

Abstract
Neurodegenerative diseases are characterized by an irreversible and progressive loss of neuronal structure and function. While many alterations to normal cellular processes occur during neurodegeneration, a pathological accumulation of aggregated proteins constitutes a hallmark of several neurodegenerative disorders. Alzheimer's disease, specifically, is pathologically defined by the formation of amyloid plaques and tangles of hyperphosphorylated tau protein. Stress has emerged as an important factor in the development and progression of neurodegenerative diseases, including Alzheimer's. Very little is known, however, regarding the effects of stress on the mechanisms controlling abnormal protein aggregation and clearance. Chronic stress activates the hypothalamic-pituitary-adrenal (HPA) axis, causing an excessive secretion of glucocorticoids that are capable of impacting diverse physiological and cellular processes. The present review focuses on the influence of stress on a key feature of Alzheimer's disease pathology, emphasizing the relationship between tau phosphorylation and accumulation and its connection to HPA axis dysfunction.
Keywords: Stress, Neurodegeneration, HPA axis, Alzheimer's disease, Tau, Hyperphosphorylation
1. Introduction
Stress can be broadly defined as a disruption to the homeostasis of an organism. In the context of human physiology, stress is a highly complex phenomenon that involves environmental and psychosocial stimuli that elicit a series of self-regulated nervous system responses which are collectively referred to as “the stress response” (Esch et al., 2002a; Chrousos, 2009). Stress is an inevitable component of the modern lifestyle, and although certain levels of stress are normal and some stressors can even be beneficial, experiencing excessive amounts of stress over extended periods of time (chronic stress) can alter the self-regulating capacity of the stress response, ultimately causing detrimental effects on the overall physiology of the body (Andersen et al., 1994; Payne, 2013). Furthermore, several variables such as genetics, sex, environment, and early life experiences are known to impact an individual's stress sensitivity (Kapoor et al., 2006; Zavala et al., 2011; Chen and Baram, 2016; Bale and Epperson, 2015). The early life period is of particularly noteworthy importance because this is the time when brain anatomy is primarily determined. Early life stress (ELS) is thus critical in influencing responses to stress and vulnerability to disease (Lenroot and Giedd, 2006; Seifan et al., 2015).
The stress response is mediated by two distinct and tightly-controlled neuroendocrine signaling cascades, the sympathetic-adrenal-medullary (SAM) pathway and the hypothalamic-pituitary-adrenal (HPA) axis. The SAM system, which underlies the classic “fight or flight” response, is generally short-lived and involves autonomic nervous system activation which leads to increased circulating levels of epinephrine from the adrenal medulla and norepinephrine from sympathetic nerve terminals. The resulting physiological response is geared toward countering or escaping the stressor, with increased blood flow, heart and respiratory rates, and energy mobilization displayed by the stressed individual (Ulrich-Lai and Herman, 2009). The second stress response system, the HPA axis, begins in the hypothalamus and descends to the adrenal cortex where the release of glucocorticoid (GC) hormones (i.e., cortisol, corticosterone) occurs. Unlike the SAM cascade, activation of the HPA axis by stress is slower and its physiological effects have a more prolonged duration. Chronic stress exposure can lead to altered HPA axis hormone levels and adversely affect the regulation of the stress response, which can ultimately have negative impacts on the overall physiology of the body (Vanitallie, 2002; Glaser and Kiecolt-Glaser, 2005). In this way, stress can exert detrimental effects on human health and contribute to pathological conditions as far-ranging as cancer, cardiovascular dysfunction, and neurodegenerative disease (Esch et al., 2002b; Grippo and Johnson, 2009; Moreno-Smith et al., 2010).
Stress can have profound effects on brain development and function, inducing neurochemical changes and disrupting normal neuronal circuitry. A wide variety of fundamental physiological processes are influenced by stress, in fact, through the HPA axis and GC action. This knowledge underlies our critical need to more fully understand the neurological impacts of stress that may lead to pathological conditions or neuropsychiatric disorders such as AD (Meyer et al., 2001; Esch et al., 2002a,b). One of the hallmarks of several neurodegenerative diseases, including AD, is the accumulation of abnormal protein aggregates in neurons, which alters their structure and function, causes neurotoxicity, and ultimately leads to neuronal death (Cairns et al., 2004; Lim and Yue, 2015). Neurodegeneration is a process that is characterized by this type of progressive and irreversible loss in specific areas of the nervous system (Jellinger, 2010), and is accompanied by several alterations to normal cellular processes including protein misfolding and aggregation, mitochondrial dysfunction, impaired intracellular trafficking, genotoxicity, increased oxidative stress, and cytoskeletal alterations (Jellinger, 2009). This phenomenon occurs in the brain of patients with AD, the most common neurodegenerative disease worldwide, as well as those with other neurological disorders. The present review discusses cellular and molecular mechanisms through which chronic stress exposure may cause or increase risk for neurodegeneration, with a focus on the effects of stress in tau-mediated pathologies. This work compliments another recent review (Justice, 2018) in which the impacts of stress on the amyloid system in AD and other neurodegenerative or neuropsychiatric conditions was described, including clinical and behavioral perspectives.
2. Stress and physiology: the HPA axis
The HPA axis (Fig. 1) mediates part of the stress response through a complex cascade of neuroendocrine signals. Processing stressful stimuli in the brain involves the activation of neurons of the paraventricular nucleus of the hypothalamus (PVH), which release corticotrophin-releasing factor (CRF) and arginine vasopressin. These two hormones in turn stimulate the anterior pituitary gland to secrete adrenocorticotropic hormone (ACTH) into the general circulation, ultimately resulting in GC production by the adrenal cortex. Cortisol is the main GC in humans, with corticosterone playing the same role in many rodents. Proper function of the HPA axis is tightly modulated by negative feedback loops provided by cortisol and ACTH at the hypothalamic and pituitary levels (McEwen, 2007; Stephens and Wand, 2012). Furthermore, activation of the HPA axis can be influenced by a variety of hormones and neurotransmitters, including GABA, serotonin, norepinephrine, and opioids (Stephens and Wand, 2012).
Fig. 1.
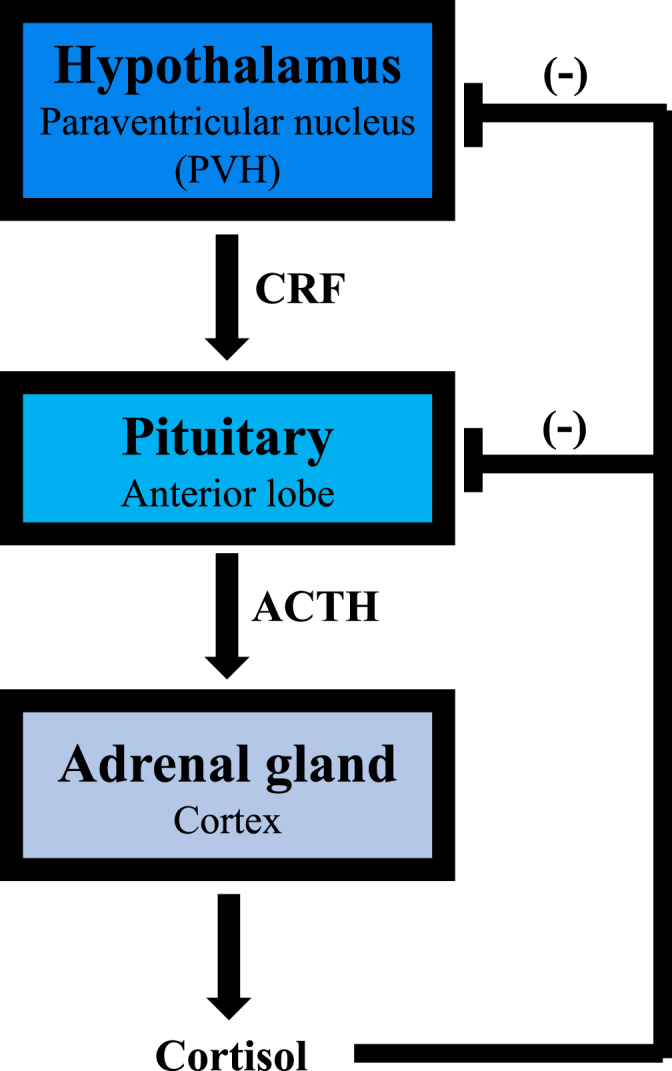
The hypothalamic-pituitary-adrenal (HPA) axis. In response to stress, the PVH produces CRF that stimulates pituitary release of ACTH, which in turn causes the release of cortisol from the adrenal gland. The system is shut down via negative feedback provided by cortisol at both pituitary and hypothalamic levels.
GCs exert broad physiological effects, modifying sugar, fat, and protein metabolism as well as influencing other processes such as cardiovascular function and immune responses (De Kloet et al., 2005). On a molecular level, GCs act through two types of receptors, the mineralocorticoid (MR) and glucocorticoid (GR) receptors, with both genomic and non-genomic effects. Genomic actions are described as slow or delayed and involve binding of cortisol to the GR (or the MR in some cases), with the resulting complex acting as a transcription factor and altering gene expression. In contrast, non-genomic actions occur faster and involve the interaction of GCs with other factors at the plasma membrane of the target cells (De Kloet et al., 1998; Ulrich-Lai and Herman, 2009).
It has become clear that GCs can affect behavioral and cognitive processes such as emotion, learning, and memory through GR and MR expressed in the brain (McEwen et al., 1986; Ahima et al., 1991; Dedovic et al., 2009; Medina et al., 2013). Stress-induced disruption of the HPA axis can cause excessive synthesis and secretion of GCs, which can lead to altered neuronal connectivity, synaptic loss, and neuronal atrophy (Watanabe et al., 1992; Sousa and Almeida, 2012). Modifications such as these can significantly alter brain function, and likely contribute to the development and progression of neurodegenerative diseases including AD (Dhikav and Anand, 2007).
3. Alzheimer's disease, stress, and tau
3.1. Alzheimer's disease
AD is the most prevalent neurodegenerative disease worldwide, estimated to affect approximately 10% of the population over 70 years old and with increasing incidence expected in the coming years (Plassman et al., 2007). Loss of memory is one of the early symptoms of AD, later progressing into other behavioral manifestations such as disorientation, aggressiveness, and dementia, culminating in complete mental collapse. AD is an incurable and irreversible condition for which there are currently no effective treatments (McKhann et al., 1984; Albert et al., 2011; Mendiola-Precoma et al., 2016). AD is characterized by two major neuropathological markers, extracellular amyloid beta (Aβ) plaques and intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau protein, which accumulate in brain regions responsible for emotion, learning, memory, and spatial navigation. Other lesions such as Hirano bodies and granulovacuolar degeneration can also be present (Ittner and Götz, 2011; Serrano-Pozo et al., 2011). A small number of AD cases (∼2%) are known to have a genetic basis; mutations in the amyloid precursor protein, presenilin-1 and presenilin-2 genes can cause familial AD. The etiology of sporadic AD cases which make up ∼98% of diagnoses remains largely unknown (Van Cauwenberghe et al., 2016).
3.2. Stress and Alzheimer's disease
Accumulating epidemiological evidence suggests that adverse environmental factors such as chronic emotional stress can play a role in the development and progression of AD, given that stress-susceptible individuals have a greater risk of manifesting AD (Wilson et al., 2003, 2005; 2006; Pardon, 2011). Furthermore, in familial cases of AD, the occurrence of a major stressful event such as loss of spouse was shown to accelerate the onset of the disease (Mejía et al., 2003). Exposure to stress early in the lifespan may also play a role in the later development of AD. Reduced quality of environment as a result of advanced maternal age, poor nutrition, low socioeconomic status, low level of education, or rural living during the early life periods of childhood and adolescence constitutes a risk factor for cognitive decline and AD as these individuals age (Stern et al., 1994; Räihä et al., 1998; Moceri et al., 2001; Borenstein et al., 2006; Miller and O'Callaghan, 2008). Elevated levels of circulating cortisol have been found in AD and dementia patients. Importantly, cortisol levels appear to be correlated with disease progression, given that higher plasma cortisol has been linked with accelerated AD outcomes and severity of dementia (Davis et al., 1986; Umegaki et al., 2000; Armanini et al., 2003; Csernansky et al., 2006; Huang et al., 2009). The results from these studies therefore provide a connection between stress, overactivation of the HPA axis, and AD. Subsequent studies in animal models have aimed to further explore the mechanisms underlying the relationship between stress and AD. For the purpose of this review, focus will be placed on the tau-related pathogenesis of AD.
3.3. Tau protein function
Tau is a highly soluble cytoskeletal protein that plays a prominent role in maintaining neuronal structure and integrity. It belongs to the family of microtubule-associated proteins (MAPs) that bind to and stabilize axonal microtubules, facilitating such diverse processes as axonal transport, synaptogenesis, and neurite outgrowth, with tau phosphorylation being critical for proper function (Drechsel et al., 1992; Nunez and Fischer, 1997; Stamer et al., 2002; Hanger et al., 2009). Although tau is widely known for this cellular physiological role, recent studies have identified additional functions for tau including the regulation of actin polymerization, recruitment of signaling molecules, control of mitochondrial dynamics, and even the control of gene expression (Dehmelt and Halpain, 2005; Baloyannis, 2006; Frost et al., 2014). Under pathophysiological conditions, however, tau can be hyperphosphorylated or misfolded, and aggregate into insoluble structures. In fact, the term “tauopathy” is used to encompass diseases that feature a pathological accumulation of tau in neuronal cells such as AD, Pick's disease, and corticobasal degeneration, among others (Spillantini et al., 1997; Ferrer et al., 2014; Orr et al., 2017). Tau hyperphosphorylation impairs its ability to bind to microtubules, decreases its solubility, and causes its aggregation into fibrillar structures known as paired helical filaments, which can then bundle into the highly neurotoxic NFTs found in AD (Wischik et al., 1988; Johnson and Stoothoff, 2004; Mietelska-Porowska et al., 2014; Arendt et al., 2016).
3.4. Stress-induced tau hyperphosphorylation
The first indications that stress can induce changes in tau phosphorylation came from studies in normal non-transgenic rodent models. Cold water stress has been shown to increase the level of phosphorylated tau (p-tau) in rat and mouse brain, including tau epitopes that are known to be hyperphosphorylated in the AD brain (Korneyev et al., 1995; Korneyev, 1998; Okawa et al., 2003; Feng et al., 2005; Yoshida et al., 2006). Importantly, these studies show that the accumulation of p-tau following cold water stress occurs rapidly, peaks, and then decreases, indicating that it may be a transient and reversible phenomenon. Other forms of stress also affect p-tau expression in rodents. Chronic exposure to restraint stress increases the levels of p-tau in the rat brain, although not to the degree where it accumulates into NFT-like p-tau (Yan et al., 2010), and rat models of chronic unpredictable mild stress have also resulted in increased tau hyperphosphorylation (Briones et al., 2011; Cuadrado-Tejedor et al., 2011; Yang et al., 2014; AbdAlla et al., 2015). These effects of restraint and chronic unpredictable stress have also been seen in mice, with increased expression of a multitude of p-tau epitopes as well as changing tau solubility (Rissman et al., 2007; Carroll et al., 2011; Sotiropoulos et al., 2011). In addition, metabolic stressors such as food deprivation and hypoxia can also increase p-tau in the mouse brain, including tau variants commonly seen in AD (Yanagisawa et al., 1999; Zhang et al., 2014).
3.5. Hormone-mediated tau hyperphosphorylation caused by stress
While non-transgenic models have provided valuable data on the ability of different stressors to induce hyperphosphorylation and abnormal accumulation of tau, their insights into the connection between the physiological response to stress (i.e. the HPA axis) and the underlying cellular and molecular mechanisms are limited. In this regard, genetic and pharmacological studies have proven useful in expanding the current knowledge in the field. Given that GC (e.g. cortisol) secretion is the key feature of HPA axis activation by stress, GC signaling has been targeted as a potential effector of stress-induced AD pathogenesis. Clinical evidence supports such a link, and GCs have been shown to cause neurotoxicity and neuronal cell atrophy and death (Watanabe et al., 1992; Behl et al., 1997; Crochemore et al., 2005; Cerqueira et al., 2007). Furthermore, GC administration has been shown to accelerate Aβ deposition in a transgenic mouse model of AD (Green et al., 2006), and both chronic and acute stress are able to induce Aβ pathology and cognitive impairment in AD mouse models (Jeong et al., 2006; Lee et al., 2009; Baglietto-Vargas et al., 2015).
Tau knock-out mice do not display hippocampal and cortical neuronal atrophy or cognitive deficits as a consequence of chronic stress, underscoring the importance of tau as an effector of stress-induced neuronal pathogenesis (Lopes et al., 2016, 2017). The administration of dexamethasone to a genetic model of AD that develops NFT pathology caused increased expression and mislocalization of tau without changing the phosphorylation status of tau (Green et al., 2006). Another study found that chronic GC administration in an AD transgenic mouse model caused a decrease in certain p-tau epitopes, but not others. The authors of this work further suggest that tau hyperphosphorylation and accumulation maybe a sequential process, with specific p-tau epitopes appearing during different stages of pathology, and could depend on other factors such as Aβ levels or kinase activity (Joshi et al., 2012). Stress has also been seen to induce the formation of abnormally truncated and insoluble tau proteins in transgenic mice (Sotiropoulos et al., 2015). More specifically, GC administration in combination with chronic unpredictable mild stress in normal rats increases the expression of p-tau epitopes commonly associated with AD (Sotiropoulos et al., 2011), whereas pharmacological enhancement of GC signaling has also been demonstrated to increase NFT-like p-tau in the rat brain (Yi et al., 2017).
The evidence linking increased GC with AD pathogenesis appears to be well established. Given the multiple regulatory mechanisms that govern the HPA axis, however, the possibility exists for other players within this signaling cascade to be involved in stress-induced tau hyperphosphorylation and accumulation (Rissman, 2009; Vyas et al., 2016). Evidence in support of this idea was first seen in the fact that in adrenalectomized mice that are devoid of endogenous GCs, cold water stress is still able to induce transient elevated levels of p-tau (Korneyev et al., 1995). The CRF system has therefore emerged as an additional potential candidate for mediating the effects of stress on tau. CRF is the hypothalamic peptide hormone that initiates HPA axis activation (Fig. 1), exerting its effects through two different G protein-coupled receptors, CRFR1 and CRFR2, which are both expressed in the brain (Bale and Vale, 2004; Power and Schulkin, 2006). Importantly, multiple sources of experimental evidence support a role for CRF in the pathogenesis of AD (Dong et al., 2014; Futch et al., 2017; Zhang and Rissman, 2017). Animal studies exploring the role of CRF signaling in stress-induced tau phosphorylation also strongly implicate CRF in AD-related tau pathogenesis. A study using CRFR1 and CRFR2 knock-out mice in combination with restraint stress demonstrated that these receptors exert differential effects on tau phosphorylation and aggregation, with CRFR1-deficient animals displaying attenuated levels of p-tau in response to stress, and stressed CRFR2 knock-out mice showing exacerbated p-tau levels (Rissman et al., 2007). A follow-on study further demonstrated that CRF signaling is involved in tau hyperphosphorylation and aggregation, since CRFR2 knock-out mice accumulated insoluble p-tau in response to restraint stress, whereas CRFR1-knockout and double CRFR knock-out mice did not. These findings indicate a prominent role for CRFR1 in mediating stress-induced tau pathogenesis (Rissman et al., 2012). An investigation using a mouse model of CRF overexpression that confers sustained GC signaling, in contrast, confirmed these observations and further supports the significance of CRF signaling in this system, with these animals showing increases in abnormal p-tau variants when compared to wild type animals (Campbell et al., 2015). A transgenic mouse model of AD in which a mutant form of tau is expressed was also used to study its relationship to stress and CRFR1, with findings demonstrating that stress-induced tau pathology is at least partially mediated by CRF signaling (Carroll et al., 2011). Finally, a study using acute immobilization stress in CRF-deficient mice showed that wild-type mice displayed higher levels of AD-associated p-tau in response to the stressor, but this effect was partially ameliorated in animals lacking CRF (Filipcik et al., 2012).
3.6. Beneficial effects of stress on tau phosphorylation and accumulation
The different forms of stress we have discussed to this point (temperature, restraint, immobilization, food deprivation) exert negative effects on tau-mediated pathogenesis and suggest a worsening pathology. However, certain stressors such as exercise and caloric restriction (CR) have been found to have beneficial health effects including decreased risk for developing neurodegenerative diseases, improving cognition, and extending lifespan (Stranahan and Mattson, 2008; van Praag, 2009; Rothman and Mattson, 2010; Lautenschlager et al., 2012). Interestingly, both exercise and CR are also known to activate the HPA axis (Droste et al., 2009; Kenny et al., 2014).
In the case of AD, human studies indicate that exercise intervention may lower the risk of developing the disease (Lautenschlager et al., 2008; Scarmeas et al., 2009; Nation et al., 2011). Exercise has been shown to improve behavioral parameters such as learning and memory, and diminish neuropathological markers in several animal models of AD (Adlard et al., 2005; Parachikova et al., 2008; Yuede et al., 2009). Regarding the effect of exercise on p-tau levels, several studies report that exercise can attenuate tau hyperphosphorylation in rodent models. Long-term treadmill exercise has been seen to dramatically reduce p-tau levels in several transgenic mouse models of AD, and this outcome is accompanied by other beneficial effects such as increases in the anti-oxidant response, improved cognitive function, and decreased Aβ deposition (Leem et al., 2009; Liu et al., 2013; Ohia-Nwoko et al., 2014; Kang and Cho, 2015). These data indicate that exercise provides a neuroprotective effect against AD pathology, even in genetically predisposed animals.
Beneficial effects of CR include increased neurogenesis, improved cognitive function, decreased anxiety, and extended lifespan (Weindruch et al., 1986; Stewart et al., 1989; Lee et al., 2002; Levay et al., 2007; Kuhla et al., 2013). In addition, CR has been shown to reduce neuronal loss and attenuate the accumulation of Aβ plaques in animal models of AD (Patel et al., 2005; Qin et al., 2006; Mouton et al., 2009), as well as decreasing tau phosphorylation in vitro (Bele et al., 2015). However, in terms of abnormal p-tau accumulation induced by stress, conflicting results have been found. A mouse model of tauopathy subjected to long-term CR was shown to have decreased p-tau accumulation (Rühlmann et al., 2016), and similar effects were observed in a transgenic model of AD that also underwent long-term CR (Halagappa et al., 2007). In a mouse model of tauopathy exposed to shorter and gradually increasing periods of CR, in contrast, no observable differences in p-tau levels were found (Brownlow et al., 2014). In yet another study, mice comprising another tauopathy model were first fed a high-calorie diet for two months and then placed on CR. These mice showed that restricted calorie intake actually aggravated tau pathology by increasing the levels of several p-tau variants (Gratuze et al., 2017). Interestingly, when the high-calorie fed animals were subjected to exercise rather than CR, p-tau accumulation was significantly reduced, thus highlighting the differential effects that two stressors can exert, as well as the importance of type and timing of stress.
3.7. Mechanisms of stress-induced neuronal damage by tau
The experimental findings described above strongly support the notion that stress-induced HPA axis dysfunction, or at least consistent and excessive HPA axis activation, can cause abnormal tau processing and lead to the type of neuropathology seen in AD. However, the specific cellular and molecular mechanisms that underlie the tau alterations in response to stress are only beginning to emerge.
3.7.1. Abnormal tau degradation: role of protein degradation pathways
An important feature in the regulation of p-tau by stress that was observed in many of the studies reviewed here is the transient nature of tau hyperphosphorylation and accumulation. This suggests that these responses may be a normal component of stress processing and that mechanisms exist to protect neuronal cells from stress-induced injury. Specifically, abnormal p-tau needs to be properly processed and degraded before it becomes neurotoxic. The proteostasis network (PN) constitutes a highly complex quality control system that ensures proper protein expression and function. The PN is composed of a multitude of signaling mechanisms and cellular machinery components that regulate protein transcription, modification, sorting, trafficking, and localization (Balch et al., 2008; Díaz-Villanueva et al., 2015). In the normal brain, chaperone proteins are a crucial defense against abnormally folded and potentially neurotoxic proteins. Chaperones assist in proper protein folding and are capable of correcting abnormal protein conformations (Kim et al., 2013). Proteolysis is a critical component of the PN, and it includes two main degradative pathways in charge of clearing misfolded and aggregated proteins: the ubiquitin-proteasome system (UPS) and the autophagy-lysosomal pathway (ALP). The UPS targets soluble, short-lived proteins for degradation by the proteasome, a multicatalytic protein complex that uses ubiquitin as a signal. The ALP is a bulk degradative pathway that involves the formation of an autophagosome which engulfs cellular components and fuses with the lysosome for destruction and nutrient recycling (Tanaka and Matsuda, 2014; Lim and Yue, 2015). The activity of the PN is known to decrease with age, and accumulating evidence indicates that PN impairment occurs in neurodegenerative diseases, allowing abnormal protein aggregation to occur and leading to irreversible neuronal damage and ultimately neuronal death (Hipp et al., 2014). However, very few studies have addressed the influence of stress on the PN and subsequent contributions to neurodegeneration. Chronic stress has been shown to alter the levels of molecular chaperones in a transgenic mouse model of tauopathy (Sotiropoulos et al., 2015). A study using non-transgenic rats demonstrated increased expression of LC3 (an autophagy marker) in response to chronic unpredictable mild stress, while another study using wild-type rats subjected to acute restraint stress also showed increased autophagic activity (Hou et al., 2015; Jevtić et al., 2016). While exercise has been shown to induce autophagy in the mouse and rat brain (He et al., 2012; Marques-Aleixo et al., 2015), tauopathy mouse models subjected to treadmill exercise have shown conflicting results. Two studies using a transgenic mouse model expressing human tau demonstrated no changes in levels of autophagic markers (Kang and Cho, 2015; Gratuze et al., 2017), while another study using a different tauopathy model subjected to long-term treadmill exercise showed increased autophagic activity (Ohia-Nwoko et al., 2014). In all of these studies, exercise was employed as an intervention strategy against tau pathogenesis, and although positive effects were observed on behavioral parameters such as locomotion and cognitive performance, it appears that the influence of stress on the PN varies and its effects on tau-dependent pathology rely on several factors such as timing, animal model, and the stressor itself.
3.7.2. Tau hyperphosphorylation by stress: role of kinases
As noted, phosphorylation is a key feature of tau, since it is both critical for proper function and is the key factor in the expression of the pathological properties of tau. In agreement with its importance in regulating tau function, more than 80 tau phosphorylation sites have been identified (Mietelska-Porowska et al., 2014). Kinases capable of phosphorylating tau include glycogen synthase kinase 3β (GSK3β), cyclin-dependent kinase 5 (cdk5), c-Jun amino terminal kinase (JNK), calmodulin-dependent kinase II (CaMKII), adenosine-monophosphate activated protein kinase (AMPK), cyclic AMP-dependent protein kinase (PKA), and several others. Accordingly, several phosphatases also participate in the proper maintenance of the tau phosphorylation/dephosphorylation balance. These include protein phosphatases 2A and 2B (PP2A and PP2B) and protein phosphatases 1 and 5 (PP1 and PP5), among others (Stoothoff and Johnson, 2005; Wang et al., 2007; Dolan and Johnson, 2010). In a study exploring cold water stress-induced tau phosphorylation, GSK3β, cdk5, and JNK were found to be potential candidates for the observed increase in p-tau levels (Okawa et al., 2003). Decreased GSK3β activity, but not other kinases such as cdk5, accompanied by selective decreases in specific p-tau epitopes has also been observed following chronic GC exposure in an AD model, suggesting specificity in the GC-modulated tau kinase activity (Joshi et al., 2012). GSK3β, cdk5, JNK, and ERK were also identified as important factors contributing to stress-induced tau hyperphosphorylation in CRFR knock-out animals, whereas JNK was found to be important in tau pathology in CRF-overexpressing mice (Rissman et al., 2007; Campbell et al., 2015). A study employing chronic unpredictable mild stress, however, identified cdk5 as the main contributor to tau hyperphosphorylation, while exercise was found to differentially affect both the GSK3β and cdk5 pathways, suggesting that different stressors may elicit specific kinase responses (Cuadrado-Tejedor et al., 2011; Liu et al., 2013).
3.7.3. Tau missorting and cytoskeletal alterations
Tau is predominantly localized to neuronal axons, where it regulates cytoskeletal organization by interacting with microtubules and actin filaments, participates in axonal transport, and contributes to neurotrophic signaling (Gustke et al., 1994; Sharma et al., 2007; Majounie et al., 2013). Missorting of tau into somatodendritic compartments is believed to be a key step in AD pathogenesis, since this is the predominant location of NFTs (Götz et al., 1995; Zempel and Mandelkow, 2014). Furthermore, tau is also known to be mislocalized to synapses in AD, thus contributing to cognitive dysfunction (Zempel et al., 2010). Chronic unpredictable stress is now known to play a role in tau phosphorylation and missorting, causing accumulation of tau in dendrites and synapses. Displacement of tau from its normal axonal location, coupled with hyperphosphorylation, could then lead to cytoskeletal instability and axonal damage; its missorting into other subcellular locations could contribute to AD pathogenesis (Sotiropoulos and Sousa, 2016; Lopes et al., 2016; Vyas et al., 2016).
4. Conclusion
Given the multifactorial nature of AD and the many physiological processes that can be affected by stress, a relationship between stress and an individual's vulnerability to development or progression of neurodegeneration has been difficult to establish. Stress appears to be a critical factor that influences tau-mediated pathogenesis in AD and possibly other tauopathies. The experimental evidence reviewed here indicates that the effects of stress on tau hyperphosphorylation and accumulation could be mediated by the activation of the HPA axis and subsequent GC action. However, the specific cellular and molecular mechanisms through which stress influences tau-mediated pathology remain poorly understood, and the specific links to GC action are yet to be described. Nevertheless, it is clear that stress is able to affect the brain and activate the HPA axis, with the subsequent GC release being able to impact a host of subcellular mechanisms that influence normal tau proteostasis, ultimately disrupting neuronal structure and function and leading to neuronal cell death, which is the ultimate outcome of neurodegeneration (Fig. 2). This process can be further aggravated by mutations in key genes, and aging which is in itself an important risk factor for neurodegenerative diseases. In addition, the experience of other stressors can further aggravate the conditions leading to neurodegeneration, and the consequences of cumulative stress exposure are currently under investigation. Interestingly, intervention by means of exercise or caloric restriction can have a beneficial effect, ameliorating or slowing stress-induced pathology even though these stimuli are also classified as stressors. Future studies should focus on further deciphering the underlying mechanisms of stress-mediated protein aggregation, with particular emphasis on GC modes of action, stress exposure in the early life period when deleterious effects on development may be manifested, and the interactions of cumulative stress and aging.
Fig. 2.

Tau pathogenesis induced by stress is mediated through HPA axis dysfunction. The proposed model shows that stress activates the HPA axis and triggers GC release. GC-mediated signaling then impacts tau proteostasis, causing misfolding, truncation, hyperphosphorylation, and abnormal accumulation and aggregation. This in turn causes neuronal damage, ultimately leading to neurodegeneration. Interventions such as exercise and caloric restriction can ameliorate tau pathogenesis, whereas other factors such as further stress, mutations, and age can worsen the pathology.
Acknowledgements
Research in the authors' laboratory is funded by grant G12MD007592 (NIMHD-NIH) awarded to the Border Biomedical Research Center, and by grant DA029989 (NIDA-NIH).
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.ynstr.2018.08.009.
Conflicts of interest
The authors declare no conflict of interest.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- AbdAlla S., El Hakim A., Abdelbaset A., Elfaramawy Y., Quitterer U. Inhibition of ACE retards tau hyperphosphorylation and signs of neuronal degeneration in aged rats subjected to chronic mild stress. BioMed Res. Int. 2015;2015:917156. doi: 10.1155/2015/917156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adlard P.A., Perreau V.M., Pop V., Cotman C.W. Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer's disease. J. Neurosci. 2005;25:4217–4221. doi: 10.1523/JNEUROSCI.0496-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahima R., Krozowski Z., Harlan R. Type I corticosteroid receptor-like immunoreactivity in the rat CNS: distribution and regulation by corticosteroids. J. Comp. Neurol. 1991;313:522–538. doi: 10.1002/cne.903130312. [DOI] [PubMed] [Google Scholar]
- Albert M.S., DeKosky S.T., Dickson D., Dubois B., Feldman H.H., Fox N.C., Gamst A., Holtzman D.M., Jagust W.J., Petersen R.C., Snyder P.J., Carrillo M.C., Thies B., Phelps C.H. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen B.L., Kiecolt-Glaser J.K., Glaser R. A biobehavioral model of cancer stress and disease course. Am. Psychol. 1994;49:389–404. doi: 10.1037//0003-066x.49.5.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T., Stieler J.T., Holzer M. Tau and tauopathies. Brain Res. Bull. 2016;126:238–292. doi: 10.1016/j.brainresbull.2016.08.018. [DOI] [PubMed] [Google Scholar]
- Armanini D., Vecchio F., Basso A., Milone F.F., Simoncini M., Fiore C., Mattarello M.J., Sartorato P., Karbowiak I. Alzheimer's disease: pathophysiological implications of measurement of plasma cortisol, plasma dehydroepiandrosterone sulfate, and lymphocytic corticosteroid receptors. Endocrine. 2003;22:113–118. doi: 10.1385/ENDO:22:2:113. [DOI] [PubMed] [Google Scholar]
- Baglietto-Vargas D., Chen Y., Suh D., Ager R.R., Rodriguez-Ortiz C.J., Medeiros R., Myczek K., Green K.N., Baram T.Z., LaFerla F.M. Short-term modern life-like stress exacerbates Aβ-pathology and synapse loss in 3xTg-AD mice. J. Neurochem. 2015;134:915–926. doi: 10.1111/jnc.13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch W.E., Morimoto R.I., Dillin A., Kelly J.W. Adapting proteostais for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- Bale T.L., Epperson C.N. Sex differences and stress across the lifespan. Nat. Neurosci. 2015;18:1413–1420. doi: 10.1038/nn.4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale T.L., Vale W.W. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu. Rev. Pharmacol. Toxicol. 2004;44:525–557. doi: 10.1146/annurev.pharmtox.44.101802.121410. [DOI] [PubMed] [Google Scholar]
- Baloyannis S.J. Mitochondrial alterations in Alzheimer's disease. J. Alzheimers Dis. 2006;9:119–126. doi: 10.3233/jad-2006-9204. [DOI] [PubMed] [Google Scholar]
- Bele M.S., Gajare K.A., Deshmukh A.A. Caloric restriction mimetic 2-deoxyglucose maintains cytoarchitecture and reduces tau phosphorylation in primary culture of mouse hippocampal pyramidal neurons. In Vitro Cell. Dev. Biol. Anim. 2015;51:546–555. doi: 10.1007/s11626-015-9867-1. [DOI] [PubMed] [Google Scholar]
- Behl C., Lezoualc'h F., Trapp T., Widmann M., Skutella T., Holsboer F. Glucocorticoids enhance oxidative stress-induced cell death in hippocampal neurons in vitro. Endocrinology. 1997;13:101–106. doi: 10.1210/endo.138.1.4835. [DOI] [PubMed] [Google Scholar]
- Borenstein A.R., Copenhaver C.I., Mortimer J.A. Early-life risk factors for Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2006;20:63–72. doi: 10.1097/01.wad.0000201854.62116.d7. [DOI] [PubMed] [Google Scholar]
- Briones A., Gagno S., Martisova E., Dobarro M., Aisa B., Solas M., Tordera R., Ramírez M. Stress-induced anhedonia is associated with an increase in Alzheimer's disease-related markers. Br. J. Pharmacol. 2011;165:897–907. doi: 10.1111/j.1476-5381.2011.01602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlow M.L., Joly-Amado A., Azam S., Elza M., Selenica M.L., Pappas C., Small B., Engelman R., Gordon M.N., Morgan D. Partial rescue of memory deficits induced by calorie restriction in a mouse model of tau deposition. Behav. Brain Res. 2014;271:79–88. doi: 10.1016/j.bbr.2014.06.001. [DOI] [PubMed] [Google Scholar]
- Cairns N.J., Lee V.M., Trojanowski J.Q. The cytoskeleton in neurodegenerative diseases. J. Pathol. 2004;204:438–449. doi: 10.1002/path.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell M.S.N., Zhang C., Monte L., Roe A.D., Rice K.C., Taché Y., Masliah E., Rissman R.A. Increased tau phosphorylation and aggregation in the hippocampus of mice overexpressing corticotropin-releasing factor. J. Alzheimers Dis. 2015;43:967–976. doi: 10.3233/JAD-141281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll J.C., Iba M., Bangasser D.A., Valentino R.J., James M.J., Brunden K.R., Lee V.M., Trojanowski J.Q. Chronic stress exacerbates tau pathology, neurodegeneration, and cognitive performance through a corticotropin-releasing factor receptor-dependent mechanism in a transgenic mouse model of tauopathy. J. Neurosci. 2011;31:11436–11449. doi: 10.1523/JNEUROSCI.3836-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerqueira J.J., Taipa R., Uylings H.B., Almeida O.F., Sousa N. Specific configuration of dendritic degeneration in pyramidal neurons of the medial prefrontal cortex induced by differing corticosteroid regimens. Cerebr. Cortex. 2007;17:1998–2006. doi: 10.1093/cercor/bhl108. [DOI] [PubMed] [Google Scholar]
- Chen Y., Baram T.Z. Toward understanding how early-life stress reprograms cognitive and emotional brain networks. Neuropsychopharmacology. 2016;41:197–206. doi: 10.1038/npp.2015.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrousos G. Stress and disorders of the stress system. Nat. Rev. Endocrinol. 2009;5:374–381. doi: 10.1038/nrendo.2009.106. [DOI] [PubMed] [Google Scholar]
- Crochemore C., Lu J., Wu Y., Liposits Z., Sousa N., Holsboer F., Almeida O.F. Direct targeting of hippocampal neurons for apoptosis by glucocorticoids is reversible by mineralocorticoid receptor activation. Mol. Psychiatr. 2005;10:790–798. doi: 10.1038/sj.mp.4001679. [DOI] [PubMed] [Google Scholar]
- Csernansky J.G., Dong H., Fagan A.M., Wang L., Xiong C., Holtzman D.M., Morris J.C. Plasma cortisol and progression of dementia in subjects with Alzheimer-type dementia. Am. J. Psychiatr. 2006;163:2164–2169. doi: 10.1176/appi.ajp.163.12.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado-Tejedor M., Ricobaraza A., Del Río J., Frechilla D., Franco R., Pérez-Mediavilla A., Garcia-Osta A. Chronic mild stress in mice promotes cognitive impairment and CDK5-dependent tau hyperphosphorylation. Behav. Brain Res. 2011;220:338–343. doi: 10.1016/j.bbr.2011.01.005. [DOI] [PubMed] [Google Scholar]
- Davis K.L., Davis B.M., Greenwald B.S., Mohs R.C., Mathé A.A., Johns C.A., Horvath T.B. Cortisol and Alzheimer's disease, I: basal studies. Am. J. Psychiatr. 1986;143:300–305. doi: 10.1176/ajp.143.3.300. [DOI] [PubMed] [Google Scholar]
- De Kloet E.R., Joëls M., Holsboer F. Stress and the brain: from adaptation to disease. Nat. Rev. Neurosci. 2005;6:463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- De Kloet E.R., Vreugdenhil E., Oitzl M.S., Joëls M. Brain corticosteroid receptor balance in health and disease. Endocr. Rev. 1998;19:269–301. doi: 10.1210/edrv.19.3.0331. [DOI] [PubMed] [Google Scholar]
- Dedovic K., Duchesne A., Andrews J., Engert V., Pruessner J.C. The brain and the stress axis: the neural correlates of cortisol regulation in response to stress. Neuroimage. 2009;47:864–871. doi: 10.1016/j.neuroimage.2009.05.074. [DOI] [PubMed] [Google Scholar]
- Dehmelt L., Halpain S. The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 2005;6:204. doi: 10.1186/gb-2004-6-1-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhikav V., Anand K.S. Glucocorticoids may initiate Alzheimer's disease: a potential therapeutic role for mifepristone (RU-486) Med. Hypotheses. 2007;68:1088–1092. doi: 10.1016/j.mehy.2006.09.038. [DOI] [PubMed] [Google Scholar]
- Díaz-Villanueva J.F., Díaz-Molina R., García-González V. Protein folding and mechanisms of proteostasis. Int. J. Mol. Sci. 2015;16:17193–18230. doi: 10.3390/ijms160817193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan P.J., Johnson G.V. The role of tau kinases in Alzheimer's disease. Curr. Opin. Drug Discov. Dev. 2010;13:595–603. [PMC free article] [PubMed] [Google Scholar]
- Dong H., Wang S., Zeng Z., Li F., Montalvo-Ortiz J., Tucker C., Akhtar S., Shi J., Meltzer H.Y., Rice K.C., Csernansky J.G. Effects of corticotrophin-releasing factor receptor 1 antagonists on amyloid-β and behavior in Tg2576 mice. Psychopharmacology. 2014;23:4711–4722. doi: 10.1007/s00213-014-3629-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drechsel D.N., Hyman A.A., Cobb M.H., Kirschner M.W. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell. 1992;3:1141–1154. doi: 10.1091/mbc.3.10.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droste S.K., Collins A., Lightman S.L., Linthorst A.C., Reul J.M. Distinct, time-dependent effects of voluntary exercise on circadian and ultradian rhythms and stress responses of free corticosterone in the rat hippocampus. Endocrinology. 2009;150:4170–4179. doi: 10.1210/en.2009-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esch T., Stefano G.B., Fricchione G.L., Benson H. The role of stress in neurodegenerative diseases and mental disorders. Neuroendocrinol. Lett. 2002;23:199–2008. [PubMed] [Google Scholar]
- Esch T., Stefano G.B., Fricchione G.L., Benson H. Stress in cardiovascular diseases. Med. Sci. Mon. Int. Med. J. Exp. Clin. Res. 2002;8:93–101. [PubMed] [Google Scholar]
- Feng Q., Cheng B., Yang R., Sun F.Y., Zhu C.Q. Dynamic changes of phosphorylated tau in mouse hippocampus after cold water stress. Neurosci. Lett. 2005;388:13–16. doi: 10.1016/j.neulet.2005.06.022. [DOI] [PubMed] [Google Scholar]
- Ferrer I., López-González I., Carmona M., Arregui L., Dalfó E., Torrejón-Escribano B., Diehl R., Kovacs G.G. Glial and neuronal tau pathology in tauopathies: characterization of disease-specific phenotypes and tau pathology progression. J. Neuropathol. Exp. Neurol. 2014;73:81–97. doi: 10.1097/NEN.0000000000000030. [DOI] [PubMed] [Google Scholar]
- Filipcik P., Novak P., Mravec B., Ondicova K., Krajciova G., Novak M., Kvetnansky R. Tau protein phosphorylation in diverse brain areas of normal and CRH deficient mice: up-regulation by stress. Cell. Mol. Neurobiol. 2012;32:837–845. doi: 10.1007/s10571-011-9788-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B., Hemberg M., Lewis J., Feany M.B. Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 2014;17:357–366. doi: 10.1038/nn.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futch H.S., Croft C.L., Truong V.Q., Krause E.G., Golde T.E. Targeting psychologic stress signaling pathways in Alzheimer's disease. Mol. Nerodegener. 2017;12:49. doi: 10.1186/s13024-017-0190-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser R., Kiecolt-Glaser J.R. Stress-induced immune dysfunction: implications for health. Nat. Rev. Immunol. 2005;5:243–251. doi: 10.1038/nri1571. [DOI] [PubMed] [Google Scholar]
- Götz J., Probst A., Spillantini M.G., Schäfer T., Jakes R., Bürki K., Goedert M. Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J. 1995;14:1301–1313. doi: 10.1002/j.1460-2075.1995.tb07116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratuze M., Julien J., Morin F., Marette A., Planel E. Differential effects of voluntary treadmill exercise and caloric restriction on tau pathogenesis in a mouse model of Alzheimer's disease-like tau pathology fed with Western diet. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2017;79:452–461. doi: 10.1016/j.pnpbp.2017.08.001. [DOI] [PubMed] [Google Scholar]
- Green K.N., Billings L.M., Roozendaal B., McGaugh J.L., LaFerla F.M. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer's disease. J. Neurosci. 2006;26:9047–9056. doi: 10.1523/JNEUROSCI.2797-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grippo A.J., Johnson A.K. Stress, depression and cardiovascular dysregulation: a review of neurobiological mechanisms and the integration of research from preclinical disease models. Stress. 2009;12:1–21. doi: 10.1080/10253890802046281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustke N., Trinczek B., Biernat J., Mandelkow E.M., Mandelkow E. Domains of tau protein and interactions with microtubules. Biochemistry. 1994;33:9511–9522. doi: 10.1021/bi00198a017. [DOI] [PubMed] [Google Scholar]
- Halagappa V.K., Guo Z., Pearson M., Matsuoka Y., Cutler R.G., Laferla F.M., Mattson M.P. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer's disease. Neurobiol. Dis. 2007;26:212–220. doi: 10.1016/j.nbd.2006.12.019. [DOI] [PubMed] [Google Scholar]
- Hanger D.P., Anderton B.H., Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009;15:112–119. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- He C., Sumpter R., Jr., Levine B. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy. 2012;8:1548–1551. doi: 10.4161/auto.21327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipp M.S., Park S.H., Hartl F.U. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 2014;24:506–514. doi: 10.1016/j.tcb.2014.05.003. [DOI] [PubMed] [Google Scholar]
- Hou G., Zhao Y., Yang X., Yuan T.F. Autophagy does not lead to the asymmetrical hippocampal injury in chronic stress. Physiol. Behav. 2015;144:1–6. doi: 10.1016/j.physbeh.2015.03.011. [DOI] [PubMed] [Google Scholar]
- Huang C.W., Lui C.C., Chang W.N., Lu C.H., Wang Y.L., Chang C.C. Elevated basal cortisol level predicts lower hippocampal volume and cognitive decline in Alzheimer's disease. J. Clin. Neurosci. 2009;16:1283–1286. doi: 10.1016/j.jocn.2008.12.026. [DOI] [PubMed] [Google Scholar]
- Ittner L.M., Götz J. Amyloid-β and tau--a toxic pas de deux in Alzheimer's disease. Nat. Rev. Neurosci. 2011;12:65–72. doi: 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- Jellinger K.A. Recent advances in our understanding of neurodegeneration. J. Neural. Transm. 2009;116:1111–1166. doi: 10.1007/s00702-009-0240-y. [DOI] [PubMed] [Google Scholar]
- Jellinger K.A. Basic mechanisms of neurodegeneration: a critical update. J. Cell Mol. Med. 2010;14:457–487. doi: 10.1111/j.1582-4934.2010.01010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong Y.H., Park C.H., Yoo J., Shin K.Y., Ahn S.M., Kim H.S., Lee S.H., Emson P.C., Suh Y.H. Chronic stress accelerates learning and memory impairments and increases amyloid deposition in APPV717I-CT100 transgenic mice, an Alzheimer's disease model. Faseb. J. 2006;20:729–731. doi: 10.1096/fj.05-4265fje. [DOI] [PubMed] [Google Scholar]
- Jevtić G., Nikolić T., Mirčić A., Stojković T., Velimirović M., Trajković V., Marković I., Trbovich A.M., Radonjić N.V., Petronijević N.D. Mitochondrial impairment, apoptosis and autophagy in a rat brain as immediate and long-term effects of perinatal phencyclidine treatment - influence of restraint stress. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2016;66:87–96. doi: 10.1016/j.pnpbp.2015.11.014. [DOI] [PubMed] [Google Scholar]
- Johnson G.V., Stoothoff W.H. Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 2004;117:5721–5729. doi: 10.1242/jcs.01558. [DOI] [PubMed] [Google Scholar]
- Joshi Y.B., Chu J., Praticò D. Stress hormone leads to memory deficits and altered tau phosphorylation in a model of Alzheimer's disease. J. Alzheimers Dis. 2012;31:167–176. doi: 10.3233/JAD-2012-120328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice N.J. The relationship between stress and Alzheimer's disease. Neurobiol. Stress. 2018;8:127–133. doi: 10.1016/j.ynstr.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang E.B., Cho J.Y. Effect of treadmill exercise on PI3K/AKT/mTOR, autophagy, and Tau hyperphosphorylation in the cerebral cortex of NSE/htau23 transgenic mice. J. Exerc. Nutrition Biochem. 2015;19:199–209. doi: 10.5717/jenb.2015.15090806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A., Dunn E., Kostaki A., Andrews M.H., Matthews S.G. Fetal programming of hypothalamo-pituitary-adrenal function: prenatal stress and glucocorticoids. J. Physiol. (London) 2006;572:31–44. doi: 10.1113/jphysiol.2006.105254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny R., Dinan T., Cai G., Spencer S.J. Effects of mild calorie restriction on anxiety and hypothalamic-pituitary-adrenal axis responses to stress in the male rat. Phys. Rep. 2014;2 doi: 10.1002/phy2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.E., Hipp M.S., Bracher A., Hayer-Hartl M., Hartl F.U. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013;82:323–355. doi: 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- Korneyev A. Stress-induced tau phosphorylation in mouse strains with different brain Erk 1 + 2 immunoreactivity. Neurochem. Res. 1998;23:1539–1543. doi: 10.1023/a:1020980004539. [DOI] [PubMed] [Google Scholar]
- Korneyev A., Binder L., Bernardis J. Rapid reversible phosphorylation of rat brain tau proteins in response to cold water stress. Neurosci. Lett. 1995;191:19–22. doi: 10.1016/0304-3940(95)11546-3. [DOI] [PubMed] [Google Scholar]
- Kuhla A., Lange S., Holzmann C., Maass F., Petersen J., Vollmar B., Wree A. Lifelong caloric restriction increases working memory in mice. PLoS One. 2013;8 doi: 10.1371/journal.pone.0068778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautenschlager N.T., Cox K., Cyarto E.V. The influence of exercise on brain aging and dementia. Biochim. Biophys. Acta. 2012;1822:474–481. doi: 10.1016/j.bbadis.2011.07.010. [DOI] [PubMed] [Google Scholar]
- Lautenschlager N.T., Cox K.L., Flicker L., Foster J.K., van Bockxmeer F.M., Xiao J., Greenop K.R., Almeida O.P. Effect of physical activity on cognitive function in older adults at risk for Alzheimer disease: a randomized trial. J. Am. Med. Assoc. 2008;300:1027–1037. doi: 10.1001/jama.300.9.1027. [DOI] [PubMed] [Google Scholar]
- Lee J., Duan W., Mattson M.P. Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J. Neurochem. 2002;82:1367–1375. doi: 10.1046/j.1471-4159.2002.01085.x. [DOI] [PubMed] [Google Scholar]
- Lee K.W., Kim J.B., Seo J.S., Kim T.K., Im J.Y., Baek I.S., Kim K.S., Lee J.K., Han P.L. Behavioral stress accelerates plaque pathogenesis in the brain of Tg2576 mice via generation of metabolic oxidative stress. J. Neurochem. 2009;108:165–175. doi: 10.1111/j.1471-4159.2008.05769.x. [DOI] [PubMed] [Google Scholar]
- Leem Y.H., Lim H.J., Shim S.B., Cho J.Y., Kim B.S., Han P.L. Repression of tau hyperphosphorylation by chronic endurance exercise in aged transgenic mouse model of tauopathies. J. Neurosci. Res. 2009;87:2561–2570. doi: 10.1002/jnr.22075. [DOI] [PubMed] [Google Scholar]
- Lenroot R.K., Giedd J.N. Brain development in children and adolescents: insights from anatomical magnetic resonance imaging. Neurosci. Biobehav. Rev. 2006;30:718–729. doi: 10.1016/j.neubiorev.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Levay E.A., Govic A., Penman J., Paolini A.G., Kent S. Effects of adult-onset calorie restriction on anxiety-like behavior in rats. Physiol. Behav. 2007;92:889–896. doi: 10.1016/j.physbeh.2007.06.018. [DOI] [PubMed] [Google Scholar]
- Lim J., Yue Z. Neuronal aggregates: formation, clearance and spreading. Dev. Cell. 2015;32:491–501. doi: 10.1016/j.devcel.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.L., Zhao G., Zhang H., Shi L.D. Long-term treadmill exercise inhibits the progression of Alzheimer's disease-like neuropathology in the hippocampus of APP/PS1 transgenic mice. Behav. Brain Res. 2013;256:261–272. doi: 10.1016/j.bbr.2013.08.008. [DOI] [PubMed] [Google Scholar]
- Lopes S., Teplytska L., Vaz-Silva J., Dioli C., Trindade R., Morais M., Webhofer C., Maccarrone G., Almeida O.F.X., Turck C.W., Sousa N., Sotiropoulos I., Filiou M.D. Tau deletion prevents stress-induced dendritic atrophy in prefrontal cortex: role of synaptic mitochondria. Cerebr. Cortex. 2017;27:2580–2591. doi: 10.1093/cercor/bhw057. [DOI] [PubMed] [Google Scholar]
- Lopes S., Vaz-Silva J., Pinto V., Dalla C., Kokras N., Bedenk B., Mack N., Czisch M., Almeida O.F., Sousa N., Sotiropoulos I. Tau protein is essential for stress-induced brain pathology. Proc. Natl. Acad. Sci. Unit. States Am. 2016;113:E3755–E3763. doi: 10.1073/pnas.1600953113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majounie E., Cross W., Newsway V., Dillman A., Vandrovcova J., Morris C.M., Nalls M.A., Ferrucci L., Owen M.J., O'Donovan M.C., Cookson M.R., Singleton A.B., de Silva R., Morris H.R. Variation in tau isoform expression in different brain regions and disease states. Neurobiol. Aging. 2013;34:1922e1912–1922e1927. doi: 10.1016/j.neurobiolaging.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Aleixo I., Santos-Alves E., Balça M.M., Rizo-Roca D., Moreira P.I., Oliveira P.J., Magalhães J., Ascensão A. Physical exercise improves brain cortex and cerebellum mitochondrial bioenergetics and alters apoptotic, dynamic and auto(mito)phagy markers. Neuroscience. 2015;301:480–495. doi: 10.1016/j.neuroscience.2015.06.027. [DOI] [PubMed] [Google Scholar]
- McEwen B.S. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol. Rev. 2007;87 doi: 10.1152/physrev.00041.2006. 873-804. [DOI] [PubMed] [Google Scholar]
- McEwen B.S., De Kloet E.R., Rostene W. Adrenal steroid receptors and action in the nervous system. Physiol. Rev. 1986;66:1121–1188. doi: 10.1152/physrev.1986.66.4.1121. [DOI] [PubMed] [Google Scholar]
- McKhann G., Drachman D., Folstein M., Katzman R., Price D., Stadlan E.M. Clinical diagnosis of alzheimer's disease: report of the NINCDS-ADRDAWork group under the auspices of department of health and human services task force on alzheimer's disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Medina A., Seasholtz A.F., Sharma V., Burke S., Bunney W., Jr., Myers R.M., Schatzberg A., Akil H., Watson S.J. Glucocorticoid and mineralocorticoid receptor expression in the human hippocampus in major depressive disorder. J. Psychiatr. Res. 2013;47:307–314. doi: 10.1016/j.jpsychires.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejía S., Giraldo M., Pineda D., Ardila A., Lopera F. Nongenetic factors as modifiers of the age of onset of familial Alzheimer's disease. Int. Psychogeriatr. 2003;15:337–349. doi: 10.1017/s1041610203009591. [DOI] [PubMed] [Google Scholar]
- Mendiola-Precoma J., Berumen L.C., Padilla K., Garcia-Alcocer G. Therapies for prevention and treatment of Alzheimer's disease. BioMed Res. Int. 2016;2016:2589276. doi: 10.1155/2016/2589276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer S.E., Chrousos G.P., Gold P.W. Major depression and the stress system: a life span perspective. Dev. Psycopathol. 2001;13:565–580. doi: 10.1017/s095457940100308x. [DOI] [PubMed] [Google Scholar]
- Mietelska-Porowska A., Wasik U., Goras M., Filipek A., Niewiadomska G. Tau protein modifications and interactions: their role in function and dysfunction. Int. J. Mol. Sci. 2014;15:4671–4713. doi: 10.3390/ijms15034671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D.B., O'Callaghan J.P. Do early-life insults contribute to the late-life development of Parkinson and Alzheimer diseases? Metabolism. 2008;57:S44–S49. doi: 10.1016/j.metabol.2008.07.011. [DOI] [PubMed] [Google Scholar]
- Moceri V.M., Kukull W.A., Emanual I., van Belle G., Starr J.R., Schellenberg G.D., McCormick W.C., Bowen J.D., Teri L., Larson E.B. Using census data and birth certificates to reconstruct the early-life socioeconomic environment and the relation to the development of Alzheimer's disease. Epidemiology. 2001;12:383–389. doi: 10.1097/00001648-200107000-00007. [DOI] [PubMed] [Google Scholar]
- Moreno-Smith M., Lutgendorf S.K., Sood A.K. Impact of stress on cancer metastasis. Future Oncol. 2010;6:1863–1881. doi: 10.2217/fon.10.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouton P.R., Chachich M.E., Quigley C., Spangler E., Ingram D.K. Caloric restriction attenuates amyloid deposition in middle-aged dtg APP/PS1 mice. Neurosci. Lett. 2009;464:184–187. doi: 10.1016/j.neulet.2009.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nation D.A., Hong S., Jak A.J., Delano-Wood L., Mills P.J., Bondi M.W., Dimsdale J.E. Stress, exercise, and Alzheimer's disease: a neurovascular pathway. Med. Hypotheses. 2011;76:846–854. doi: 10.1016/j.mehy.2011.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez J., Fischer I. Microtubule-associated proteins (MAPs) in the peripheral nervous system during development and regeneration. J. Mol. Neurosci. 1997;8:207–222. doi: 10.1007/BF02736834. [DOI] [PubMed] [Google Scholar]
- Ohia-Nwoko O., Montazari S., Lau Y.S., Eriksen J.L. Long-term treadmill exercise attenuates tau pathology in P301S tau transgenic mice. Mol. Neurodegener. 2014;9:54. doi: 10.1186/1750-1326-9-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okawa Y., Ishiguro K., Fujita S.C. Stress-induced hyperphosphorylation of tau in the mouse brain. FEBS Lett. 2003;535:183–189. doi: 10.1016/s0014-5793(02)03883-8. [DOI] [PubMed] [Google Scholar]
- Orr M.E., Sullivan A.C., Frost B. A brief overview of tauopathy: causes, consequences, and therapeutic strategies. Trends Pharmacol. Sci. 2017;38:637–648. doi: 10.1016/j.tips.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parachikova A., Nichol K.E., Cotman C.W. Short-term exercise in aged Tg2576 mice alters neuroinflammation and improves cognition. Neurobiol. Dis. 2008;30:121–129. doi: 10.1016/j.nbd.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardon M.C. Therapeutic potential of some stress mediators in early Alzheimer's disease. Exp. Gerontol. 2011;46:170–173. doi: 10.1016/j.exger.2010.09.006. [DOI] [PubMed] [Google Scholar]
- Patel N.V., Gordon M.N., Connor K.E., Good R.A., Engelman R.W., Mason J., Morgan D.G., Morgan T.E., Finch C.E. Caloric restriction attenuates Abeta-deposition in Alzheimer transgenic models. Neurobiol. Aging. 2005;26:995–1000. doi: 10.1016/j.neurobiolaging.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Payne J.K. State of the science: stress, inflammation, and cancer. Oncol. Nurs. Forum. 2013;41:533–540. doi: 10.1188/14.ONF.533-540. [DOI] [PubMed] [Google Scholar]
- Plassman B.L., Langa K.M., Fisher G.G., Heeringa S.G., Weir D.R., Ofstedal M.B., Burke J.R., Hurd M.D., Potter G.G., Rodgers W.L., Steffens D.C., Willis R.J., Wallace R.B. Prevalence of dementia in the United States: the aging, demographics, and memory study. Neuroepidemiology. 2007;29:125–132. doi: 10.1159/000109998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power M.L., Schulkin J. Functions of corticotropin-releasing hormone in anthropoid primates: from brain to placenta. Am. J. Hum. Biol. 2006;18:431–447. doi: 10.1002/ajhb.20521. [DOI] [PubMed] [Google Scholar]
- Qin W., Chachich M., Lane M., Roth G., Bryant M., de Cabo R., Ottinger M.A., Mattison J., Ingram D., Gandy S., Pasinetti G.M. Calorie restriction attenuates Alzheimer's disease type brain amyloidosis in Squirrel monkeys (Saimiri sciureus) J. Alzheimers Dis. 2006;10:417–422. doi: 10.3233/jad-2006-10411. [DOI] [PubMed] [Google Scholar]
- Räihä I., Kaprio J., Koskenvuo M., Rajala T., Sourander L. Environmental differences in twin pairs discordant for Alzheimer's disease. J. Neurol. Neurosurg. Psychiatry. 1998;65:785–787. doi: 10.1136/jnnp.65.5.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissman R.A. Stress-induced tau phosphorylation: functional neuroplasticity or neuronal vulnerability? J. Alzheimers Dis. 2009;18:453–457. doi: 10.3233/JAD-2009-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissman R.A., Lee K.F., Vale W., Sawchenko P.E. Corticotropin-releasing factor receptors differentially regulate stress-induced tau phosphorylation. J. Neurosci. 2007;27:6552–6562. doi: 10.1523/JNEUROSCI.5173-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissman R.A., Staup M.A., Lee A.R., Justice N.J., Rice K.C., Vale W., Sawchenko P.E. Corticotropin-releasing factor receptor-dependent effects of repeated stress on tau phosphorylation, solubility, and aggregation. Proc. Natl. Acad. Sci. Unit. States Am. 2012;109:6277–6282. doi: 10.1073/pnas.1203140109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman S.M., Mattson M.P. Adverse stress, hippocampal networks, and Alzheimer's disease. NeuroMolecular Med. 2010;12:56–70. doi: 10.1007/s12017-009-8107-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rühlmann C., Wölk T., Blümel T., Stahn L., Vollmar B., Kuhla A. Long-term caloric restriction in ApoE-deficient mice results in neuroprotection via Fgf21-induced AMPK/mTOR pathway. Aging (N Y) 2016;8:2777–2789. doi: 10.18632/aging.101086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarmeas N., Luchsinger J.A., Schupf N., Brickman A.M., Cosentino S., Tang M.X., Stern Y. Physical activity, diet, and risk of Alzheimer disease. J. Am. Med. Assoc. 2009;302:627–637. doi: 10.1001/jama.2009.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifan A., Schelke M., Obeng-Aduasare Y., Isaacson R. Early life epidemiology of Alzheimer's disease--a critical review. Neuroepidemiology. 2015;45:237–254. doi: 10.1159/000439568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pozo A., Frosch M.P., Masliah E., Hyman B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011;1:a008169. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V.M., Litersky J.M., Bhaskar K., Lee G. Tau impacts on growth-factor-stimulated actin remodeling. J. Cell Sci. 2007;20:748–757. doi: 10.1242/jcs.03378. [DOI] [PubMed] [Google Scholar]
- Sotiropoulos I., Catania C., Pinto L.G., Silva R., Pollerberg G.E., Takashima A., Sousa N., Almeida O.F. Stress acts cumulatively to precipitate Alzheimer's disease-like tau pathology and cognitive deficits. 2011;31:7840–7847. doi: 10.1523/JNEUROSCI.0730-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiropoulos I., Silva J., Kimura T., Rodrigues A.J., Costa P., Almeida O.F., Sousa N., Takashima A. Female hippocampus vulnerability to environmental stress, a precipitating factor in Tau aggregation pathology. J. Alzheimers Dis. 2015;43:763–774. doi: 10.3233/JAD-140693. [DOI] [PubMed] [Google Scholar]
- Sotiropoulos I., Sousa N. Tau as the converging protein between chronic stress and Alzheimer's disease synaptic pathology. Neurodegener. Dis. 2016;16:22–25. doi: 10.1159/000440844. [DOI] [PubMed] [Google Scholar]
- Sousa N., Almeida O.F. Disconnection and reconnection: the morphological basis of (mal)adaptation to stress. Trends Neurosci. 2012;35:742–751. doi: 10.1016/j.tins.2012.08.006. [DOI] [PubMed] [Google Scholar]
- Spillantini M.G., Goedert M., Crowther R.A., Murrell J.R., Farlow M.R., Ghetti B. Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc. Natl. Acad. Sci. Unit. States Am. 1997;94:4113–4118. doi: 10.1073/pnas.94.8.4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamer K., Vogel R., Thies E., Mandelkow E., Mandelkow E.M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002;156:1051–1063. doi: 10.1083/jcb.200108057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M.A., Wand G. Stress and the HPA axis: role of glucocorticoids in alcohol dependence. Alcohol Res. 2012;34:468–483. [PMC free article] [PubMed] [Google Scholar]
- Stern Y., Gurland B., Tatemichi T.K., Tang M.X., Wilder D., Mayeux R. Influence of education and occupation on the incidence of Alzheimer's disease. J. Am. Med. Assoc. 1994;271:1004–1010. [PubMed] [Google Scholar]
- Stewart J., Mitchell J., Kalant N. The effects of life-long food restriction on spatial memory in young and aged Fischer 344 rats measured in the eight-arm radial and the Morris water mazes. Neurobiol. Aging. 1989;10:669–675. doi: 10.1016/0197-4580(89)90003-1. [DOI] [PubMed] [Google Scholar]
- Stoothoff W.H., Johnson G.V. Tau phosphorylation: physiological and pathological consequences. Biochim. Biophys. Acta. 2005;1739:280–297. doi: 10.1016/j.bbadis.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Stranahan A.M., Mattson M.P. Impact of energy intake and expenditure on neuronal plasticity. NeuroMolecular Med. 2008;10:209–218. doi: 10.1007/s12017-008-8043-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K., Matsuda N. Proteostasis and neurodegeneration: the roles of proteasomal degradation and autophagy. Biochim. Biophys. Acta. 2014;1843:197–204. doi: 10.1016/j.bbamcr.2013.03.012. [DOI] [PubMed] [Google Scholar]
- Umegaki H., Ikari H., Nakahata H., Endo H., Suzuki Y., Ogawa O., Nakamura A., Yamamoto T., Iguchi A. Plasma cortisol levels in elderly female subjects with Alzheimer's disease: a cross-sectional and longitudinal study. Brain Res. 2000;881:241–243. doi: 10.1016/s0006-8993(00)02847-x. [DOI] [PubMed] [Google Scholar]
- Ulrich-Lai Y.M., Herman J.P. Neural regulation of endocrine and autonomic stress responses. Nat. Rev. Neurosci. 2009;10:397–409. doi: 10.1038/nrn2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Cauwenberghe C., Van Broeckhoven C., Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet. Med. 2016;18:421–430. doi: 10.1038/gim.2015.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Praag H. Exercise and the brain: something to chew on. Trends Neurosci. 2009;32:283–290. doi: 10.1016/j.tins.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanitallie T.B. Stress: a risk factor for serious illness. Metabolism. 2002;51:40–45. doi: 10.1053/meta.2002.33191. [DOI] [PubMed] [Google Scholar]
- Vyas S., Rodrigues A.J., Silva J.M., Tronche F., Almeida O.F., Sousa N., Sotiropoulos I. Chronic stress and glucocorticoids: from neuronal plasticity to neurodegeneration. Neural Plast. 2016;2016 doi: 10.1155/2016/6391686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.Z., Grundke-Iqbal I., Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur. J. Neurosci. 2007;25:59–68. doi: 10.1111/j.1460-9568.2006.05226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y., Gould E., McEwen B.S. Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res. 1992;588:341–345. doi: 10.1016/0006-8993(92)91597-8. [DOI] [PubMed] [Google Scholar]
- Weindruch R., Walford R.L., Fligiel S., Guthrie D. The retardation of aging in mice by dietary restriction: longevity, cancer, immunity and lifetime energy intake. J. Nutr. 1986;116:641–654. doi: 10.1093/jn/116.4.641. [DOI] [PubMed] [Google Scholar]
- Wilson R.S., Arnold S.E., Schneider J.A., Kelly J.F., Tang Y., Bennett D.A. Chronic psychological distress and risk of Alzheimer's disease in old age. Neuroepidemiology. 2006;27:143–153. doi: 10.1159/000095761. [DOI] [PubMed] [Google Scholar]
- Wilson R.S., Barnes L.L., Bennett D.A., Li Y., Bienias J.L., Mendes de Leon C.F., Evans D.A. Proneness to psychological distress and risk of Alzheimer disease in a biracial community. Neurology. 2005;64:380–382. doi: 10.1212/01.WNL.0000149525.53525.E7. [DOI] [PubMed] [Google Scholar]
- Wilson R.S., Evans D.A., Bienias J.L., Mendes de Leon C.F., Schneider J.A., Bennett D.A. Proneness to psychological distress is associated with risk of Alzheimer's disease. Neurology. 2003;61:1479–1485. doi: 10.1212/01.wnl.0000096167.56734.59. [DOI] [PubMed] [Google Scholar]
- Wischik C.M., Novak M., Thøgersen H.C., Edwards P.C., Runswick M.J., Jakes R., Walker J.E., Milstein C., Roth M., Klug A. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. Unit. States Am. 1988;85:4506–4510. doi: 10.1073/pnas.85.12.4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J., Sun X.B., Wang H.Q., Zhao H., Zhao X.Y., Xu Y.X., Guo J.C., Zhu C.Q. Chronic restraint stress alters the expression and distribution of phosphorylated tau and MAP2 in cortex and hippocampus of rat brain. Brain Res. 2010;1347:132–141. doi: 10.1016/j.brainres.2010.05.074. [DOI] [PubMed] [Google Scholar]
- Yanagisawa M., Planel E., Ishiguro K., Fujita S.C. Starvation induces tau hyperphosphorylation in mouse brain: implications for Alzheimer's disease. FEBS Lett. 1999;461:329–333. doi: 10.1016/s0014-5793(99)01480-5. [DOI] [PubMed] [Google Scholar]
- Yang C., Guo X., Wang G.H., Wang H.L., Liu Z.C., Liu H., Zhu Z.X., Li Y. Changes in tau phosphorylation levels in the hippocampus and frontal cortex following chronic stress. Braz. J. Med. Biol. Res. 2014;47:237–244. doi: 10.1590/1414-431X20133275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J.H., Brown C., Whitehead G., Piers T., Lee Y.S., Perez C.M., Regan P., Whitcomb D.J., Cho K. Glucocorticoids activate a synapse weakening pathway culminating in tau phosphorylation in the hippocampus. Pharmacol. Res. 2017;121:42–51. doi: 10.1016/j.phrs.2017.04.015. [DOI] [PubMed] [Google Scholar]
- Yoshida S., Maeda M., Kaku S., Ikeya H., Yamada K., Nakaike S. Lithium inhibits stress-induced changes in tau phosphorylation in the mouse hippocampus. J. Neural. Transm. 2006;113:1803–1814. doi: 10.1007/s00702-006-0528-0. [DOI] [PubMed] [Google Scholar]
- Yuede C.M., Zimmerman S.D., Dong H., Kling M.J., Bero A.W., Holtzman D.M., Timson B.F., Csernansky J.G. Effects of voluntary and forced exercise on plaque deposition, hippocampal volume, and behavior in the Tg2576 mouse model of Alzheimer's disease. Neurobiol. Dis. 2009;35:426–432. doi: 10.1016/j.nbd.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Rissman R.A. Corticotropin-releasing factor receptor-1 modulates biomarkers of DNA oxidation in Alzheimer's disease mice. PLoS One. 2017;12 doi: 10.1371/journal.pone.0181367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.E., Yang X., Li L., Sui X., Tian Q., Wei W., Wang J., Liu G. Hypoxia-induced tau phosphorylation and memory deficits in rats. Neurodegener. Res. 2014;14:107–116. doi: 10.1159/000362239. [DOI] [PubMed] [Google Scholar]
- Zavala J.K., Fernandez A.A., Gosselink K.L. Female responses to acute and repeated restraint stress differ from those in males. Physiol. Behav. 2011;104:215–221. doi: 10.1016/j.physbeh.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H., Mandelkow E. Lost after translation: missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014;37:721–732. doi: 10.1016/j.tins.2014.08.004. [DOI] [PubMed] [Google Scholar]
- Zempel H., Thies E., Mandelkow E., Mandelkow E.M. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J. Neurosci. 2010;30:11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.