

Polyglutamine (polyQ) aggregates are associated with pathology in protein-folding diseases and with toxicity in the yeast Saccharomyces cerevisiae. Protection from polyQ toxicity in yeast by human DnaJB6 coincides with sequestration of aggregates.
KEYWORDS: DnaJB6, Huntington's disease, J-protein, molecular chaperone, heat shock protein (HSP), amyloid
ABSTRACT
Polyglutamine (polyQ) aggregates are associated with pathology in protein-folding diseases and with toxicity in the yeast Saccharomyces cerevisiae. Protection from polyQ toxicity in yeast by human DnaJB6 coincides with sequestration of aggregates. Gathering of misfolded proteins into deposition sites by protein quality control (PQC) factors has led to the view that PQC processes protect cells by spatially segregating toxic aggregates. Whether DnaJB6 depends on this machinery to sequester polyQ aggregates, if this sequestration is needed for DnaJB6 to protect cells, and the identity of the deposition site are unknown. Here, we found DnaJB6-driven deposits share characteristics with perivacuolar insoluble protein deposition sites (IPODs). Binding of DnaJB6 to aggregates was necessary, but not enough, for detoxification. Focal formation required a DnaJB6-Hsp70 interaction and actin, polyQ could be detoxified without sequestration, and segregation of aggregates alone was not protective. Our findings suggest DnaJB6 binds to smaller polyQ aggregates to block their toxicity. Assembly and segregation of detoxified aggregates are driven by an Hsp70- and actin-dependent process. Our findings show sequestration of aggregates is not the primary mechanism by which DnaJB6 suppresses toxicity and raise questions regarding how and when misfolded proteins are detoxified during spatial segregation.
INTRODUCTION
Human proteopathies, such as Alzheimer's, Huntington's, and Parkinson's diseases and type 2 diabetes, are characterized by the aggregation of misfolded proteins into amyloids, which are highly ordered and insoluble protein fibers of a single type of protein (1). While the presence of amyloid is almost always associated with disease pathology, it remains unclear if amyloid accumulation itself is toxic or instead represents an inert and possibly cytoprotective process. Particular attention has been placed on oligomers, low-molecular-weight multimers, as a causative agent of cell death (2–4). Amyloid generation is believed to occur through a two-step nucleation mechanism that can produce oligomers at both stages. Primary nucleation is the initial generation of aggregates from monomers, with oligomers arising as likely on-pathway intermediates. Secondary nucleation produces smaller protein structures from the surfaces of amyloid that in turn catalyze formation of new fibers (5).
Protein chaperones, which help other proteins fold into and maintain their native conformations, can prevent amyloid formation and toxic protein accumulation (6). Human DnaJB6, an Hsp70 cochaperone, protects yeast and mammalian systems from Huntington's disease-related polyglutamine (polyQ) toxicity that is caused by expressing a disease-related version of Huntingtin exon 1 containing 103 to 119 glutamines and green fluorescent protein (here HttQ103-GFP) (7, 8). DnaJB6 is the J-protein most closely related to the yeast Sis1, which also protects cells from toxicity due to protein misfolding (9–12).
DnaJB6 has a signature N-terminal J-domain that mediates interaction with Hsp70, an adjacent glycine-phenylalanine (GF)-rich region that, when mutated, leads to inherited myopathies, a serine-threonine (ST)-rich region that binds amyloid, and a small C-terminal region (CTD) of unknown function (7, 13, 14). Studies of polyQ and Aβ peptide aggregation in vitro show that DnaJB6 blocks both primary and secondary nucleation processes by binding to peptides and oligomers (15, 16). The ST region of DnaJB6 is critical for antiamyloid activity, but the J-domain is not needed for inhibiting amyloid formation or protecting cells from polyQ toxicity. These observations imply that DnaJB6 confers protection independently of Hsp70 (7, 8), which is unlike Sis1.
A broad aspect of cellular protein quality control (PQC) is the partitioning of misfolded proteins into various deposition sites that are defined by their locations in the cell and the factors involved in their formation (9, 17–22). These sites include the perinuclear aggresomes and StiF (Sti1 foci), the juxtanuclear JUNQ and intranuclear INQ, the cytoplasmic CytoQ (also called Q-bodies), and the perivacuolar IPOD (insoluble protein deposit). These studies have led to the widely held view that spatial segregation is a universal PQC process that protects cells by sequestering small, disperse toxic aggregates into these deposition sites, where they cause less harm. Correspondingly, studies of polyQ toxicity in yeast show that toxic forms of polyQ produce smaller, heterogeneously sized, and disperse aggregates, whereas nontoxic versions form one or only a few larger aggregates (23, 24).
Similarly, when DnaJB6 protects yeast cells from a toxic version of HttQ103-GFP, smaller disperse aggregates of HttQ103-GFP are collected into a single large structure (8). Using DnaJB6 and the yeast system, we set out to identify this structure and to more clearly define characteristics of the species of aggregates that cause toxicity. We also aimed to determine if this distinctive deposition site has a functional role in the protection process or if DnaJB6 requires other PQC factors to protect cells or drive formation of this structure. As DnaJB6 is unique in its ability to bind amyloid and alter its self-propagating character, we also assessed how the various functional regions of DnaJB6 contribute to the binding, shuttling, and detoxification of HttQ103-GFP aggregates. We found binding of DnaJB6 to HttQ103-GFP was essential but not enough to provide protection, and cooperation of DnaJB6 with Hsp70 and actin was needed for collecting and partitioning of HttQ103-GFP aggregates. This spatial segregation alone was not enough for DnaJB6 to protect cells from HttQ103-GFP toxicity. Our findings show that sequestration of toxic aggregates is not the primary means by which DnaJB6 suppresses polyQ toxicity and raise questions about spatial segregation alone as a general process to protect cells.
RESULTS
Bmh1 and Sti1 are not involved in toxicity prevention or HttQ103-GFP focus formation.
When HttQ103-GFP is expressed in prion-free yeast, it generally remains diffuse in the cytoplasm and is not toxic. In yeast propagating amyloid-based prions, which presumably act as amyloid “seeds,” HttQ103-GFP assembles into multiple disperse aggregates and is toxic (25) (Fig. 1A and B). Coexpressing DnaJB6 prevents the toxicity and causes these aggregates to be collected into a single larger structure (8) (Fig. 1B). Galactose-induced expression of DnaJB6 was itself somewhat toxic in our strains (Fig. 1A, compare growth of cells expressing DnaJB6 with that of cells carrying only empty vectors). The reason for this effect is unclear, but as versions of DnaJB6 lacking the J-domain do not have a similar effect, it could involve impairment of important Hsp70 functions if DnaJB6 competes with endogenous J-proteins for binding to Hsp70.
FIG 1.
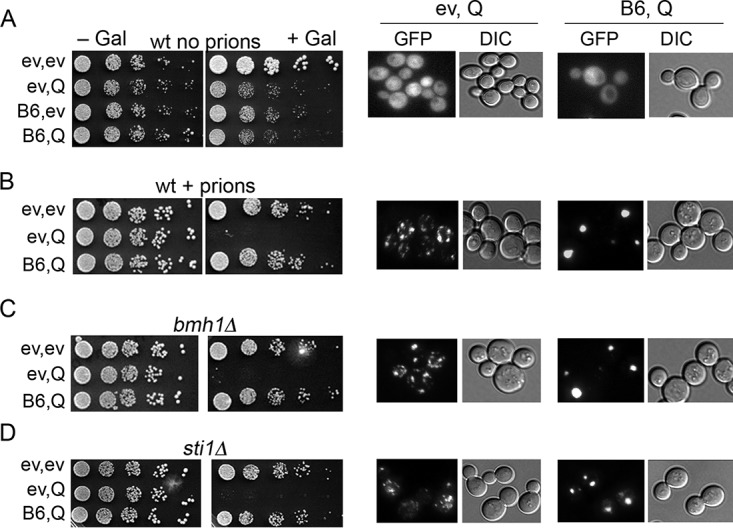
Neither Bmh1 nor Sti1 is needed for DnaJB6 to block HttQ103-GFP toxicity. Wild-type cells lacking prions (strain NK104) and expressing galactose-inducible proteins (ev, empty vector; Q, HttQ103-GFP; B6, DnaJB6) were grown overnight in SRaf medium and 5-fold serially diluted, and 5-μl drops were inoculated onto noninducing (−Gal) or inducing (+Gal) medium as indicated. Panels on the left show growth inhibition caused by HttQ103-GFP, and panels on the right show aggregation status of HttQ103-GFP in the same overnight cultures used for the toxicity assay after shifting to liquid SGal medium for 4 h. (A) Cells lacking prions; HttQ103-GFP does not form toxic aggregates. (B) As described for panel A, except cells propagate both [PSI+] and [PIN+] prions (strain NK101). HttQ103-GFP forms multiple disperse aggregates that are toxic in wild-type cells. Coexpression of DnaJB6 prevents toxicity and drives the smaller aggregates into single large foci. (C and D) As described for panel B, except cells lack Bmh1 (C) or Sti1 (D). DnaJB6 retains the ability to protect cells and assemble disperse aggregates into large foci when Bmh1 or Sti1 is absent.
Given the broad involvement of J-proteins and Hsp70s in protein quality control, the HttQ103-GFP structure formed in cells expressing DnaJB6 could represent any of several subcellular protein deposition compartments (21, 22).
Toxicity of HttQ103-GFP in yeast is muted if it contains the proline-rich region of Huntingtin exon 1 (HttQP103-GFP). Earlier work showed that HttQP103-GFP is gathered into a single perinuclear structure representing an aggresome, a collecting site for misfolded proteins at the microtubule organizing center (24). Bmh1 is necessary for aggresome formation in yeast, and in bmh1Δ cells HttQP103-GFP remains in smaller disperse aggregates and is toxic. When we expressed HttQ103-GFP and DnaJB6 in bmh1Δ cells, we found Bmh1 was not required for prevention of toxicity by DnaJB6 or for formation of the large aggregate (Fig. 1C). Thus, the HttQ103-GFP focus does not represent an aggresome, indicating that DnaJB6 does not promote sequestration of polyQ lacking the proline-rich region to the aggresome, and that the ability to form an aggresome is not necessary for DnaJB6-mediated protection.
Deleting Sti1, a cochaperone of Hsp70 and Hsp90, sensitizes cells to toxic effects of HttQ103-GFP and increases both the number and dispersion of small HttQ103-GFP aggregates in yeast cells (9). In contrast, elevating Sti1 protects cells from HttQ103-GFP toxicity and causes HttQ103-GFP aggregates to assemble into one or two perinuclear foci. These foci are called StiF (Sti1 foci), as they are distinct from aggresomes JUNQ and INQ. We found that in cells lacking Sti1, DnaJB6 still neutralized toxicity (Fig. 1D) and directed formation of HttQ103-GFP foci. Thus, Sti1 has no role in the ability of DnaJB6 to protect cells from HttQ103-GFP toxicity or to drive focus formation, and the foci formed by DnaJB6 are not StiF.
Hsp42, Btn2, and Cur1 are not needed for toxicity prevention or HttQ103-GFP focus formation.
Under conditions of stress, Hsp42 directs misfolded proteins to CytoQ sites, while Btn2 and the structurally related Cur1 direct them to the INQ (17, 19, 20, 22, 26, 27). Under nonstress conditions, elevating expression of Hsp42, Btn2, or Cur1 can cure cells of amyloid-based [URE3] prions by collecting disperse protein aggregates into one or a few larger aggregates (18, 28, 29). DnaJB6 also cures cells of [URE3] but seems to do so by arresting amyloid formation and causing eventual disappearance of prion aggregates (8, 29). There is complicated cross talk among these factors, as curing of [URE3] by Btn2 depends on Hsp42, curing by Cur1 is partially dependent on Hsp42, and curing by Hsp42 requires Cur1 but not Btn2 (28).
To determine if these factors are involved in the neutralization of HttQ103-GFP toxicity or the collecting of the smaller aggregates into one place, we tested effects of DnaJB6 in cells lacking Hsp42, Btn2, Cur1, or both Btn2 and Cur1 (Fig. 2A to D). HttQ103-GFP was slightly less toxic in cells lacking Btn2 or Cur1, and cells lacking both grew more slowly, even in the absence of HttQ103-GFP. These results suggest Btn2 and Cur1 enhance polyQ toxic effects slightly. Alternatively, depletion of Btn2 and Cur1 might cause a stress that induces other PQC factors, which provide modest protection. Regardless, in all of the mutants HttQ103-GFP was clearly toxic, and DnaJB6 clearly neutralized this toxicity and caused HttQ103-GFP to form one or two large foci per cell. Thus, none of these three proteins are needed for DnaJB6 to protect cells from HttQ103-GFP toxicity or to drive formation of the large foci. These results suggest the large foci are different from CytoQ or INQ.
FIG 2.
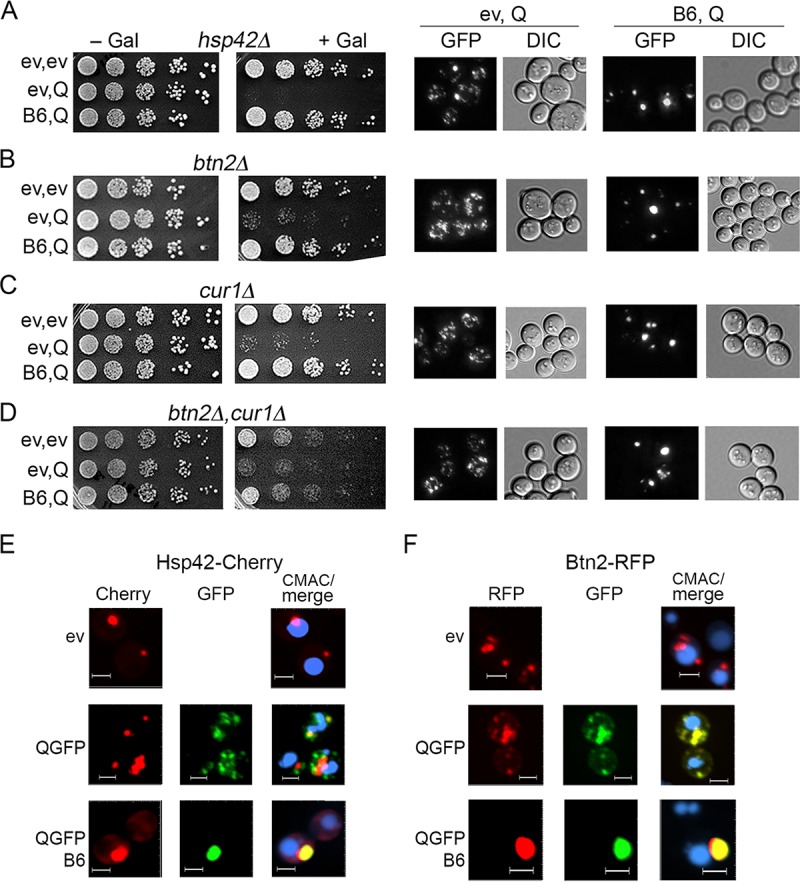
Hsp42, Btn2, and Cur1 are not needed for DnaJB6 to block HttQ103-GFP toxicity. (A to D) [PSI+] [PIN+] cells were grown and plated as described in the legend to Fig. 1. Cells lacking Hsp42, Btn2, or Cur1 (as indicated) respond to HttQ103-GFP like the wild type. DnaJB6 similarly reduces toxicity and causes assembly of the disperse aggregates into large foci. (E and F) Cells carrying plasmids encoding Hsp42-Cherry (E) or Btn2-RFP (F) were grown in SRaf, transferred to SGal, and visualized by confocal imaging after 4 h. (E) Hsp42-Cherry colocalizes with single foci of HttQ103-QGFP in the presence of DnaJB6 (88% of cells, n = 100) but only partially with smaller foci when DnaJB6 is absent (51% of cells, n = 150). (F) Btn2-RFP colocalizes with HttQ103-GFP foci in 86% of cells (n = 55) without DnaJB6 and in 78% of cells (n = 89) coexpressing DnaJB6. Qualitative confocal microscopy was used to monitor these features. CMAC (blue) stains vacuoles. Panels are sized for consistency; all scale bars are 3 μm.
Hsp42 is required for proteins to be collected into CytoQ deposition sites after cells are exposed to stress and proteins in these sites are solubilized or degraded as cells recover under nonstress conditions (19, 20, 27). In contrast, HttQ103-GFP aggregates form in the absence of Hsp42 and remain small and disperse (Fig. 2A), which also indicates the small aggregates are different from CytoQ. Accordingly, Hsp42-mCherry only partially colocalized with the smaller aggregates, although it did colocalize almost entirely with the large foci in cells expressing DnaJB6 (Fig. 2E). Additionally, although Btn2 needs Hsp42 to colocalize with CytoQ sites, we found Btn2-RFP colocalized strongly with the disperse HttQ103-GFP punctae in cells without DnaJB6. It also colocalized with the large GFP foci in cells coexpressing DnaJB6 (Fig. 2F). These results are consistent with Hsp42 and Btn2 recognizing HttQ103-GFP aggregates as misfolded protein, but they are not needed for formation of these small foci and they do not detoxify them.
Large HttQ103-GFP foci resemble IPOD.
In stressed cells HttQ97-GFP can be targeted to the IPOD, as are other proteins prone to forming amyloid (e.g., yeast prion proteins Rnq1 and Ure2) (17, 27). The amyloid-forming region of Sup35 fused to GFP also collects at the IPOD, and this process relies on the actin/myosin machinery (30). It was possible that DnaJB6-directed sequestration of HttQ103-GFP aggregates by a similar process under nonstress conditions when stress-induced chaperones are not highly expressed and proteins in general are not aggregating.
To determine if the large foci of HttQ103-GFP reside at a specific site near the vacuole or nucleus, we stained these organelles and monitored fluorescence in cells expressing HttQ103-GFP. We also monitored red fluorescent protein (RFP)-tagged wild-type and modified versions of DnaJB6 in the same cells. When HttQ103-GFP was expressed alone, the multiple aggregates it formed were dispersed randomly in the cytoplasm (Fig. 3A, upper left). When DnaJB6-RFP was expressed alone it was diffuse in the cytoplasm with a noticeable concentration in the nucleus (Fig. 3A, lower left). When both were expressed in the same cells, DnaJB6-RFP caused HttQ103-GFP to form single large structures, indicating the RFP tag did not inhibit DnaJB6 function, and DnaJB6-RFP colocalized with them (Fig. 3A, right). In the absence of prions, both HttQ103-GFP and DnaJB6-RFP remained diffuse (Fig. 3B). These results do not establish whether or not DnaJB6 binds to soluble HttQ103-GFP in vivo, but they are consistent with the ability of DnaJB6 to bind polyQ oligomers in vitro and suggest it binds directly to aggregates of HttQ103-GFP in vivo.
FIG 3.
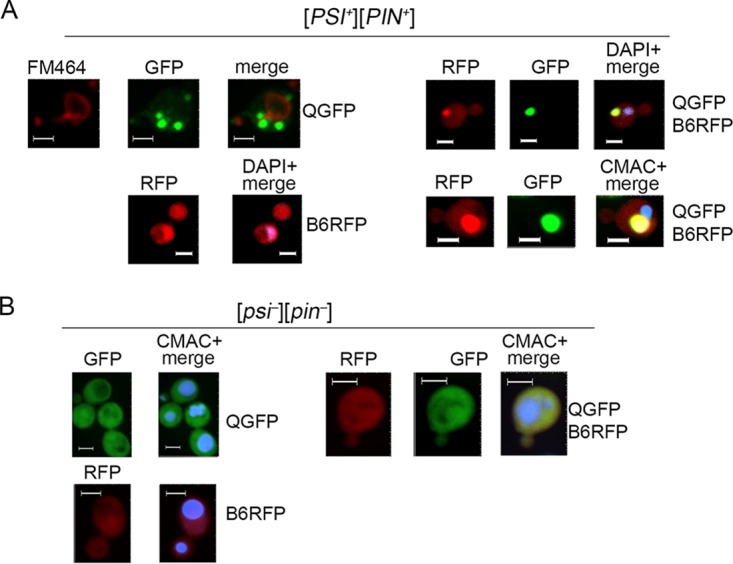
DnaJB6 colocalizes with HttQ103-GFP foci adjacent to vacuole but not nucleus. (A) [PSI+] [PIN+] cells carrying plasmids encoding the indicated galactose-inducible proteins (ev, empty vector; QGFP, HttQ103-GFP; B6RFP, DnaJB6-RFP) grown in SRaf were transferred to SGal, grown for 4 h, and visualized by confocal imaging. The nucleus was visualized by DAPI and the vacuole was stained with FM464 (red) or CMAC (blue). Panels on the left show separate expression of HttQ103-GFP (upper) and DnaJB6-RFP (lower). HttQ103-GFP forms multiple foci scattered in the cytoplasm rather than adjacent to the vacuole (FM464, red). DnaJB6-RFP is diffuse in the cytoplasm. Panels on right show cells expressing both HttQ103-GFP and DnaJB6. Multiple HttQ103-GFP foci are collected into one location mostly coincident (89% cells, n = 62) with DnaJB6. (B) As described for panel A but using cells lacking prions ([psi−] [pin−]). Panels are sized for consistency; all scale bars are 3 μm.
These single structures were not consistently located adjacent to the nucleus (Fig. 3A, upper right), which confirms they are not aggresomes, StiFs, JUNQ, or INQ, but they were observed consistently adjacent to vacuoles (Fig. 3A, lower right). Together our results suggest that DnaJB6 neutralizes toxicity of HttQ103-GFP by interacting with it directly and that it facilitates sequestration of disperse HttQ103-GFP aggregates to a site near the vacuole.
The perivacuolar IPOD forms adjacent to the preautophagosomal structure (PAS; also called the phagophore assembly site), where formation of autophagosomes is initiated (17, 30). The autophagy-related protein Atg8 concentrates at the PAS and therefore colocalizes strongly near the IPOD. In addition, accumulation of amyloidogenic proteins at the IPOD does not depend on Hsp42 (26, 27). As our observations point to the large DnaJB6-driven HttQ103-GFP foci observed in our cells as residing at the IPOD, we expected to find RFP-Atg8 concentrated very near these foci in cells expressing DnaJB6. We expressed RFP-Atg8 in cells expressing both HttQ103-GFP and untagged DnaJB6 and found it formed bright foci near the vacuole, presumably at the PAS (Fig. 4A). In greater than 80% of cells, however, the RFP-Atg8 foci were well separated from the DnaJB6-driven perivacuolar HttQ103-GFP foci, indicating that these foci were not localized primarily adjacent to the PAS. A detectable amount of more diffuse RFP-Atg8 did overlap more consistently with the HttQ103-GFP foci, however, indicating a smaller portion of perivacuolar RFP-Atg8 colocalized with the large foci.
FIG 4.
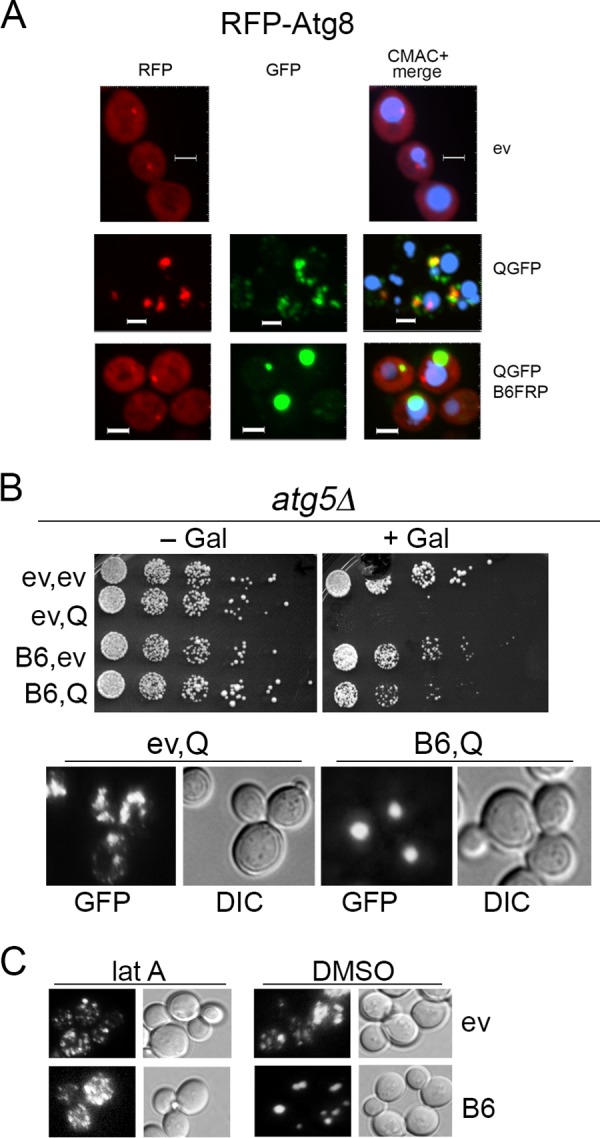
Autophagy pathway is not involved in DnaJB6-driven HttQ103-GFP focus formation or toxicity prevention, but latrunculin A blocks focus formation. (A) Cells expressing RFP-Atg8 and empty vector (ev), HttQ103-GFP (QGFP), or nontagged DnaJB6 (B6) were visualized using confocal microscopy. RFP-Atg8 localizes at the PAS (phagophore assembly site) adjacent to the vacuole (CMAC blue) (uppermost images). In cells expressing both proteins, most foci of RFP-Atg8 (76%, n = 51) overlap HttQ103-GFP aggregates, but most HttQ103-GFP foci do not overlap RFP-Atg8. When DnaJB6 is also expressed, HttQ103-GFP forms single foci well separated from RFP-Atg8 foci in most (82%, n = 58) cells. Panels are sized for consistency; all scale bars are 3 μm. (B) An atg5 deletion strain expressing the indicated galactose-inducible proteins was grown and plated as described in the legend to Fig. 1. DnaJB6 protects cells and assembles disperse aggregates into large foci when Atg5 is absent. (C) Latrunculin A (lat A) or DMSO was added to cells expressing galactose-inducible HttQ103-GFP without (ev) or with coexpression of DnaJB6 (B6), and cells were monitored microscopically after inducing for 4 h.
In cells expressing HttQ103-GFP without DnaJB6, a portion of HttQ103-GFP did localize with Atg8 near the vacuole (Fig. 4A). Much of the HttQ103-GFP fluorescence in these cells resided in smaller cytoplasmic punctae, only some of which overlapped with RFP-Atg8 that presumably represent Atg8-associated autophagic vesicles. Under conditions of stress, transport of proteins to the IPOD relies on a vesicular actin/myosin-directed process that can be similarly associated with Atg8 (19, 30). The presence of Atg8 vesicles in our cells suggests the smaller dispersed HttQ103-GFP aggregates cause enough of a stress to induce an autophagic response. Although some polyQ aggregates appeared to be associated with these vesicles, they were not processed efficiently enough to overcome the toxicity or to be collected in one place. Thus, a stronger stress might be needed to process polyQ aggregates efficiently or for IPODs to form at the PAS.
We did not observe such cytoplasmic RFP-Atg8 punctae in cells expressing DnaJB6, which is consistent with the nonstress conditions we use and the detoxification and collection of HttQ103GFP aggregates before they cause an Atg8-associated response. The sequestration of polyQ aggregates by DnaJB6 is therefore much more effective than the Atg8-associated pathway in the absence of exogenous environmental stress. Together with the observation that DnaJB6 causes foci to form distantly from the PAS, it seems DnaJB6 prevents polyQ from causing a stress and collects polyQ aggregates to an IPOD-like structure by a process separate from the autophagy system.
In agreement with this interpretation, we found atg5Δ cells were killed by HttQ103-GFP and that DnaJB6 both protected them from this HttQ103-GFP toxicity and caused formation of single foci (Fig. 4B). Thus, autophagy does not seem to be involved in the toxicity or the protection and focus formation provided by DnaJB6.
Since the localization of DnaJB6-driven HttQ103-GFP foci with the PAS was noticeably different from earlier observations, some uncertainty remained about whether the foci represent a characteristically defined IPOD. We therefore tested if sequestration of HttQ103-GFP aggregates depended on actin filaments by monitoring formation of HttQ103-GFP foci in cells treated with latrunculin A, which depolymerizes actin filaments. We found that latrunculin A efficiently blocked DnaJB6-driven formation of the large foci (Fig. 4C). Thus, actin filaments were needed for sequestration of polyQ aggregates by DnaJB6, which is a characteristic of deposition of amyloidogenic protein to the IPOD.
STC regions of DnaJB6 are enough to protect from toxicity, showing Hsp70 independence.
The ST region of DnaJB6 (defined here as amino acid residues 132 to 195) (Fig. 5A) binds polyQ to prevent amyloid formation in vitro, and it is required for prevention of HttQ103-GFP toxicity in mammalian cells (7, 31). Mutations or deletions in this region, particularly from residues 155 to 195, impair the protein's protective functionality (7, 13).
FIG 5.

STC regions are necessary and sufficient for protection from toxicity but cannot gather disperse aggregates. (A) Domain structure of DnaJB6 is diagrammed with amino acid residues delineating domains indicated above (J, J-domain; GF, glycine-phenylalanine rich; ST, serine-threonine rich; CTD, C-terminal domain). (B, left) Cells expressing galactose-inducible HttQ103-GFP together with wild-type and mutant versions of DnaJB6 as indicated (C, CTD; R, RFP). Cells carrying plasmids encoding the indicated galactose-inducible proteins (ev, empty vector; Q, HttQ103-GFP; B6, DnaJB6; D33N, point mutation in J-domain) were grown and plated as described for Fig. 1. (Right, upper) Versions of DnaJB6 without coexpressing HttQ103-GFP. (Lower) Independent replicate of the experiment shown on the left but done on a different lot of medium. (C) Microscopic observation of aggregation status of HttQ103-GFP 4 h after inducing cultures used for the toxicity assay shown in panel B. (D) Cells expressing HttQ103-GFP- and RFP-tagged versions of DnaJB6 mutants ST (top), STC (middle), and D33N (bottom) were monitored by confocal microscopy. All mutant versions of DnaJB6 are diffuse when expressed alone. All also colocalize completely with multiple HttQ103-GFP foci (91%, n = 149; 94%, n = 189; 92%, n = 153, respectively) but fail to collect them. Panels are sized for consistency; all scale bars are 3 μm.
To assess the contribution of this and other structural domains to the ability of DnaJB6 to protect cells, we tested effects of mutating or deleting the J-domain, ST, or CTD (Fig. 5B). The D33N mutation, which disrupts the ability of the DnaJB6 J-domain to interact with and regulate Hsp70, only partially reduces its ability to protect mammalian cells from polyQ toxicity and does not affect its ability to cure cells of [URE3] prions (7, 8). In addition to being crucial for protecting mammalian cells from polyQ toxicity, the ST region is necessary for curing of [URE3]. As we earlier found the CTD of DnaJB6 (residues 196 to 241) strongly influenced curing yeast of amyloid-based prions (8), we sought to determine if this region also was important for protecting yeast from HttQ103-GFP.
In our toxicity assay DnaJB6-D33N protected cells from HttQ103-GFP like wild-type DnaJB6 (Fig. 5B), suggesting that protection from HttQ103-GFP toxicity did not depend on interaction of DnaJB6 with Hsp70. In contrast, DnaJB6 lacking only its ST (ΔST; residues 155 to 195) or its CTD (ΔCTD; residues 196 to 241) failed to protect cells, indicating both of these regions are important for detoxifying HttQ103-GFP.
To determine if the ST region alone was enough to provide cytoprotective functions or if it needed to cooperate with the CTD, we assessed effects of expressing only the ST region (residues 132 to 195) or the combined ST and CTD regions (STC; residues 132 to 241). We found that ST alone provided little protection, but the STC consistently protected cells from HttQ103-GFP toxicity (Fig. 5B). Thus, the CTD cooperates with the ST region to provide protection. Since STC lacks the entire J-domain that mediates interaction with Hsp70, these results further support the conclusion that DnaJB6 does not need to cooperate with Hsp70 to protect yeast from HttQ103-GFP toxicity. Moreover, the GF region, mutations of which can cause human myopathies (32–34), is also dispensable for protection.
It was possible that the CTD residues simply stabilized conformation of the ST region, which we presumed was binding directly to the aggregates rather than contributing a specific function. We therefore tested effects of tagging RFP to the ST region in place of the CTD and to the C-terminal end of STC. Although there were modest differences among experiments, probably due to variations in batches of media or cell-to-cell expression levels of the Gal-induced proteins, we found that ST-RFP behaved like ST and STC-RFP behaved like STC (Fig. 5B). Thus, appending RFP to the ST or STC did not alter their cytoprotective properties. These results show that the combined STC is necessary and sufficient for protecting cells from HttQ103-GFP toxicity and support the conclusion that CTD has more than a stabilizing role in this DnaJB6 function.
Focus formation is not needed for DnaJB6 to detoxify HttQ103-GFP.
We observed that the HttQ103-GFP aggregates in cells coexpressing STC or DanJB6-D33N remained numerous and disperse (Fig. 5C). Thus, although these mutants protected cells from HttQ103-GFP toxicity, they lost ability to drive the small HttQ103-GFP aggregates into large foci. Together, our data show that neither interaction with Hsp70 nor collecting the smaller aggregates of HttQ103-GFP into one place is needed for DnaJB6 to protect cells from toxicity and suggest that the assembly of the small disperse aggregates of HttQ103-GFP into large foci by DnaJB6 is an Hsp70-dependent process that occurs separately from neutralization of HttQ103-GFP toxicity.
The ST region is the part of DnaJB6 that possesses amyloid-binding properties, so we presumed ST-RFP and STC-RFP both would bind HttQ103-GFP aggregates in cells. We observed ST-RFP and STC-RFP each did indeed colocalize predominantly with the numerous, smaller HttQ103-GFP aggregates that remain dispersed in the cytoplasm (Fig. 5D). Thus, the ST alone was enough to bind to the HttQ103-GFP aggregates, and the CTD must provide a function beyond specifying this binding to protect from toxicity.
Notably, although DnaJB6ΔCTD failed to protect cells from HttQ103-GFP toxicity, it collected smaller aggregates into larger foci (Fig. 5C). Thus, the CTD was dispensable for DnaJB6 to sequester HttQ103-GFP aggregates into one place, and spatial segregation alone did not provide protection.
Redundant and divergent functions of DnaJB6 and DnaJB7.
DnaJB7, which is structurally related to DnaJB6, sequesters polyQ aggregates very inefficiently and provides less protection from polyQ toxicity (8). Our findings here suggest the J-domain and CTD of DnaJB6 possess partially independent modular activities, raising the question of whether these regions of DnaJB7 possess similar functions. To test this idea, we separately replaced the J-domain and CTD of DnaJB6 with those from DnaJB7. Since nuclear localization of DnaJB7 via a nuclear localization signal at its C terminus probably limits cytoplasmic DnaJB7, we used only the parts of its CTD with the closest homology to DnaJB6b (Fig. 6A). We also tested complementary hybrid proteins with the CTD and J-domains of DnaJB7 replaced by those of DnaJB6 to assess if the functions were transferable.
FIG 6.

Redundant and diverse functions of DnaJB6 and DnaJB7 domains. (A) Amino acid sequence of homologous regions of DnaJB6 and DnaJB7 J-domains (differences dotted) and CTDs. Residues 245 to 309 of DnaJB7, which include a nuclear localization signal, are not shown. (B) Focus formation in cells expressing HttQ103-GFP and the indicated versions of DnaJB6 (JB6), DnaJB7 (JB7; lacks residues 227 to 234, underlined, and 245 to 309), and DnaJB6/7 hybrid proteins. JB6-7J is DnaJB6 with residues 1 to 70 replaced by those of DnaJB7; JB6-7C is DnaJB6 with residues 196 to 241 replaced by residues 192 to 244 (lacking 227 to 234) of DnaJB7. JB7-6J is JB7 with residues 1 to 70 replaced by those of DnaJB6. JB7-6C is JB7 with residues 192 to 244 replaced by residues 196 to 241 of DnaJB6. Cells were grown and images captured as described in the legend to Fig. 1.
In cells expressing DnaJB6 with the J-domain or CTD of DnaJB7 (designated DnaJB6-7J and DnaJB6-7C, respectively), aggregates of HttQ103-GFP were collected into single foci, which shows redundancy of J-domain function and is consistent with the CTD being dispensable for sequestration. In contrast, both DnaJB7-6J and DnaJB7-6C failed to sequester HttQ013-GFP aggregates, indicating something more than Hsp70 interaction is needed for this process.
DnaJB6-7C was able to protect cells, but not to the same degree as DnaJB6. Thus, the CTD of DnaJB7 exhibits cytoprotective activity that partially overlaps that of DnaJB6. The reduced cytoprotection could reflect a difference in intrinsic activity of the CTD of DnaJB7 or its ability to cooperate with the adjacent ST region. Alternatively, differences between the DnaJB6 and DnaJB7 CTDs could determine differences in interactions with other PQC factors that are needed for protection.
In addition to failing to drive formation of foci, DnaJB7-6J did not protect cells and DnaJB7-6C protected only marginally. Thus, a specific DnaJB6 function in addition to a general J-domain function was needed to confer spatial segregation activity, and the DnaJB6 CTD required other DnaJB6 activity to confer effective protection.
Our data show that cooperation of the ST and CTD regions of DnaJB6 was important for cytoprotection, and we suspected the ST region of DnaJB7 was defective either in cooperating with the CTD of DnaJB6 or simply in binding the polyQ aggregates. We replaced ST residues 155 to 195 of DnaJB6 with homologous residues 151 to 191 from DnaJB7 and found this DnaJB6-7ST protein sequestered aggregates but did not protect cells very well from polyQ toxicity. These results suggest that the ST of DnaJB7 binds polyQ aggregates, which is needed to collect them, but cannot cooperate with the DnaJB6 CTD to provide effective protection. Together with our other findings, they also suggest the GF region of DnaJB6 is involved in the sequestration process, and the GF of DnaJB7 lacks this function.
Unexpectedly, DnaJB6-7J was much less effective at protecting cells than DnaJB6. As the J-domain mutant DnaJB6-D33N protected cells like DnaJB6, these results suggest that activity of the DnaJB7 J-domain actually interferes with the ability of the rest of DnaJB6 to protect cells from polyQ toxicity. How this inhibitory effect is mediated is as yet uncertain.
Table 1 summarizes the abilities of the different DnaJB proteins to protect cells from polyQ toxicity and to drive spatial segregation of polyQ aggregates. The hybrid proteins again showed that sequestering of polyQ aggregates was separable from protection from polyQ toxicity.
TABLE 1.
Summary of protection from polyQ toxicity and spatial segregation of polyQ aggregates by DnaJB proteinsa
| Protein | Toxicity protection | Spatial segregation |
|---|---|---|
| DnaJB6 | + | + |
| JB6-D33N | + | − |
| JB6-STC only | + | − |
| JB6-ST only | − | − |
| JB6ΔST | − | − |
| JB6ΔC | − | + |
| DnaJB6-7J | − | + |
| DnaJB6-7C | +/− | + |
| DnaJB6-7ST | − | + |
| DnaJB7 | − | − |
| DnaJB7-6J | − | − |
| DnaJB7-6C | −/+ | − |
DISCUSSION
Our data suggest that DnaJB6 protects yeast cells from HttQ103-GFP toxicity through a direct interaction with HttQ103-GFP aggregates, which agrees with earlier work showing DnaJB6 can bind polyQ peptides and amyloid directly (7, 15, 31). Consistent with this conclusion, the ability of DnaJB6 to cure yeast of amyloid-based prions correlates with its ability to bind the prion proteins directly and block their amyloid nucleation and assembly (8). If such a direct interaction of DnaJB6 with HttQ103-GFP aggregates does confer protection, then some aspect of amyloid formation, growth, or replication must underlie the toxicity.
We did not identify particular species of polyQ aggregates that cause toxicity, so the properties that make aggregates toxic remain uncertain; however, the ability of DnaJB6 to bind polyQ aggregates at various stages of assembly could counteract toxic effects in a number of ways. DnaJB6 does not prevent polyQ aggregates from forming in yeast cells, but it does neutralize their damaging properties, perhaps by disrupting amyloid-forming properties. As polyglutamine aggregates interact with many cellular proteins (10, 24, 35–37), it is possible that binding of DnaJB6 to polyQ protects cells simply by masking surfaces that would otherwise bind and deplete cellular factors. Alternatively, by binding directly to oligomeric polyQ species that cause the toxicity, DnaJB6 might disrupt their ability to maintain or propagate structures that confer harmful properties. Direct binding to less toxic small or large aggregates might also allow DnaJB6 to prevent them from developing into or nucleating more toxic species.
These explanations are consistent with both a widespread view that smaller aggregates or oligomers cause the toxicity and that DnaJB6 can bind substoichiometrically to polyQ and Aβ peptides and oligomers, which block further assembly into amyloid (15, 16, 31). This direct neutralization of HttQ103-GFP toxicity by DnaJB6 is qualitatively different from that by Btn2, which we find binds the small multiple polyQ aggregates in vivo but does not protect cells.
The way DnaJB6 acts on Ure2 amyloid to cure cells of [URE3] is also different from Btn2 and Hsp42. Btn2 and Hsp42 act as aggregases (20) that collect Ure2 amyloid into large cytosolic clumps, which can be restricted from spreading between dividing cells to produce prion-free cells (18, 28, 29). We do not evaluate it here, but increasing expression of Hsp70 system chaperones can also protect cells from polyQ toxicity without diminishing aggregation, leading to a similar view that driving lower-molecular-weight oligomers or other on-pathway intermediates into possibly more inert amyloid forms is protective (38–40). In contrast, and in line with its ability to disrupt amyloid propagation, DnaJB6 binds to Ure2 amyloid and blocks continued assembly and secondary nucleation (8), which results in gradual loss of [URE3], presumably as the amyloid is continually processed by the cellular disaggregation machinery (29).
Unlike the way it acts on [URE3], DnaJB6 drives assembly of the detoxified polyQ aggregates into a large perivacuolar IPOD-like structure. These findings suggest DnaJB6 similarly disrupts propagation of Ure2 and polyQ amyloid in yeast cells, but DnaJB6-bound polyQ is subsequently acted on by PQC factors that process it for spatial segregation. The properties of polyQ and Ure2 aggregates that underlie their differences in how they are processed are not yet known but might be connected to the requirement of Hsp70 specifically for polyQ sequestration. The J-domain of DnaJB6 is not required for curing of [URE3] or for protection from polyQ toxicity, but it is required for segregation of polyQ aggregates.
As sequestration by DnaJB6 still occurs when its J-domain or ST region is replaced by those of DnaJB7, the GF region of DnaJB6 seems to be specifically required for spatial segregation of HttQ103-GFP aggregates. Functional specificity of J-proteins is influenced by GF regions (41, 42), and alterations in this part of DnaJB6 cause various inherited myopathies (32–34), underscoring its physiological importance. The requirement of the GF region for spatial segregation of polyQ aggregates is probably mediated through a specific effect on Hsp70, but how GF regions regulate functions of this machinery remains an open question.
Just as transport of amyloidogenic proteins to the IPOD is dependent on an actin-myosin process, we find formation of the large HttQ103-GFP foci driven by DnaJB6 relies on actin. This collecting process also depends on Hsp70 in an apparently early step. J-proteins are considered obligate cochaperones of Hsp70 (43). As J-proteins, particularly the yeast Sis1, are involved in spatial segregation and detoxification of several types of protein aggregates (9–12), we suspect that Hsp70 has a similar general role in such segregation processes.
Several earlier studies show that, aside from simply preventing spreading of aggregates, similar assembly and spatial segregation of aggregates of polyQ and other misfolded proteins by other PQC factors is crucial for protection from toxicity (9, 12, 17, 19, 20, 23, 24, 44). These and other studies support a view that such sequestration of toxic aggregates is a general response by the cell to defend against toxicity of misfolded proteins.
Here, however, we find spatial segregation is not needed for DnaJB6 to detoxify HttQ103-GFP aggregates, which suggests DnaJB6 neutralizes them before they are recognized and processed as substrates for sequestration. Moreover, we find that spatial segregation of HttQ103-GFP aggregates driven by DnaJB6 does not itself protect cells from polyQ toxicity, which suggests toxic polyQ aggregates are too small to be detected by our methods and the CTD is needed to direct them to the segregation process. Alternatively, aggregates within the deposit remain toxic and the CTD somehow suppresses this property. These findings raise questions of whether sequestration of other harmful misfolded proteins by other PQC processes is itself providing the protection or if it can occur separately from detoxification. It is possible that in some other instances the collecting of misfolded proteins to segregated deposition sites is a secondary process that consolidates and segregates debris that is not so harmful or has already been detoxified.
The way DnaJB6 protects mammalian cells from polyQ toxicity relies on similar DnaJB6 functions but differs somewhat from what we observe here. The CTD region as we define it here is dispensable for protecting mammalian cells, but we find it was necessary for protecting yeast. Although the CTD is not needed to link DnaJB6-bound polyQ to the segregation machinery, its role in blocking toxicity of polyQ aggregates is so far unknown. The CTD might recruit other PQC factors or be needed for DnaJB6 dimerization (14), which could influence functional consequences of DnaJB6 binding to polyQ. Additionally, formation of variably sized insoluble aggregates in mammalian cells is associated with toxicity, and DnaJB6 markedly reduces the accumulation of aggregates, resulting instead in a diffuse polyQ phenotype (7). DnaJB6 seems to prevent initial assembly of polyQ into the aggregates that would be recognized by mammalian PQC systems as targets to be sequestered. Thus, it likely protects mammalian cells by binding oligomers at early stages of assembly that could be toxic or by preventing them from subsequently producing or developing into more toxic species of aggregates.
Because of its potent antiamyloid activity, DnaJB6 is considered to have realistic potential in therapeutic approaches to amyloid diseases. Our work supports a view that direct neutralization of amyloid toxicity by DnaJB6 could provide a relatively straightforward approach.
MATERIALS AND METHODS
Yeast strains and growth conditions.
Yeast strains used are listed in Table 2. All are isogenic to NK101, which propagates prions [PSI+] and [PIN+]. [PSI+] is a prion of Sup35, and [PIN+] (also known as [RNQ1+]) is a prion of Rnq1 (46, 47). Deletion mutants were generated by standard methods (48). To obtain strain NK104 lacking prions, cells were grown in YPAD (1% yeast extract, 2% peptone, 2% dextrose, and 400 mg/liter adenine) containing 3 mM guanidine for 16 to 24 h and then spread onto 1/2 YPD (0.5% yeast extract, 2% peptone, 2% dextrose). [PSI+]/[psi−] status was then assessed by white/red colony color (8) and [PIN+]/[pin−] status by transforming cells with a plasmid encoding Rnq1-GFP and monitoring GFP aggregation by fluorescence microscopy.
TABLE 2.
Yeast strains used in this studya
| Strain | Genotype |
|---|---|
| NK101 | MATa kar1-1 ade2-1 SUQ5 his3Δ202 leu2Δ1 trp1Δ63 ura3-52 [PSI+] [PIN+] |
| NK104 | NK101 [psi−] [pin−] |
| 1611 | NK101 MATα sti1::HIS3 |
| 1673 | NK101 hsp42::KanMX4 |
| 1778 | NK101 bmh1::KanMX4 |
| 1780 | NK101 btn2::TRP1 |
| 1782 | NK101 cur1::KanMX4 |
| 1784 | NK101 btn2::TRP1 cur1::KanMX4 |
| 1341 | NK101 atg5::KanMX4 |
All strains are isogenic to NK101, a confirmed [PSI+] [PIN+] version of strain 779-6A (45).
Minimal media were used for plasmid selection. Synthetic dextrose (SD) medium contains 2% dextrose and 0.7% yeast nitrogen base supplemented with required amino acids. SRaf is similar but has 2% raffinose in place of dextrose. SGal is SRaf plus 2% galactose. Solid media are the same but contain 2% agar (Difco). Cells were grown at 30°C unless indicated otherwise. From a 4 mM stock solution in dimethyl sulfoxide (DMSO), latrunculin A was used at a final concentration of 200 μM.
Plasmids.
Plasmids used are listed in Table 3. Most yeast expression plasmids are derived from pRU4, a centromeric LEU2-marked plasmid with the GAL1 promoter and CYC1 terminator. DnaJB6 and DnaJB7 cDNA were gifts from H. Kampinga. They were subcloned as described previously (8). Truncated gene JB6-ST, JB6-STC, ST-RFP, and STC-RFP plasmids were synthesized by Genewiz, Inc. Hybrid DnaJB6/JB7 plasmids were made by using QuikChange lightning site-directed mutagenesis kit (Agilent) and an overlapping PCR approach and then cloned into plasmid pRU4.
TABLE 3.
Plasmids used in this study
| Name | Description | Promoter | Marker | Source or reference |
|---|---|---|---|---|
| pYES2-Q103GFP | Q103-GFP | GAL1 | URA3 | 24 |
| pRU14 | DnaJB6b | GAL1 | LEU2 | 8 |
| pJE201 | DnaJB6b | GAL1 | TRP1 | This study |
| pJE202 | DnaJB6b | GAL1 | HIS3 | This study |
| pMR360 | DnaJB6-RFP | GAL1 | LEU2 | 8 |
| pNK104 | JB6-ST only | GAL1 | LEU2 | This study |
| pNK105 | JB6-STC only | GAL1 | LEU2 | This study |
| pNK106 | ST-RFP | GAL1 | LEU2 | This study |
| pNK107 | STC-RFP | GAL1 | LEU2 | This study |
| pRU29 | DnaJB6ΔST | GAL1 | LEU2 | This study |
| pJE195 | DnaJB6ΔCTD | GAL1 | LEU2 | This study |
| pRU22 | DnaJB6-D33N | GAL1 | LEU2 | This study |
| pJE204 | DnaJB6D33N-RFP | GAL1 | LEU2 | This study |
| pDK58 | Btn2-RFP | GAL1 | LEU2 | 49 |
| pHsp42-Cherry | Hsp42-Cherry | GAL1 | LEU2 | 29 |
| pJB6-7J | JB6 + JB7 J-domain | GAL1 | LEU2 | This study |
| pJB6-7C | JB6 + JB7 CTDa | GAL1 | LEU2 | This study |
| pJB7 | DnaJB7a | GAL1 | LEU2 | This study |
| pJB7-6J | JB7a + JB6 J-domain | GAL1 | LEU2 | This study |
| pJB7-6C | JB7 + JB6 CTD | GAL1 | LEU2 | This study |
| pRS315RNQ1-GFP | Rnq1-GFP | RNQ1 | LEU2 | 50 |
| pCUP1-DuDre-Atg8-404 | RFP-Atg8 | CUP1 | TRP1 | 51 |
CTD of DnaJB7 in these constructs lacks residues 227 to 234 and 245 to 309 (Fig. 6A).
HttQ103-GFP toxicity assay.
Transformants of strain NK101 carrying plasmids for expressing HttQ103-GFP and DnaJB6b under the control of the GAL1 promoter or control vectors were grown overnight first in SD and then in SRaf. Cultures were diluted to an optical density at 600 nm (OD600) of 0.2, and 5-μl aliquots of 5-fold serial dilutions were dropped onto noninducing (SRaf) and inducing (SGal) plates, which were incubated at 30°C for 3 days and then scanned.
Fluorescence microscopy.
Transformants grown in SD overnight were diluted in SRaf and grown overnight. Cells were then subcultured in SGal liquid medium for 4 h. For monitoring RFP-Atg8, copper sulfate was added to 100 μM. Images were captured with a Nikon Eclipse Ni microscope equipped with a Q-Imaging Retiga Exi digital camera, Plan APO VC 60×/100× oil immersion differential interference contrast (DIC) optics, and GFP filter.
Confocal microscopy.
Confocal images were taken on a Nikon Eclipse TE-2000U spinning disk confocal microscope with a 100×/1.4 numerical aperture objective and a C9100-13 electron-multiplying charge-coupled device camera by Hamamatsu. HttQ103-GFP was imaged with a 491-nm laser at 40% intensity, and RFP-tagged plasmids were imaged with a 561-nm laser. 4′,6-Diamidino-2-phenylindole (DAPI) and 7-amino-4-chloromethylcoumarin (CMAC) were imaged using a DAPI filter. z stacks were taken at 0.2-μm intervals. Images were analyzed using Imaris software 8.4.1 and prepared for display using Adobe Photoshop. Briefly, images were opened in Surpass view of Imaris and intensity adjusted while maintaining a gamma value of 1.0. Single cells were cropped using the Crop 3D option.
In all cultures, we observed broad variability among individual cells with regard to expression of the different proteins. Many cells expressed neither or only one of the two expected proteins. Examples of representative fields of view and how the data were processed for presentation are shown as images in the supplemental material. Data were quantified as indicated in the text or figure legends.
DAPI staining.
Log-phase cells (OD600 of 0.4 to 0.6) growing in SRaf were transferred to SGal for 4 h and then fixed with 4% formaldehyde (28906; ThermoFisher) at 23°C for 1 h. Fixation was quenched by using 2.5 M glycine, and then cells were washed with 1× phosphate buffer saline (PBS), suspended in 1× PBS, mixed with DAPI (1 mg/ml stock), and incubated on a roller drum at 23°C for 1 h in the dark. Cells were harvested at 3,000 rpm, washed thrice with 1× PBS, and mounted in Vectashield (Vector Laboratories) on slides with 3% agarose pads.
Vacuole staining.
Vacuoles were stained with CellTracker blue CMAC (Y-7531; ThermoFisher), which selectively stains the yeast vacuole lumen. One ml of cells (106 cells/ml) was collected and suspended in 10 mM HEPES buffer, pH 7.4, containing 5% glucose. CMAC was added to a final concentration of 100 μM for 25 min at room temperature in the dark. Cells were harvested, washed, and suspended in 1× PBS.
ACKNOWLEDGMENTS
We thank our NIH and NIDDK colleagues for helpful discussions, Ruchika Sharma for plasmids, and Mike Reidy for valuable technical and intellectual support. Plasmid pCUP1-DuDre-Atg8-404 was a gift from Zhiping Xie (69201; Addgene).
This work was supported by the Intramural Program of the National Institutes of Health, National Institute of Diabetes, and Digestive and Kidney Diseases.
N.L.K. initiated the project and, with J.K. and D.C.M., conceived ideas for experimental approaches, analyzed data, and interpreted results. D.C.M. constructed strains and some plasmids; J.K. and N.L.K. conducted experiments and wrote the paper with D.C.M.
We have no conflicts of interest to declare.
REFERENCES
- 1.Knowles TP, Vendruscolo M, Dobson CM. 2014. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 15:384–396. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- 2.Marshall KE, Marchante R, Xue WF, Serpell LC. 2014. The relationship between amyloid structure and cytotoxicity. Prion 8:28860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verma M, Vats A, Taneja V. 2015. Toxic species in amyloid disorders: oligomers or mature fibrils. Ann Indian Acad Neurol 18:138–145. doi: 10.4103/0972-2327.144284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sengupta U, Nilson AN, Kayed R. 2016. The role of amyloid-beta oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 6:42–49. doi: 10.1016/j.ebiom.2016.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen SI, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM, Knowles TP. 2013. Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci U S A 110:9758–9763. doi: 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakahira H, Breuer P, Hayer-Hartl MK, Hartl FU. 2002. Molecular chaperones as modulators of polyglutamine protein aggregation and toxicity. Proc Natl Acad Sci U S A 99(Suppl 4):S16412–S16418. doi: 10.1073/pnas.182426899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hageman J, Rujano MA, van Waarde MA, Kakkar V, Dirks RP, Govorukhina N, Oosterveld-Hut HM, Lubsen NH, Kampinga HH. 2010. A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol Cell 37:355–369. doi: 10.1016/j.molcel.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Reidy M, Sharma R, Roberts BL, Masison DC. 2016. Human J-protein DnaJB6b cures a subset of Saccharomyces cerevisiae prions and selectively blocks assembly of structurally related amyloids. J Biol Chem 291:4035–4047. doi: 10.1074/jbc.M115.700393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolfe KJ, Ren HY, Trepte P, Cyr DM. 2013. The Hsp70/90 cochaperone, Sti1, suppresses proteotoxicity by regulating spatial quality control of amyloid-like proteins. Mol Biol Cell 24:3588–3602. doi: 10.1091/mbc.e13-06-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park SH, Kukushkin Y, Gupta R, Chen T, Konagai A, Hipp MS, Hayer-Hartl M, Hartl FU. 2013. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell 154:134–145. doi: 10.1016/j.cell.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 11.Park SK, Hong JY, Arslan F, Kanneganti V, Patel B, Tietsort A, Tank EMH, Li X, Barmada SJ, Liebman SW. 2017. Overexpression of the essential Sis1 chaperone reduces TDP-43 effects on toxicity and proteolysis. PLoS Genet 13:e1006805. doi: 10.1371/journal.pgen.1006805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park SK, Arslan F, Kanneganti V, Barmada SJ, Purushothaman P, Verma SC, Liebman SW. 2018. Overexpression of a conserved HSP40 chaperone reduces toxicity of several neurodegenerative disease proteins. Prion 12:16–22. doi: 10.1080/19336896.2017.1423185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kakkar V, Mansson C, de Mattos EP, Bergink S, van der Zwaag M, van Waarde MA, Kloosterhuis NJ, Melki R, van Cruchten RT, Al-Karadaghi S, Arosio P, Dobson CM, Knowles TP, Bates GP, van Deursen JM, Linse S, van de Sluis B, Emanuelsson C, Kampinga HH. 2016. The S/T-rich motif in the DNAJB6 chaperone delays polyglutamine aggregation and the onset of disease in a mouse model. Mol Cell 62:272–283. doi: 10.1016/j.molcel.2016.03.017. [DOI] [PubMed] [Google Scholar]
- 14.Soderberg CAG, Mansson C, Bernfur K, Rutsdottir G, Harmark J, Rajan S, Al-Karadaghi S, Rasmussen M, Hojrup P, Hebert H, Emanuelsson C. 2018. Structural modelling of the DNAJB6 oligomeric chaperone shows a peptide-binding cleft lined with conserved S/T-residues at the dimer interface. Sci Rep 8:5199. doi: 10.1038/s41598-018-23035-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mansson C, Arosio P, Hussein R, Kampinga HH, Hashem RM, Boelens WC, Dobson CM, Knowles TP, Linse S, Emanuelsson C. 2014. Interaction of the molecular chaperone DNAJB6 with growing amyloid-beta 42 (Abeta42) aggregates leads to sub-stoichiometric inhibition of amyloid formation. J Biol Chem 289:31066–31076. doi: 10.1074/jbc.M114.595124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mansson C, Kakkar V, Monsellier E, Sourigues Y, Harmark J, Kampinga HH, Melki R, Emanuelsson C. 2014. DNAJB6 is a peptide-binding chaperone which can suppress amyloid fibrillation of polyglutamine peptides at substoichiometric molar ratios. Cell Stress Chaperones 19:227–239. doi: 10.1007/s12192-013-0448-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaganovich D, Kopito R, Frydman J. 2008. Misfolded proteins partition between two distinct quality control compartments. Nature 454:1088–1095. doi: 10.1038/nature07195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kryndushkin DS, Shewmaker F, Wickner RB. 2008. Curing of the [URE3] prion by Btn2p, a Batten disease-related protein. EMBO J 27:2725–2735. doi: 10.1038/emboj.2008.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Specht S, Miller SB, Mogk A, Bukau B. 2011. Hsp42 is required for sequestration of protein aggregates into deposition sites in Saccharomyces cerevisiae. J Cell Biol 195:617–629. doi: 10.1083/jcb.201106037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller SB, Ho CT, Winkler J, Khokhrina M, Neuner A, Mohamed MY, Guilbride DL, Richter K, Lisby M, Schiebel E, Mogk A, Bukau B. 2015. Compartment-specific aggregases direct distinct nuclear and cytoplasmic aggregate deposition. EMBO J 34:778–797. doi: 10.15252/embj.201489524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sontag EM, Samant RS, Frydman J. 2017. Mechanisms and functions of spatial protein quality control. Annu Rev Biochem 86:97–122. doi: 10.1146/annurev-biochem-060815-014616. [DOI] [PubMed] [Google Scholar]
- 22.Miller SB, Mogk A, Bukau B. 2015. Spatially organized aggregation of misfolded proteins as cellular stress defense strategy. J Mol Biol 427:1564–1574. doi: 10.1016/j.jmb.2015.02.006. [DOI] [PubMed] [Google Scholar]
- 23.Duennwald ML, Jagadish S, Muchowski PJ, Lindquist S. 2006. Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc Natl Acad Sci U S A 103:11045–11050. doi: 10.1073/pnas.0604547103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Meriin AB, Zaarur N, Romanova NV, Chernoff YO, Costello CE, Sherman MY. 2009. Abnormal proteins can form aggresome in yeast: aggresome-targeting signals and components of the machinery. FASEB J 23:451–463. doi: 10.1096/fj.08-117614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meriin AB, Zhang X, He X, Newnam GP, Chernoff YO, Sherman MY. 2002. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J Cell Biol 157:997–1004. doi: 10.1083/jcb.200112104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malinovska L, Kroschwald S, Munder MC, Richter D, Alberti S. 2012. Molecular chaperones and stress-inducible protein-sorting factors coordinate the spatiotemporal distribution of protein aggregates. Mol Biol Cell 23:3041–3056. doi: 10.1091/mbc.e12-03-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Escusa-Toret S, Vonk WI, Frydman J. 2013. Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Nat Cell Biol 15:1231–1243. doi: 10.1038/ncb2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wickner RB, Bezsonov E, Bateman DA. 2014. Normal levels of the antiprion proteins Btn2 and Cur1 cure most newly formed [URE3] prion variants. Proc Natl Acad Sci U S A 111:E2711–E2720. doi: 10.1073/pnas.1409582111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao X, Lanz J, Steinberg D, Pease T, Ahearn JM, Bezsonov EE, Staguhn E, Eisenberg E, Masison DC, Greene LE. 2018. Real-time imaging of yeast cells reveals several distinct mechanisms of curing of the [URE3] prion. J Biol Chem 293:3104–3117. doi: 10.1074/jbc.M117.809079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar R, Nawroth PP, Tyedmers J. 2016. Prion aggregates are recruited to the insoluble protein deposit (IPOD) via myosin 2-based vesicular transport. PLoS Genet 12:e1006324. doi: 10.1371/journal.pgen.1006324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gillis J, Schipper-Krom S, Juenemann K, Gruber A, Coolen S, van den Nieuwendijk R, van Veen H, Overkleeft H, Goedhart J, Kampinga HH, Reits EA. 2013. The DNAJB6 and DNAJB8 protein chaperones prevent intracellular aggregation of polyglutamine peptides. J Biol Chem 288:17225–17237. doi: 10.1074/jbc.M112.421685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harms MB, Sommerville RB, Allred P, Bell S, Ma D, Cooper P, Lopate G, Pestronk A, Weihl CC, Baloh RH. 2012. Exome sequencing reveals DNAJB6 mutations in dominantly-inherited myopathy. Ann Neurol 71:407–416. doi: 10.1002/ana.22683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarparanta J, Jonson PH, Golzio C, Sandell S, Luque H, Screen M, McDonald K, Stajich JM, Mahjneh I, Vihola A, Raheem O, Penttila S, Lehtinen S, Huovinen S, Palmio J, Tasca G, Ricci E, Hackman P, Hauser M, Katsanis N, Udd B. 2012. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat Genet 44:450–455. doi: 10.1038/ng.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruggieri A, Brancati F, Zanotti S, Maggi L, Pasanisi MB, Saredi S, Terracciano C, Antozzi C, MR DA, Sangiuolo F, Novelli G, Marshall CR, Scherer SW, Morandi L, Federici L, Massa R, Mora M, Minassian BA. 2015. Complete loss of the DNAJB6 G/F domain and novel missense mutations cause distal-onset DNAJB6 myopathy. Acta Neuropathol Commun 3:44. doi: 10.1186/s40478-015-0224-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCampbell A, Taylor JP, Taye AA, Robitschek J, Li M, Walcott J, Merry D, Chai Y, Paulson H, Sobue G, Fischbeck KH. 2000. CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet 9:2197–2202. doi: 10.1093/hmg/9.14.2197. [DOI] [PubMed] [Google Scholar]
- 36.Zhao X, Park YN, Todor H, Moomau C, Masison D, Eisenberg E, Greene LE. 2012. Sequestration of Sup35 by aggregates of huntingtin fragments causes toxicity of [PSI+] yeast. J Biol Chem 287:23346–23355. doi: 10.1074/jbc.M111.287748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim YE, Hosp F, Frottin F, Ge H, Mann M, Hayer-Hartl M, Hartl FU. 2016. Soluble oligomers of PolyQ-expanded Huntingtin target a multiplicity of key cellular factors. Mol Cell 63:951–964. doi: 10.1016/j.molcel.2016.07.022. [DOI] [PubMed] [Google Scholar]
- 38.Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM. 1999. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet 23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- 39.Muchowski PJ, Schaffar G, Sittler A, Wanker EE, Hayer-Hartl MK, Hartl FU. 2000. Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc Natl Acad Sci U S A 97:7841–7846. doi: 10.1073/pnas.140202897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Douglas PM, Treusch S, Ren HY, Halfmann R, Duennwald ML, Lindquist S, Cyr DM. 2008. Chaperone-dependent amyloid assembly protects cells from prion toxicity. Proc Natl Acad Sci U S A 105:7206–7211. doi: 10.1073/pnas.0802593105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yan W, Craig EA. 1999. The glycine-phenylalanine-rich region determines the specificity of the yeast Hsp40 Sis1. Mol Cell Biol 19:7751–7758. doi: 10.1128/MCB.19.11.7751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu HY, Ziegelhoffer T, Osipiuk J, Ciesielski SJ, Baranowski M, Zhou M, Joachimiak A, Craig EA. 2015. Roles of intramolecular and intermolecular interactions in functional regulation of the Hsp70 J-protein co-chaperone Sis1. J Mol Biol 427:1632–1643. doi: 10.1016/j.jmb.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Craig EA, Huang P, Aron R, Andrew A. 2006. The diverse roles of J-proteins, the obligate Hsp70 co-chaperone. Rev Physiol Biochem Pharmacol 156:1–21. [DOI] [PubMed] [Google Scholar]
- 44.Chafekar SM, Wisen S, Thompson AD, Echeverria A, Walter GM, Evans CG, Makley LN, Gestwicki JE, Duennwald ML. 2012. Pharmacological tuning of heat shock protein 70 modulates polyglutamine toxicity and aggregation. ACS Chem Biol 7:1556–1564. doi: 10.1021/cb300166p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jung G, Jones G, Wegrzyn RD, Masison DC. 2000. A role for cytosolic hsp70 in yeast [PSI(+)] prion propagation and [PSI(+)] as a cellular stress. Genetics 156:559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Derkatch IL, Bradley ME, Hong JY, Liebman SW. 2001. Prions affect the appearance of other prions: the story of [PIN(+)]. Cell 106:171–182. doi: 10.1016/S0092-8674(01)00427-5. [DOI] [PubMed] [Google Scholar]
- 47.Wickner RB. 2016. Yeast and fungal prions. Cold Spring Harb Perspect Biol 8:a023531. doi: 10.1101/cshperspect.a023531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sherman F. 2002. Getting started with yeast. Methods Enzymol 350:3–41. doi: 10.1016/S0076-6879(02)50954-X. [DOI] [PubMed] [Google Scholar]
- 49.Kryndushkin D, Ihrke G, Piermartiri TC, Shewmaker F. 2012. A yeast model of optineurin proteinopathy reveals a unique aggregation pattern associated with cellular toxicity. Mol Microbiol 86:1531–1547. doi: 10.1111/mmi.12075. [DOI] [PubMed] [Google Scholar]
- 50.Kirkland PA, Reidy M, Masison DC. 2011. Functions of yeast Hsp40 chaperone Sis1p dispensable for prion propagation but important for prion curing and protection from prion toxicity. Genetics 188:565–577. doi: 10.1534/genetics.111.129460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li D, Song JZ, Shan MH, Li SP, Liu W, Li H, Zhu J, Wang Y, Lin J, Xie Z. 2015. A fluorescent tool set for yeast Atg proteins. Autophagy 11:954–960. doi: 10.1080/15548627.2015.1040971. [DOI] [PMC free article] [PubMed] [Google Scholar]