

Deficiency of huntingtin-interacting protein 1 (Hip1) results in degenerative phenotypes. Here we generated a Hip1 deficiency allele where a floxed transcriptional stop cassette and a human HIP1 cDNA were knocked into intron 1 of the mouse Hip1 locus.
KEYWORDS: GDPD3, GDE7, phosphocholine, endocytosis, HIP1, kypholordosis, Cre recombinase
ABSTRACT
Deficiency of huntingtin-interacting protein 1 (Hip1) results in degenerative phenotypes. Here we generated a Hip1 deficiency allele where a floxed transcriptional stop cassette and a human HIP1 cDNA were knocked into intron 1 of the mouse Hip1 locus. CMV-Cre-mediated germ line excision of the stop cassette resulted in expression of HIP1 and rescue of the Hip1 knockout phenotype. Mx1-Cre-mediated excision led to HIP1 expression in spleen, kidney and liver, and also rescued the phenotype. In contrast, hGFAP-Cre-mediated, brain-specific HIP1 expression did not rescue the phenotype. Metabolomics and microarrays of several Hip1 knockout tissues identified low phosphocholine (PC) levels and low glycerophosphodiester phosphodiesterase domain containing 3 (Gdpd3) gene expression. Since Gdpd3 has lysophospholipase D activity that results in the formation of choline, a precursor of PC, Gdpd3 downregulation could lead to the low PC levels. To test whether Gdpd3 contributes to the Hip1 deficiency phenotype, we generated Gdpd3 knockout mice. Double knockout of Gdpd3 and Hip1 worsened the Hip1 phenotype. This suggests that Gdpd3 compensates for Hip1 loss. More-detailed knowledge of how Hip1 deficiency leads to low PC will improve our understanding of HIP1 in choline metabolism in normal and disease states.
INTRODUCTION
Huntingtin-interacting protein 1 (HIP1) was originally discovered as a protein that interacts with huntingtin, the protein product of the gene mutated in Huntington's disease (1, 2). HIP1 was later found to be an endocytic protein that binds clathrin, AP2 (3–6), inositol lipids (7–9) and actin (10). Knockout of Hip1 alone (11, 12) or with its only known mammalian relative, Hip1-related (Hip1r) (13, 14), results in a degenerative phenotype in mice. This phenotype, which is more severe in double-knockout (DKO) mice, includes testicular degeneration, spinal defects, and lowered body weight. Although it has been shown that endocytosis of AMPA receptors in neurons is disrupted in knockout mice (11), the mechanisms leading to most of the knockout phenotypes are unclear, as are the roles of HIP1 in normal and neoplastic tissues.
HIP1 was first linked to receptor tyrosine kinase (RTK) signaling when it was identified as a chromosomal translocation partner with the platelet-derived growth factor β receptor (PDGFβR) gene in leukemia (15). This HIP1/PDGFβR translocation is a member of a large family of chromosomal translocations involving the PDGFβR gene (16–19). Recently, others have identified HIP1 as a partner in chromosomal translocations involving the anaplastic lymphoma kinase (ALK) gene in non-small-cell lung cancer (20–22). Like HIP1/PDGFβR, the HIP1/ALK translocation is a member of a large family of chromosomal translocations that involve the ALK gene.
In addition to the HIP1/PDGFβR and HIP1/ALK mutations, the HIP1 protein itself is expressed at high levels in several other cancers (23–25), and overexpression of HIP1 transforms fibroblasts (24) and prostate epithelial cells (26). This transforming activity is also linked to RTKs, as HIP1-transformed cells display increased levels of epidermal growth factor receptor (EGFR) (24). Indeed, HIP1 prolongs the half-lives of RTKs, such as EGFR and PDGFβR (8). Additionally, treatment of the HIP1-transformed cells with an EGFR inhibitor reverses the transformed phenotype (24). One possible explanation for EGFR overexpression in these cancers is that HIP1-dependent stabilization of RTKs occurs via clathrin or membrane vesicle sequestration. Low levels of clathrin or altered membrane dynamics could decrease endocytosis-mediated RTK degradation. This would then prolong RTK signaling and promote transformation.
Because our past attempt to generate a conditional knockout of Hip1 (12, 27) was thwarted by an unexpected cryptic splicing event hidden by the complexity of the large Hip1 gene (32 exons spread over 220 kb), we generated Hip1-deficient mice with the option to conditionally express a single copy of human HIP1 in specific tissues to investigate how Hip1 deficiency leads to degeneration. With this new model, we discovered that the expression of Hip1 in most neurons and glia in the brain is not required for normal physiology but that Hip1 expression in spleen, liver, and kidney may be important for normal physiology. We also assayed a variety of Hip1-deficient tissues for alterations in metabolism and gene expression and discovered that low phosphocholine (PC) levels and glycerophosphodiester phosphodiesterase domain containing 3 (Gdpd3) gene expression were associated with Hip1 deficiency. We also observed doubly Gdpd3/Hip1-deficient mice to have spinal degeneration at younger ages than singly Hip1-deficient mice, suggesting that Gdpd3 partially compensates for the Hip1 deficiency. Low PC levels in Hip1-deficient tissue is opposite to the elevated levels of HIP1 and PC found in cancers (28).Thus, HIP1 may, via endocytosis of lipids or via effects on gene expression, lead to normal or abnormal levels of choline-related metabolites in both healthy and diseased tissues.
RESULTS
Generation of a knock-in allele of human HIP1.
Here, we report the generation of a novel Hip1 knockout allele (Hip1LSL) (Fig. 1A). This allele also has the human HIP1 cDNA knocked into the mouse Hip1 locus downstream of a loxP-flanked transcriptional stop (Lox-stop-Lox) (LSL) cassette. Having previously found that a multicopy-transgenic HIP1 cDNA allele was able to rescue the Hip1 and Hip1r double-knockout degenerative phenotype (13), we hypothesized that HIP1 as a single-copy allele may also substitute for Hip1 (or Hip1r) deficiency phenotypes. This conditional knock-in allele allows for use of specific Cre alleles to guide tissue-specific expression together with the endogenous mouse Hip1 promoter to regulate the expression of human HIP1. In addition to the conditional nature of this allele that allows for expression of HIP1 in specific tissues at specific times, there were other potential advantages of this targeting strategy. First, since HIP1 is expressed at high levels in many cancers (24, 25, 29, 30), humanization of a mouse allele could be of future use for preclinical in vivo HIP1-targeted drug testing. Second, this type of humanization addresses the question of whether the many introns of the large, 250-kb Hip1 locus are required for regulation of the Hip1 gene. We reasoned that if the expression of HIP1 from a single knocked-in copy does not restore normalcy to the Hip1-deficient mice, the complex genomic structure of the allele is necessary for HIP1 expression.
FIG 1.

Generation of Hip1-deficient and conditionally HIP1-humanized mice. (A) Schematic of the first 3 exons of the 32-exon murine Hip1 locus (Hip1+), the targeted knock-in allele with a floxed stop cassette (LSL), and a human HIP1 cDNA inserted into the murine Hip1 locus by homologous recombination (Hip1LSL) and the Cre-mediated recombined allele with the excised stop cassette (Hip1HIP1). The following features are indicated: stop cassette bracketed by loxP recombination sequences (LSL), partial mouse exon 2 fused to a human HIP1 cDNA-IRES-eGFP poly(A) tail (gray box), and 5′ genomic hybridization probe. (B) Southern blot confirmation of successfully targeted ES cell lines. Shown are ES cell lines carrying wild-type (Hip1+/+) or Hip1LSL targeted alleles (A8, B4, and C7). Genomic DNA was digested with EcoRI or HindIII and Southern blotted either with the 5′ probe to yield a 15.7-kb band corresponding to the wild-type allele and a 13.4-kb band corresponding to the recombined allele or with the 3′ neomycin probe to yield a 15.5-kb band corresponding to the recombined allele. The neomycin expression cassette was then excised by transfection of the cells with an Flp-expressing plasmid. (C) A CMV-Cre transgene was used to generate a germ line Cre-mediated recombined allele, Hip1HIP1, as delineated in panel A. Here GFP expression in peripheral blood white blood cells (WBCs) was quantitated to confirm that excision of the stop cassette leads to GFP expression in Hip1HIP1/HIP1 mouse tissue. Data represent mean ± standard error of the mean (SEM) (n = 14 per group). (D) Top, expression of human HIP1 from the Hip1 locus prevents the weight loss of Hip1-deficient mice. Data represent mean ± SEM (n = 8 to 16). ***, P < 0.005; *, P < 0.05; n.s., not significant. Ages ranged between 5 and 6.5 months. Bottom, Western blot analysis confirmed that expression of human HIP1 protein replaces mouse Hip1 protein in lung tissue from Hip1HIP1/HIP1 mice. The top panel represents Western blot with a mouse-specific monoclonal antibody (UM1B11) and the bottom panel a human-specific polyclonal antibody (UM323). (E) Expression of human HIP1 from the Hip1 locus prevents kypholordotic and weight loss phenotypes in all mice. Top, the Kaplan-Meier curves depict the percentage of mice with a normal phenotype as a function of time (in days) since birth. Hip1-deficient mice (Hip1LSL/LSL) are represented by the dotted line, and the fully humanized HIP1 mice (Hip1HIP1/HIP1) are represented by the solid line (n = 11 per genotype). Bottom, representative mice at 6 months of age with and without the Hip1 deficiency-associated kypholordosis and diminished weight. Note that the HIP1-humanized mouse (Hip1HIP1/HIP1) displays a slight kyphosis that is also observed in wild-type mice. Without the lordosis displayed by the Hip1LSL/LSL mouse, this slight kyphosis is normal. (F) The kypholordotic phenotype of Hip1 and Hip1r double knockout mice is rescued by a single copy of human HIP1. Top, the Kaplan-Meier curves depict the percentage of mice with a normal phenotype as a function of time (in days) since birth. Rescued Hip1null/HIP1; Hip1r−/− mice are represented by the solid line and double-knockout Hip1null/null; Hip1r−/− mice by the dotted line. Bottom, representative photographs of 2-month-old Hip1null/null; Hip1r−/− mouse with kypholordosis and an age- and gender-matched Hip1null/HIP1; Hip1r−/− rescue mouse with no phenotype.
We successfully targeted embryonic stem (ES) cells (Fig. 1B) and generated three independent mouse lines carrying the Hip1LSL allele (the “knockout” allele). Homozygous and heterozygous knockout mice were born at predicted Mendelian frequencies. To generate the germ line “humanized” allele, Hip1LSL/+ mice were crossed with transgenic CMV-Cre mice (31). The cytomegalovirus (CMV) promoter is activated in all tissues including germ cells. The resulting Hip1HIP1 humanized allele was used to generate fully humanized, Hip1HIP1/HIP1 mice (the CMV-Cre allele was crossed out). Hip1HIP1/HIP1 mice could be distinguished from Hip1 knockout (Hip1LSL/LSL) mice by green fluorescent protein (GFP) expression in peripheral blood (Fig. 1C) or by the presence of human HIP1 protein, rather than mouse Hip1 protein, in mouse tissues (Fig. 1D, bottom).
Rescue of the Hip1 deficiency phenotype by a knocked-in human HIP1 cDNA.
Although indistinguishable at birth from their wild-type or heterozygous littermates, by 6 months of age, all Hip1 knockout mice (Hip1LSL/LSL) were afflicted with lowered body weight and severe spinal defects. In contrast, the HIP1 humanized mice (Hip1HIP1/HIP1) remained normal in weight (Fig. 1D, top) and were without a kypholordotic spine (Fig. 1E). These data indicate that a single copy of human HIP1 cDNA is able to fully substitute for the mouse Hip1 gene. This was also supported by the ability of the Hip1HIP1 allele to fully rescue the Hip1/Hip1r double-knockout phenotype, which was more severe than in the single-knockout mice (Fig. 1F).
Brain-specific HIP1 expression does not prevent the Hip1 knockout phenotype.
Because Hip1 is expressed at high levels in the brain and the Hip1 protein interacts with huntingtin, the product of the gene mutated in Huntington's syndrome (1, 2), we next asked whether restricted expression of HIP1 in neural tissues would rescue the Hip1 deficiency phenotypes. Because Hip1 has also been shown to be required for AMPA receptor trafficking in hippocampal neurons (11), we predicted that HIP1 expression in the brain would rescue the Hip1 deficiency phenotypes. To restrict HIP1 expression to neural tissue, we crossed the Hip1LSL/LSL mice with transgenic hGFAP-Cre mice (32). The human glial fibrillary acidic protein (hGFAP) promoter in these mice has been shown to be active as early as embryonic day 13.5 and induces recombination in multipotent neural progenitor cells. This hGFAP-Cre line therefore targets many neuronal populations (including hippocampal neurons) and glial cell types. As expected, Western blot analysis of human HIP1 expression in hGFAP-Cre; Hip1LSL /LSL brains demonstrated levels similar to those in the germ line humanized Hip1HIP1/HIP1 mouse brains, and HIP1 protein was not detected in spleen, lung, kidney or liver (Fig. 2B). To our surprise, the degenerative phenotypes associated with Hip1 deficiency were not rescued by hGFAP-Cre-mediated expression of HIP1 (Fig. 2A). These hGFAP-Cre data suggest that Hip1 deficiency in the brain is unlikely to contribute to the Hip1 deficiency phenotype, although a possibility remains that the small number of cell types that did not have fully Cre-recombined alleles may contribute to the phenotype.
FIG 2.

Tissue-specific rescue of Hip1 deficiency with human HIP1. (A) The Hip1 deficiency phenotype was not rescued by expression of human HIP1 in the nervous system with the hGFAP-Cre transgene. Representative photographs of 6-month-old hGFAP-Cre; Hip1LSL/LSL and hGFAP-Cre; Hip1+/+ mice are shown. (B) Western blotting for tissue-specific expression analysis of human HIP1 in brain from hGFAP-Cre; Hip1LSL/LSL mice. Tissue extracts from representatives of each genotype were analyzed. The human-specific HIP1 antibody UM323 was used to detect HIP1 in the brain and showed no detectable HIP1 in spleen, liver, kidney, and lung tissues. (C) At 6 weeks of age, Hip1LSL/LSL and Mx1-cre; Hip1LSL/LSL mice were treated with pIpC to induce Mx1-Cre-mediated expression of human HIP1 in the hematopoietic system, kidney, and liver. Expression of Mx1-Cre prevented the weight loss observed in Hip1-deficient mice (Hip1LSL/LSL). All mice were between 5.5 and 6 months of age, as this is when the weight loss was most apparent. Data represent mean ± SEM (n = 5). n.s., not significant; *, P < 0.05. (D) The kypholordotic spinal curvature in Hip1-deficient mice is rescued by Mx1-Cre-mediated expression of human HIP1. Representative photographs of 4-month-old pIpC-treated Hip1LSL/LSL and Mx1-cre; Hip1LSL/LSL mice are shown. (E) Western blotting for tissue-specific expression analysis of human HIP1 in spleen, liver, and kidney tissues. Tissue extracts from three mice of each genotype were analyzed. The human-specific HIP1 antibody UM323 was used to detect HIP1 in the spleen, liver, and kidney, with no detectable HIP1 in brain and lung tissues. (F) Kaplan-Meier curves depicting the lack of or the progression of the development of the kypholordotic phenotype in bone marrow-transplanted mice. Transplantation of Hip1-deficient (Hip1LSL/LSL) bone marrow into irradiated wild-type (Hip1+/+) mice does not lead to the development of the kypholordotic phenotype. Transplantation of wild-type (Hip1+/+) bone marrow into irradiated Hip1-deficient (Hip1LSL/LSL) mice does not prevent the development of the kypholordotic phenotype (n = 5 per group).
Mx1-Cre-mediated HIP1 expression rescues Hip1-deficient mice.
Next, we crossed Hip1LSL/LSL mice to mice carrying a Mx1-Cre transgene in order to express HIP1 in adult liver, kidney, and the hematopoietic system (33). We did this because these tissues express high Hip1 levels and the Hip1 deficiency phenotype develops only in adulthood. The Mx1 promoter is activated by interferon, which can be induced by synthetic double-stranded RNA, poly(I)-poly(C) (pIpC) (33). Mice were induced with pIpC at 6 weeks of age prior to the onset of the degenerative phenotype. The induced Mx1-Cre; Hip1LSL/LSL mice (n = 5) were of normal weight (Fig. 2C) and without spinal defects at 4 months of age (Fig. 2D), whereas induced Hip1LSL/LSL littermates were affected with the degenerative phenotype.
The pIpC-induced Hip1LSL/LSL mice and Mx1-Cre; Hip1LSL/LSL rescued mice were evaluated for human HIP1 expression to confirm specificity of the Cre allele. Western blot analyses for human HIP1 protein demonstrated that pIpC-induced recombination occurred most effectively in spleen and liver. Kidney did not show as robust HIP1 expression. As expected, HIP1 was not detected in brain or lung tissues (Fig. 2E). These Mx1-Cre data suggest that Hip1 deficiency in adult liver and/or hematopoietic tissues (and less likely kidney) contributes to development of Hip1 deficiency phenotypes.
Bone marrow transplant of wild-type bone marrow does not prevent the Hip1 knockout phenotype.
We transplanted wild-type bone marrow into Hip1-deficient mice to test whether replacement of the Hip1-deficient bone marrow with normal bone marrow could rescue the kypholordotic phenotype. Bone marrow cells from 6-week-old wild-type (CD45.1+) or Hip1LSL/LSL mice were transplanted into 6-week-old lethally irradiated Hip1LSL/LSL mice (CD45.2+) (n = 5 per group). These mice all developed a kypholordotic phenotype, indicating that irradiation, wild-type bone marrow (confirmed by the replacement of the CD45.2+ cells by CD45.1+ cells), or the transplant process in general does not prevent the phenotype (Fig. 2F).
We also transplanted Hip1-deficient bone marrow into wild-type mice to test whether Hip1 deficiency in bone marrow can cause the development of the Hip1 mutant kypholordotic phenotype. Whole bone marrow cells from 6-week-old Hip1LSL /LSL (CD45.2+) mice were transplanted into 6-week-old lethally irradiated wild-type (CD45.1+) mice (n = 18). These mice did not develop the phenotype despite living to 1 year of age. Replacement of wild-type bone marrow with Hip1-deficient bone marrow was confirmed by replacement of CD45.1+ cells by CD45.2+ cells (data not shown). In sum, Hip1 deficiency in bone marrow is insufficient for the development of the phenotype, and Hip1 expression in the bone marrow is insufficient for the rescue of the phenotype.
Hip1-deficient tissues have low PC levels.
The adult-onset weight loss, generalized weakness, and spinal defects of Hip1-deficient mice raise the possibility of an endocrine or progressive metabolic defect. A prior search for endocrine defects (e.g., thyroid or other hormone changes) in Hip1/Hip1r double-knockout mice, which have more severe phenotypes than the Hip1 single-knockout mice (Fig. 1F), did not identify abnormalities (13). Abnormalities were also not found in bone-forming (osteoblast) or resorbing (osteoclast) cells of these mice. Specifically, osteoblast and osteoclast frequencies in bone sections from the femurs and vertebrae were normal. Osteoclast differentiation in vivo and ex vivo was intact as evidenced by normal levels of serum tartrate-resistant acid phosphatase (TRAP) and normal differentiation of bone marrow cells to osteoclasts. Serum osteocalcin levels were also normal, suggesting normal osteoblast function (13). We therefore used mass spectrometry to compare levels of >100 metabolites in HIP1 rescue versus Hip1-deficient tissues. Deletion of Hip1 resulted in altered concentrations of multiple choline-related metabolites, although total choline levels were not changed in any tissues tested (brain, kidney, lung, liver, and spleen). The most striking change was in phosphocholine (PC), which is generated by phosphorylation of choline by choline kinase. PC was significantly decreased in all Hip1-deficient tissues tested except brain, where it still trended low (Fig. 3). The largest observed decrease in PC was in the liver (by 0.41-fold [P = 0.0003; variable importance in projection {VIP} = 1.8343]), a tissue where choline metabolism is particularly relevant (34). Because PC is a sentinel choline metabolite that reflects overall phosphatidylcholine pathway homeostasis (35), these data suggest a general change in choline metabolism that results from loss of Hip1 expression. That choline metabolism is perturbed in Hip1-deficient tissues was further supported by additional alterations observed in acetylcholine, betaine aldehyde, trimethylglycine/betaine, and dimethylglycine (see Data Set S1 in the supplemental material). However, no changes in lysophosphatidylcholine or glycerophosphocholine (GPC) were detected. There was no difference in PC levels between liver tissues from wild-type and HIP1 rescue mice (fold change for Hip1+/+/Hip1HIP1/HIP1 = 1.231 [P = 0.446; VIP = 0.823]; n = 4 per genotype) or in any other metabolites that showed alterations in liver with Hip1 deficiency compared to human HIP1 rescue. This confirms not only that human HIP1 restores Hip1 functions to wild-type levels as observed by the normal weight and spine curvature in HIP1 rescue animals (Fig. 1D and E) but that HIP1 rescue mice are appropriate controls for these experiments.
FIG 3.

Hip1-deficient mice have low phosphocholine levels. (A) Of the 120 metabolites that were measured with LC-MS/MS, a robust and consistent metabolic change was decreased phosphocholine (PC) levels. All tissues except brain from Hip1-deficient mice (Hip1LSL/LSL) showed significantly decreased PC levels compared to those in HIP1-rescued mice (Hip1HIP1/HIP1). The units are peak areas normalized to total ion current (a marker of total metabolite abundance).The VIP (variable importance in projection) number is a measure of statistical significance. When it is above 1.0, the metabolite change is considered statistically significant. (B) The same data in panel A are shown as dot plots to visualize variability between samples. Data are expressed as relative to the average of levels in HIP1-rescued tissues (Hip1HIP1/HIP1). Data represent mean ± SEM (n = 7 per group). n.s., not significant; *, P < 0.05; ***, P < 0.0001.
There were other intriguing metabolite changes observed in Hip1-deficient tissues (see Data Set S3 in the supplemental material). One robust change was increased homocysteine in several Hip1-deficient tissues. Of the tissues tested, homocysteine was increased the most in the liver (13.41-fold [P = 0.002; VIP = 1.8]) (see Data Set S2 in the supplemental material). The concomitant decreases in pyridoxamine (a vitamer of vitamin B6) and cystathionine suggested that the increased homocysteine could be a result of a defective trans-sulfation pathway (pyridoxamine aids conversion of homocysteine to cysteine, which is then converted to cystathionine). However, vitamin B6 levels were normal in the sera of Hip1 deficient mice (data not shown). Increased homocysteine could also be due to an altered methionine cycle. In fact, S-adenosylhomocysteine (SAH) levels were increased and S-adenosylmethionine (SAM) levels were decreased in several tissues of Hip1-deficient mice (Data Set S2), suggesting changes in steady-state methylation. However, we did not detect changes in histone methylation by Western blot analysis in tissues analyzed by mass spectrometry (data not shown). Another possibility is that the increased homocysteine was simply due to altered choline metabolism. Choline is metabolized to betaine, which acts as a methyl donor in the conversion of homocysteine to methionine. Since the liver showed the greatest decreases in PC and acetylcholine and the greatest increase in homocysteine of all tissues tested, this hypothesis is plausible. It is possible that high homocysteine redirects choline for betaine synthesis at the expense of acetylcholine and PC in order to maintain methionine homeostasis. In sum, these data suggest that Hip1 deficiency results in altered choline metabolism that includes elevated homocysteine levels.
Hip1-deficient mice lose Gdpd3 expression.
In order to identify potential mediators of the Hip1 deficiency phenotypes and the altered metabolome, we compared gene expression profiles from tissues of Hip1+/+, Hip1LSL/LSL, and Hip1HIP1/HIP1 mice by microarray analysis. As expected, mouse Hip1 was not expressed in either Hip1LSL/LSL knockout or Hip1HIP1/HIP1 rescued mice but was expressed in Hip1+/+ wild-type mice (data not shown). We identified Gdpd3 (also called Gde7) as a gene with a robust and reproducible decrease in expression in all tested tissues (see Data Set S4 in the supplemental material). Gdpd3 is a gene whose protein product is a member of a glycerophosphodiester phosphodiesterase domain (Gdpd) family of enzymes, also known as the Gde family (36, 37). Expression patterns of the six other known members of the Gdpd family were not changed in Hip1-deficient mice (Fig. 4A). These decreases of Gdpd3 expression were confirmed with quantitative PCR (qPCR) (Fig. 4B). Decreased expression of Gdpd3 is not a consequence of the kypholordotic phenotype, since young, phenotypically “normal” Hip1-deficient mice show reduced Gdpd3 expression levels similarly to old, kypholordotic mice (data not shown).
FIG 4.

Decreased Gdpd3 expression in Hip1-deficient mice. (A) Heat map of RNA expression of the seven members of the GDPD family in Hip1-deficient and HIP1-rescued mice. Dark blue is the lowest and orange is the highest expression. Only Gdpd3 (Gde7) expression is lost in Hip1-deficient mouse tissues (asterisk). (B) Real-time PCR analysis of Gdpd3 expression. Data were normalized to Gapdh and expressed as relative to the average of Gdpd3 levels in HIP1-rescued tissues (Hip1HIP1/HIP1). Data represent mean ± SEM (n = 6). **, P < 0.005; ***, P < 0.0001.
Generation and analysis of Gdpd3 knockout allele.
To understand the physiological importance of Gdpd3 and to test whether Gdpd3 contributes to the Hip1 deficiency phenotypes, we generated a Gdpd3 knockout allele by clustered regularly interspaced short palindromic repeat (CRISPR)-mediated deletion of exons 2 to 7 (Fig. 5A and B). Homozygous and heterozygous knockout mice were born at predicted Mendelian frequencies and were indistinguishable at birth from wild-type littermates. Levels of Gdpd3 mRNA in liver, lung, and spleen tissues reflected the heterozygous and homozygous states of the knockout mutation (Fig. 5C). Heterozygous knockout tissues expressed approximately half of the amount of Gdpd3 message expressed in wild-type tissue, and homozygous knockout tissue had no detectable Gdpd3 message. In contrast, Gdpd3 deficiency did not alter Hip1 mRNA levels, indicating that Hip1 expression is not regulated by Gdpd3.
FIG 5.

Acceleration of kypholordosis onset in Hip1 and Gdpd3 DKO mice compared to Hip1 single-knockout mice. (A) Schematic of the targeting strategy used to generate the Gdpd3 knockout allele. (B) PCR analysis of genomic DNA isolated from tail biopsy specimens of Gdpd3 wild-type (Gdpd3+/+), heterozygous (Gdpd3+/−), and homozygous (Gdpd3−/−) mice. The wild-type Gdpd3 allele generates a 501-bp band and the knockout allele a 290-bp band with the use of oligonucleotides 1 and 3 and oligonucleotides 1 and 2, respectively. (C) qPCR analysis of Gdpd3 mRNA expression levels in liver, lung, and spleen tissue of Gdpd3+/+, Gdpd3+/−, and Gdpd3−/− mice. Data were normalized to Gapdh and expressed as relative to the average of Gdpd3 or Hip1 levels in Gdpd3+/+ tissue (n = 3 per genotype). (D) Kaplan-Meier curves of phenotype onset in Hip1 knockout mice (dotted line) and Hip1 and Gdpd3 DKO mice (solid line). The log-rank test was used to calculate significance. Mice were phenotyped for kypholordosis as described in Materials and Methods without prior knowledge of mouse genotypes. (E) Representative 10-month-old male Hip1LSL/LSL single-knockout and Hip1LSL/LSL; Gdpd3−/− DKO mice.
Both male and female Gdpd3-deficient mice had normal fertility and generated normal litter sizes and gender distributions. Unlike Hip1 knockout mice, which were afflicted with male infertility as well as weight loss and a kypholordotic spinal defect by 6 months of age, Gdpd3 knockout mice displayed no consistent defects even at 2 years of age (data not shown). Analysis of hematoxylin- and eosin-stained tissues showed no abnormalities in Gdpd3 knockout mice compared to age- and gender-matched controls.
Gdpd3 knockout mice show no changes in PC.
Gdpd3 has lysophosphatidylcholine (LPC)-specific phospholipase D (lysoPLD) activity that can generate lysophosphatidic acid (LPA) and choline (30, 34). In the first step in the biosynthesis of phosphatidylcholine, choline is phosphorylated to generate phosphocholine (PC). We hypothesized, that the decrease in Gdpd3 in Hip1-deficient mice could lead to the observed low PC levels (Fig. 6). As with Hip1-deficient tissues, we used mass spectrometry to profile metabolites in wild-type and Gdpd3-deficient tissues. Samples were prepared from age- and gender-matched Gdpd3+/+ and Gdpd3−/− mice. To our surprise, choline and related metabolites (including PC) were at normal levels in Gdpd3-deficient mice (see Data Set S5 in the supplemental material). Unlike with Hip1-deficient tissues, deletion of Gdpd3 resulted in fewer perturbations in the metabolic profile, where only a small number of metabolites showed only modest changes in concentration (see Data Set S6 in the supplemental material). These data suggest that Hip1 is required for a normal spine and normal PC levels via unknown mechanisms which could include, but are unlikely to be limited to, Gdpd3 activity.
FIG 6.
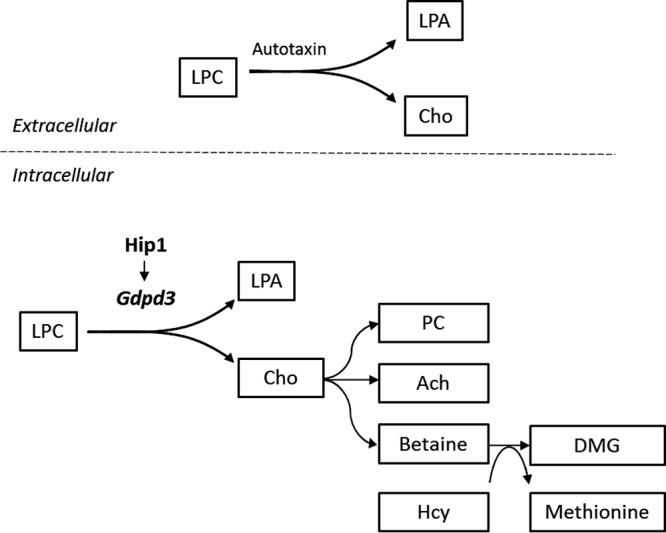
Schematic depicting a potential link between reduced Gdpd3 and alterations in choline-related metabolites in Hip1-deficient mice. Extracellularly, LPC is metabolized to LPA and choline by the lysoPLD autotaxin. Hip1 deficiency results in decreased Gdpd3. LysoPLD activity of Gdpd3 predicts that alterations seen in choline-related metabolites with Hip1 deficiency are due to reduced Gdpd3 levels. Changes in choline-related metabolites in liver of Hip1-deficient mice are depicted. LPC, lysophosphatidylcholine; LPA, lysophosphatidic acid; Cho, choline; PC, phosphocholine; Ach, acetylcholine; DMG, dimethylglycine; Hcy, homocysteine.
Hip1/Gdpd3 DKO mice.
Gdpd3 deficiency hastened the onset and severity of the degenerative phenotype caused by Hip1 deficiency. The time of onset of the kypholordosis as detected by palpation in Hip1/Gdpd3 double-knockout (DKO) mice was significantly sooner than that in Hip1 single-knockout mice (Fig. 5D). All phenotyping was performed by an investigator (G. L. Perez) blinded to genotype. There was no difference in the time of onset of phenotype between Hip1LSL/LSL and Hip1LSL/LSL; Gdpd3+/− mice (data not shown). In a majority of cases, by the time of sacrifice due to failure to thrive, the angle of the thoracic curvature was smaller (more severe) in DKO mice than in age- and gender-matched Hip1 single-knockout mice (Fig. 5E). These data indicate that Gdpd3 and Hip1 compensate for one another to maintain spinal homeostasis. These data also suggest that the diminished Gdpd3 expression in the Hip1 knockout mouse tissues (Fig. 4) partially contributes to the Hip1 knockout phenotype.
DISCUSSION
The role of HIP1 in leukemia (15) and lung cancer (20–22) as an oncogenic fusion partner and the necessity of Hip1 for mouse homeostasis are well established (11, 12). Additionally, HIP1 itself is upregulated in many cancers (24, 25, 29, 30) and is prognostic in prostate cancer (25). However, mechanisms for how HIP1 overexpression contributes to transformation (24, 26) and how its deficiency in mice leads to degeneration are not known. Because HIP1 binds to clathrin and AP2 (3–6) and alters endocytosis in cultured cells when perturbed (38), disrupted endocytosis that results from HIP1 abnormalities is a reasonable hypothesis for possible mechanisms leading to cancer, as well as mechanisms leading to degeneration (39).
HIP1 is a large 250-kb, 32-exon gene, and its second intron is a rare regulatory ATAC intron (27); its complex gene structure suggests that the introns and potential alternative splice products are important for maintenance of its “normal” levels. However, here we generated a Hip1-targeted constitutive knockout with the option of conditional reexpression from a humanized allele. We showed that a single-copy human HIP1 cDNA (intron free with the exception of intron 1), when present in all cells of the mouse (germ line), was able to fully rescue Hip1 deficiency phenotypes. Therefore, alternative splicing and most of the intervening sequences are not necessary for normal HIP1 levels and functions.
Our novel conditional Hip1 allele allowed us to use multiple Cre alleles to express Hip1 in a tissue-specific manner to understand the role of Hip1 in spinal homeostasis. To our surprise, reexpression of HIP1 specifically in the adult hematopoietic system, kidney, and liver by induction of Mx1-Cre with pIpC was sufficient to prevent development of the Hip1 deficiency phenotypes. Using bone marrow transplantation of Hip1 knockout bone marrow into lethally irradiated wild-type mice, we found that Hip1 deficiency in the bone marrow did not result in development of the Hip1 deficiency phenotype. In contrast, robust, but restricted, expression of HIP1 in the central nervous system (in a large population of both neurons and glia) using an hGFAP-Cre transgene was insufficient to prevent the Hip1 deficiency phenotypes. These data suggest that expression of a HIP1 cDNA in spleen, kidney, and/or liver is required for mouse homeostasis.
To explore mechanisms for how Hip1 is required for homeostasis, we analyzed the metabolomes and transcriptomes of Hip1-deficient tissues. Hip1 deficiency was associated with reduced PC levels and reduced Gdpd3 expression levels. Specifically, Hip1 deficiency was associated with low liver, kidney, and spleen PC levels. PC levels in the brain were normal. This correlates with the rescue of the Hip1 deficiency degenerative phenotypes with HIP1 expression in adult liver, kidney and spleen but not with HIP1 expression in the brain. The lack of PC depletion in the brain implies either that Hip1 is not involved in maintaining brain PC levels or that redundant metabolic mechanisms exist to ensure choline homeostasis in the brain. In fact, the capacity of neurons to synthesize choline de novo is limited, and most neuronal choline comes from influx through a high-affinity choline transporter, CHT1 (40). Lack of Hip1 may therefore be less likely to lead to decreased steady-state PC levels in the brain than in other tissues.
Gdpd3 (Gde7) is a member of the mammalian glycerophosphodiester phosphodiesterase domain containing (Gdpd) gene family (Fig. 4A) (36, 37). The common GDPD domain suggests potential functions for this family in phospholipid metabolism. However, the substrate specificity and specific enzymatic activity, as well as the physiological functions of proteins encoded by this gene family, are not well understood. As recombinant proteins, both Gdpd3 (Gde7) and Gdpd1 (Gde4; Gdpd3's closest relative) have been shown to have lysoPLD activity that catalyzes the formation of LPA and choline (41, 42). LysoPLD activity of Gdpd3 makes it plausible that low Gdpd3 expression in Hip1-deficient mice led to some of the perturbations in choline-related metabolites, as discussed in Results.
The lysoPLD activity of Gdpd3 predicts that LPA may be decreased in Hip1-deficient mice. We did, in fact, measure LPA levels in serum and tissue but found no changes with the exception of a slight elevation in Hip1-deficient liver tissue (data not shown). However, serum LPA (which is considered tumorigenic [43]) is generated by autotaxin, a PLD located on the outside surface of the cell (44). Because GDPD3 is intracellular (37), it is less likely to contribute to serum LPA levels, and it is not surprising that we do not see changes in serum LPA in Hip1-deficient mice (Fig. 6). The high abundance of extracellular/serum LPA can complicate the measuring of tissue/intracellular LPA. Any changes in intracellular LPA levels that may occur due to loss of Gdpd3 may be confounded by serum/extracellular LPA and therefore may require the use of cell culture systems.
Because HIP1 is expressed at high levels in many cancers (25) and can directly transform cells (24), these newly discovered alterations in choline-related metabolite levels in the Hip1-deficient mice may not only help unravel the mechanism of degeneration associated with Hip1 deficiency but also help explain how HIP1 overexpression transforms cells, especially since abnormal choline phospholipid metabolism (elevated PC and diminished GPC) is an emerging hallmark of cancers (28, 45). In fact, prior published data converge on a possible connection between HIP1, GDPD3, cancer, and choline. Specifically, Gdpd3 has been found to be upregulated in mouse multiple myeloma (46, 47), HIP1 is required for growth of multiple myeloma cells (48), and transgenic overexpression of HIP1 induces myeloma-like neoplasms in mice (49).
At this time, the loss of Gdpd3 in the Hip1-deficient mice does not indicate that loss of Gdpd3 expression is the cause of the Hip1 knockout phenotype. In fact, since the knockout of Gdpd3 did not show a decrease in PC levels or display degeneration (Fig. 5), the evidence for a singular role of Gdpd3 in the degenerative phenotype associated with Hip1 deficiency is absent. Knowing whether the Hip1/Gdpd3 double-knockout mice have lower PC levels than the single Hip1-deficient mice will help address this question.
In addition to Gdpd3, other GDPDs have been implicated in cancers and choline pathways. For example, GDPD5 (GDE2) is elevated in breast cancer and has been correlated with elevated PC levels (50). GDPD5 has GPC-phosphodiesterase activity that generates PC. Dysregulation of choline metabolism may therefore be a shared mechanism for how altered GDPD family members could contribute to cancer development. However, each member of the GDPD family of enzymes is thought to have unique activities and unique substrate specificities; our understanding of their activities and metabolic effects will be increased only with additional, detailed in vivo studies.
Since HIP1 is a member of a group of endocytic oncoproteins that are hypothesized to usurp normal endocytic pathways to transform cells by increasing tumor-promoting signals (51), the decreased PC levels in the knockout mice add a new layer of complexity to the endocytosis and cancer hypothesis. By altering membrane trafficking, aberrant endocytic factors such as HIP1 could simultaneously elevate levels of several growth factor receptors by altering their endocytosis (39, 51) and also by influencing membrane lipid composition. It has already been shown that inhibition of endocytosis can lead to changes in membrane lipid composition (52) and that changes in membrane lipid composition effect endocytosis of membrane receptors (53). These changes in membrane lipid composition and altered receptor signaling could then influence or be influenced by choline pathway enzymes, such as Gdpd3, through feedback loops that increase PC levels in cancer and decrease PC levels in degeneration.
MATERIALS AND METHODS
Generation of Hip1-deficient mice and conditional HIP1 knock-in mice.
The targeting vector was constructed to generate a knock-in mouse allele that conditionally expresses human HIP1 and enhanced GFP (eGFP) (Fig. 1A). The objective of this project has been to create a knock-in mouse model conditionally expressing human Hip1 and eGFP using homologous recombination in mouse C57BL/6 embryonic stem cells and subsequent blastocyst injection of the appropriate targeted ES cells to create the gene-targeted mice.
The mouse Hip1 genomic DNA sequence was retrieved from mouse chromosome 5 from the Ensembl database and used as reference in this project. The mouse bacterial artificial chromosome (BAC) DNA containing the Hip1 gene was used as the template for generating the homology arms and the Southern probes for screening targeted events. The 5′ homology arm (∼5.4 kb) (LHA) and the 3′ homology arm (∼3.6 kb) (SHA) were generated by PCR using high-fidelity Taq DNA polymerase. The Lox-stop-Lox (LSL) cassette (∼1.5 kb) was amplified from the LSL-pZero-2 vector. These fragments were cloned in the FtNwCD or pCR4.0 vector and were confirmed by restriction digestion and sequencing. The hHip1 cDNA (∼3 kb) was amplified from the pcDNA3-hHip1 plasmid. The internal ribosome entry site (IRES) (∼0.6-kb) and eGFP (∼0.9-kb) sequences were amplified from the pIRES2-AcGFP1 and pEGFP-N1 plasmids, respectively.
The final vector was obtained by standard molecular cloning. Aside from the homology arms, the final vector also contains a Lox-stop-Lox cassette (∼1.5 kb), mHip1 partial intron1 plus partial exon 2, hHip1 cDNA, IRES, eGFP plus poly(A), Frt sequences flanking the Neo expression cassette (the Neo cassette was used for positive selection of the electroporated ES cells), and a diphtheria toxin A (DTA) expression cassette (for negative selection of the ES cells). Unique restriction sites SacII and PacI were added to flank the hHip1 cDNA for easy swapping in mutant cDNA in the future. The final vector was confirmed by both restriction digestion and sequencing analysis. NotI was used for linearizing the final vector prior to electroporation.
ES cell gene targeting.
Thirty micrograms of NotI-linearized final gene-targeting vector DNA was electroporated into ∼107 C57BL/6 ES cells and selected with 200 μg/ml G418. Two plates of G418-resistant ES clones (∼192) were selected for screening. The primary ES screening was performed with 3′ PCR. Approximately 40 potential targeted clones were identified from one plate. Six clones (A8, B4, C7, C11, E6, and F4) were expanded for further analysis.
Upon completion of the ES clone expansion, additional Southern confirmation analysis was performed. Based on this analysis, five out of the six expanded clones (B4, C7, C11, E6, and F4) were confirmed for homologous recombination with single Neo integration. Clones C7 and C11 were then transfected with a Flp-expressing plasmid. Eight G418-sensitive clones (C7/A1, C7/A10, C7/B7, C11/B5, C11/B9, C11/C8, C11/D7, and C11/E8) were identified and confirmed as Neo-deleted clones by PCR.
Southern blotting.
The 5′ and 3′ external probes were generated by PCR and were tested by genomic Southern blot analysis for screening of the ES cells. The probes were cloned in the pCR4.0 backbone and confirmed by sequencing. Southern blotting to distinguish the targeted Hip1LSL allele and the wild-type allele was performed with both probes as described previously (12).
Generation of Gdpd3-deficient mice.
The targeting strategy for the generation of the Gdpd3 knockout allele was based on NCBI transcript NC_000073_NM_024228_2 (Fig. 5A). The constitutive Gdpd3 knockout mouse was generated with the assistance of Taconic Biosciences and CRISPR/Cas9-mediated genome editing. Of all potential single guide RNAs (sgRNAs), two were selected for their position and minimal potential off-target effects. Guide RNAs target sequences before exon 2 (proximal sgRNA, 5′-GAG AGG CCA GTT CAA CTG AT-3′) and after exon 6 (distal sgRNA, 5′GAC GGG GAT TCG ACA ATT GC-3′). The resulting alteration via nonhomologous-end joining (NHEJ) deleted the functionally critical exons (exons 2 to 7), generated a frameshift mutation from exon 1 to all downstream exons, and introduced a premature stop codon in exon 7. In addition, the resulting transcript was predicted to be a target for non-sense-mediated RNA decay and therefore not to be expressed at a significant level. The guide RNAs and Cas9 mRNA/protein were injected into single-cell zygotes. C57BL/6J mice were used to generate these mice. Mice were tail genotyped using the following primers: oligonucleotide 1, 5′-TCC ATG TAG GGT GGA GTG AGC-3′; oligonucleotide 2, 5′-AAG GGG CTC GTA GGG GAA G-3′; and oligonucleotide 3, 5′-ACA CAG GAA GGA CCA GGG C-3′. The amplicon size for the wild-type allele was 501 bp (with oligonucleotides 1 and 3), and that for the deletion allele was 295 bp (with oligonucleotides 1 and 2). These PCR products were cloned and sequenced. Indel modifications were distinguished from unmodified wild-type sequences by heteroduplex analysis via capillary electrophoresis.
Mice.
CMV-Cre; Hip1LSL/LSL, Mx1-Cre; Hip1LSL/LSL or hGFAP-Cre; Hip1LSL/LSL mice were generated by crossing the Hip1LSL/LSL mice with mice transgenic for the CMV-Cre (31), Mx1-Cre (33), or hGFAP-Cre gene (32), respectively. Expression of Cre recombinase from the Mx1-Cre transgene was induced by intraperitoneal injection of mice with 250 μg pIpC (Sigma, St. Louis, MO) in 100 μl phosphate-buffered saline (PBS) at 2-day intervals for 2 weeks. Mice were housed in the Unit for Laboratory Animal Medicine at the University of Texas Southwestern Medical School under specific-pathogen-free conditions. All mouse experiments were conducted after approval of the University of Texas Southwestern Medical Center Committee on the Use and Care of Animals.
Genotyping.
The Hip1LSL conditional allele and the Gdpd3 knockout allele were genotyped from tail snips using real-time PCR assays designed by Transnetyx. For Hip1, assays were designed to detect wild-type Hip1 and the Cre-recombined (excised LSL cassette) and unrecombined mutant alleles. For Gdpd3, an assay was designed to detect the wild-type Gdpd3 allele.
Western blotting.
Tissues and cells were extracted in lysis buffer containing protease and phosphatase inhibitors (50 mM Tris [pH 7.4], 150 mM NaCl, 1% Triton X-100, protease inhibitors [Roche], 30 mM sodium pyrophosphate, 50 mM NaF, 100 μM sodium orthovanadate), cleared of unbroken cells by centrifugation, and diluted to a protein concentration of 0.5 to 2.0 mg/ml. Whole-cell lysates in Laemmli buffer were separated on 6% or 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Membranes were probed with the following antibodies: mouse monoclonal anti-mouse Hip1 1B11 (1:1,000) and rabbit polyclonal anti-human HIP1 UM323 (1:5,000). The blots were then incubated with horseradish peroxidase (HRP)-conjugated mouse or rabbit secondary antibodies (1:10,000; GE Health Care) and developed using chemiluminescence (Pierce).
Metabolomics.
Mouse tissues from age-matched wild-type (n = 4), Hip1-deficient (n = 7), HIP1-rescued (n = 11), Gdpd3+/+ (n = 5), and Gdpd3−/− (n = 5) mice were rapidly removed (within 2 min of sacrifice) and snap-frozen. Wild-type, HIP1-rescued, and Hip1-deficient mice were <3 months of age. Gdpd3 knockout mice were 2 to 6 months of age. Frozen tissues (∼100 mg each) were homogenized in ice-cold 80% methanol. Following vigorous vortexing, cell debris was removed by centrifugation. The supernatant was evaporated with a SpeedVac to generate a metabolite pellet which was reconstituted in 0.03% formic acid in analytical-grade water before liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. LC-MS/MS was performed as described previously (54).
To gain an initial overview of the results, a multivariate principal-component analysis of all 120 metabolites that were measured was performed. All analyses were carried out using SIMCA-P (version 13.0.1; Umetrics). The variable importance in projection (VIP) values were calculated with the R statistical package. All other statistical analyses of the mass spectrometry data were performed with GraphPad Prism 5.0c software. P values were calculated with an unpaired Student t test.
Microarray analysis.
Total RNA was prepared from various tissues using TRIzol (Invitrogen) extraction and treated with DNase (Qiagen) to remove genomic DNA contamination. RNAs from four (two 1- to 3-month-old and two 5- to 6-month-old) mice from each genotype (Hip1LSL/LSL, Hip1HIP1/HIP1, or wild-type Hip1) were used. Microarray analysis was carried out using the Illumina MouseWG-6 V2 BeadChip whole-genome expression array (Illumina, Inc.). Each RNA sample was amplified using the Ambion TotalPrep RNA amplification kit with biotin UTP (Enzo) labeling. The Ambion Illumina RNA amplification kit used a T7 oligo(dT) primer to generate single-stranded cDNA followed by a second-strand synthesis to generate double-stranded cDNA, which was then column purified. In vitro transcription with T7 RNA polymerase generated biotin-labeled cRNA. The cRNA was then column purified and checked for size and yield using the Bio-Rad Experion system, and 1.5 μg of cRNA was hybridized to each array using standard Illumina protocols. Streptavidin-Cy3 (Amersham) was used for detection. Slides were scanned and fluorescence intensity captured with Illumina HiScan. Expression values were extracted using GenomeStudio2010.2. The data were background subtracted and quantile normalized using the MBCB algorithm (55–57).
Real-time PCR for Gdpd3 expression.
Total RNA (2 μg) was used for reverse transcription using iScript Advanced cDNA synthesis kit (Bio-Rad) according to the manufacturer's instructions. Real-time PCR was then performed using the StepOnePlus real-time PCR system (Applied Biosystems) with TaqMan gene expression master mix and specific TaqMan gene expression assays (mGdpd3 [Ms00470321_m1] and mGapdh [Ms99999915_g1]; Life Technologies). Each sample was measured in triplicate, and all data were normalized to the housekeeping glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene as a control for RNA quantity and sample processing.
Supplementary Material
ACKNOWLEDGMENTS
We thank Travis Laxson, Katherine Oravecz-Wilson, Alanna Coughran, Gunjan Singh, and other members of the Ross lab for their technical assistance and intellectual contributions. We thank Xialolei Shi and Ralph Deberardinis for assistance with mass spectrometry and the Genomics Shared Resource core at the Harold C. Simmons Comprehensive Cancer Center for their expertise in RNA expression analysis.
The Genomics Shared Resource is supported in part by an NCI Cancer Center Support Grant, 1P30 CA142543. This work was supported by National Cancer Institute grants to T.S.R. (R01 CA82363-03 and R01 CA098730-01) and by a Burroughs Wellcome Fund Clinical Scientist Award in Translational Research.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00385-18.
REFERENCES
- 1.Kalchman MA, Koide HB, McCutcheon K, Graham RK, Nichol K, Nishiyama K, Kazemi-Esfarjani P, Lynn FC, Wellington C, Metzler M, Goldberg YP, Kanazawa I, Gietz RD, Hayden MR. 1997. HIP1, a human homologue of S. cerevisiae Sla2p, interacts with membrane-associated huntingtin in the brain. Nat Genet 16:44–53. doi: 10.1038/ng0597-44. [DOI] [PubMed] [Google Scholar]
- 2.Wanker EE, Rovira C, Scherzinger E, Hasenbank R, Walter S, Tait D, Colicelli J, Lehrach H. 1997. HIP-I: a huntingtin interacting protein isolated by the yeast two-hybrid system. Hum Mol Genet 6:487–495. doi: 10.1093/hmg/6.3.487. [DOI] [PubMed] [Google Scholar]
- 3.Metzler M, Legendre-Guillemin V, Gan L, Chopra V, Kwok A, McPherson PS, Hayden MR. 2001. HIP1 functions in clathrin-mediated endocytosis through binding to clathrin and adaptor protein 2. J Biol Chem 276:39271–39276. doi: 10.1074/jbc.C100401200. [DOI] [PubMed] [Google Scholar]
- 4.Mishra SK, Agostinelli NR, Brett TJ, Mizukami I, Ross TS, Traub LM. 2001. Clathrin- and AP-2-binding sites in HIP1 uncover a general assembly role for endocytic accessory proteins. J Biol Chem 276:46230–46236. doi: 10.1074/jbc.M108177200. [DOI] [PubMed] [Google Scholar]
- 5.Rao DS, Chang JC, Kumar PD, Mizukami I, Smithson GM, Bradley SV, Parlow AF, Ross TS. 2001. Huntingtin interacting protein 1 is a clathrin coat binding protein required for differentiation of late spermatogenic progenitors. Mol Cell Biol 21:7796–7806. doi: 10.1128/MCB.21.22.7796-7806.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waelter S, Scherzinger E, Hasenbank R, Nordhoff E, Lurz R, Goehler H, Gauss C, Sathasivam K, Bates GP, Lehrach H, Wanker EE. 2001. The huntingtin interacting protein HIP1 is a clathrin and alpha-adaptin-binding protein involved in receptor-mediated endocytosis. Hum Mol Genet 10:1807–1817. doi: 10.1093/hmg/10.17.1807. [DOI] [PubMed] [Google Scholar]
- 7.Ford MG, Pearse BM, Higgins MK, Vallis Y, Owen DJ, Gibson A, Hopkins CR, Evans PR, McMahon HT. 2001. Simultaneous binding of PtdIns(4,5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science 291:1051–1055. doi: 10.1126/science.291.5506.1051. [DOI] [PubMed] [Google Scholar]
- 8.Hyun TS, Rao DS, Saint-Dic D, Michael LE, Kumar PD, Bradley SV, Mizukami IF, Oravecz-Wilson KI, Ross TS. 2004. HIP1 and HIP1r stabilize receptor tyrosine kinases and bind 3-phosphoinositides via epsin N-terminal homology domains. J Biol Chem 279:14294–14306. doi: 10.1074/jbc.M312645200. [DOI] [PubMed] [Google Scholar]
- 9.Itoh T, Koshiba S, Kigawa T, Kikuchi A, Yokoyama S, Takenawa T. 2001. Role of the ENTH domain in phosphatidylinositol-4,5-bisphosphate binding and endocytosis. Science 291:1047–1051. doi: 10.1126/science.291.5506.1047. [DOI] [PubMed] [Google Scholar]
- 10.Senetar MA, Foster SJ, McCann RO. 2004. Intrasteric inhibition mediates the interaction of the I/LWEQ module proteins Talin1, Talin2, Hip1, and Hip12 with actin. Biochemistry 43:15418–15428. doi: 10.1021/bi0487239. [DOI] [PubMed] [Google Scholar]
- 11.Metzler M, Li B, Gan L, Georgiou J, Gutekunst CA, Wang Y, Torre E, Devon RS, Oh R, Legendre-Guillemin V, Rich M, Alvarez C, Gertsenstein M, McPherson PS, Nagy A, Wang YT, Roder JC, Raymond LA, Hayden MR. 2003. Disruption of the endocytic protein HIP1 results in neurological deficits and decreased AMPA receptor trafficking. EMBO J 22:3254–3266. doi: 10.1093/emboj/cdg334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oravecz-Wilson KI, Kiel MJ, Li L, Rao DS, Saint-Dic D, Kumar PD, Provot MM, Hankenson KD, Reddy VN, Lieberman AP, Morrison SJ, Ross TS. 2004. Huntingtin interacting protein 1 mutations lead to abnormal hematopoiesis, spinal defects and cataracts. Hum Mol Genet 13:851–867. doi: 10.1093/hmg/ddh102. [DOI] [PubMed] [Google Scholar]
- 13.Bradley SV, Hyun TS, Oravecz-Wilson KI, Li L, Waldorff EI, Ermilov AN, Goldstein SA, Zhang CX, Drubin DG, Varela K, Parlow A, Dlugosz AA, Ross TS. 2007. Degenerative phenotypes caused by the combined deficiency of murine HIP1 and HIP1r are rescued by human HIP1. Hum Mol Genet 16:1279–1292. doi: 10.1093/hmg/ddm076. [DOI] [PubMed] [Google Scholar]
- 14.Hyun TS, Li L, Oravecz-Wilson KI, Bradley SV, Provot MM, Munaco AJ, Mizukami IF, Sun H, Ross TS. 2004. Hip1-related mutant mice grow and develop normally but have accelerated spinal abnormalities and dwarfism in the absence of HIP1. Mol Cell Biol 24:4329–4340. doi: 10.1128/MCB.24.10.4329-4340.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ross TS, Bernard OA, Berger R, Gilliland DG. 1998. Fusion of Huntingtin interacting protein 1 to platelet-derived growth factor beta receptor (PDGFbetaR) in chronic myelomonocytic leukemia with t(5;7)(q33;q11.2). Blood 91:4419–4426. [PubMed] [Google Scholar]
- 16.Golub TR, Barker GF, Lovett M, Gilliland DG. 1994. Fusion of PDGF receptor beta to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell 77:307–316. doi: 10.1016/0092-8674(94)90322-0. [DOI] [PubMed] [Google Scholar]
- 17.Grand FH, Burgstaller S, Kuhr T, Baxter EJ, Webersinke G, Thaler J, Chase AJ, Cross NC. 2004. p53-Binding protein 1 is fused to the platelet-derived growth factor receptor beta in a patient with a t(5;15)(q33;q22) and an imatinib-responsive eosinophilic myeloproliferative disorder. Cancer Res 64:7216–7219. doi: 10.1158/0008-5472.CAN-04-2005. [DOI] [PubMed] [Google Scholar]
- 18.Jones AV, Cross NC. 2004. Oncogenic derivatives of platelet-derived growth factor receptors. Cell Mol Life Sci 61:2912–2923. doi: 10.1007/s00018-004-4272-z. [DOI] [PubMed] [Google Scholar]
- 19.Tefferi A, Vardiman JW. 2008. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia 22:14–22. doi: 10.1038/sj.leu.2404955. [DOI] [PubMed] [Google Scholar]
- 20.Hong M, Kim RN, Song JY, Choi SJ, Oh E, Lira ME, Mao M, Takeuchi K, Han J, Kim J, Choi YL. 2014. HIP1-ALK, a novel fusion protein identified in lung adenocarcinoma. J Thorac Oncol 9:419–422. doi: 10.1097/JTO.0000000000000061. [DOI] [PubMed] [Google Scholar]
- 21.Fang DD, Zhang B, Gu Q, Lira M, Xu Q, Sun H, Qian M, Sheng W, Ozeck M, Wang Z, Zhang C, Chen X, Chen KX, Li J, Chen SH, Christensen J, Mao M, Chan CC. 2014. HIP1-ALK, a novel ALK fusion variant that responds to crizotinib. J Thorac Oncol 9:285–294. doi: 10.1097/JTO.0000000000000087. [DOI] [PubMed] [Google Scholar]
- 22.Ou SH, Klempner SJ, Greenbowe JR, Azada M, Schrock AB, Ali SM, Ross JS, Stephens PJ, Miller VA. 2014. Identification of a novel HIP1-ALK fusion variant in non-small-cell lung cancer (NSCLC) and discovery of ALK I1171 (I1171N/S) mutations in two ALK-rearranged NSCLC patients with resistance to Alectinib. J Thorac Oncol 9:1821–1825. doi: 10.1097/JTO.0000000000000368. [DOI] [PubMed] [Google Scholar]
- 23.Bradley SV, Holland EC, Liu GY, Thomas D, Hyun TS, Ross TS. 2007. Huntingtin interacting protein 1 is a novel brain tumor marker that associates with epidermal growth factor receptor. Cancer Res 67:3609–3615. doi: 10.1158/0008-5472.CAN-06-4803. [DOI] [PubMed] [Google Scholar]
- 24.Rao DS, Bradley SV, Kumar PD, Hyun TS, Saint-Dic D, Oravecz-Wilson KI, Kleer CG, Ross TS. 2003. Altered receptor trafficking in Huntingtin interacting protein 1-transformed cells. Cancer Cell 3:471–482. doi: 10.1016/S1535-6108(03)00107-7. [DOI] [PubMed] [Google Scholar]
- 25.Rao DS, Hyun TS, Kumar PD, Mizukami IF, Rubin MA, Lucas PC, Sanda MG, Ross TS. 2002. Huntingtin-interacting protein 1 is overexpressed in prostate and colon cancer and is critical for cellular survival. J Clin Invest 110:351–360. doi: 10.1172/JCI0215529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J, Yu W, Cai Y, Ren C, Ittmann MM. 2008. Altered fibroblast growth factor receptor 4 stability promotes prostate cancer progression. Neoplasia 10:847–856. doi: 10.1593/neo.08450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Graves CW, Philips ST, Bradley SV, Oravecz-Wilson KI, Li L, Gauvin A, Ross TS. 2008. Use of a cryptic splice site for the expression of huntingtin interacting protein 1 in select normal and neoplastic tissues. Cancer Res 68:1064–1073. doi: 10.1158/0008-5472.CAN-07-5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glunde K, Bhujwalla ZM, Ronen SM. 2011. Choline metabolism in malignant transformation. Nat Rev Cancer 11:835–848. doi: 10.1038/nrc3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ames HM, Bichakjian CK, Liu GY, Oravecz-Wilson KI, Fullen DR, Verhaegen ME, Johnson TM, Dlugosz AA, Ross TS. 2011. Huntingtin-interacting protein 1: a Merkel cell carcinoma marker that interacts with c-Kit. J Investig Dermatol 131:2113–2120. doi: 10.1038/jid.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marghalani S, Feller JK, Mahalingam M, Mirzabeigi M. 2015. Huntingtin interacting protein 1 as a histopathologic adjunct in the diagnosis of Merkel cell carcinoma. Int J Dermatol 54:640–647. doi: 10.1111/ijd.12454. [DOI] [PubMed] [Google Scholar]
- 31.Dupe V, Davenne M, Brocard J, Dolle P, Mark M, Dierich A, Chambon P, Rijli FM. 1997. In vivo functional analysis of the Hoxa-1 3′ retinoic acid response element (3′RARE). Development 124:399–410. [DOI] [PubMed] [Google Scholar]
- 32.Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. 2001. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis 31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
- 33.Kuhn R, Schwenk F, Aguet M, Rajewsky K. 1995. Inducible gene targeting in mice. Science 269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 34.Corbin KD, Zeisel SH. 2012. Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr Opin Gastroenterol 28:159–165. doi: 10.1097/MOG.0b013e32834e7b4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Z, Agellon LB, Vance DE. 2005. Phosphatidylcholine homeostasis and liver failure. J Biol Chem 280:37798–37802. doi: 10.1074/jbc.M508575200. [DOI] [PubMed] [Google Scholar]
- 36.Yanaka N. 2007. Mammalian glycerophosphodiester phosphodiesterases. Biosci Biotechnol Biochem 71:1811–1818. doi: 10.1271/bbb.70062. [DOI] [PubMed] [Google Scholar]
- 37.Corda D, Mosca MG, Ohshima N, Grauso L, Yanaka N, Mariggio S. 2014. The emerging physiological roles of the glycerophosphodiesterase family. FEBS J 281:998–1016. doi: 10.1111/febs.12699. [DOI] [PubMed] [Google Scholar]
- 38.Chen CY, Brodsky FM. 2005. Huntingtin-interacting protein 1 (Hip1) and Hip1-related protein (Hip1R) bind the conserved sequence of clathrin light chains and thereby influence clathrin assembly in vitro and actin distribution in vivo. J Biol Chem 280:6109–6117. doi: 10.1074/jbc.M408454200. [DOI] [PubMed] [Google Scholar]
- 39.Hyun TS, Ross TS. 2004. HIP1: trafficking roles and regulation of tumorigenesis. Trends Mol Med 10:194–199. doi: 10.1016/j.molmed.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 40.Lecomte MJ, De Gois S, Guerci A, Ravassard P, Faucon Biguet N, Mallet J, Berrard S. 2005. Differential expression and regulation of the high-affinity choline transporter CHT1 and choline acetyltransferase in neurons of superior cervical ganglia. Mol Cell Neurosci 28:303–313. doi: 10.1016/j.mcn.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 41.Ohshima N, Kudo T, Yamashita Y, Mariggio S, Araki M, Honda A, Nagano T, Isaji C, Kato N, Corda D, Izumi T, Yanaka N. 2015. New members of the mammalian glycerophosphodiester phosphodiesterase family: GDE4 and GDE7 produce lysophosphatidic acid by lysophospholipase D activity. J Biol Chem 290:4260–4271. doi: 10.1074/jbc.M114.614537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsuboi K, Okamoto Y, Rahman IA, Uyama T, Inoue T, Tokumura A, Ueda N. 2015. Glycerophosphodiesterase GDE4 as a novel lysophospholipase D: a possible involvement in bioactive N-acylethanolamine biosynthesis. Biochim Biophys Acta 1851:537–548. doi: 10.1016/j.bbalip.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 43.Mills GB, Moolenaar WH. 2003. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer 3:582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 44.Murph M, Tanaka T, Liu S, Mills GB. 2006. Of spiders and crabs: the emergence of lysophospholipids and their metabolic pathways as targets for therapy in cancer. Clin Cancer Res 12:6598–6602. doi: 10.1158/1078-0432.CCR-06-1721. [DOI] [PubMed] [Google Scholar]
- 45.Trousil S, Lee P, Pinato DJ, Ellis JK, Dina R, Aboagye EO, Keun HC, Sharma R. 2014. Alterations of choline phospholipid metabolism in endometrial cancer are caused by choline kinase alpha overexpression and a hyperactivated deacylation pathway. Cancer Res 74:6867–6877. doi: 10.1158/0008-5472.CAN-13-2409. [DOI] [PubMed] [Google Scholar]
- 46.Fowler JA, Lwin ST, Drake MT, Edwards JR, Kyle RA, Mundy GR, Edwards CM. 2011. Host-derived adiponectin is tumor-suppressive and a novel therapeutic target for multiple myeloma and the associated bone disease. Blood 118:5872–5882. doi: 10.1182/blood-2011-01-330407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boylan KL, Gosse MA, Staggs SE, Janz S, Grindle S, Kansas GS, Van Ness BG. 2007. A transgenic mouse model of plasma cell malignancy shows phenotypic, cytogenetic, and gene expression heterogeneity similar to human multiple myeloma. Cancer Res 67:4069–4078. doi: 10.1158/0008-5472.CAN-06-3699. [DOI] [PubMed] [Google Scholar]
- 48.Tiedemann RE, Zhu YX, Schmidt J, Shi CX, Sereduk C, Yin H, Mousses S, Stewart AK. 2012. Identification of molecular vulnerabilities in human multiple myeloma cells by RNA interference lethality screening of the druggable genome. Cancer Res 72:757–768. doi: 10.1158/0008-5472.CAN-11-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bradley SV, Smith MR, Hyun TS, Lucas PC, Li L, Antonuk D, Joshi I, Jin F, Ross TS. 2007. Aberrant Huntingtin interacting protein 1 in lymphoid malignancies. Cancer Res 67:8923–8931. doi: 10.1158/0008-5472.CAN-07-2153. [DOI] [PubMed] [Google Scholar]
- 50.Cao MD, Dopkens M, Krishnamachary B, Vesuna F, Gadiya MM, Lonning PE, Bhujwalla ZM, Gribbestad IS, Glunde K. 2012. Glycerophosphodiester phosphodiesterase domain containing 5 (GDPD5) expression correlates with malignant choline phospholipid metabolite profiles in human breast cancer. NMR Biomed 25:1033–1042. doi: 10.1002/nbm.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mosesson Y, Mills GB, Yarden Y. 2008. Derailed endocytosis: an emerging feature of cancer. Nat Rev Cancer 8:835–850. doi: 10.1038/nrc2521. [DOI] [PubMed] [Google Scholar]
- 52.Mato JM, Pencev D, Vasanthakumar G, Schiffmann E, Pastan I. 1983. Inhibitors of endocytosis perturb phospholipid metabolism in rabbit neutrophils and other cells. Proc Natl Acad Sci U S A 80:1929–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weber U, Eroglu C, Mlodzik M. 2003. Phospholipid membrane composition affects EGF receptor and Notch signaling through effects on endocytosis during Drosophila development. Dev Cell 5:559–570. doi: 10.1016/S1534-5807(03)00273-9. [DOI] [PubMed] [Google Scholar]
- 54.Mullen AR, Hu Z, Shi X, Jiang L, Boroughs LK, Kovacs Z, Boriack R, Rakheja D, Sullivan LB, Linehan WM, Chandel NS, DeBerardinis RJ. 2014. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep 7:1679–1690. doi: 10.1016/j.celrep.2014.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ding LH, Xie Y, Park S, Xiao G, Story MD. 2008. Enhanced identification and biological validation of differential gene expression via Illumina whole-genome expression arrays through the use of the model-based background correction methodology. Nucleic Acids Res 36:e58. doi: 10.1093/nar/gkn234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Allen JD, Chen M, Xie Y. 2009. Model-based background correction (MBCB): R methods and GUI for Illumina bead-array data. J Cancer Sci Ther 1:25–27. doi: 10.4172/1948-5956.1000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xie Y, Wang X, Story M. 2009. Statistical methods of background correction for Illumina BeadArray data. Bioinformatics 25:751–757. doi: 10.1093/bioinformatics/btp040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.