

Abstract
Mortality rates in influenza appear to have been shaped by evolution. During the 1918 pandemic, mortality rates were lower in children compared with adults. This mortality difference occurs in a wide variety of infectious diseases. It has been replicated in mice and might be due to greater tolerance of infection, not greater resistance. Importantly, combination treatment with inexpensive and widely available generic drugs (e.g. statins and angiotensin receptor blockers) might change the damaging host response in adults to a more tolerant response in children. These drugs might work by modifying endothelial dysfunction, mitochondrial biogenesis and immunometabolism. Treating the host response might be the only practical way to reduce global mortality during the next influenza pandemic. It might also help reduce mortality due to seasonal influenza and other forms of acute critical illness. To realize these benefits, we need laboratory and clinical studies of host response treatment before and after puberty.
Keywords: pandemic influenza, immunomodulatory treatment, mortality in children and adults, global public health, generic drugs
INTRODUCTION
An evolutionary perspective in public health has been important in explaining associations between different human phenotypes and chronic diseases [1]. The same perspective might help us understand many forms of acute critical illness. It might also suggest better ways to manage critically ill patients.
Two recent studies of influenza virus infection and endotoxemia in mice have shown that survival is better before puberty than after puberty. These studies help explain the lower mortality in children compared with adults seen in the 1918 influenza pandemic and in many other types of acute illness. This difference is probably the heritage of human evolution. Understanding the scientific basis for this difference suggests an alternative way to respond to the next pandemic. Instead of relying on vaccination and antiviral treatment, we might be able to treat patients with inexpensive generic drugs that modify the host response to acute critical illness. Unlike pandemic vaccines and antiviral treatments, these drugs will be available in any country with a basic healthcare system. If laboratory and clinical research convincingly demonstrates this approach works, it would benefit people everywhere. This idea has been discussed several times in the past decade [2–9].
MORTALITY IN CHILDREN AND ADULTS WAS DIFFERENT DURING THE 1918 INFLUENZA PANDEMIC
The 1918 influenza pandemic is remembered because it killed as many as 50–100 million people worldwide: an estimated 2.5% or more of the global population [9]. Remarkably, the mortality rate was much higher in younger adults than it was in children, giving rise to the familiar W-shaped pandemic mortality curve (Fig. 1) [6, 10]. A similar mortality pattern has not been described for subsequent pandemics (1957, 1968 and 2009).
Figure 1.
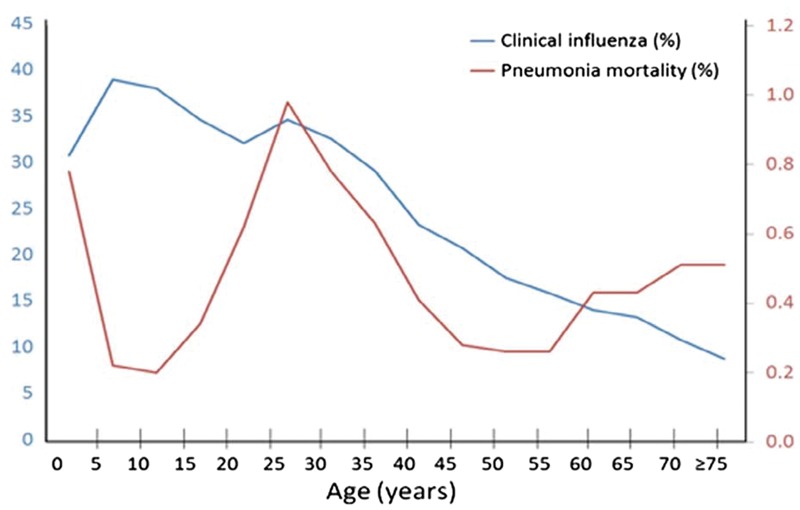
Clinical influenza illness (blue) and pneumonia mortality (red) during the 1918 influenza pandemic. From Ref. [6] with permission
Some investigators have attributed high pandemic mortality in young adults to secondary bacterial pneumonia [11]. This explanation is incomplete and unsatisfactory for several reasons. Children were infected with the 1918 virus more frequently than adults (Fig. 1) and they were almost certainly colonized with the same bacteria that were associated with bacterial pneumonia in adults, yet their pneumonia mortality rates were much lower [6, 12]. More important, lower mortality among children compared with adults was not unique to the 1918 pandemic. Children have lower mortality than adults due to infections caused by many bacteria (e.g. Group A Streptococcus, S. pneumoniae, S. aureus and M. tuberculosis) and viruses (e.g. mumps, varicella, poliomyelitis, Epstein Barr virus, hepatitis E, yellow fever, SARS and smallpox). Similar mortality differences have been seen in several other conditions (e.g. disseminated C. albicans infection, acute lung injury accompanying severe malaria, sickle cell chest syndrome, multi-organ failure following severe trauma, severe burn injury and febrile neutropenia) [6, 10]. In short, lower mortality rates among children were not unique to the 1918 pandemic.
Most influenza scientists have sought to explain the 1918 mortality difference by studying the virus. Numerous reports have shown that the virulence of individual influenza viruses differs markedly in laboratory models of infection. For example, influenza A (H3N2) viruses are generally more virulent than H1N1 viruses, and the 1918 virus (H1N1) was more virulent than the ordinary seasonal H1N1 influenza viruses seen today. Antigenic changes in human influenza viruses are frequently reported over the course of a single influenza season [13], and in 1918 the mortality impact of the second pandemic wave in the fall was much greater than it was during the first wave the preceding spring [10].
ORIGINAL ANTIGENIC SIN AND ANTIGENIC IMPRINTING
Several investigators have sought to explain the greater mortality among adults in 1918 by studying influenza disease in human populations. During the 1918 influenza pandemic, children 5–14 years of age who were living in remote, isolated communities were often the only ones to survive [14]. There is general agreement that pandemic mortality peaked in young adults, but it was much lower in adults ≥45 years of age (Fig. 1). Their lower mortality is thought due to the protective effect of residual immunity following exposure to H1N1-like viruses that circulated before an H3N2-like virus (Russian influenza) appeared in the early 1890s [10]. Influenza virologists agree that antigenic priming following infection with these earlier H1N1viruses (known as ‘original antigenic sin’) provided some measure of long-lasting protection against the highly virulent H1N1 virus that emerged in 1918 [15]. Accordingly, individuals born after the early 1890s were exposed only to H3N2-like viruses and would have missed antigenic priming with pre-1890 H1N1-like viruses. As young adults in 1918, they were susceptible to infection with the new pandemic H1N1 virus.
Influenza A viruses are categorized into two groups. Group 1 includes H1, H2 and H5 subtypes, whereas Group 2 includes H3 and H7 subtypes (Figure 1A in Ref. [16]). This phylogenetic understanding has informed recent age-specific analyses (based on individual birth year) of mortality patterns before, during and after the 1918 pandemic [17–21]. Worobey et al. have used a host-specific molecular clock approach to demonstrate that high mortality in young adults may have been due to childhood exposure to a doubly heterosubtypic putative H3N8 virus that circulated from 1889 to 1900 [19]. They think that young children (but not infants) were protected by childhood exposure to a newly emerged (post-1900) H1variant or N1 antigens. Miller et al. interpret the historical data more cautiously [18, 20]. They agree with Worobey et al that early life antigenic imprinting might have led to a dysregulated T-cell response that increased the risk of death following infection in 1918 with a new and antigenically dissimilar influenza virus. However, they question whether a new H1 virus emerged in the early 1900s to replace the H3 virus that first appeared in 1890.
Worobey et al.have also analyzed cases and deaths due to H5N1 and H7N9 influenza. They have shown that HA imprinting was “the dominant explanatory factor for observed incidence and mortality patterns for both H5N1 and H7N9” [21]. For example, individuals born during the period when H1N1 viruses circulated (1918–1957) were protected against H5N1 infection (both are Group 1 viruses) but were at increased risk of H7N9 infection (a Group 2 virus) [21]. However, individuals born during the 1968–2015 period (the H3N2 era) were protected against H7N9 infection (both are Group 2 viruses), whereas those born before 1957 (the H1N1 era) were not.
The importance of antigenic imprinting was also shown for the 2009 H1N1 pandemic. Individuals born before 1957 were exposed to seasonal H1N1 viruses before the emergence of the new H2N2 pandemic virus in 1957. As older individuals, they experienced a lower incidence of pH1N1 disease in 2009 than those born after 1957 [22]. However, Miller et al. have argued that early life H2N2 virus infection may have actually increased the risk of death during the heterotypic 2009 H1N1 pandemic [23].
The importance of antigenic imprinting for human influenza is undeniable. ‘First flu’ may indeed be ‘forever’ [24], but whether antigenic imprinting is helpful or harmful for every individual is still an open question. A similar uncertainty has arisen about influenza vaccination. The doctrine of ‘original antigenic sin’ was developed following observations that influenza vaccination led to the anamnestic recall of antibodies to earlier influenza viruses [15]. Recent epidemiologic studies, however, suggest that repeated influenza vaccinations may lead to reduced vaccination effectiveness [15, 23]. Moreover, repeated infection of ferrets with H3N2 viruses affects both the quantity and quality of their antibody responses [25]. Thus, there appear to be two sides to the host response following repeated influenza virus infections and vaccination. Moreover, these observations leave open the larger question of whether antigenic interaction among influenza viruses is the only determinant of how an individual will respond to influenza virus infection.
MOUSE MODELS OF INFLUENZA AND ENDOTOXEMIA DEMONSTRATE DIFFERENT MORTALITY BEFORE AND AFTER PUBERTY
Writing about the 1918 pandemic, Ahmed et al. observed that “children were not protected from infection, but, for reasons that are as mysterious today as they were in 1918, they were able to cope with the disease much better than their adult counterparts” [10]. They added, “… this change in disease susceptibility occurs around the time of puberty, and it is possible that sex-associated hormones are involved in this transition” [10].
In trying to understand the ‘mystery’ of greater mortality among young adults during the 1918 pandemic, scientists have studied influenza viruses and the human response to previous infection. In essence, they have asked ‘why did young adults die?’ They could also have asked ‘why did children live?’ [6, 9, 12].
Influenza scientists have never created an experimental model of the mortality experience seen in the 1918. Recently, however, Suber and Kobzik experimentally replicated the different susceptibility of children and adults to influenza-related mortality [26]. They infected groups of C57BL/6 mice with influenza A(H1N1) (PR8) virus. Male and female mice were infected on either postnatal day 25 (P25, prepubertal) or postnatal day 28 (P28, pubertal). (In C57BL/6 mice, puberty usually starts on postnatal day P27 or P28.) Mortality was much greater in pubertal (P28) than in prepubertal (P25) mice (Fig. 2a and b). Deaths began to occur 9 days following infection. By this time, pulmonary virus titers had fallen to levels much lower than they were on days 3 and 6, and they were similar in prepubertal and pubertal mice (Fig. 2c).
Figure 2.
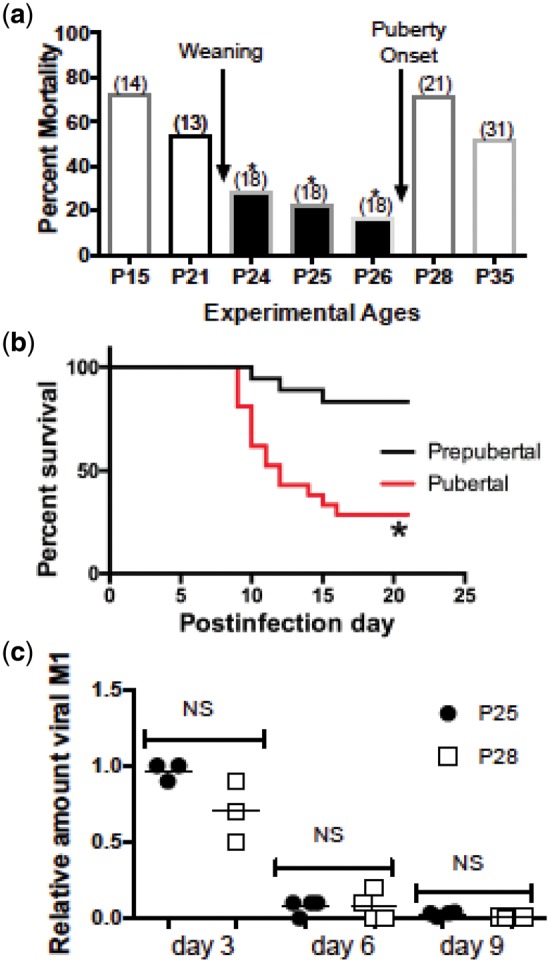
Prepubertal mice infected with influenza virus experience lower mortality compared with pubertal mice. Mice were inoculated intranasally with PR8 H1N1 influenza virus (1 HAU). (a) Prepubertal mice (postnatal days P24 and P25) experienced lower mortality compared with pubertal mice (P28). The number of mice per group is shown in parentheses. Mice were monitored for 21 days after infection. Survival studies were compared by the Mantel–Cox log-rank test; P ≤ 0.01 for P24, P25 and P26 mice versus P28 mice compared individually and for the pool of P24–P26 mice compared with either P28 or P35 mice. (b) Summary of three trials showing survival of groups (n = 25 each) of P26 (prepubertal) compared with P28 (pubertal) mice; *P = 0.006. (c) Virus load in the lungs of P25 (prepubertal) and P28 (pubertal) mice; qPCR for M1 mRNA on postinfection days 3, 6 and 9. Results are shown for duplicate samples from at least four mice per time point per group in a single trial. Differences between P25 and P28 mice were not significant by the Kruskal–Wallis with Dunn’s multiple comparisons test. From Ref. [26] with permission
High-dose, exogenous estrogen treatment is known to protect adult female mice infected with influenza virus [27]. To determine the role of sex hormones in the prepubertal/pubertal mortality difference, Suber and Kobzik castrated male and female mice on day P21 and infected them on day P28 [26]. Over the next 3 weeks, mortality was reduced in castrated but not in sham-operated mice. They then blocked the onset of puberty in P28-infected female mice by pretreating them (starting on day P21) with leuprolide, a gonadotropin-releasing hormone (GnRH) analog. Leuprolide desensitizes GnRH receptors and decreases the secretion of gonadotropins, and this blocks the normal pubertal increase in estrogen. They also pretreated male and female P28-infected mice with acyline, a GnRH antagonist. Both leuprolide and acyline pretreatment improved survival [26]. In addition, when P21-ovariectomized mice were infected on day P28, estrogen treatment given 0–2 or 6–8 days following infection abrogated the protective effect of ovariectomy. Protection was also reversed in castrated males by treatment with both estrogen and testosterone (normally, testosterone is converted to estrogen by aromatase).
Transcriptome profiling over the course of infection showed marked enrichment of estrogen, β-estradiol and estrogen receptor 1 [26]. For this reason, estrogen receptor blockade was carried out using fulvestrant. Pretreating P28-infected male and female mice reduced subsequent mortality, and when fulvestrant was given to females 3 days following infection, survival was greatly improved. In addition, in older postpubertal male and female mice infected on day P42, survival also improved when fulvestrant treatment was started 3 days following infection [26].
Transcriptome analysis also showed increased expression of IL-1β in the lungs and blood leukocytes of pubertal (P28) mice 9 days after infection [26]. When pubertal mice were infected on day P28 and then treated with anti-IL-1β blocking antibody 5 and 9 days later, survival was greatly improved. Early treatment on the day of infection, however, was not beneficial, suggesting that IL-1β activity was expressed differently at different stages of disease [26].
As discussed above mortality rates in many infectious diseases are lower in children than they are in adults [6, 10], For this reason, Joachim and Kobzik studied mice with endotoxin (LPS)-induced sepsis before and after puberty [28]. The conditions for these experiments differed slightly from those in the influenza experiments [26]; all mice were female and postpubertal mice were given LPS on postnatal days 33–35, not earlier. Prepubertal mice injected intraperitoneally with LPS on day P24–26 had significantly better survival than postpubertal mice 20 h after injection (Fig. 3a), although endotoxin levels in the blood were similar in both groups (Fig. 3b) [28]. As in the influenza experiments, survival in prepubertal mice improved when the onset of puberty was delayed by pretreatment with estrogen (3 days before and on the day of LPS injection). Similarly, in postpubertal mice injected with LPS, blocking the onset of puberty by pretreatment with leuprolide (administered daily from prepubertal day P24 to postpubertal day P35) substantially improved survival. In addition, fulvestrant was administered to pre- and postpubertal mice to determine whether the increase in LPS-induced mortality was specifically due to the onset of puberty or the lack of estrogen activity, but the results were indeterminate. Finally, in LPS-treated postpubertal mice, adoptive transfer of peritoneal cells (macrophages and B and T cells) harvested from LPS-naive pre- and postpubertal mice had different effects on survival: mice that received cells from prepubertal mice had significantly lower mortality than those treated with postpubertal cells (Fig. 3c) [28].
Figure 3.

Prepubertal mice with endotoxin (LPS)-induced sepsis experience lower mortality compared with postpubertal mice. Female mice were inoculated intraperitoneally with a dose of E. coli LPS known to cause 80–90% mortality in control mice. (a) Prepubertal C57BL/6 mice (postnatal days P24–26) experienced lower mortality compared with postpubertal mice (postnatal days P33–35; N ≥ 56 in each group). Typical experiments lasted 72 h. (b) Endotoxin levels (EU/ml) at 20 h were similar in pre- and postpubertal mice. (c) Naïve peritoneal cells were collected from pre- and postpubertal mice by peritoneal lavage and administered to recipient postpubertal mice by intraperitoneal injection. Following incubation of donor cells for 30–60 min, mice were injected intraperitoneally with LPS (Salmonella enterica) and followed for 5 days. Mortality in mice injected with prepubertal peritoneal cells was significantly lower than it was in control mice or those that received postpubertal peritoneal cells. From Ref. [28] with permission
Investigators have presented many arguments for and against the use of murine models to explain aspects of acute critical illness in humans [29, 30]. For comparative studies of puberty, mice present special problems; humans live much longer than mice and human puberty extends over several years, not a day or two as in mice. Nonetheless, murine studies before and after puberty can be of considerable value. For example, estrogens have an important role in puberty and they can also modify the response to acute critical illness. Kobzik et al. have shown that unlike other studies, estrogen treatment reversed protection against influenza mortality in prepubertal and castrated mice [26]. These apparently contradictory findings might reflect the known pro-inflammatory activities of low-dose estrogens and their anti-inflammatory effects when higher doses are used [26, 27].
Suber and Kobzik have shown that the different influenza mortality rates in pre- and postpubertal mice have nothing to do with previous exposure to influenza viruses and/or control of virus replication. This observation is critically important. Although antigenic imprinting clearly influences human outcomes in both pandemic and seasonal influenza [15, 18–20, 22, 23], differences in antigenic imprinting might not be the only explanation for better childhood survival during the 1918 pandemic.
In attempting to understand the ‘mystery’ of better survival among children compared with adults during the 1918 pandemic, investigators have focused exclusively on infection with influenza viruses and ignored the better survival of children with other infectious diseases and noninfectious critical illnesses [6, 10]. The better survival of prepubertal mice following endotoxin treatment shown by Kobzik et al. suggests that this is a general phenomenon. Long-lasting, age-specific antigenic imprinting seen with influenza is not known to occur with most other forms of acute critical illness [6, 10]. Instead, changes in the host response to critical illness associated with increased mortality appear to begin with the onset of puberty. It follows that factors associated with prepuberty might somehow contribute to the better survival of children compared with adults.
THE HOST RESPONSE TO INFLUENZA
Infection with influenza viruses initially targets respiratory epithelial cells [31, 32]. In response, myeloid and lymphoid cells mount a brisk pro-inflammatory response, often called a ‘cytokine storm’. Patients who develop severe illness are unable to control what becomes a systemically dysregulated immune response. After several days (usually a week or more), they develop evidence of immunosuppression [6, 32]. Death occurs in those who are unable to resolve their illness and restore homeostasis.
The pathogenesis of acute lung injury, including severe influenza, involves (among other things) mitochondrial dysfunction [33], oxidative stress [34, 35], endothelial dysfunction [36, 37] and molecular mechanisms (e.g. specific lipid mediators) that initiate the resolution of pulmonary and systemic injury and the restoration of homeostasis [38, 39]. The evolutionarily conserved process of autophagy is central to the host response [40]; it contributes to both influenza virus replication [41] and the evolution of influenza-related lung injury [42, 43].
The host response to infection may involve mechanisms that enhance resistance (which reduces pathogen burden) or tolerance (which reduces the impact of infection) [44, 45]. Both resistance and tolerance are driven by a multiplicity of metabolic changes in immune and other host cells [46–49], some of which include estrogen signaling [50]. Some of these immunometabolic changes have been documented in mice with experimental influenza virus infection [51].
The molecular mechanisms that account for the difference in the mortality rates of children and adults with different forms of acute critical illness (seen in humans and now replicated in mice) are largely unknown. Working together, the endocrine, nervous and immune systems integrate and regulate the availability of energy. Evolutionary biologists have developed the theory of life history, which emphasizes trade-offs in how energy is allocated to storage, activity, maintenance, and the anabolic activities of growth and reproduction [52]. According to life history theory, the transition to puberty is accompanied by an overall switch in the allocation of energy from growth to reproduction. Nonetheless, although this theory has given us a better understanding of changes in energy metabolism that occur over extended periods of time, it has yet to explain the sudden and intense increase in energy expenditure that accompanies the host response to acute critical illness or whether tradeoffs in energy allocation in critical illness are different before and after puberty.
GENETICS, EPIGENETICS, AND THE INDIVIDUAL HOST RESPONSE
In outbreaks of seasonal and pandemic influenza, only a small number of individuals who are infected develop severe or fatal illness. This was true during the 1918 pandemic; although approximately one-third of the human population was infected, only a small proportion died. Some of this protection is due to CD8+ T-cell immunity, especially immunity directed against the evolutionarily conserved NP antigen [53]. This immunity reflects previous exposure of populations to influenza virus antigens, but importantly it encompasses both Group 1 and Group 2 viruses, unlike antigenic imprinting discussed above. T-cell immunity does not prevent the occurrence of infection, but it modifies the course of illness, reducing virus shedding and in some instances limiting or preventing the occurrence of symptoms [53]. Inborn genetic variants can also account for life-threatening infections [54], but the susceptibility of most individuals probably depends more on variations in host defense mechanisms that are expressed only after infection has occurred [53, 55].
The importance of these post-infection variations was demonstrated in 25 HA-seronegative healthy young adults who were experimentally infected with influenza H3N2 virus [56]. (A similar study was undertaken with H1N1 challenge infection [57].) Following infection, peripheral blood cytokine responses were determined every six hours for the next five days. In nine subjects who developed symptomatic illness, there were early increases in cytokines associated with fever, leucocyte recruitment and markers of innate antiviral immunity, and some of these increases appeared as early as two days before the onset of symptoms [56]. These cytokine findings were also demonstrated in a parallel genomic analysis. In contrast, the 17 subjects who remained asymptomatic showed early and persistent down regulation of the same inflammatory markers. Symptomatic subjects developed cytokine profiles similar to those that have been seen in patients who develop severe illness, while those who remained asymptomatic showed host responses indicating rapid control of infection. These findings suggested that the “inflammatory pathway an individual will follow is probably determined at (a) very early, even presymptomatic time” [56]. Which pathway is followed is probably determined (at least in part) by epigenetic factors [58–60].
EVOLUTION, ENERGY METABOLISM AND INFLUENZA OUTCOMES IN CHILDREN AND ADULTS
In a study of populations in Sweden and Japan during the early and late years of the 20th century, evolutionary biologists showed that the increase in the annual probability of death due to all causes was greatest during the second decade of life, not in later years (Fig. 4) [61]. Other studies have shown that stress differentially allocates energy resources between reproduction and immune function [62], and estrogens contribute to the energy trade-offs that help maintain homeostasis [52, 63]. At least some of the mechanisms responsible for maintaining homeostasis during puberty are epigenetically regulated [64].
Figure 4.
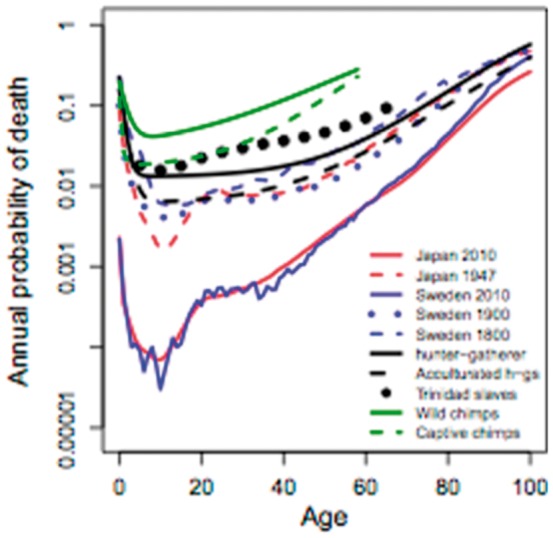
Annual probability of death for several human populations over time. The hunter gatherer curve approximates the typical human mortality profile over almost all of evolutionary time. The curves for Japan in 1947 and Japan and Sweden in 2010 demonstrate the steep rise in the modern all-cause mortality profile during the second decade of life. From reference 61, with permission
Numerous laboratory and clinical studies have described biological pathways that are associated with the susceptibility of neonates and the elderly to acute critical illnesses, but very few studies have compared host responses before and after puberty [65, 66]. One such study documented the responses of weanling and adult ferrets following infection with the 2009 H1N1 pandemic virus [65]. Compared with adult ferrets, weanlings developed much milder clinical illnesses and had less evidence of pulmonary damage, yet rapid virus clearance from the respiratory tract was seen in both groups. Like adults, the immune responses of weanlings to infection were robust, but they were different. Pro-inflammatory cytokine responses in the two groups were similar, but regulatory response genes for IL-10 and TGF-1 were more highly expressed in weanlings [65]. Because influenza in ferrets closely mimics the disease in humans, this study suggests that the milder response to influenza in children compared with adults is due to a more strongly expressed regulatory response.
Very few studies have directly compared the cell signaling responses of children and adults to acute critical illness. Nonetheless, in 2004, surgeons reported two studies that directly compared the inflammatory responses of peritoneal macrophages harvested from the sterile abdomens of children and adults [67, 68]. Pro- and anti-inflammatory responses were elicited by exposing the macrophages ex vivo to endotoxin and IL-1. Unlike adults, responses in children were dominated by an IL-10 anti-inflammatory pattern. Recent research has shown that DNA methylation stably reduces the expression of IL-10 in Th1 cells [69]. Short-term reversal of this epigenetic mechanism can bring about an increase in IL-10 gene expression.
TREATING THE HOST RESPONSE TO PANDEMIC INFLUENZA WITH INEXPENSIVE GENERIC DRUGS
A decade ago, surgeons who were involved in liver transplantation in children and adults sought to better understand inflammatory responses in their patients by studying hepatic ischemic-reperfusion injury in mice of different ages [70]. They found evidence of less inflammation but greater autophagy in the livers of younger (4–5-week old) mice compared with older (10–12-week old) mice, and the response of younger mice was associated with greater nuclear retention of PPARγ activity. Following pre-treatment (for three days) of older mice with the PPARγ agonist rosiglitazone, their highly inflammatory response was changed to the less inflammatory response seen in younger mice, and this change was associated with the autophagy pattern seen in younger mice. It is important to recognize that in this study younger mice were not clearly shown to be pre-pubertal and treatment was given before, not after the ischemic episode. Nonetheless, pre-treatment with rosiglitazone was able to “roll back” the damaging response of “adults” to the more benign response of “children”.
PPARγ agonists have important effects on energy metabolism, and there is considerable “crosstalk” among these agents (glitazones) and other drugs that also have immunometabolic activities (e.g., statins [71], ACE inhibitors and angiotensin receptor blockers [72], metformin [73]) [5–9]. These findings suggest that many if not all of these drugs could be used to change the host response of adults to that seen in children. The drugs would probably work better if given in combination rather than by themselves [8]. For a severe infection like pandemic influenza, treatment with these drugs could improve an adult's tolerance of infection [44–47] and might improve survival. The drugs might also have similar effects in children who develop life-threatening illness.
ALTERNATIVE RESPONSES TO THE NEXT INFLUENZA PANDEMIC
Ever since the emergence of highly virulent avian H5N1 influenza in 1997, virologists have warned of the possibility of a new and devastating influenza pandemic. In 2006, epidemiologists provided an estimate of what global mortality might be if the next pandemic is like the one in 1918 [74]. This estimate (51 to 81 million deaths) seems low because in 1918 pandemic mortality is thought to have been 50–100 million and today the global population is four times larger. A recent study from the Institute for Disease Modeling estimates that during the first six months of a 1918-like pandemic, almost 33 million people could die [75]. Moreover, if the next pandemic is caused by an H5N1-like virus, which has a high case fatality rate, its impact on global mortality could be much worse [3–6]. Influenza virologists are now concerned about the possibility of an H7N9 pandemic [9, 76]. Nonetheless, even in the absence of pandemics, yearly outbreaks of seasonal influenza cause appreciable mortality worldwide [77]. Most of these influenza-related deaths (pandemic and seasonal) occur in developing countries [74, 77].
Current strategies for national and global pandemic preparedness focus on influenza vaccination for populations and antiviral treatment for individuals [9]. Influenza virologists hope to develop universal influenza vaccines that will provide long-lasting protection, making it unnecessary to vaccinate against seasonal influenza each year [78]. Vaccination with a universal vaccine might conceivably provide protection against infection with a future pandemic virus. Recent virologic studies, however, raise important questions about whether this strategy will work [79, 80]. Moreover, much of the world will lack the human infrastructure to guarantee administration of a universal vaccine [9]. This means that for the foreseeable future, health officials responsible for pandemic preparedness will have to count on using conventional pandemic vaccines. Unfortunately, none of these vaccines will be available during the first six pandemic months [9]. Moreover, when they eventually become available, it is unlikely they will be equitably distributed to low- and middle-income countries that don't produce their own influenza vaccines [81].
Antiviral treatment of individual pandemic patients could also be problematic. Supplies of one of the drugs (oseltamivir) are limited and the drug is not widely used. There is also concern about the development of antiviral resistance. Moreover, a recent report on 1220 patients hospitalized in China with laboratory-confirmed H7N9 influenza showed that although 70% of all patients were treated with oseltamivir, case fatality rates were still 40% [82].
An alternative strategy for reducing pandemic mortality would be to develop effective treatments that target the host response of patients who develop severe illness [2–9]. These drugs might have some effect on the development of symptomatic illness [83], but their potential impact on pandemic mortality would be far more important. Agendas for laboratory and clinical research to evaluate their potential have been published in several articles (Table 6 in reference 8, Table 1 in reference 9, and more generally in references 2 and 4–6). Although influenza scientists often regard host response treatment as an adjunct to antivirals [84], some of this research must be limited to generic drugs that target the host response because most of the world's people won't have access to antivirals but will have access to generics. These studies must go beyond documenting cytokine responses following infection and examine immunometabolic and epigenetic factors that affect (among other things) cellular immunity, endothelial function and energy metabolism [8, 9]. All of these studies should include comparisons before and after puberty.
Treating the host response holds promise for not only reducing pandemic mortality but also for reducing the appreciable mortality associated with seasonal influenza and other forms of acute critical illness (e.g., Ebola virus disease [8]). Many of the candidate drugs are produced as generics in developing countries, and supply chains for their worldwide delivery are already in place [5–9]. Physicians are familiar with these drugs because they use them every day. If this treatment strategy were shown to work, it could be used in any country with a basic healthcare system. For a pandemic, treatment could start in all countries on the first pandemic day.
CONCLUSION: EVOLUTION SUGGESTS A PRACTICAL RESPONSE TO A GLOBAL INFLUENZA PANDEMIC
Influenza virologists have expanded our understanding of the molecular biology and epidemiology of influenza viruses. Laboratory and clinical investigators have deepened our understanding of the host response to critical illness. Evolutionary biologists have suggested that evolution provides insights that could help public health. All of these developments should shape the way we respond to the next influenza pandemic.
Charles Darwin wrote “… observation must be for or against some view if it is to be of any service” [85]. His view – the hypothesis that evolution is guided by natural selection – was supported by extensive observations made over several decades, and its explanatory power (i.e., ‘service’) has withstood challenge for almost 160 years. The idea (hypothesis) of treating the host response to pandemic influenza was introduced in 2004 [2]. Its potential explanatory power has also been supported by experimental and clinical observations, although treating the host response has received little attention from scientists and health officials [4–9]. Yet in a practical sense this approach to treatment could be of great service (in Darwin's word) if it could reduce global mortality during the next pandemic.
Kobzik and colleagues have shown age-specific differences in influenza mortality in mice before and after puberty that are not affected by previous infection (antigenic imprinting) with influenza viruses [26]. Considered with evidence from endotoxemic mice [28] and other studies [4–9], their findings suggest that the mortality impact of pandemic and seasonal influenza and other forms of acute critical illness might be reduced by treating the host response. By reducing the damage caused by influenza in adults to the more tolerant response seen in children, treatment could in effect “roll back” evolution.
Physicians will inevitably be called upon to manage seriously ill patients during the next pandemic, but there is a real risk they will relive the experiences of physicians 100 years ago [9, 86]. This is sure to occur if influenza scientists and public health officials continue to reject the possibility that host response treatment could reduce pandemic mortality [7–9, 87]. If we are to take seriously the challenge of preparing for the next pandemic, it is self-evident that a “top down” approach based on vaccination and antiviral treatment, driven by the decisions of elite scientists, health officials and corporate executives, will not meet the world's needs [2–9]. Instead, an effective response must include a “bottom up” approach to individual patient treatment by ordinary doctors working in ordinary healthcare systems who use ordinary, widely available and inexpensive generic drugs that modify the host response.
The studies reviewed here suggest that the biological basis for treating the host response reflects our evolutionary heritage. This idea might not be revolutionary [88], but its practical implications for health, equity and security during the next pandemic could be immense. Consequently, investigators must undertake laboratory and clinical research to convincingly show whether it will be effective. If it is effective and is put into practice, it would represent a striking application of the idea of evolutionary public health [1].
Conflict of interest: None declared.
REFERENCES
- 1. Wells JCK, Nesse RM, Sear R, Johnstone RA, Stearns SC.. Evolutionary public health: introducing the concept. Lancet 2017;390:500-9. doi: 10.1016/S0140-6736(17)30572-X. [DOI] [PubMed] [Google Scholar]
- 2. Fedson DS. Pandemic influenza: a potential role for statins in treatment and prophylaxis. Clin Infect Dis 2006;43:199-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fedson DS, Dunnill P.. Commentary: From scarcity to abundance: pandemic vaccines and other agents for “have not” countries. J Public Health Policy 2007;28:322-40. [DOI] [PubMed] [Google Scholar]
- 4. Fedson DS. Meeting the challenge of influenza pandemic preparedness in developing countries. Emerg Infect Dis 2009;15:365-71. doi: 10.3201/eid1503.080857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fedson DS. Confronting the next influenza pandemic with anti-inflammatory and immunomodulatory agents: why they are needed and how they might work. Influenza Other Respir Viruses 2009;3:129-42. doi: 10.1111/j.1750-2659.2009.00090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fedson DS. Treating influenza with statins and other immunomodulatory agents. Antiviral Res 2013;99:417-35. doi: 10.1016/j.antiviral.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 7. Fedson DS. How will physicians respond to the next influenza pandemic? Clin Infect Dis 2014;58:233-7. doi: 10.1093/cid/cit695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fedson DS. Treating the host response to emerging virus diseases: lessons learned from sepsis, pneumonia, influenza and Ebola. Ann Transl Med 2016;4:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fedson DS. Clinician-initiated research on treating the host response to pandemic influenza. Hum Vaccin Immunother 2018;14:790–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ahmed R, Oldstone MBA, Palese P.. Protective immunity and susceptibility to infectious diseases: lessons from the 1918 influenza pandemic. Nat Immunol 2007;8:1188-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morens DM, Taubenberger JK, Fauci AS.. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis 2008;198962-70. doi: 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fedson DS. Was bacterial pneumonia the predominant cause of death in the 1918-1919 influenza pandemic? J Infect Dis 2009;199:1408-9; author reply 1409-10. doi: 10.1086/597621. [DOI] [PubMed] [Google Scholar]
- 13. Nelson MI, Edelman L, Spiro DJ, et al. Molecular epidemiology of A/H3N2 and A/H1N1 influenza virus during a single epidemic season in the United States. PLoS Pathog 2008;4:e1000133. doi: 10.1371/journal.ppat.1000133. Erratum in PLoS Pathog 2008;4. doi.org//10.1371/annotation/1391941e-93d3-48d3-8c9a-b7c6d98f9527. Sengamalay, Naomi [added]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mamelund SE. Geography may explain adult mortality from the 1918-20 influenza pandemic. Epidemics 2011;3:46-60. doi: 10.1016/j.epidem.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 15. Monto AS, Malosh RE, Petrie JG, Martin ET.. The doctrine of original antigenic sin: separating good from evil. J Infect Dis 2017;215:1782-8. doi: 10.1093/infdis/jix173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Joyce MG, Wheatley AK, Thomas PV, et al. Vaccine-induced antibodies that neutralize group 1 and group 2 influenza A viruses. Cell 2016;166:609-23. doi: 10.1016/j.cell.2016.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Viboud C, Eisenstein J, Reid AH, Janczewski TA, Morens DM, Taubenberger JK, Age- and sex-specific mortality associated with the 1918-1919 influenza pandemic in Kentucky. J Infect Dis 2013;207:721-9. doi: 10.1093/infdis/jis745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gagnon A, Miller MS, Hallman SA, et al. Age-specific mortality during the 1918 influenza pandemic: unravelling the mystery of high young adult mortality. PLoS One 2013;8:e69586. doi: 10.1371/journal.pone.0069586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Worobey M, Han GZ, Rambaut A.. Genesis and pathogenesis of the 1918 pandemic H1N1 influenza A virus. Proc Natl Acad Sci USA 2014;111:8107-12. doi: 10.1073/pnas.1324197111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gagnon A, Madrenas J, Miller MS.. Is antigenic sin always "original?" Re-examining the evidence regarding circulation of a human H1 influenza virus immediately prior to the 1918 Spanish flu. PLoS Pathog 2015;11:e1004615. doi: 10.1371/journal.ppat.1004615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gostic KM, Ambrose M, Worobey M, Lloyd-Smith JO.. Potent protection against H5H1 and H7N9 influenza via childhood hemagglutinin imprinting. Science 2016;354: 722-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jacobs JH, Archer BN, Baker MG, et al. Searching for sharp drops in the incidence of pandemic A/H1N1 influenza by single year of age. PLoS One 2012;7:e42328. doi: 10.1371/journal.pone.0042328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gagnon A, Acosta, Hallman E, S, et al. Pandemic paradox: early life H2N2 pandemic influenza infection enhanced susceptibility to death during the 2009 H1N1 pandemic. MBio 2018;9. pii: e02091-17. doi: 10.1128/mBio.02091-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Viboud C, Epstein SL.. First flu is forever. Science 2016;354:706-707. doi: 10.1126/science.aak9816. [DOI] [PubMed] [Google Scholar]
- 25. Kosikova M, Li L, Radvak, Ye P, Wan Z, Xie XF, H.. Imprinting of repeated influenza A/H3 exposures on antibody quantity and antibody quality: implications on seasonal vaccine strain selection and vaccine performance. Clin Infect Dis 2018. doi: 10.1093/cid/ciy327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Suber F, Kobzik L.. Childhood tolerance of severe influenza: a mortality analysis in mice. Am J Physiol Lung Cell Mol Physiol 2017;313:L1087-95. doi: 10.1152/ajplung.00364.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Robinson DP, Lorenzo ME, Jian W, Klein SL.. Elevated 17β-estradiol protects females from influenza A virus pathogenesis by suppressing inflammatory responses. PLoS Pathog 2011;7:e1002149. doi: 10.1371/journal.ppat.1002149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Joachim R, Suber F, Kobzik L.. Characterising pre-pubertal resistance to death from endotoxemia. Sci Rep 2017;7:16541. doi:10.10138/s41598-017-16743-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Takao K, Miyakawa T.. Genomic responses in mouse models greatly mimic human inflammatory diseases. Proc Natl Acad Sci U S A 2015;112:1167-72. doi: 10.1073/pnas.1401965111. Erratum in: Proc Natl Acad Sci U S A. 2015 Mar 10;112(10):E1163-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Efron PA, Mohr AM, Moore FA, Moldawer LL.. The future of murine sepsis and trauma research models. J Leukoc Biol 2015. ;98:945-52. doi: 10.1189/jlb.5MR0315-127R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim KS, Jung H, Shin IK, Choi BR, Kim DH.. Induction of interleukin-1 beta (IL-1β) is a critical component of lung inflammation during influenza A (H1N1) virus infection. J Med Virol 2015;87:1104-12. doi: 10.1002/jmv.24138. [DOI] [PubMed] [Google Scholar]
- 32. Peteranderl C, Herold S, Schmoldt C.. Human influenza virus infections. Semin Respir Crit Care Med 2016:487-500. doi: 10.1055/s-0036-1584801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Piantadosi CA, Suliman HB.. Mitochondrial dysfunction in lung pathogenesis. Annu Rev Physiol 2017;79:495-515. doi: 10.1146/annurev-physiol-022516-034322. [DOI] [PubMed] [Google Scholar]
- 34. Imai Y1, Kuba K, Neely GG, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008;133:235-49. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu M, Chen F, Liu T, Chen F, Liu S, Yang J.. The role of oxidative stress in influenza virus infection. Microbes Infect 2017. Dec;19(12):580-586. doi: 10.1016/j.micinf.2017.08.008. [DOI] [PubMed] [Google Scholar]
- 36. Teijaro JR, Walsh KB, Cahalan S, et al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 2011;146:980-91. doi: 10.1016/j.cell.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Armstrong SM, Mubareka S, Lee WL.. The lung microvascular endothelium as a therapeutic target in severe influenza. Antiviral Res 2013;99:113-8. doi: 10.1016/j.antiviral.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 38. Tam VC, Quehenberger O, Oshansky CM, et al. Lipidomic profiling of influenza infection identifies mediators that induce and resolve inflammation. Cell 2013;154:213-27. doi: 10.1016/j.cell.2013.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Serhan CN. Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. FASEB J 2017;31:1273-88. doi: 10.1096/fj.201601222R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sachdev U, Lotze MT.. Perpetual change: autophagy, the endothelium, and response to vascular injury. J Leukoc Biol 2017;102:221-35. doi: 10.1189/jlb.3RU1116-484RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Feizi N, Mehrbod P, Romani B, et al. Autophagy induction regulates influenza virus replication in a time-dependent manner. J Med Microbiol 2017;66:536-41. doi: 10.1099/jmm.0.000455. [DOI] [PubMed] [Google Scholar]
- 42. Sun Y, Li C, Shu Y, et al. Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Sci Signal 2012. Feb 21;5(212):ra16. doi: 10.1126/scisignal.2001931. [DOI] [PubMed] [Google Scholar]
- 43. Chung MT, Lee YM, Shen HH, et al. Activation of autophagy is involved in the protective effect of 17β-oestradiol on endotoxaemia-induced multiple organ dysfunction in ovariectomized rats. J Cell Mol Med 2017;21:3705-17. doi: 10.1111/jcmm.13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Medzhitov R, Schneider DS, Soares MP.. Disease tolerance as a defense strategy. Science 2012;335:936-41. doi: 10.1126/science.1214935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Soares MP, Teixeira L, Moita LF.. Disease tolerance and immunity in host protection against infection. Nat Rev Immunol 2017;17:83-96. doi: 10.1038/nri.2016.136. [DOI] [PubMed] [Google Scholar]
- 46. Meunier I, Kaufmann E, Downey J, Divangahi M.. Unravelling the networks dictating host resistance versus tolerance during pulmonary infections. Cell Tissue Res 2017;367:525-36. doi: 10.1007/s00441-017-2572-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Crane MJ, Lee, FitzGerald KM, E. S1 Jamieson AM. Surviving deadly lung infections: innate host tolerance mechanisms in the pulmonary system. Front Immunol 2018;9:1421. doi: 10.3389/fimmu.2018.01421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. O'Neill LA, Kishton RJ, Rathmell J.. A guide to immunometabolism for immunologists. Nat Rev Immunol 2016;16:553-65. doi: 10.1038/nri.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rambold AS, Pearce EL.. Mitochondrial dynamics at the interface of immune cell metabolism and function. Trends Immunol 2018;39:6-18. doi: 10.1016/j.it.2017.08.006. [DOI] [PubMed] [Google Scholar]
- 50. Kovats S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol 2015;294:63-9. doi: 10.1016/j.cellimm.2015.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chandler JD, Hu X, Ko EJ, et al. Metabolic pathways of lung inflammation revealed by high-resolution metabolomics (HRM) of H1N1 influenza virus infection in mice. Am J Physiol Regul Integr Comp Physiol 2016;311:R906-R916. doi: 10.1152/ajpregu.00298.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ellison PT. Endocrinology, energetics, and human life history: a synthetic model. Horm Behav 2017;91:97-106. doi: 10.1016/j.yhbeh.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 53. Hayward AC, Wang L, Goonetilleke N, et al. Natural T cell-mediated protection against seasonal and pandemic influenza. Results of the Flu Watch Cohort Study. Am J Respir Crit Care Med 2015;191:1422-31. doi: 10.1164/rccm.201411-1988OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Casanova JL. Human genetic basis of interindividual variability in the course of infection. Proc Natl Acad Sci U S A 2015;112:E7118-27. doi: 10.1073/pnas.1521644112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Piasecka B, Duffy D, Urrutia A, et al. Distinctive roles of age, sex, and genetics in shaping transcriptional variation of human immune responses to microbial challenges. Proc Natl Acad Sci U S A 2017. Dec 27. pii: 201714765. doi: 10.1073/pnas.1714765115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McClain MT, Henao R, Williams J, et al. Differential evolution of peripheral cytokine levels in symptomatic and asymptomatic responses to experimental influenza virus challenge. Clin Exp Immunol 2016;183:441-51. doi: 10.1111/cei.12736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Huang Y, Zaas AK, Rao A, et al. Temporal dynamics of host molecular responses differentiate symptomatic and asymptomatic influenza a infection. PLoS Genet 2011;7:e1002234. doi: 10.1371/journal.pgen.1002234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Matilainen O, Quirós PM, Auwerx J.. Mitochondria and epigenetics - crosstalk in homeostasis and stress. Trends Cell Biol 2017;27:453-63. doi: 10.1016/j.tcb.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 59. Pociask DA, Robinson KM, Chen K, et al. Epigenetic and transcriptomic regulation of lung repair during recovery from influenza infection. Am J Pathol 2017;187:851-63. doi: 10.1016/j.ajpath.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Feinberg AP. The key role of epigenetics in human disease prevention and mitigation. N Engl J Med 2018;378:1323-34. doi: 10.1056/NEJMra1402513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Burger O, Baudisch A, Vaupel JW.. Human mortality improvement in evolutionary context. Proc Natl Acad Sci U S A 2012;109:18210-4. doi: 10.1073/pnas.1215627109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Carlton ED, Cooper CL, Demas GE.. Metabolic stressors and signals differentially affect energy allocation between reproduction and immune function. Gen Comp Endocrinol 2014;208:21-9. doi: 10.1016/j.ygcen.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. López M, Tena-Sempere M.. Estrogens and the control of energy homeostasis: a brain perspective. Trends Endocrinol Metab 2015;26:411-21. doi: 10.1016/j.tem.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 64. Lomniczi A, Ojeda SR.. The emerging role of epigenetics in the regulation of female puberty. Endocr Dev 2016;29:1-16. doi: 10.1159/000438840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Huang SSH, Banner D, Degousee N, et al. Differential pathological and immune responses in newly weaned ferrets are associated with a mild clinical outcome of pandemic 2009 H1N1 infection. J Virol 2012;86:13187-201. doi: 10.1128/JVI.01456-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yi X, Yuan Y, Li N, et al. A mouse model with age-dependent immune response and immune-tolerance for HBV infection. Vaccine 2018;36:794-801. doi.org/10.1016/j.vaccine.2017.12.071. [DOI] [PubMed] [Google Scholar]
- 67. Barsness KA, Bensard DD, Partrick DA, Calkins CM, Hendrickson RJ, McIntyre RC Jr.. Endotoxin induces an exaggerated interleukin-10 response in peritoneal macrophages of children compared with adults. J Pediatr Surg 2004;39:912-5; discussion 912-5. [DOI] [PubMed] [Google Scholar]
- 68. Barsness KA, Bensard DD, Partrick DA, et al. IL-1beta induces an exaggerated pro- and anti-inflammatory response in peritoneal macrophages of children compared with adults. Pediatr Surg Int 2004;20:238-42. [DOI] [PubMed] [Google Scholar]
- 69. Hwang W, Lee CG, Lee C, et al. Locus-specific reversible DNA methylation regulates transient IL-10 expression in Th1 cells. J Immunol 2018;200:1865-75. doi: 10.4049/jimmunol.1701162. [DOI] [PubMed] [Google Scholar]
- 70. Shin T, Kuboki S, Huber N, et al. Activation of peroxisome proliferator-activated receptor-gamma during hepatic ischemia is age-dependent. J Surg Res 2008;147:200-5. doi: 10.1016/j.jss.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Oesterle 68., Laufs A, Liao UJK. Pleiotropic effects of statins on the cardiovascular system. Circ Res 2017;120:229-43. doi: 10.1161/CIRCRESAHA.116.308537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Di Raimondo D, Tuttolomondo A, Butta C, Miceli S, Licata G, Pinto A.. Effects of ACE-inhibitors and angiotensin receptor blockers on inflammation. Curr Pharm Des 2012; 18:4385-413. [DOI] [PubMed] [Google Scholar]
- 73. Saisho Y. Metformin and inflammation: its potential beyond glucose-lowering effect. Endocr Metab Immune Disord Drug Targets 2015;15:196-205. [DOI] [PubMed] [Google Scholar]
- 74. Murray CJ, Lopez AD, Chin B, Feehan D, Hill KH.. Estimation of potential global pandemic influenza mortality on the basis of vital registry data from the 1918-20 pandemic: a quantitative analysis. Lancet 2006;368:2211-8. [DOI] [PubMed] [Google Scholar]
- 75. Gates B. Innovation for pandemics. N Engl J Med 2018;378:2057-9. [DOI] [PubMed] [Google Scholar]
- 76. Shi J, Deng G, Kong H, et al. H7N9 virulent mutants detected in chickens in China pose an increased threat to humans. Cell Res 2017:1409-21. doi: 10.1038/cr.2017.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Iuliano AD, Roguski KM, Chang HM, et al. Estimates of global seasonal influenza-associated respiratory mortality: a modeling study .Lancet 2017. Dec 13. pii: S0140-6736(17)33293-2. doi.org/10.1016/S0140-6736(17)33293-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Paules CI, Marston HD, Eisinger RW, Baltimore D, Fauci AS.. The pathway to a universal influenza vaccine. Immunity 2017;47:599-603. doi: 10.1016/j.immuni.2017.09.007. [DOI] [PubMed] [Google Scholar]
- 79. Anderson CS, Ortega S, Chaves FA, et al. Natural and directed antigenic drift of the H1 influenza virus hemagglutinin stalk domain .Sci Rep 2017;7:14614. doi: 10.1038/s41598-017-14931-7. Erratum in: Sci Rep 2018;8:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Park JK, Han A, Czajkowski L, et al. Evaluation of pre-existing anti-hemagglutinin stalk antibody as a correlate of protection in a healthy volunteer challenge with influenza A/H1N1pdm virus. MBio 2018;9. pii: e02284-17. doi: 10.1128/mBio.02284-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Eccleston-Turner M. The pandemic influenza preparedness framework: a visible procurement option for developing states? Med Law Int 2017;17:227-48. doi: 10.1177/0968533217723683. [Google Scholar]
- 82. Wang X, Jiang H, Wu P, et al. Epidemiology of avian influenza A H7N9 virus in human beings across five epidemics in mainland China, 2013-17: an epidemiological study of laboratory-confirmed case series. Lancet Infect Dis 2017; 17: 822-32. doi: 10.1016/S1473-3099(17)30323-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Fedson DS. Statins, influenza vaccination and influenza. J Infect Dis 2017;215:484-5. doi: 10.1093/infdis/jiw537. [DOI] [PubMed] [Google Scholar]
- 84. Hui DS, Lee N, Chan PK, Beigel JH.. The role of adjuvant immunomodulatory agents for treatment of severe influenza. Antiviral Res 2018;150:202-16. doi: 10.1016/j.antiviral.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ayala FJ. Darwin and the scientific method. Proc Natl Acad Sci USA 2009;106 Suppl 1:10033-9. doi: 10.1073/pnas.0901404106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Starr I. Influenza in 1918: recollections of the epidemic in Philadelphia. 1976. Ann Intern Med 2006;145:138-40. [DOI] [PubMed] [Google Scholar]
- 87. Baddeley M. Herding, social influences and behavioural bias in scientific research: Simple awareness of the hidden pressures and beliefs that influence our thinking can help to preserve objectivity. EMBO Rep 2015;16:902-5. doi: 10.15252/embr.201540637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Casadevall A, Fang FC.. Revolutionary science. MBio 2016;7:e00158-16. doi:10.1128/mbio00158-16. [DOI] [PMC free article] [PubMed] [Google Scholar]