholar]; c) Gehringer M, Laufer SA, Angew. Chem., Int. Ed. 2017, Ahead of Print: 10.1002/anie.201710195. [DOI] [Google Scholar]
- [9].a) Steiner T, Angew. Chem. Int. Ed. 2002, 41, 48–76; [Google Scholar]; b) Jeffrey GA, Crystallography Reviews 2003, 9, 135–176; [Google Scholar]; c) Pihko PM, Hydrogen bonding in organic synthesis, Wiley-VCH, Weinheim, 2009. [Google Scholar]
- [10].a) Liu W, Wang H, Li C-J, Org. Lett. 2016, 18, 2184–2187; [DOI] [PubMed] [Google Scholar]; b) Colomer I, Batchelor-McAuley C, Odell B, Donohoe TJ, Compton RG, J. Am. Chem. Soc. 2016, 138, 8855–8861; [DOI] [PubMed] [Google Scholar]; c) Tian Y, Xu X, Zhang L, Qu J, Org. Lett. 2016, 18, 268–271; [DOI] [PubMed] [Google Scholar]; d) Berkessel A, Adrio JA, J. Am. Chem. Soc. 2006, 128, 13412–13420; [DOI] [PubMed] [Google Scholar]; e) Wencel-Delord J, Colobert F, Org. Chem. Front. 2016, 3, 394–400; [Google Scholar]; f) Zeng X, Liu S, Shi Z, Xu B, Org. Lett. 2016, 18, 4770–4773. [DOI] [PubMed] [Google Scholar]
- [11].a) Kim DW, Song CE, Chi DY, J. Am. Chem. Soc. 2002, 124, 10278–10279; [DOI] [PubMed] [Google Scholar]; b) Lee J-W, Oliveira MT, Jang HB, Lee S, Chi DY, Kim DW, Song CE, Chem. Soc. Rev. 2016, 45, 4638–4650; [DOI] [PubMed] [Google Scholar]; c) Kim DW, Ahn D-S, Oh Y-H, Lee S, Kil HS, Oh SJ, Lee SJ, Kim JS, Ryu JS, Moon DH, Chi DY, J. Am. Chem. Soc. 2006, 128, 16394–16397; [DOI] [PubMed] [Google Scholar]; d) Egli M, Pallan PS, Allerson CR, Prakash TP, Berdeja A, Yu J, Lee S, Watt A, Gaus H, Bhat B, Swayze EE, Seth PP, J. Am. Chem. Soc. 2011, 133, 16642–16649; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kim DW, Jeong, Lim ST, Sohn M-H, Katzenellenbogen JA, Chi DY, J. Org. Chem. 2008, 73, 957–962; [DOI] [PubMed] [Google Scholar]; f) Kim DW, Jeong H-J, Lim ST, Sohn M-H, Angew. Chem. Int. Ed. 2008, 47, 8404–8406; [DOI] [PubMed] [Google Scholar]; g) Engle KM, Pfeifer L, Pidgeon GW, Giuffredi GT, Thompson AL, Paton RS, Brown JM, Gouverneur V, Chem. Sci. 2015, 6, 5293–5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gray EE, Nielsen MK, Choquette KA, Kalow JA, Graham TJA, Doyle AG, J. Am. Chem. Soc. 2016, 138, 10802–10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) DeKorver KA, Li H, Lohse AG, Hayashi R, Lu Z, Zhang Y, Hsung RP, Chem. Rev. 2010, 110, 5064–5106; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Evano G, Coste A, Jouvin K, Angew. Chem. Int. Ed. 2010, 49, 2840–2859; [DOI] [PubMed] [Google Scholar]; c) Wang X-N, Yeom H-S, Fang L-C, He S, Ma Z-X, Kedrowski BL, Hsung RP, Acc. Chem. Res. 2014, 47, 560–578; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hu L, Xu S, Zhao Z, Yang Y, Peng Z, Yang M, Wang C, Zhao J, J. Am. Chem. Soc. 2016, 138, 13135–13138; [DOI] [PubMed] [Google Scholar]; e) Xu S, Liu J, Hu D, Bi X, Green Chem. 2015, 17, 184–187; [Google Scholar]; f) Zhang Y, Tetrahedron Lett. 2005, 46, 6483–6486; [Google Scholar]; g) Zhang Y, Tetrahedron 2006, 62, 3917–3927; [Google Scholar]; h) Wu W, Jiang H, Acc. Chem. Res. 2014, 47, 2483–2504; [DOI] [PubMed] [Google Scholar]; i) Li J, Yang W, Yang S, Huang L, Wu W, Sun Y, Jiang H, Angew. Chem. Int. Ed. 2014, 53, 7219–7222; [DOI] [PubMed] [Google Scholar]; j) Liu L-P, Malhotra D, Paton RS, Houk KN, Hammond GB, Angew. Chem. Int. Ed. 2010, 49, 9132–9135; [DOI] [PubMed] [Google Scholar]; k) Li Y, Liu X, Jiang H, Liu B, Chen Z, Zhou P, Angew. Chem. Int. Ed. 2011, 50, 6341–6345; [DOI] [PubMed] [Google Scholar]; l) Li J, Yang S, Huang L, Chen H, Jiang H, RSC Adv. 2013, 3, 11529–11532; [Google Scholar]; m) Siva Reddy A, Kumara Swamy KC, Angew. Chem., Int. Ed. 2017, 56, 6984–6988. [DOI] [PubMed] [Google Scholar]
- [14].a) Xi Y, Zhu G, Tang L, Ma S, Zhang D, Zhang R, He G, Zhu H, Org. Biomol. Chem. 2017, 15, 7218–7226; [DOI] [PubMed] [Google Scholar]; b) Zhu G, Qiu S, Xi Y, Ding Y, Zhang D, Zhang R, He G, Zhu H, Org. Biomol. Chem. 2016, 14, 7746–7753; [DOI] [PubMed] [Google Scholar]; c) He G, Qiu S, Huang H, Zhu G, Zhang D, Zhang R, Zhu H, Org. Lett. 2016, 18, 1856–1859; [DOI] [PubMed] [Google Scholar]; d) Metayer B, Compain G, Jouvin K, Martin-Mingot A, Bachmann C, Marrot J, Evano G, Thibaudeau S, J. Org. Chem. 2015, 80, 3397–3410; [DOI] [PubMed] [Google Scholar]; e) Che J, Li Y, Zhang F, Zheng R, Bai Y, Zhu G, Tetrahedron Lett. 2014, 55, 6240–6242; [Google Scholar]; f) Compain G, Jouvin K, Martin-Mingot A, Evano G, Marrot J, Thibaudeau S, Chem. Commun. 2012, 48, 5196–5198. [DOI] [PubMed] [Google Scholar]
- [15].Liu S, Chin FT, Cheng Z, Chen X, in Radiochemical Syntheses, John Wiley & Sons, Inc., 2012, pp. 51–60. [Google Scholar]
- [16].Li Z-B, Wu Z, Chen K, Chin FT, Chen X, Bioconjugate Chem. 2007, 18, 1987–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Stang PJ, Summerville R, J. Am. Chem. Soc. 1969, 91, 4600–4601; [Google Scholar]; b) Zhang S-L, Wan H-X, Deng Z-Q, Org. Biomol. Chem. 2017, 15, 6367–6374. [DOI] [PubMed] [Google Scholar]
- [18].Métayer B, Compain G, Jouvin K, Martin-Mingot A, Bachmann C, Marrot J, Evano G, Thibaudeau S, J. Org. Chem. 2015, 80, 3397–3410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.