

Abstract

Although single-cell mRNA sequencing has been a powerful tool to explore cellular heterogeneity, the sequencing of small RNA at the single-cell level (sc-sRNA-seq) remains a challenge, as these have no consensus sequence, are relatively low abundant, and are difficult to amplify in a bias-free fashion. We present two methods of single-cell-lysis that enable sc-sRNA-seq. The first method is a chemical-based technique with overnight freezing while the second method leverages on-chip electrical lysis of plasma membrane and physical extraction and separation of cytoplasmic RNA via isotachophoresis. We coupled these two methods with off-chip small RNA library preparation using CleanTag modified adapters to prevent the formation of adapter dimers. We then demonstrated sc-sRNA-seq with single K562 human leukemic cells. Our approaches offer a relatively short hands-on time of 6 h and efficient generation of on-target reads. The sc-sRNA-seq with our approaches showed detection of miRNA with various abundances ranging from 16 000 copies/cell to about 10 copies/cell. We anticipate this approach will create a new opportunity to explore cellular heterogeneity through small RNA expression.
Single-cell RNA sequencing (scRNA-seq) is a growing field with a wide range of associated methods.1 scRNA-seq offers unique advantages of exploring heterogeneity among cells leveraging high-dimensional transcriptomic data.2 The methods also enable assaying rare cells such as circulating tumor cells (CTCs)3 or fine needle aspirate biopsies4 and help the development of an understanding of tissue composition, transcription dynamics, and gene regulation.5
A variety of microfluidic devices have been applied to single-cell transcriptomics as they offer a range of advantages including reducing the need for highly trained personnel,6 reducing measurement uncertainty by improving reproducibility, saving time and resources, and enabling higher throughput and automation.7 Devices and processes range from cell sorting to systems with high degrees of automation including scRNA-seq library preparation.8 Further, microfluidics has been applied to the study of subcellular transcriptomic distribution by separating the cytoplasmic RNA content from the nuclear content.9−12 Recently, Abdelmoez et al.13 were able to physically separate the nucleus from the cytoplasmic content by using a microfluidic chip which traps the cell, electrically lyses the plasma membrane, and then electrophoretically transports the cytoplasmic content away from the trapped nucleus using isotachophoresis (ITP).
An important exception to these advances in scRNA-seq, and the main focus of the current work, is the processing, isolation, and deep sequencing of small RNA (sRNA-seq) of single cells. To date, sRNA profiling has been restricted to a large number of cells.14−17 This limitation is due mostly to the relatively low abundance of sRNAs compared to mRNAs, and also the large variation in sRNA expression depending on the type and state of the cells. Another important challenge in single-cell sRNA-seq (sc-sRNA-seq) is the formation of adapter dimers during library preparation. sRNA library preparation involves ligating adapters to the sRNA to enable amplification of the library for downstream sequencing, but low input RNA amounts imply that adapters often ligate to themselves and create empty library components known as adapter dimers.
To our knowledge, the only successful attempt of sc-sRNA-seq was the work by Faridani et al.18 The work reported sequencing of micro RNAs (miRNAs), sno-derived sRNAs(sdRNAs), and tRNA derived sRNAs (tsRNAs) with single naïve and primed human embryonic stem cells and cancer cells. The work also demonstrated segregating different cell types with sRNA expression. Shore et al.19 reported a method for sRNA-seq library preparation using chemically modified adaptors, which we here term as CleanTag. CleanTag enables sRNA-seq with single-cell-quantity RNA (10 pg) by suppressing the formation of adaptor dimers and offers a simple workflow with a relatively short hands-on time of about 6 h. However, we know of no report that couples CleanTag with single-cell lysis and demonstrates sc-sRNA-seq.
In this work, we report two single-cell lysing techniques that enable sc-sRNA-seq coupled with CleanTag-based library construction. The first technique uses a mild detergent with temperature variation for single-cell lysing. The second uses a microfluidic device which electrically lyses the plasma membrane and extracts the cytoplasmic nucleic acids of a single cell. Each technique robustly enables sc-sRNA-seq with CleanTag for the first time and provides sRNA expressions that correlate well with published studies. To develop our sc-sRNA-seq techniques, we explored four different lysis approaches coupling with CleanTag and critically benchmarked the yield of on-target reads, sensitivity, and reproducibility. Our approach offers a much simpler work flow, shorter hands-on time (6 h versus 18 h by Faridani et al.18), and superiority regarding a greater library yield and reduced amounts of adapter dimers.
Materials and Methods
Chemicals
We purchased Tris, HEPES, Imidazole, and HCl from Thermo Fisher Scientific and polyvinylpyrrolidone (PVP, MW 1 MDa) from Sigma-Aldrich. The leading electrolyte (LE) buffer was composed of 50 mM Tris and 25 mM HCl containing 0.4% PVP at pH 8.1. The trailing electrolytes (TE) buffer was 50 mM Imidazole and 25 mM HEPES containing 0.4% PVP at pH 7.6. All solutions were prepared in DNase-/RNase-free deionized (DI) water (Life Technologies).
Cell Culture
We used K562 (chronic myelogenous leukemia) cells in our single-cell experiments. K562 cells were cultured in Dulbecco’s modified eagle medium with 4.5 g/L glucose, l-glutamine, and sodium pyruvate (Thermo Fisher). Fetal bovine serum and penicillin-streptomycin-glutamine were added to the media with 10% and 1% of volumetric concentrations, respectively. The cells were cultured at a temperature of 37 °C with 5% CO2.
Microfluidic Chip
We used the microfluidic chip design reported by Abdelmoez et al.13 Briefly, the microchannel included a T-junction layout with a hydrodynamic trap immediately upstream the junction. The trap had a 3 μm-wide gap and was 5 μm long. The main channel for ITP had a width of 50 μm and depth of 25 μm. The three microchannels were, respectively, connected to 3 mm-diameter wells: an inlet, an outlet, and a waste. The device was made of polydimethylsiloxane (Sylgard 184, Dow Corning) using soft lithography and sealed with a glass slide by plasma bonding.
Single-Cell Lysis with a Chemical Agent
To lyse a single cell in Triton X-100, 1 μL of cell suspension containing a single cell was added to 3 μL of lysis buffer composed of 0.13% Triton X-100 with 4 units of RNase inhibitor (SUPERaseIn RNase inhibitor, Invitrogen) in a PCR tube and stored at −80 °C at least overnight. In our preliminary experiments, we also examined lyses with the same Triton X-100 chemistry but without freezing overnight.
In addition, we examined an off-the-shelf single-cell lysis kit (Ambion Single-Cell Lysis Kit) for comparison. A volume of 1 μL of cell suspension containing a single cell was added to 5 μL of the lysis buffer and incubated for 5 min after which 1 μL of the kit’s stop buffer was added.
Single-Cell Lysis Using Microfluidics Based Approach
Each microfluidic chip was used once to avoid introducing any contamination. At the start of each experiment, the microchannels were washed with 1 M NaOH supplemented with 0.1% Triton X-100 for 1 min, then with 1 M HCl supplemented with 0.1% Triton X-100 for 1 min, and deionized (DI) water supplemented with 0.1% Triton X-100 for 1 min. This was accomplished by filling the inlet and outlet wells with the aforementioned washing solution then applying a vacuum at the waste well. We note that Triton X-100 was used to prevent bubble formation in the microchannel and was removed completely by exchanging the solution in the microchannel with LE and TE.
After washing, 10 μL of LE was loaded in the outlet well and 10 μL of TE was loaded in the inlet well and we briefly applied vacuum to the waste well. A 1 μL aliquot of cell suspension manually observed to contain a single cell (with a microscope during its pipetting from a Petri dish) was added to the inlet well. The capture of the single cell at the trap via a pressure-driven flow from the inlet to the waste was visualized using an optical microscope. Then, 10 μL of TE was loaded into the waste well to reduce the pressure-driven flow. Platinum wire electrodes were inserted into the three wells and two source meters (2410, Keithley) were used to apply, respectively, −150, −170, and 0 V to the electrodes at the inlet, waste, and outlet wells. The dc voltage caused the plasma membrane lysis within 0.1 s and initiated ITP extracting the cytoplasmic nucleic acid content away from the lysed cell, focusing the nucleic acids contents into the TE/LE interface, and transporting it toward the outlet well within 2 min. Detailed description of a similar protocol and chip, together with a narrated video description, were reported by Kuriyama et al.11
Library Preparation and Sequencing
The library preparation protocol was as follows: CleanTag Small RNA Library Preparation Kit (TriLink BioTechnologies, LLC) (catalog no. L-3206) was used to prepare all 24 sRNA libraries. Manufacturer’s recommended protocol19 was followed with a few changes (see Table S-1). Briefly, the first step was ligation of the 3′ adapter onto the RNA of interest with incubation for 1 h at 28 °C and heat deactivation at 65 °C for 20 min. Next step was ligation of the 5′ adapter on the RNA library for 1 h at 28 °C followed by heat deactivation at 65 °C for 20 min. We synthesized cDNA (1 h at 50 °C) and amplified it via PCR (98 °C for 30 s; temperature cycles of 98 °C for 10 s, 60 °C for 30 s, and 72 °C for 15 s; and 72 °C at 10 min) with Illumina standard barcoding index primers (no. L-3204 and no. L-3205 TriLink BioTechnologies, LLC). Note that the number of cycles can be found in Table S-1. FirstChoice Human Leukemia (K562) Total RNA (Life Technologies) was used in control experiments at input amounts of 100 ng or 10 pg. All PCR products (80–86 μL) were purified using 144 μL of Ampure XP (Beckman Coulter) magnetic beads to eliminate products shorter than 100 base pairs. No size-selection via Ampure XP was performed to avoid loss of material. Purified libraries were analyzed by a 2100 Agilent Bioanalyzer (Agilent Technologies) where the samples were diluted 1:3. Libraries were also quantified by a Qubit 3.0 fluorometer (Thermo Fisher) dsDNA HS Assay kit. Twenty-four samples were pooled to occupy equal space on the flow cell. Based on the concentration of the sample, a final mixture of 10 nM with each sample comprising 0.416 nM was prepared. High-throughput sequencing was performed on an Illumina HiSeq 2500 with single-end 50 nt by GENWIZ (La Jolla, CA).
Data Analysis
To compare the protocols at a similar sequencing depth, some of the reads were first down-sampled to make the average depth of the individual protocol 9.58 M reads using Seqtk20 (see Table S-2). Next, the adapter sequence (TGGAATTCTCGGGTGCCAAGG) was trimmed and reads shorter than 15 nt were discarded by cutadapt (version 1.14) with the following parameters: -e 0.1 -O 1 -u 2 −minimum-length 15. Afterward, reads aligned to rRNA (rRNA) sequences (human_all_rRNA.fasta21) were discarded using STAR (version 2.5.3a).22 The remaining reads were then aligned to the human genome (hg38) also using STAR with parameters of ENCODE miRNA-seq project.23 Finally, expressions of transcripts in counts per million reads (CPM) were estimated with the following databases (UCSC Genome Browser for tRNA and ENSEMBL for other transcripts). The multiple-aligned reads were assigned to the multiple genomic locations by a weighing approach, dividing the number of reads by the number of annotated locations.18,24
Note that the RNA length cutoff value used when analyzing the sc-sRNA-seq data in the manuscript was 15 nt, with the exception in the section entitled Quality of Sequencing Reads which used a cutoff at 18 nt in order to compare our results with the available data on sc-sRNA-seq.18 Differential RNA analysis was performed using Monocole software.25
Results and Discussion
Overview of sc-sRNA-seq with CleanTag
In this work, we report two approaches of single-cell lyses for sc-sRNA-seq coupled with CleanTag small RNA library preparation. The first technique depends on the chemical lysis of single cells with the mild detergent Triton X-100 and freezing overnight at −80 °C (Figure 1A). The second technique uses a microfluidic chip that selectively lyses the plasma membrane extracting the cytoplasmic RNA through the application of electric current as described in the methods and previous publications (Figure 1B).13 Both methods take about 6 h of hands-on time for the library construction, while the microfluidic approach offers a significantly faster protocol for lysis and extraction of cytoplasmic RNA (∼5 min) than lysis with Triton X-100 (∼12 h). Furthermore, the microfluidic approach facilitates the separation of nuclear and cytoplasmic RNA (including small RNA) with minimal cross-contamination.13 The chip can save and recover the nucleus of each cell (see also the work of Abdelmoez et al.,13 although the cell’s nuclei were not analyzed in the current work.) We demonstrated sc-sRNA-seq coupled with CleanTag19 with these two methods and critically benchmarked the sensitivity and repeatability of sRNA quantification.
Figure 1.
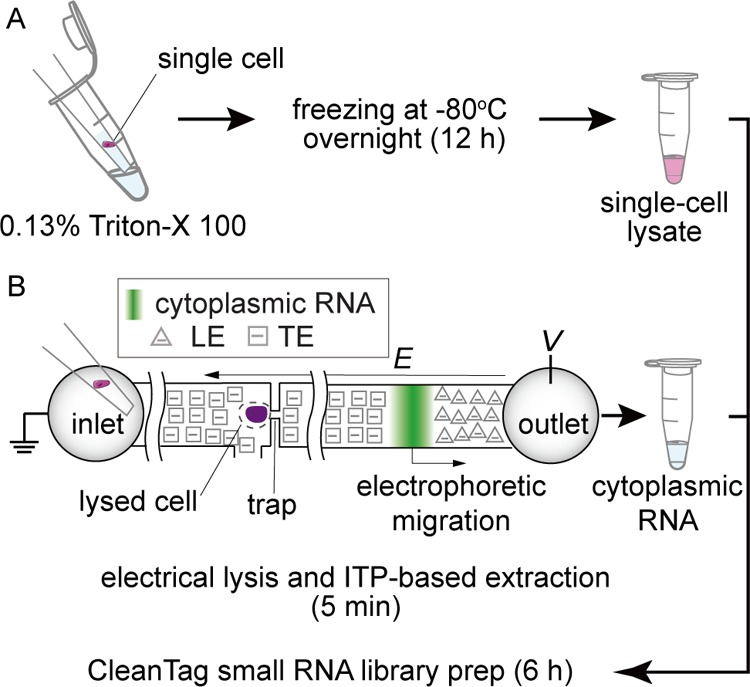
Single-cell lyses for sc-sRNA-seq with CleanTag library preparation: (A) lysis via 0.13% Triton X-100 and freezing overnight (∼12 h) and (B) electrical lysis and ITP-based extraction of cytoplasmic RNA (∼5 min). Both samples are processed with CleanTag small RNA library prep (6 h).
Single-Cell Lyses for CleanTag
To find an appropriate cell-lysis approach, we explored various methods of single-cell lyses including off-the-shelf single-cell lysis kit from Ambion, Triton X-100, and the microfluidic approach. We examined two conditions of Triton-based lysis: with and without an overnight incubation at −80 °C. Among these approaches, we observed successful library construction using two approaches, Triton X-100 with an overnight incubation at −80 °C and using the microfluidic approach (Figure 2). The electropherogram in Figure 2 showed that a variety of RNA products were detected including miRNA around 140 nt and adapter dimers around 120 nt. Single-cell samples generated between 5 ng/μL and 60 ng/μL of purified DNA with the CleanTag library preparation kit (cf. Figure S-1 in the Supporting Information). In contrast, we observed no detectable library peaks with the other two single-cell lysis approaches as shown in Figure S-2. In this paper, we thus used the overnight freezing for Triton-based lysis unless we explicitly specify the condition. For reference, we included electropherograms of 10 pg and 100 ng of bulk samples in Figure S-2C,D, respectively.
Figure 2.

Capillary gel electropherograms using the Agilent Bioanalyzer for products of the CleanTag library preparation. (A) Electropherogram for single K562 cell lysed via Triton X-100 with overnight-freezing and for (B) on-chip ITP extracted single K562 cell cytoplasmic RNA. Both electropherograms show abundant content ranging from 120 to 150 nt, consistent with ligated mature small RNA molecules.
Quality of Sequencing Reads
To elucidate the quality of the sRNA-seq reads, we analyzed the composition of raw reads as shown in Figure 3. We here summarized the off-target reads for sRNA-seq as three categories: too short reads (<18 nt), unmapped reads, and reads aligned on rRNAs. We also divided the on-target reads for sRNA-seq into uniquely mapped reads and multiple mapped reads. We observed the Triton-based and the microfluidics-based lyses, respectively, yielded 17% and 4% of on-target reads. We hypothesize that the difference in the fraction of the on-target reads is due to the different reaction volumes, resulting in slightly different buffer conditions. As summarized in Table S-1, our protocols output different volumes respectively as 4 μL, 8 μL, and 2 μL (including 1 μL of nuclease-free water) with Triton, microfluidics, and 10 pg of bulk samples, which resulted in 12, 16, and 10 μL of reaction volumes by adding 8 μL of the adapter ligation master mix at the 3′ adaptor ligation step. To compare our approach to the protocol by Faridani et al.,18 we applied the same in silico analyses to the sRNA-seq data and obtained 5% of on-target reads as shown in Figure 3C. This comparison showed that CleanTag with Triton-based lysis yielded the highest fraction of on-target reads among the three methods. (Note the data by Faridani et al.18 were created with different cells, HEK293FT, than our K562 cells.) We also compared the performance of the CleanTag-based sRNA-seq with 10 pg and 100 ng of total RNA versus those by Faridani et al.18 as shown in Figure S-3. We again observed a better performance with the CleanTag-based approach, yielding more fractions of on-target reads. Notably, we observed that CleanTag increased the on-target reads by reducing too short reads, which we attribute to the suppression of adapter dimer formation. These quality control results could suggest that the use of the CleanTag sc-sRNA-seq has an advantage over the method described by Faridani et al.18 in efficient generation of on-target reads.
Figure 3.

Composition of sequencing reads obtained with two lyses protocols, (A) Triton and (B) microfluidics-based approaches (single K562 cells), and (C) that obtained with data from Faridani et al.18 (single HEK293FT cells).
Detection of Small RNAs
Our sc-sRNA-seq protocols consistently detected both pre-sRNAs and mature sRNAs as shown in Figure 4A,B, respectively. Both Triton and microfluidics-based lyses showed similar numbers of detected sRNAs with control experiments using 10 pg of total RNA. Our approaches detected similar numbers of sRNAs per cell to those of Faridani et al.18 However, the quantitative comparison is difficult due to the different cell types. We confirmed the detection of the sRNAs assessing the length of the aligned reads as shown in Figure 4C–E. As similar to the results by Faridani et al.,18 we observed peaks in the distributions of read length at 22 nt, 33 nt, and 39 nt, respectively, with miRNA, tsRNA, and sdRNA(<40 nt). Interestingly, our method detected specifically mature miRNAs more than their precursors whereas our method detected other precursors of tRNAs and snoRNAs. For reference, we included the distribution of length of mapped reads with 100 ng of bulk in Figure S-4. The distributions of aligned reads on respective precursors further evidenced detection of sRNAs showing that the mature RNAs derived from specific loci of precursors (Figure S-5). We note that our protocol also consistently detected other types of sRNAs as shown in Figure S-6, while we mainly discuss miRNA, tsRNA, and sdRNA in detail in this work.
Figure 4.

Characterization of sc-sRNA-seq approaches using single K562 cells: (A) number of precursor RNAs (pre-miRNAs, tRNAs, snoRNAs) and (B) number of mature sRNAs (miRNAs, tsRNAs, and sdRNAs) detected with Triton and microfluidic-based lyses and single-cell-quantity total RNA (10 pg) extracted from K562 cells. (C–E) Length distribution of aligned reads on miRNA, tRNA, and snoRNA. (F) Pearson correlation in all combinations of replicate sample pairs prepared with the same method. (G) Pearson correlation of sRNA expression to control experiments with 100 ng of total RNA.
To assess reproducibility of the sRNAs detection, we compared the number of sRNAs detected in all combinations of replicate sample pairs prepared with the same method to the mean total number of sRNAs detected using that method. Single-cell samples showed reproducibility between 63% and 76% and no significant difference from 10 pg of bulk samples, whereas the 100 ng of bulk samples showed higher reproducibility between 90% and 96% (see Figure S-7). We thus hypothesize that the main source of variability in single-cell samples is due to the small amount of RNA.
Repeatability of Small RNA Quantification
We next assessed the repeatability of the sRNAs quantification by analyzing the Pearson correlation in all combinations of replicate sample pairs prepared with the same method as shown in Figure 4F. In contrast to Faridani et al.,18 in which they reported significantly low coefficients of correlation with miRNAs, we observed that coefficients of correlation ranged from 0.6 to 0.9 with all three sRNAs and no apparent difference among the protocols. In this analysis, we included control experiments with single-cell RNA quantities, 10 pg, to discern the increase in the technical uncertainty due to the lysis. The similar coefficients of correlation with the control experiments supported the repeatability of our single-cell lysis. We also examined the correlation of the sRNA expression to control experiments with 100 ng of total RNA as shown in Figure 4G and observed that coefficients of correlation roughly ranged from 0.5 to 0.8. These results suggest that neither the Triton nor the microfluidics-based approaches introduced adverse effects on the CleanTag chemistry. In Figure S-8, we showed the overall coefficients of correlation including those across different protocols using all of the sRNA species. The hierarchical analysis showed that Triton (scTriton) and microfluidics-based (scITP) sc-sRNA-seq data are closer to each other than to the data with 10 pg of total RNA.
Difference in Triton vs Microfluidics Methods
To elucidate the difference between Triton and microfluidic-based methods, we performed differential expression (DE) analysis comparing both sc-sRNA-seq data versus 10 pg of total RNA shown in Figure 5. DE analysis showed more numbers of DE sRNAs in the microfluidics-based method than Triton-based method. As expected, we observed that DE sRNAs, which are supposed to be enriched in the nuclei, e.g., sdRNA and small nuclear RNA (snRNA), were under expressed in microfluidics-based sc-sRNA-seq as we only used the cytoplasmic fractions here. Triton-based method also showed under-expression with some nuclear enriched sRNAs, which may imply incomplete lysis of nuclear membrane.9 We also examined the length bias due to the difference in the lysis as shown in Figure S-9. However, we observed no clear evidence for length bias.
Figure 5.

Scatter plots of sRNA expression comparing two lysing and extraction methods to 10 pg of K562 prepurified RNA sample. (A) Plotted are correlation of sRNA expression for Triton vs 10 pg total RNA for small RNA types (miRNA, tsRNA, sdRNA, lincRNA, and snRNA). (B) Correlation of sRNA expression of microfluidics-based approach vs 10 pg total RNA for the same small RNA types. The Triton samples were better correlated with 10 pg total RNA especially when comparing nuclear enriched sRNA such as sdRNA and snRNA since the microfluidics-based approach used only cytoplasmic fraction. Note that DEG is a database of essential genes.
miRNA Expression Pattern
To benchmark the quantification of sRNAs with our protocol, we compared the measured expression of 13 miRNAs, known to be expressed in K562 cells, to those measured by RT-qPCR (Figure 6).26,27 We think this comparison can provide a better assessment than comparing to Faridani et al.’s data18 for two reasons: First, Faridani et al. used different cell lines than ours. Second, the sRNA-seq protocols have a significant bias,28 which makes the comparison difficult between one protocol and another. Overall, the trend across the samples was consistent with the bulk 100 ng samples. Further, it was also consistent with the microfluidics-based RT-qPCR reported by White et al.26 For example, miR192 families, miR192-1 and miR-192-2, which were reported as the most abundant miRNA (about 16 000 copies/cell) among these miRNAs, showed the highest expression with K562 cells. miR-16 (miR-16-1 and miR-16-2), which was reported as 804 ± 261 (S.D.) copies/cell in K562 cell,26 was detected in all the single-cell samples. In contrast, miR-223, which was reported as 513 ± 406 copies/cell,26 was detected in two samples out of n = 3 with Triton-based approach and ten samples out of n = 12 with the microfluidics-based approach despite the similar expression levels. We partially attribute the difference in the detection of miR-16 vs miR-223 to the difference in their regulation. miR-16 is known to be tightly regulated and has a unimodal expression distribution, whereas miR-223 has large cell-to-cell variation ranging from 10 to 1 000 copies/cell and multimodal distribution.26 However, we also note that limit of detection of our approach was roughly 10–100 copies/cell, which we hypothesize from the stochastic detection of miRNA with abundance of about 10 copies/cell, e.g., miR-181A and miR-196A families.26 We thus hope in the future to improve the limit of detection and the system’s throughput to analyze large numbers of single cells and clearly dissect this cell-to-cell variation.
Figure 6.

Expression pattern of miRNAs known to be expressed in K562 cells for the following samples: (A) single K562 cells prepared with Triton X-100-based lysis, (B) those prepared with microfluidics-base lysis and extraction, (C) 10 pg of total RNA of K562 cells, and (D) 100 ng of total RNA of K562 cells. miRNA sequences are listed in the order of increasing median abundance as observed in the 100 ng of total RNA data (see also White et al.26 for comparison). The 100 ng of total RNA showed the least variation as expected.
Conclusions
Traditional genetic analyses have relied on measurements in bulk cell populations. However, these measurements are the average of events stochastically occurring in thousands or millions of cells. Hence, there has been increased interest in single-cell analyses and the development of methods which efficiently allow the sequencing of small amounts of RNA. However, there remain challenges associated with the single-cell analysis of sRNA molecules, due to their very small abundance and difficulties in sample preparation.
In this work, we demonstrated two approaches of lysing single cells compatible with CleanTag library preparation for sRNA sequencing: Triton X-100 (overnight-frozen) lysis and electrical lysis using microfluidics. The latter method used a device which separated the nucleus from the cytoplasmic content of a single cell by selectively lysing the plasma membrane of a single cell in a trap via electrical lysis and then quickly and selectively extracting cytoplasmic nucleic acid contents with ITP.
The RNA samples were processed using the CleanTag Small RNA Library Preparation kit that leveraged chemically modified adapters to disfavor adapter dimer formation, the main obstacle to the sequencing of very low input samples.19 To our knowledge, the present work is the second publication demonstrating sequencing of sRNAs from single cells, the first being by Faridani et al.18 The CleanTag methodology coupled with the microfluidics-based lyses and extraction have important advantages over the method described by Faridani et al. The microfluidic device performs rapid, automated lysing and then conducts fractionation of cytoplasmic nucleic acids from nucleic acids from cell nucleus. After dispensing and trapping the cell, the on-chip process takes 2 min to extract the cytoplasmic RNA content using the device. When comparing the Triton-based lysis vs the microfluidics-based lysis, the microfluidics-based lysis uniquely offered a quicker protocol and better correlation with 100 ng of the bulk sample and RT-qPCR results. On the other hand, the Triton-based lysis offered more efficient production of on-target reads and an easier workflow using standard equipment for lysis. The CleanTag methodology is considerably streamlined, with library preparation accomplished in 6 h. Despite a relatively small number of samples analyzed, we were able to sequence sRNAs from single-cell samples. Our results were slightly improved regarding the sequencing quality when compared to those of Faridani et al.; however, we hypothesize there is significant room for improvement regarding the quality of the reads and the information that can be obtained from single-cell small RNA sequencing data.
Lastly, in this paper we have striven to benchmark the sensitivity and repeatability of the proposed protocols as well as compare the sRNA-seq results to (albeit very limited) published work, to shed light on what may be biological signal over the noise. The results suggest that sc-sRNA-seq is possible and provides reproducible results which at least in part capture cell-to-cell heterogeneity.
Acknowledgments
We gratefully thank Kei Iida of Medical Research Support Center of Kyoto University for a script counting reads. R.K. acknowledges support from the Jordanian-American Commission for Educational Exchange (the Binational Fulbright Commission in Jordan) through the Jordanian Visiting Post-Doctoral Scholar Fellowship. C.M.H. acknowledges support from the National Institute of Standards and Technology (NIST) NRC Postdoctoral Associateship Program. C.M.H., S.A.M., and J.G.S. further acknowledge support from the NIST Joint Initiative for Metrology in Biology at Stanford. H.S. acknowledges grants from ImPACT Program of Council for Science, Technology, and Innovation (Cabinet Office, Government of Japan) and also by Japan Society for the Promotion of Science under Grants 26289035 and 26630052. Research for CleanTag was funded with a Small Business Innovation Research (SBIR) grant through the National Institutes of Health (NIH), Grant Number: 1R43HG006820-01A1. Certain commercial equipment, instruments or materials are identified in this paper in order to specify the experimental procedure adequately. Such identification is not intended to imply recommendation or endorsement by the National Institute of Standards and Technology (NIST) nor is it intended to imply that the materials or equipment identified are necessarily the best available for the purpose.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b02773.
Table S-1, modified conditions of individual samples in CleanTag small RNA library preparation; Table S-2, summary of sequencing reads for the various sample types; Figure S-1, quantification of purified library yields observed for the various sample types; Figure S-2, capillary gel electropherograms using the Agilent Bioanalyzer for products of the CleanTag library preparation; Figure S-3, proportion of on-target reads and off-target reads from sRNA-seq library prepared with 10 pg and 100 ng of K562 total RNA; Figure S-4, length distribution of aligned reads with 100 ng of bulk samples; Figure S-5, distribution of aligned reads on respective precursors; Figure S-6, measured abundance of sRNAs (<40 nt); Figure S-7, reproducibility, as evaluated by the percentage of sRNAs detected in pairs of replicate samples out of the mean total number of sRNAs detected in this pair of samples; Figure S-8, heatmap summarizing correlations among the sequenced samples and protocol types; and Figure S-9, Violin plot showing total measured small RNA length distributions (PDF)
Author Contributions
× R.K. and S.S. contributed equally to this work.
The authors declare no competing financial interest.
Notes
Original FASTQ sequencing files are deposited in the NCBI Short Reads Archive (SRA) under SRA study number SRP140975. In the comparison to Faridani et al., we downloaded SRR3495667–SRR3495684 and SRR3495694–SRR3495696 and applied the same in silico analyses used in this study.
Supplementary Material
References
- Saliba A. E.; Westermann A. J.; Gorski S. A.; Vogel J. Nucleic Acids Res. 2014, 42, 8845–8860. 10.1093/nar/gku555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedlund E.; Deng Q. Mol. Aspects Med. 2018, 59, 36–46. 10.1016/j.mam.2017.07.003. [DOI] [PubMed] [Google Scholar]
- Miyamoto D. T.; Ting D. T.; Toner M.; Maheswaran S.; Haber D. A. Cold Spring Harbor Symp. Quant. Biol. 2016, 81, 269–274. 10.1101/sqb.2016.81.031120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K.; Takeda H.; Nishijima N.; Orito E.; Joko K.; Uchida Y.; Izumi N.; Nishio K.; Osaki Y. Oncotarget 2015, 6, 21636–21644. 10.18632/oncotarget.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziejczyk A. A.; Kim J. K.; Svensson V.; Marioni J. C.; Teichmann S. A. Mol. Cell 2015, 58, 610–620. 10.1016/j.molcel.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Streets A. M.; Zhang X.; Cao C.; Pang Y.; Wu X.; Xiong L.; Yang L.; Fu Y.; Zhao L.; Tang F.; Huang Y. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 7048–7053. 10.1073/pnas.1402030111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S.; Qing T.; Zheng Y.; Jin L.; Shi L. Oncotarget 2017, 8, 53763–53779. 10.18632/oncotarget.17893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilionis R.; Nainys J.; Veres A.; Savova V.; Zemmour D.; Klein A. M.; Mazutis L. Nat. Protoc. 2017, 12, 44–73. 10.1038/nprot.2016.154. [DOI] [PubMed] [Google Scholar]
- Han L.; Zi X.; Garmire L. X.; Wu Y.; Weissman S. M.; Pan X.; Fan R. Sci. Rep. 2015, 4, 6485. 10.1038/srep06485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyama K.; Shintaku H.; Santiago J. G. Electrophoresis 2015, 36, 1658–1662. 10.1002/elps.201500040. [DOI] [PubMed] [Google Scholar]
- Kuriyama K.; Shintaku H.; Santiago J. G. Bio-protocol 2016, 6, e1844. 10.21769/BioProtoc.1844. [DOI] [Google Scholar]
- Shintaku H.; Nishikii H.; Marshall L. A.; Kotera H.; Santiago J. G. Anal. Chem. 2014, 86, 1953–1957. 10.1021/ac4040218. [DOI] [PubMed] [Google Scholar]
- Abdelmoez M. N.; Iida K.; Oguchi Y.; Nishikii H.; Yokokawa R.; Kotera H.; Uemura S.; Santiago J. G.; Shintaku H. Genome Biol. 2018, 19, 66. 10.1186/s13059-018-1446-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi Y.; Ohkubo T.; Kawaji H.; Machida A.; Miyata H.; Goda S.; Roy S.; Hayashizaki Y.; Suzuki H.; Yokota T. Neurosci. Lett. 2017, 636, 48–57. 10.1016/j.neulet.2016.10.042. [DOI] [PubMed] [Google Scholar]
- Li Y.; Zeng A.; Han X. S.; Li G.; Li Y. Q.; Shen B.; Jing Q. Genomics Data 2017, 14, 114–125. 10.1016/j.gdata.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law P. T.; Qin H.; Ching A. K.; Lai K. P.; Co N. N.; He M.; Lung R. W.; Chan A. W.; Chan T. F.; Wong N. J. Hepatol. 2013, 58, 1165–1173. 10.1016/j.jhep.2013.01.032. [DOI] [PubMed] [Google Scholar]
- Chen C. J.; Heard E. Methods 2013, 63, 76–84. 10.1016/j.ymeth.2013.05.001. [DOI] [PubMed] [Google Scholar]
- Faridani O. R.; Abdullayev I.; Hagemann-Jensen M.; Schell J. P.; Lanner F.; Sandberg R. Nat. Biotechnol. 2016, 34, 1264–1266. 10.1038/nbt.3701. [DOI] [PubMed] [Google Scholar]
- Shore S.; Henderson J. M.; Lebedev A.; Salcedo M. P.; Zon G.; McCaffrey A. P.; Paul N.; Hogrefe R. I. PLoS One 2016, 11, e0167009. 10.1371/journal.pone.0167009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W.; Le S.; Li Y.; Hu F. PLoS One 2016, 11, e0163962. 10.1371/journal.pone.0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://sourceforge.net/projects/rseqflow/files.
- Dobin A.; Davis C. A.; Schlesinger F.; Drenkow J.; Zaleski C.; Jha S.; Batut P.; Chaisson M.; Gingeras T. R. Bioinformatics 2013, 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://www.encodeproject.org/microrna/microrna-seq.
- Sakuma M.; Iida K.; Hagiwara M. BMC Mol. Biol. 2015, 16, 16. 10.1186/s12867-015-0044-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C.; Cacchiarelli D.; Grimsby J.; Pokharel P.; Li S.; Morse M.; Lennon N. J.; Livak K. J.; Mikkelsen T. S.; Rinn J. L. Nat. Biotechnol. 2014, 32, 381–386. 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White A. K.; VanInsberghe M.; Petriv O. I.; Hamidi M.; Sikorski D.; Marra M. A.; Piret J.; Aparicio S.; Hansen C. L. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 13999–14004. 10.1073/pnas.1019446108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White A. K.; Heyries K. A.; Doolin C.; Vaninsberghe M.; Hansen C. L. Anal. Chem. 2013, 85, 7182–7190. 10.1021/ac400896j. [DOI] [PubMed] [Google Scholar]
- Giraldez M. D.; Spengler R. M.; Etheridge A.; Godoy P. M.; Barczak A. J.; Srinivasan S.; De Hoff P. L.; Tanriverdi K.; Courtright A.; Lu S.; Khoory J.; Rubio R.; Baxter D.; Driedonks T. A. P.; Buermans H. P. J.; Nolte-’t Hoen E. N. M.; Jiang H.; Wang K.; Ghiran I.; Wang Y. E.; Van Keuren-Jensen K.; Freedman J. E.; Woodruff P. G.; Laurent L. C.; Erle D. J.; Galas D. J.; Tewari M. Nat. Biotechnol. 2018, 36, 746–757. 10.1038/nbt.4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.