

In Brief
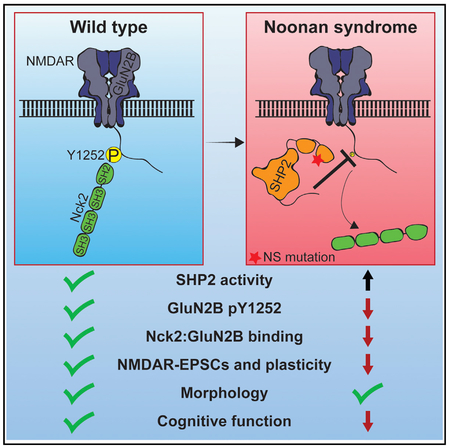
Noonan syndrome (NS) is caused by hyperactive SHP2 and is associated with cognitive deficits. Levy et al. find that NMDA receptor (NMDAR)-mediated currents are disrupted in NS and identify GluN2B Y1252 as a neural substrate of SHP2. Y1252 in turn binds the actin regulator Nck2 to control spine actin and regulate NMDAR currents.
SUMMARY
Hyperactivating mutations in the non-receptor tyro-sine phosphatase SHP2 cause Noonan syndrome (NS). NS is associated with cognitive deficits, but how hyperactivation of SHP2 in NS changes neuron function is not well understood. We find that mice bearing an NS-associated SHP2 allele (NS mice) have selectively impaired Schaffer collateral-CA1 NMDA (N-methyl-D-aspartate) receptor (NMDAR)-mediated neurotransmission and that residual NMDAR-mediated currents decay faster in NS mice because of reduced contribution of GluN1:GluN2B diheteromers. Consistent with altered GluN2B function, we identify GluN2B Y1252 as an NS-associated SHP2 substrate both in vitro and in vivo. Mutation of Y1252 does not alter recombinant GluN1:GluN2B receptor kinetics. Instead, phospho-Y1252 binds the actin-regulatory adaptor protein Nck2, and this interaction is required for proper NMDAR function. These results establish SHP2 and Nck2 as NMDAR regulatory proteins and strongly suggest that NMDAR dysfunction contributes to NS cognitive deficits.
Graphical Abstract
INTRODUCTION
Autosomal dominant mutations in PTPN11, which encodes the Src homology 2 (SH2) domain-containing non-receptor protein tyrosine phosphatase SHP2, account for 50% of cases of Noonan syndrome (NS) (Tartaglia et al., 2001, 2011). NS is associated with congenital heart defects, short stature, and craniofacial abnormalities (Noonan, 1968; Tartaglia et al., 2011), as well as an array of cognitive deficits (Duenas et al., 1973; Verhoeven et al., 2008). Although SHP2 is abundant in the brain and present at synapses (Reeves et al., 1996), how its mutation in NS disrupts neuronal function is not well understood.
NS-associated mutations disrupt autoinhibition of SHP2 phosphatase activity by its N-terminal SH2 domain (N-SH2), both hyperactivating SHP2 and causing it to aberrantly bind other proteins (Araki et al., 2004; Paardekooper Overman et al., 2014; Tartaglia et al., 2001). Mutation of N-SH2 D61 in NS causes greater phosphatase hyperactivation and is associated with more severe cognitive symptoms than another common NS mutation, N308D (Keilhack et al., 2005; Pierpont et al., 2009). In mice, heterozygous knockin of D61G or brain-specific overexpression of SHP2 D61G or D61Y causes learning and memory deficits (Altmüller et al., 2017; Lee et al., 2014), whereas these phenotypes are milder in N308D mutants (Lee et al., 2014). These results suggest that increased SHP2 phosphatase activity itself underlies cognitive problems in NS patients.
N-methyl-D-aspartate (NMDA) receptors (NMDARs) are critical for synaptic plasticity and learning and memory (Traynelis et al., 2010), and tyrosine phosphorylation (pY) of GluN2 subunits regulates NMDAR function (Chen and Roche, 2007). GluN2B in particular is tyrosine phosphorylated at several sites (Nakazawa et al., 2001), and although roles for pY1070 and pY1472 in surface trafficking of GluN2B have been established (Lu et al., 2015; Prybylowski et al., 2005), the functions of other GluN2B tyrosine phosphorylation sites are not understood.
We find that the Ptpn11D61G/+ mouse model of NS exhibits defects in behavior, synaptic plasticity, and GluN2B-mediated neurotransmission, and we identify GluN2B Y1252 as a direct substrate of SHP2 in vitro and in vivo. Charge at Y1252 does not alter GluN1:GluN2B receptor properties. Instead, we find pY1252 acts as a binding site for the SH2-domain-containing protein Nck2, and this interaction is required for NMDAR function. Our data establish that SHP2 regulates NMDAR function by controlling GluN2B Y1252 phosphorylation and Nck2 binding, and implicate NMDAR dysfunction as a likely contributor to cognitive deficits in NS.
RESULTS
NS Mice Exhibit Deficits in Behavior and Synaptic Plasticity
NS patients have cognitive and learning deficits (Duenas et al., 1973; Verhoeven et al., 2008). We tested whether Ptpn11D61G/+ mice (NS mice; Araki et al., 2004) exhibited defects in a cortex-and hippocampus-dependent novel object recognition task (Antunes and Biala, 2012). 48 hr after exploring two identical objects, wild-type (WT) mice preferred to explore a novel object, whereas NS mice explored both a novel and familiar object equally (Figures 1A and 1B). WT and NS mice spent equivalent time in object exploration (Figure 1C), indicating that NS mice did not display gross exploration deficits. These results demonstrate that NS mice are unable to discriminate between novel and familiar objects.
Figure 1. Ptpn11D61G/+ (NS) Mice Exhibit Deficits in Behavior and Synaptic Plasticity.

(A) On day 1, WT and NS mice do not prefer one of two identical objects over chance. Data in (A)–(C) are means + SEM, and symbols are exploration times from individual animals (n = 8 WT and 7 NS mice). WT: p = 0.85; NS: p = 0.28 (one-sample t test of object 1 exploration time versus 15 s [dotted line]).
(B) On day 3, WT mice prefer to explore a novel object, whereas NS mice explore both objects equally. WT: **p = 0.0029; NS: p = 0.56 (one-sample t test of novel object exploration time versus 15 s).
(C) WT and NS mice spent equal time accumulating 30 s object exploration time on both days. Day 1: p = 0.62; day 3: p = 0.54 (unpaired t test).
(D) NS mice display impaired theta-burst stimulation (TBS)-induced LTP. Insets show the average of two EPSCs before (WT: 1, NS: 3) and after (WT: 2, NS: 4) TBS, and their overlay. Data are means + SEM (n = 8 neurons) normalized to the baseline amplitude. ****p < 0.0001, effects of genotype and time; interaction p = 0.57 (two-way ANOVA).
We also measured the ability of NS mice to undergo theta-burst stimulation (TBS)-induced long-term potentiation (LTP) at hippocampal Schaffer collateral (SC)-CA1 synapses. Whole-cell SC-evoked CA1 excitatory postsynaptic currents (EPSCs) in WT mice were potentiated by ~75% immediately after TBS and remained potentiated by ~40% 50 min later. By contrast, CA1 EPSCs in NS mice were initially potentiated to levels lower than WT and decayed to baseline 30 min after TBS (Figure 1D). Our findings agree with and extend previous reports that several N-SH2 D61 mutant animals fail other hippocampus-dependent memory tasks and cannot stably express TBS-induced LTP (Altmüller et al., 2017; Lee et al., 2014).
NS Mice Have Normal CA1 Pyramidal Neuron Structure
Because neurons often have altered morphology in neurological disorders (Fiala et al., 2002; Kulkarni and Firestein, 2012), we tested NS mice for possible impacts of hyperactive SHP2 on neuron structure. Total dendrite length and numbers of dendrite branches of CA1 pyramidal neuron apical and basal dendritic arbors were indistinguishable between NS and WT mice (Figures 2A–2C). Dendritic spine density on terminal dendrite segments of CA1 neurons, both proximal and distal to the cell body, was also similar in NS and WT mice (Figures 2D–2F). Finally, using electron microscopy of CA1 stratum radiatum (Figure 2G), we found that synapse density (Figure 2H), post-synaptic density length (Figure 2I), and spine head area (Figure 2J) were all similar in NS and WT mice. Together, these results demonstrate that CA1 pyramidal neuron structure appears normal in NS mice.
Figure 2. NS Mice Have Normal CA1 Pyramidal Neuron Structure.

(A) Representative reconstructions of CA1 pyramidal neuron dendrite arbors from P40–P50 WT and NS mice.
(B and C) Basal and apical dendrite length (B) and number of branch points (C) of CA1 pyramidal neurons are unchanged in NS mice versus WT. Bars show mean + SEM, and circles show data from individual neurons (napical length = 17 WT and 17 NS neurons, nbasal length = 19 WT and 16 NS neurons; napical nodes = 19 WT and 17 NS neurons, nbasal nodes = 19 WT and 17 NS neurons). Basal length: p = 0.19; apical length: p = 0.55; basal nodes: p = 0.59; apical nodes: p = 0.55 (unpaired t test).
(D) Representative images of dendrite segments from Thy1-GFP-expressing WT and NS mice. Scale bars, 5 μm.
(E and F) Spine density on oblique apical dendrites proximal (E) or distal (F) to the soma are unchanged in NS mice compared with WT (p = 0.85). Bars show mean + SEM, and circles show average spine density from individual animals (n = 5 WT and 3 NS mice). Distal: p = 0.85; proximal: p = 0.72 (unpaired t test).
(G) Representative electron micrographs from WT and NS stratum radiatum. Post-synaptic regions are pseudo-colored in cyan, and presynaptic regions in magenta. Scale bars, 500 nm.
(H) Synapse density is unchanged in NS versus WT littermates. Data are average synapse density per animal, with paired littermates connected by lines (n = 3 WT and 3 NS mice). The mean and SEM of the differences are indicated on the graph. p = 0.76 (paired t test).
(I) The average post-synaptic density (PSD) length is unchanged in NS compared with WT. Histogram shows the population of PSD lengths (n = 1,031 WT and 1,100 NS PSDs) in 40-nm bins. Inset shows box (25th percentile, median, and 75th percentile) and whisker (2.5%–97.5%) plot. p = 0.098 (Mann-Whitney test).
(J) The average spine head area is unchanged in NS mice compared with WT. Graphs are as described for (I), and the histogram bin size is 0.04 μm2. p = 0.091 (Mann-Whitney test).
NMDAR Function Is Selectively Disrupted in NS Mice
We next tested whether the behavior and plasticity deficits in NS mice were associated with altered excitatory neurotransmission. We recorded SC-evoked NMDAR- and (alpha)-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor (AMPAR)-mediated EPSCs in CA1 neurons. The NMDAR/AMPAR-EPSC ratio was significantly decreased in CA1 neurons from NS slices relative to WT (Figures 3A and 3B), suggesting that normal excitatory transmission is disrupted in NS mice.
Figure 3. NMDA Receptor Function Is Selectively Disrupted in NS Mice.

(A) Representative traces of NMDAR-EPSCs (+440 Mv holding potential) and AMPAR-EPSCs (−70 Mv) recorded from the same WT and NS neurons.
(B) The NMDAR/AMPAR-EPSC ratio is reduced in NS mice compared with WT (n = 17 WT and 21 NS neurons). In all panels, data were analyzed by un-paired t test, histograms show means + SEM, and symbols are data from individual neurons. *p =0.014.
(C) (i) Representative NMDAR-Mepsc recordings (left) and average currents (right) recorded from WT and NS neurons (n = 22 WT and 21 NS neurons). NMDAR-Mepsc amplitude (ii) is reduced (p = 0.052), frequency (iii) is unchanged (p = 0.19), and decay time (iv) is reduced (**p = 0.0066) in NS mice compared with WT.
(D) (i) Representative AMPAR-Mepsc recordings (left) and average currents (right) recorded from WT and NS neurons (n = 19 WT and 20 NS neurons). AMPAR-Mepsc amplitude (ii; p = 0.52), frequency (iii; p = 0.079), and decay time (iv, p = 0.097) are unchanged in NS mice compared with WT.
We recorded miniature EPSCs (mEPSCs) from CA1 neurons to measure whether NMDAR- or AMPAR-mediated currents were selectively altered in NS mice. NMDAR-mEPSC frequency in NS mice was unchanged compared with WT, consistent with unchanged synapse density, and there was a strong trend (p = 0.052) toward reduced NMDAR-mEPSC amplitude. NMDAR-mEPSCs also decayed significantly faster in NS neurons than WT, as shown by the lower τweighted of the decay (Figure 3C). By contrast, NS AMPAR-mEPSC amplitude, frequency, and decay time were similar to WT (Figure 3D). These data indicate that NMDAR function is selectively disrupted in NS mice.
GluN1:GluN2B Diheteromers Are Selectively Disrupted in NS Mice, while NMDAR Subunit Surface Levels Are Unaltered
GluN1:GluN2B diheteromers have slower deactivation rates than GluN1:GluN2A diheteromers or GluN1:GluN2A:GluN2B triheteromers (Hansen et al., 2014; Traynelis et al., 2010). The faster τweighted of NMDAR-mESPCs in NS mice (Figure 3Civ) suggested that GluN1:GluN2B diheteromers may contribute less to the NMDAR-EPSC τweighted in NS mice. We recorded SC-evoked NMDAR-EPSCs plus or minus ifenprodil, which selectively inhibits GluN2B-containing receptors, but does not change the τweighted of triheteromeric receptors (Hansen et al., 2014; Tovar et al., 2013). Ifenprodil significantly decreased the NMDAR-EPSC τweighted in WT neurons, consistent with removal of slow-decaying GluN1:GluN2B diheteromers from τweighted. By contrast, τweighted was already faster in NS mice and was not changed further by ifenprodil, indicating that GluN1: GluN2B NMDAR function is compromised in NS mice (Figure 4A).
Figure 4. GluN1:GluN2B Diheteromers Are Selectively Disrupted in NS Mice, whereas NMDAR Subunit Surface Levels Are Unaltered.

(A) The NMDAR-EPSC τweighted is faster in NS mice than WT and is not further sped by ifenprodil. Traces are representative peak-scaled SC-evoked NMDAR-EPSCs from indicated conditions. Histogram bars are mean τweighted, and connected circles are τweighted of individual neurons before and after ifenprodil (n = 12 WT and 12 NS neurons). Data were analyzed by two-way repeated-measures ANOVA (effects of genotype: p = 0.008; ifenprodil: p < 0.0001; and interaction: p = 0.0041) followed by post hoc Holm-Sidak test (WT-NSvehicle: adjusted p = 0.0002; WT-NSifenprodil: adjusted p = 0.67; vehicle-ifenprodilWT: adjusted p < 0.0001; vehicle-ifenprodilNS: adjusted p = 0.20). ***p < 0.001; ****p < 0.0001.
(B) Representative western blots show surface (S) and total (T) levels of the indicated proteins in surface biotinylation assay.
(C) Quantifications of blots in (B) show that surface levels of NMDAR and AMPAR subunits are not different in NS mice compared with WT. Data are surface divided by total signal for each protein normalized to the WT mean, and littermate WT and NS mice are connected by lines (n = 6 WT and 6 NS mice). The mean, SEM and p value (paired t test) of the differences are indicated on the graphs.
(D) The percent of evoked NMDAR-EPSC amplitude blocked by ifenprodil treatment is not different between WT and NS mice (n = 8 WT and 9 NS neurons). Data are shown as means + SEM; p = 0.78 (Welch’s t test).
We also measured NMDAR surface levels by western blotting of biotinylated extracellular proteins purified with NeutrAvidin. Surface levels of NMDAR subunits GluN2B, GluN2A, and GluN1, as well as of the AMPAR subunit GluA1, were not significantly altered in NS mice compared with WT. Negative control cytoplasmic proteins were never detected in surface fractions (Figures 4B and 4C). Consistent with this, the percent of NMDAR-EPSC amplitude blocked by ifenprodil was not different between genotypes, indicating an equal proportion of surface GluN2B-containing receptors (Figure 4D). These data show that faster decay of NMDAR-EPSCs is not accompanied by detectable changes in surface NMDAR subunits.
SHP2 Directly Dephosphorylates GluN2B Y1252
GluN2B is tyrosine phosphorylated at several sites (Nakazawa et al., 2001). Given the reduced GluN1:GluN2B diheteromer function in NS mice, we measured whether SHP2 could dephosphorylate GluN2B, and if so, which phosphotyrosines were targeted. We first tested whether a “substrate trapping” mutant of SHP2 (D425A/Q506A; Mercan and Bennett, 2010) could complex with phospho-GluN2B (Figures 5A and S1A). This mutant can bind, but not dephosphorylate, substrates, forming a stable enzyme-substrate complex that can be affinity purified. Glutathione-S-transferase (GST)-SHP2 DAQA pulled down 14 ± 4-fold more GluN2B from HEK cell lysate than GST-SHP2 WT, and GST alone did not interact with GluN2B. Pre-incubation of GST-SHP2 DAQA with the tyrosine phosphatase active site inhibitor sodium orthovanadate reduced the GluN2B:GST-SHP2 DAQA interaction to 6 ± 2-fold over GST-SHP2 WT, but did not completely eliminate the interaction, suggesting the SHP2 phosphatase domain may form additional contacts with phosphorylated GluN2B (Figure 5B). These results demonstrate that SHP2 and GluN2B can form an enzyme-substrate complex.
Figure 5. SHP2 Directly Dephosphorylates GluN2B Y1252.

(A) Schematic of constructs used in (B)–(F). Purifications are shown in Figures S1A–S1C. Stars indicate mutated residues.
(B) GluN2B interacts more strongly with the GSTSHP2 PTP substrate trapping mutant (DAQA) than with GST-SHP2 PTP WT or GST, and this is inhibited by pre-incubation with sodium orthovanadate (VO4). Blot shown is representative of three experiments.
(C) Representative western blots of in vitro dephosphorylation of GluN2B-tail by WT, phosphatase-dead (CS), or NS-associated SHP2 (DG).
(D) Quantification of blots in € shows SHP2 DG selectively dephosphorylates Y1252, whereas auto-inhibited WT and phosphatase-dead CS have no effect. Y1252 data are fit with a one-phase exponential decay. Data are means ± SEM from three experiments. See Figures S1D and S1E for analysis of total GluN2B-tail phosphorylation.
(E) Representative western blots of hippocampal synaptosomes probed with the indicated primary antibodies.
(F) Quantifications of blots in € show Py71252 is reduced by ~50% in NS synaptosomes, with Py1336 and Py1472 also reduced by ~28%, relative to WT. Data are phosphotyrosine signal normalized to total GluN2B, or GluN2B itself, normalized to the WT mean. Paired littermates are connected by lines (n = 7 WT and 7 NS mice). Mean, SEM, and p value (paired t test) of the differences are indicated on the graphs.
GST, glutathione-S-transferase; MBP, maltose binding protein; PTP, phosphatase domain.
We next tested whether SHP2 selectively targets one or more phosphotyrosines on the GluN2B tail in vitro. We purified and phosphorylated a recombinant GluN2B-tail fragment, then incubated it with purified WT, C459S, or D61G SHP2 (Figures 5A, S1B, and S1C). Active D61G SHP2 dephosphorylated GluN2B-tail over time, whereas neither autoinhibited WT nor phosphatase dead C459S SHP2 altered GluN2B-tail phosphorylation (Figures S1D and S1E). Interestingly, GluN2B-tail dephosphorylation plateaued at 40% of baseline, suggesting that SHP2 targets some, but not all, GluN2B phosphotyrosines. Blotting with site-specific antibodies for the three major phosphotyrosines on GluN2B (Y1472, Y1336, and Y1252; Nakazawa et al., 2001) revealed SHP2 selectively dephosphorylated GluN2B Y1252 and did not dephosphorylate Y1336 or Y1472 (Figures 5C and 5D).
To test whether GluN2B tyrosine phosphorylation was also reduced by hyperactive SHP2 in vivo, we western blotted hippocampal synaptosomes from WT and NS littermates with phospho-site-specific antibodies. Phosphorylation at all three sites was significantly reduced in NS synaptosomes compared with WT, with the most robust reduction at Y1252 (~50%; Figures 5E and 5F). Total GluN2B levels were unaffected, consistent with no change in surface GluN2B, and indicating reduced phosphorylation is not caused by receptor loss from synaptosomes. These in vitro and in vivo experiments together demonstrate that hyperactive SHP2 in NS directly dephosphorylates GluN2B Y1252 and reduces GluN2B tyrosine phosphorylation in vivo.
Substitutions of GluN2B Y1252 to F or E Do Not Alter Channel Properties of GluN1:GluN2B Receptors
Given that SHP2 dephosphorylates GluN2B Y1252 and NMDAREPSCs decay faster in NS mice, we tested whether charge at GluN2B Y1252 altered GluN1:GluN2B diheteromer channel properties. We co-expressed unphosphorylatable (Y1252F or YF) and phospho-charge-mimicking (Y1252E or YE) GluN2B mutants with GluN1 in HEK cells, excised outside-out patches, and measured currents driven by brief pulses of saturating glutamate in the continuous presence of saturating glycine (Figures 6A and 6B, and S2A). Although heterologous cells do not replicate the synaptic network of scaffolding and interacting proteins, they enable measurements of how specific mutations directly affect receptor function. Weighted time constants calculated from bi-exponential fits of the mean ensemble currents (Figures 6C and S2B) were used to estimate receptor deactivation rates, which were similar between receptor mutants (Figure 6D). There were likewise no significant differences for the time constants and relative amplitudes of the two individual exponential components (Table S1).
Figure 6. Substitutions of GluN2B Y1252 to F or E Do Not Alter Channel Properties of GluN1:GluN2B Receptors.

(A and B) Example of responses to 2-ms applications of 1 Mm glutamate (at arrows) for hosphor-dead (Y1252F; YF) (A) or hosphor-charge-mimicking (Y1252E; YE) (B) GluN1:GluN2B receptors in outside-out patches. Example WT responses can be found in Figure S2A.
(C) Mean glutamate-evoked currents from two other patches for receptors containing YF or YE GluN2B. Mean WT currents can be found in Figure S2B.
(D) Weighted deactivation time constants for all GluN2B mutants are similar (n = 4 WT, 9 YF, and 8 YE patches). Data are means + SEM and were analyzed by oneway ANOVA. Full deactivation statistics can be found in Table S1.
(E and F) Examples of unitary currents recorded during continuous application of 1 Mm glutamate for YF € or YE (F) receptors. Example WT unitary currents can be found in Figure S2C.
(G) Histograms of open times for YF (blue) and YE (red) GluN2B mutants. The smooth curves are bi-exponential fits to the distributions of individual open time values (n = 992 Y1252F and 1,273 Y1252E open times).
(H) Weighted mean open times (ms) from the bi-exponential fits and mean unitary conductance (Ps) of the main open level of WT, YF, and YE GluN2B subunit are similar (n = 2, 3, and 4 patches, respectively). Data are means + SEM and were analyzed by one-way ANOVA. Full unitary current statistics can be found in Table S2.
We also recorded glutamate-evoked single-channel currents under steady-state conditions. The patches contained more than one receptor, precluding measurements of open probability or burst durations, but the open probability was low enough that portions of the records containing multiple openings could be removed and the edited records idealized to obtain estimates of open times and unitary current amplitudes (Figures 6E and 6F, and S2B). The weighted mean open time was not different between receptor mutants (Figures 6G and 6H). Consistent with previous reports (Stern et al., 1992), each patch contained openings to a main conductance level of ~50 pS and to a smaller subconductance level of ~40 pS (~10% of all openings). The unitary conductance of the main open level was not different between mutants (Figure 6H; Table S2). These results suggest that neither lack of phosphorylation nor mimicking of a negative charge at Y1252 is sufficient to alter GluN1:GluN2B diheteromer kinetics or unitary conductance.
GluN2B pY1252 Scaffolds the Actin Regulator Nck2
Because manipulating charge at Y1252 did not change GluN1: GluN2B diheteromer properties, we tested whether pY1252 could scaffold specific phosphotyrosine-binding proteins. We screened a GST-fusion library of phosphotyrosine-interacting domains for binding to a phosphopeptide encompassing GluN2B Y1252 (Figure S3) and identified several SH2 domains that selectively bound this peptide (Figures 7A and S4; Table S3). The SH2 domain of Nck2 yielded the highest signal in this screen. The highly homologous SH2 domain of Nck1, which is expressed at lower levels in hippocampus than Nck2 (Thévenot et al., 2011), but has nearly identical phosphopeptide specificity (Frese et al., 2006), also selectively bound pY1252, and a fusion protein of three Nck1 SH2 domains bound with about three times higher signal to noise than either single Nck SH2 domain. To confirm this interaction, we immunoprecipitated GluN2B WT or Y1252F from HEK cells co-expressing Myc-Nck2 along with Src to phosphorylate NMDARs. 50% less Myc-Nck2 coimmunoprecipitated with GluN2B Y1252F than with GluN2B WT, showing that Nck2 and GluN2B form a pY1252-dependent complex (Figures 7B and S6).
Figure 7. GluN2B Py1252 Binds SH2-Domain-Containing Proteins and Scaffolds Nck2 to Regulate NMDAR-EPSC Properties.

(A) Top hits identified by SH2-domain binding screen. (Left) Signal for anti-GST (green) and avidin (red) indicate probe binding and peptide loading, respectively. (Right) Background subtracted signal (ΔpY1252–Y1252) is shown in histogram. Nck 3x contains three fused Nck1 SH2 domains. A screen schematic can be found in Figure S3, and complete screen results are in Figure S4 and Table S3.
(B) (Left) Representative western blots of GluN2B WT or Y1252F (YF) and Myc-Nck2 co-immunoprecipitations (coIPs). (Right) 50% less Myc-Nck2 coIPed with GluN2B YF than WT, whereas GluN1 coIP is unchanged. Data are means of the ratio of IPed protein to GluN2B normalized to the WT mean + SEM. Nck2: *p = 0.046; GluN1: p = 0.96 (unpaired t test). n = 7 WT and 6 YF cultures. Control IgG IP can be found in Figure S6.
(C) (Left) Representative immunofluorescence images of WT and NS neurons transfected with GFP and stained for Nck2 and phalloidin. Orange dotted lines show neuron morphology from GFP fill. (Right) Dendritic spine Nck2 and phalloidin intensity are reduced in NS neurons. ****p < 0.0001 for both (Mann-Whitney test). Graphs are box (25th percentile, median, and 75th percentile) and whisker (2.5%–97.5%) plots. n = 86 WT spines from 8 neurons and 83 NS spines from 10 neurons.
(D) WT GST-Nck2 SH2 domain reduces NMDAR-EPSC amplitude. Data are mean NMDAR-EPSC amplitudes normalized to the first amplitude, and were analyzed by one-way ANOVA of the relative amplitude at 30 min (p < 0.0001) followed by post hoc Tukey test (at 2.5 μg/mL: WT versus RK, p = 0.011, WT versus HI, p = 0.0018, RK versus HI, p = 0.99; at 5 μg/mL: WT versus RK, p < 0.0001, WT versus HI, p < 0.0001, RK versus HI, p = 0.93; at 2.5 μg/mL into NS: WT 2.5 versus WT 2.5 (in NS), p = 0.038, RK 2.5 versus WT 2.5 (in NS), p = 0.96, HI 2.5 versus WT 2.5 (in NS), p = 0.58). n (neurons) = 4 each WT 2.5, WT 5, RK 2.5, RK 5, and HI 2.5, n = 3 HI 5, and n = 6 WT 2.5 (in NS). Representative traces can be found in Figure S5A. AMPAR-EPSCs and Rs were unchanged by GST-Nck2 SH2 infusion; these data are in Figures S5B and S5C.
(E) WT GST-Nck2 SH2 domain speeds NMDAR-EPSC τweighted. WT 2.5: p = 0.0018, RK 2.5: p = 0.10, HI 2.5: p = 0.26, WT 5: p = 0.0019, RK 5: p = 0.12, HI 5: p = 0.049, WT 2.5 (in NS): p = 0.0031 (paired t test). Paired infusion start and end values are connected by lines, and bars show mean decay time. *p < 0.05; **p < 0.01. WT SH2 domain occluded the effect of ifenprodil on both amplitude and decay; these results are in Figures S5D and S5E.
Nck2 is a scaffold protein that binds phosphorylated receptors via its SH2 domain and uses its three SH3 domains to localize and activate actin regulators (Rohatgi et al., 2001). Given Nck2’s role in actin dynamics and its identification as a pY1252 binding partner, we tested whether actin structure was altered in dissociated hippocampal neurons cultured from litter-mate WT and NS mice. Nck2 staining was reduced in dendritic spines from NS neurons compared with WT, suggesting binding to GluN2B Y1252 may be required to localize Nck2 to spines. Actin filament staining was also reduced in NS spines relative to WT, demonstrating NS neurons have disrupted actin structure associated with loss of Nck2 binding (Figure 7C). These results together suggest Nck2 selectively binds phosphorylated GluN2B Y1252, and disruption of this interaction in NS reduces spine actin.
Disrupting Nck2 SH2 Domain Interactions Alters NMDAR-EPSCs Similar to NS
To assess a possible functional role of Nck2 in NMDAR regulation, we disrupted Nck2’s ability to interact with downstream partners by infusing recombinant GST-Nck2 SH2 domain into CA1 neurons through the patch pipette. If Nck2-pY1252 binding regulates NMDAR function, the recombinant protein should act in a dominant-negative manner, because the isolated SH2 domain will compete with endogenous Nck2 for binding to GluN2B, but lacks the SH3 domains that recruit effectors (Mayer, 2000). WT GST-Nck2 SH2 domain reduced NMDAR-EPSC amplitudes in a dose-dependent manner. 2.5 μg/mL WT protein reduced amplitude 42% ± 7%, significantly more than negative controls Nck2 R311K (which cannot bind phosphotyrosines) or heat-inactivated (HI) WT protein (15% ± 1% and 10% ± 4%, respectively) (Figures 7D and S5A). 5 μg/mL WT protein saturated the effect, stably reducing amplitudes by 67% ± 5% at 15 min after infusion, whereas R311K and HI protein of the same concentration had little effect (19% ± 2% and 11% ± 1%, respectively) and were not different from the same proteins at 2.5 μg/mL. By contrast, AMPAR-EPSC amplitude was not differentially reduced by WT versus R311K or HI Nck2, suggesting Nck2 specifically regulates NMDAR-EPSCs (Figure S5B). WT GST-Nck2 SH2 also sped NMDAR-EPSC τweighted in a dose-dependent manner, whereas R311K and HI protein only marginally changed decay, if at all (Figure 7E), consistent with the changes in NMDAR-EPSCs in NS mice. The small, but statistically equivalent, reductions in amplitude caused by HI and R311K protein are likely due to protein buffer components or dialysis of the neuron over long recordings. Series resistance was unchanged by protein infusion (Figure S5C).
We next used ifenprodil to test whether GST-Nck2 SH2 infusion specifically affected GluN2B-containing NMDARs, in which case ifenprodil effects on NMDAR-EPSCs should be occluded by prior SH2 infusion. Ifenprodil dramatically reduced both NMDAR-EPSC amplitude and decay when added 30 min after negative control 5 μg/mL HI GST-Nck2 SH2 infusion, as expected (Figures S5D and S5E). By contrast, there was no further effect of ifenprodil on NMDAR-EPSC amplitude when either 2.5 or saturating 5 μg/mL WT GST-Nck2 SH2 protein was previously infused. The effect of ifenprodil on NMDAR-EPSC decay was similarly occluded by 5 μg/mL WT GST-Nck2 SH2, although a small effect remained at 2.5 μg/mL protein. These results together indicate loss of Nck2 SH2 binding specifically affects GluN2B-containing NMDARs.
GluN2B pY1252 is reduced by approximately 50% in NS mice (Figure 5), suggesting some GluN2B pY1252:Nck2 interactions may remain in NS that could be further disrupted by dominant-negative Nck2. Although infusion of 2.5 μg/mL WT Nck2 into NS neurons had no effect on NMDAR-EPSC amplitude relative to HI or R311K (Figure 7D), τweighted was reduced, albeit to a lesser extent than in WT neurons (Figure 7E). These data are consistent with only partial loss of the pY1252:Nck2 interaction in NS mice and also suggest the effects of Y1252 dephosphorylation on amplitude may saturate faster than those on decay. Together, these results align with the altered NMDAR-EPSC properties in NS mice and are consistent with a model where GluN2B pY1252 scaffolds Nck2 to regulate NMDAR-EPSC properties.
DISCUSSION
SHP2 is expressed in the brain and NS is associated with cognitive deficits, but the normal synaptic functions of SHP2 and whether and how they are altered in NS are not well understood. We find that an NS model mouse exhibits deficits in behavior and plasticity, and while hippocampal neuron structure appears normal, GluN2B-mediated neurotransmission is disrupted. Consistent with alterations in GluN2B-containing receptors, we find that SHP2 directly dephosphorylates GluN2B Y1252. Altering charge at GluN2B Y1252 does not impact GluN1:GluN2B receptor properties. Instead, phosphorylated Y1252 is a binding site for the actin regulator Nck2, and disruption of this interaction impairs NMDAR properties similar to that observed in NS mice. Together, our data reveal that SHP2 modulates NMDAR function via regulation of GluN2B Y1252 phosphorylation and Nck2 binding, and strongly suggest a role of NMDAR dysfunction in NS cognitive deficits.
Contribution of Activating Mutations in SHP2 to Cognitive Deficits
Our findings of disrupted novel object recognition and NMDAR-dependent hippocampal plasticity in NS mice agree with and extend previous reports (Altmüller et al., 2017; Lee et al., 2014), and our result that LTP is absent in whole-cell recordings also confirms previous work using extracellular field recordings (Lee et al., 2014). Together, these results strongly affirm that NS-associated mutations of SHP2 D61 in mice cause behavioral and plasticity defects. Despite robust changes to plasticity and behavior, we find the morphology of CA1 pyramidal neurons of NS mice does not appear different from their WT littermates. Thus, although SHP2 signals in cytoskeletal regulatory pathways in other systems (Inagaki et al., 2000), any disruption of those pathways in NS mice is insufficient to change CA1 neuron structure. Given that the D61G knockin allele is present throughout development, it is possible that NS neurons compensate for increased SHP2 activity over time.
SHP2 Directly Regulates GluN2B-Containing NMDARs
We report here that NMDAR function is selectively disrupted in NS mice: NMDAR-EPSCs decay faster because of a greatly reduced contribution of slow-decaying GluN1:GluN2B diheteromers. Hippocampal NMDARs exist primarily as GluN1: GluN2A diheteromers, GluN1:GluN2A:GluN2B triheteromers, or GluN1:GluN2B diheteromers. Although estimates vary, triheteromers appear to be the major NMDAR species at mature hippocampal synapses, with variable contributions from different diheteromers depending on species, strain, and age (Gray et al., 2011; Rauner and Köhr, 2011; Tovar et al., 2013). GluN2B remains expressed at high levels in adult hippocampus (Monyer et al., 1994; Xiao et al., 2016), and our mice retain a relatively large proportion of GluN2B diheteromers, because a concentration of ifenprodil that nearly completely inhibits GluN1:GluN2B diheteromers greatly speeds NMDAR-EPSC decay. Although the amplitude from triheteromers is reduced ~20%–40% by ifenprodil (Hansen et al., 2014; Rauner and Köhr, 2011), their decay time is unaffected by this treatment (Tovar et al., 2013). Thus, our finding that NMDAR-EPSC decay is already faster in NS mice than WT, and is not further increased by ifenprodil, indicates that GluN1:GluN2B diheteromer function is specifically disrupted. We did not find significant changes in AMPAR-mEPSCs in NS mice, which contrasts with a previous report showing disrupted AMPAR function in NS mice and in mice overexpressing SHP2D61G in hippocampus (Lee et al., 2014). Our recordings were made at a younger age than Lee et al. (2014), suggesting SHP2 may impact different receptors at different ages. Consistent with this, a recent report showed over-expression of SHP2D61G in dissociated hippocampal neurons altered both NMDAR and AMPAR clustering at early culture ages, but only AMPARs at later times (Oh et al., 2017).
GluN2B Y1252 Is a Substrate for NS-Associated SHP2
SHP2 is associated with the NMDAR complex (Husi et al., 2000), but GluN2B has not been identified as a direct SHP2 substrate. GluN2B is tyrosine phosphorylated at several sites in vivo (Nakazawa et al., 2001; Traynelis et al., 2010), and although pY1070 and pY1472 regulate GluN2B surface trafficking (Lu et al., 2015; Prybylowski et al., 2005), the regulation and possible functions of the other tyrosine phosphorylation sites are not understood. We show here that SHP2 directly dephosphorylates GluN2B at Y1252, but not Y1336 or Y1472, in vitro, and that Y1252 phosphorylation is robustly reduced in NS mice. Y1472 and Y1336 phosphorylation are also reduced in vivo, but less robustly than Y1252. Y1472 is primarily dephosphorylated by STEP (Snyder et al., 2005). However, STEP infusion decreases NMDAR open times (Pelkey et al., 2002), and Y1472 phosphorylation alters NMDAR surface levels (Prybylowski et al., 2005). Because neither of these properties are changed when Y1252 is manipulated (Figures 4B, 4C, and 6G), STEP and pY1472 cannot account for the NMDAR-EPSC changes in NS mice. Because SHP2 only dephosphorylates Y1252 in vitro, pY1252 may help scaffold the major GluN2B kinase Fyn, which has an SH2 domain, to GluN2B, a role also ascribed to pY1070 (Lu et al., 2015). Alternatively, reduced Nck2 binding to NMDARs and actin content in spines may impact the ability of other kinases or phosphatases to regulate NMDAR activity by altering scaffold protein location.
Both aberrant substrate dephosphorylation and aberrant binding to cellular proteins by mutant SHP2 are important for NS phenotypes (Paardekooper Overman et al., 2014). However, the N308D mutation does not hyperactivate SHP2 to the same extent as D61G, and N308D is associated with less severe cognitive deficits than D61 mutations in humans and mice (Keil-hack et al., 2005; Lee et al., 2014; Pierpont et al., 2009). These observations together with our data indicate increased SHP2 phosphatase activity itself is likely a critical determinant of neuron dysfunction in NS, and dephosphorylation of GluN2B by SHP2 is a critical component. We note that ~30% of patients with the related disease Noonan syndrome with multiple lentigines (also called LEOPARD syndrome), which is associated with reduced SHP2 phosphatase activity, also have mild learning difficulty. However, these patients rarely have severe cognitive impairments (Sarkozy et al., 2008), and loss of forebrain SHP2 is not associated with major changes in NMDAR-EPSC amplitudes or decay (Yan et al., 2017).
The discovery of GluN2B as a direct brain substrate of SHP2 in NS suggests SHP2 inhibition may be an effective treatment for NS-associated cognitive symptoms. Specific allosteric SHP2 inhibitors have been identified for cancer treatment (Chen et al., 2016) that could be used to treat NS cognitive symptoms. SHP2 is likely a better target in NS than Fyn or NMDARs, because it may be possible to titrate an SHP2 inhibitor to reduce aberrant SHP2 activity to more normal levels, whereas potentiating Fyn and NMDARs may be nonspecific because of their wide variety of signaling partners.
How Does GluN2B Y1252 Phosphorylation Regulate NMDAR Currents?
Although previous work found no effect of manipulating tyrosine kinase activity on GluN1:GluN2B function in whole-cell patch-clamp experiments (Köhr and Seeburg, 1996), the dephosphorylation of GluN2B Y1252 by SHP2 and involvement of GluN2B in the altered kinetics of NMDAR-EPSCs in NS mice warranted revisiting this issue. However, consistent with previous results, we find that substitution of Y1252 to either unphosphorylatable F or to phospho-charge-mimicking E is not sufficient to directly alter kinetics or single-channel conductance of GluN2B diheteromers. An alternative explanation is that pY1252 may act as a scaffold for another protein to regulate NMDAR function. GluN2B binds an extensive list of synaptic proteins, many through interactions with phosphotyrosine or phosphoserine, and these interactions are important for NMDAR functions, including support of plasticity and transcription (Husi et al., 2000). We find that the actin-regulatory scaffold Nck2 binds selectively to GluN2B pY1252. In addition, disrupting Nck2 SH2 interactions is sufficient to reduce NMDAR-EPSC amplitude and decay time but has no effect on AMPAR-EPSC amplitude, consistent with a model where GluN2B pY1252 scaffolds Nck2 to regulate NMDAR properties.
Nck2 activates N-WASp, a nucleation promoting factor of the Arp2/3 complex, to promote actin filament branch nucleation (Rohatgi et al., 2001). Actin filaments regulate channel gating of a diverse set of transmembrane ion channels (Huang et al., 2013; Shumilina et al., 2003; Zhang et al., 2016). Inhibitors of actin polymerization are associated with reduced NMDAREPSC function, although the mechanism by which actin regulates currents is not understood (Furukawa et al., 1997; Nörenberg et al., 1999; Rosenmund and Westbrook, 1993). We find that both Nck2 and actin content are reduced in dendritic spines of NS neurons relative to WT, suggesting that Nck2 may couple NMDARs to actin regulation to maintain channel function. Although the exact mechanism by which Nck2 and actin regulate NMDAR currents is unknown, an actin-gating mechanism recently proposed for Kv3.3 channels may provide hints. The actin-regulatory protein Hax1 binds to the C-terminal tail of Kv3.3 and drives actin branching close to the channel to physically block channel inactivation. Deletion of Hax1 decouples actin polymerization from the receptor, altering Kv3.3 kinetics (Zhang et al., 2016). Future work will hopefully reveal how actin regulates EPSC characteristics for other receptor types.
In total, the data demonstrate that NS-associated SHP2 can specifically dephosphorylate GluN2B Y1252 and are consistent with a model where GluN2B Y1252 scaffolds Nck2 to regulate the activity of diheteromeric GluN1:GluN2B NMDAR, and strongly implicate NMDAR dysfunction in NS cognitive deficits.
EXPERIMENTAL PROCEDURES
Animals
All animal procedures were compliant with federal regulations and approved by the Yale University Institutional Animal Care and Use Committee. Ptpn11D61G/+ knockin mice (NS mice; Araki et al., 2004) were maintained in a mixed C57BL/6J 3 129/SvJ background and crossed with Thy-1 GFP line M (Feng et al., 2000) to visualize dendritic spines. We used only male animals to control for variation in CA1 dendritic spine density with estrus (Woolley et al., 1990), and all animals were post-natal day (P) 40–P46 at time of experiment unless otherwise specified.
Behavior
Novel object recognition was performed as described previously (Kerrisk et al., 2013). On day 1, mice explored two identical objects for a total of 30 s within 6 min total cage exploration time. The task was repeated 48 hr later, but one object was replaced with a novel object. Object exploration is defined as direct whisking or sniffing of the object.
Electrophysiology
Whole-cell recordings from hippocampal CA1 neurons were made as described previously (Xiao et al., 2016). EPSCs were evoked with 0.05-Hz stimulation of the SC. AMPAR-EPSC amplitudes were measured at the peak of the EPSCs recorded at −70 mV, and NMDAR-EPSC amplitudes were recorded from the same neurons at +40 mV 40 ms after the stimulus artifact. NMDAR-EPSC decay kinetics were measured by fitting recordings with a double exponential decay. GluN2B contribution was determined from τweighted of NMDAR-EPSCs recorded before and after addition of 3 μM ifenprodil. LTP was induced by TBS in the presence of 10 μM bicuculine methiodide (BMI). Recombinant GST-Nck2 SH2 domain was included in the patch solution where indicated at 2.5 or 5 μg/mL.
AMPAR-mEPSCs were recorded in whole-cell patch mode at −70 mV with 1 μM tetrodotoxin (TTX) and 10 μM BMI in the external solution, and NMDAR-mEPSCs were recorded at −40 mV with 1 μM TTX, 10 μM BMI, and 20 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) with 0.1 mM Mg2+ and 3.8 mM Ca2+ to partially remove the Mg2+ block.
Anatomy
Dendrite morphology was quantified using Neurolucida from neurons filled with biocytin and stained with avidin-horseradish peroxidase (HRP) and 3,3’-diaminobenzidine (DAB). Dendritic spine density was measured from mice carrying the Thy1-GFP:M transgene perfused with 4% paraformaldehyde (PFA). Spines on apical dendrites of CA1 neurons were counted in Fiji (Schindelin et al., 2012), and 3D dendrite segment length was measured using the Simple Neurite Tracer Fiji plugin (Longair et al., 2011). Spine ultrastructure was measured from mice perfused with 4% PFA/2% glutaraldehyde. Synapses were counted as an electron-dense post-synaptic density (PSD) apposed to a presynaptic terminal containing synaptic vesicles, and PSD length was measured from these synapses. Spine head area was measured to the thinnest part of the spine neck.
Surface Biotinylation
WT and NS littermate hippocampi were incubated 10 min in phosphate buffered saline plus 1 mM CaCl2 and 0.1 mM MgCl2, pH 8.0 (PBSCM) with or without 0.5 mg/mL sulfosuccinimidyl-6-[biotin-amido]hexanoate (sulfo-NHS LC)-biotin. Tissue was washed in PBSCM + 20 mM glycine, then in PBSCM, then lysed in radioimmunoprecipitation assay (RIPA) buffer. 100 μg of protein was incubated overnight with 50 μL of NeutrAvidin, with 10 μg of protein reserved for input. The resin was washed extensively with RIPA buffer, and samples were eluted/solubilized by addition of SDS-PAGE sample buffer and heating to 65°C for 10 min.
Recombinant Protein
GST-SHP2 phosphatase domains (PTPs) were produced in Rosetta E. coli by standard methods (Smith and Johnson, 1988). GST-mSHP2 was produced from baculovirus per the manufacturer’s instructions (Bac-to-Bac; Thermo). GST was cleaved with PreScission protease before its removal with fresh glutathione-agarose.
MBP-GluN2B C3-His pMAL-TEV (GluN2B-tail) was purified from bacteria as above, and eluted from Ni-NTA resin with 250 mM imidazole. 10 μM protein was tyrosine phosphorylated by incubating with 100 nM Src kinase Δ85-C, 15 mM MgCl2, 5 mM MnCl2, and 100 μM ATP for 2.5 hr at room temperature. Phosphorylated protein was re-purified on amylose resin and eluted with 10 mM maltose. GST-Nck2 SH2 domain WT and R311K were purified as described above. See Supplemental Experimental Procedures for details.
Substrate Trapping
Phoenix-AMPHO cells were transfected with Src, GluN1–1a, and YFP-GluN2B, then maintained in media supplemented with 150 μM aminophosphonovalerate (APV) and 11.25 μM MK801. Tyrosine phosphorylation was induced 48 hr after transfection with 100 μM pervanadate, and cells were lysed in buffer containing 5 mM iodoacetic acid (IAA). Excess IAA was neutralized with DTT; then lysates were incubated with GST-SHP2 PTP linked to glutathione agarose for 3 hr at 4°C. For vanadate competition, GST-SHP2 PTP resin was pre-incubated with 10 mM Na3VO4. The resin was washed 3× with lysis buffer without IAA and bound proteins eluted by boiling in SDS-PAGE sample buffer.
Dephosphorylation
MBP-GluN2B C3-His (140 nM) and full-length mSHP2 (14 nM) were mixed 1:1 in assay buffer + 5 mM DTT and incubated at 32°C for the times indicated. Reactions were stopped by addition of SDS-PAGE sample buffer and boiling. Samples were western blotted sequentially for GluN2B tyrosine phosphorylation, SHP2, and total GluN2B, and in parallel for each phosphotyrosine site.
Fractionation
Hippocampi were homogenized in buffer containing 320 mM sucrose and 10 mM HEPES (pH 7.4), and centrifuged at 900 × g to remove debris, then centrifuged 20 min at 12,000 × g to pellet synaptosomes. The pellets were washed twice in 4 mM HEPES (pH 7.4), then solubilized by boiling in SDS-PAGE sample buffer.
NMDAR Kinetics
tsA201 cells were co-transfected with GluN1, GluN2B WT, or phosphomutants and GFP (1:1:0.2), and maintained in medium containing 0.5 mM APV and 1 mM MgCl2. Currents were recorded 24–48 hr later at room temperature at −100 mV holding potential in saturating (100 μM) glycine. The external solution was applied to outside-out patches using a theta glass application pipette mounted on a piezoelectric bimorph, as described previously (Robert and Howe, 2003). Patches were positioned near the interface of the solutions flowing from the theta glass pipette. The speed of solution exchange (estimated from open tip potentials) was 300–600 μs. Deactivation decay was measured from patches subjected to saturating (1 mM) glutamate for 2 ms. Single-channel data were recorded during continuous glutamate application sampled at 50 kHz and subjected to digital low-pass filtering at 3 kHz. The filtered data were exported to QuB acquisition software (https://milesculabs.biology.missouri.edu/QuB.html) for analysis.
SH2-Domain Screen
Reverse-phase SH2 domain screening (Rosette assay) was performed as described previously (Ng and Machida, 2017).
Co-immunoprecipitation
HEK293 cells were transfected with Src, GluN1, GluN2B, and Myc-Nck2 as described above, crosslinked with dithiobis(succinimidyl propionate) (DSP) 48 hr later, then lysed in buffer containing 2% TX100. 200 μg lysate was precleared with protein A/G agarose, then incubated with 3 μg mouse anti-GluN2B or mouse immunoglobulin G (IgG) overnight at 4°C. Antibody complexes were immobilized with fresh protein A/G agarose for 1 hr, washed, and eluted with SDS-PAGE sample buffer at room temperature, then western blotted for the indicated proteins.
Primary Neuron Culture and Immunofluorescence
Primary hippocampal cultures from littermate P0–P1 WT or NS pups were made as described previously (Lin et al., 2013). Cells were transfected with EGFP-N1 at day in vitro (DIV) 12 to visualize morphology, fixed with 2% PFA at DIV15, and then stained with rabbit anti-Nck2 and goat anti-rabbit Atto594, Phalloidin-Atto647N, and a GFP-booster_Atto488 nanobody.
Statistical Analysis
Most comparisons were made with unpaired two-tailed Student’s t tests (or Welch’s test if F-test showed unequal SDs) or with paired t tests, and normality was confirmed with a D’Agostino’s/Pearson’s normality test when appropriate. For object recognition, we used one-sample t tests of object 1 or novel object exploration time against a hypothetical mean of 15 s. PSD length, spine head area, and Nck2/actin immunofluorescence were not normally distributed, so we used Mann-Whitney tests. We tested for group differences in LTP with two-way ANOVA. For evoked NMDAR-EPSC decay with ifenprodil, we used two-way repeated-measures ANOVA with post hoc Holm-Sidak test for comparisons between groups. Dephosphorylation time courses were fit with a one-phase exponential decay with Y0 = 1. For NMDAR kinetic measures, we used one-way ANOVA to test for differences between groups. To analyze the effect of GST-Nck2 SH2 domain on EPSC amplitude, we used one-way ANOVA followed by Tukey post hoc test of the EPSC amplitude 30 min after protein infusion. We tested for changes in Rs during protein infusion using two-way ANOVA. All statistical analysis was performed in Prism 7 (GraphPad).
Supplementary Material
Highlights.
NMDA receptor-mediated transmission is disrupted in Noonan syndrome (NS) model mice
NS-associated hyperactive SHP2 selectively dephosphorylates GluN2B Y1252
The actin regulatory protein Nck2 binds to phosphorylated GluN2B Y1252
Disrupting Nck2-pY interactions alters NMDAR currents similar to NS mice
ACKNOWLEDGMENTS
This work was supported by NIH grants F31-MH-105043 (to A.D.L.), T32-GM-007223 (to A.D.L. and J.E.S.), T32-NS-041228 (to S.M.K.), R01-AR-066003 and R01-HL-134166 (to A.M.B.), R01-DC-013791 and R01-DC-012441 (to C.A.G.), R01-CA-154966 (to K.M.), R01-NS-089662 (to A.J.K.), and R01-NS105640 and R01-MH115939 (to A.J.K.). Additional support was from an American Heart Association Postdoctoral Fellowship grant 16POST27250302 (to X.X.), a National Science Foundation Graduate Research Fellowship (to S.M.K.), the Yale School of Medicine (to S.P.S.D. and J.R.H.), the Leukemia and Lymphoma Society Quest for Cures R0818–14 (to K.M.), and a pilot grant P30-DA-018343 from the Yale NIDA Proteomics Center (to A.J.K.). We thank C. Kaliszewski, X. Ye, and J.-S. Yi for expert technical assistance, and Koleske lab members for critical feedback on the project.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, six figures, and three tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.07.006.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Altmüller F, Pothula S, Annamneedi A, Nakhaei-Rad S, Montenegro-Venegas C, Pina-Fernández E, Marini C, Santos M, Schanze D, Montag D, et al. (2017). Aberrant neuronal activity-induced signaling and gene expression in a mouse model of RASopathy. PLoS Genet 13, e1006684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes M, and Biala G (2012). The novel object recognition memory: neurobiology, test procedure, and its modifications. Cogn. Process 13, 93–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, Kutok JL, Yang W, Pao LI, Gilliland DG, Epstein JA, and Neel BG (2004). Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat. Med 10, 849–857. [DOI] [PubMed] [Google Scholar]
- Chen B-S, and Roche KW (2007). Regulation of NMDA receptors by phosphorylation. Neuropharmacology 53, 362–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-NP, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, Antonakos B, Chen CH-T, Chen Z, Cooke VG, et al. (2016). Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535, 148–152. [DOI] [PubMed] [Google Scholar]
- Duenas DA, Preissig S, Summitt RL, Wilroy RS, Lemmi H, and Dews JE (1973). Neurologic manifestations of the Noonan syndrome. South. Med. J 66, 193–196. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, and Sanes JR (2000). Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28, 41–51. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, and Harris KM (2002). Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res. Brain Res. Rev 39, 29–54. [DOI] [PubMed] [Google Scholar]
- Frese S, Schubert W-D, Findeis AC, Marquardt T, Roske YS, Stradal TEB, and Heinz DW (2006). The phosphotyrosine peptide binding specificity of Nck1 and Nck2 Src homology 2 domains. J. Biol. Chem 281, 18236–18245. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Fu W, Li Y, Witke W, Kwiatkowski DJ, and Mattson MP (1997). The actin-severing protein gelsolin modulates calcium channel and NMDA receptor activities and vulnerability to excitotoxicity in hippocampal neurons. J. Neurosci 17, 8178–8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JA, Shi Y, Usui H, During MJ, Sakimura K, and Nicoll RA (2011). Distinct modes of AMPA receptor suppression at developing synapses by GluN2A and GluN2B: single-cell NMDA receptor subunit deletion in vivo. Neuron 71, 1085–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Ogden KK, Yuan H, and Traynelis SF (2014). Distinct functional and pharmacological properties of Triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron 81, 1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Liang L, Liu P, Wei H, Sachs F, Niu W, and Wang W (2013). Mechanical effects on KATP channel gating in rat ventricular myocytes. PLoS ONE 8, e63337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husi H, Ward MA, Choudhary JS, Blackstock WP, and Grant SGN (2000). Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat. Neurosci 3, 661–669. [DOI] [PubMed] [Google Scholar]
- Inagaki K, Noguchi T, Matozaki T, Horikawa T, Fukunaga K, Tsuda M, Ichihashi M, and Kasuga M (2000). Roles for the protein tyrosine phosphatase SHP-2 in cytoskeletal organization, cell adhesion and cell migration revealed by overexpression of a dominant negative mutant. Oncogene 19, 75–84. [DOI] [PubMed] [Google Scholar]
- Keilhack H, David FS, McGregor M, Cantley LC, and Neel BG (2005). Diverse biochemical properties of Shp2 mutants. Implications for disease phenotypes. J. Biol. Chem 280, 30984–30993. [DOI] [PubMed] [Google Scholar]
- Kerrisk ME, Greer CA, and Koleske AJ (2013). Integrin α 3 is required for late postnatal stability of dendrite arbors, dendritic spines and synapses, and mouse behavior. J. Neurosci 33, 6742–6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhr G, and Seeburg PH (1996). Subtype-specific regulation of recombinant NMDA receptor-channels by protein tyrosine kinases of the src family. J. Physiol 492, 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni VA, and Firestein BL (2012). The dendritic tree and brain disorders. Mol. Cell. Neurosci 50, 10–20. [DOI] [PubMed] [Google Scholar]
- Lee Y-S, Ehninger D, Zhou M, Oh J-Y, Kang M, Kwak C, Ryu H-H, Butz D, Araki T, Cai Y, et al. (2014). Mechanism and treatment for learning and memory deficits in mouse models of Noonan syndrome. Nat. Neurosci 17, 1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y-C, Yeckel MF, and Koleske AJ (2013). Abl2/Arg controls dendritic spine and dendrite arbor stability via distinct cytoskeletal control pathways. J. Neurosci 33, 1846–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longair MH, Baker DA, and Armstrong JD (2011). Simple Neurite Tracer: open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics 27, 2453–2454. [DOI] [PubMed] [Google Scholar]
- Lu W, Fang W, Li J, Zhang B, Yang Q, Yan X, Peng L, Ai H, Wang J-J, Liu X, et al. (2015). Phosphorylation of tyrosine 1070 at the GluN2B subunit is regulated by synaptic activity and critical for surface expression of N-methyl-D-aspartate (NMDA) receptors. J. Biol. Chem 290, 22945–22954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer BJ (2000). Using protein-interaction domains to manipulate signaling pathways In Signaling Networks and Cell Cycle Control, Gutkind JS, ed. (Springer Nature; ), pp. 439–452. [Google Scholar]
- Mercan F, and Bennett AM (2010). Analysis of protein tyrosine phosphatases and substrates. Curr. Protoc. Mol. Biol Chapter 18, Unit 18.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, and Seeburg PH (1994). Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Komai S, Tezuka T, Hisatsune C, Umemori H, Semba K, Mishina M, Manabe T, and Yamamoto T (2001). Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. J. Biol. Chem 276, 693–699. [DOI] [PubMed] [Google Scholar]
- Ng KY, and Machida K (2017). Rosette assay: highly customizable dot-blot for SH2 domain screening. Methods Mol. Biol 1555, 437–451. [DOI] [PubMed] [Google Scholar]
- Noonan JA (1968). Hypertelorism with Turner phenotype. A new syndrome with associated congenital heart disease. Am. J. Dis. Child 116, 373–380. [DOI] [PubMed] [Google Scholar]
- Nörenberg W, Hofmann F, Illes P, Aktories K, and Meyer DK (1999). Rundown of somatodendritic N-methyl-D-aspartate (NMDA) receptor channels in rat hippocampal neurones: evidence for a role of the small GTPase RhoA. Br. J. Pharmacol 127, 1060–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J-Y, Rhee S, Silva AJ, Lee Y-S, and Kim HK (2017). Noonan syndrome-associated SHP2 mutation differentially modulates the expression of postsynaptic receptors according to developmental maturation. Neurosci. Lett 649, 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paardekooper Overman J, Yi J-S, Bonetti M, Soulsby M, Preisinger C, Stokes MP, Hui L, Silva JC, Overvoorde J, Giansanti P, et al. (2014). PZR coordinates Shp2 Noonan and LEOPARD syndrome signaling in zebrafish and mice. Mol. Cell. Biol 34, 2874–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkey KA, Askalan R, Paul S, Kalia LV, Nguyen TH, Pitcher GM, Salter MWW, and Lombroso PJ (2002). Tyrosine phosphatase STEP is a tonic brake on induction of long-term potentiation. Neuron 34, 127–138. [DOI] [PubMed] [Google Scholar]
- Pierpont EI, Pierpont ME, Mendelsohn NJ, Roberts AE, Tworog-Dube E, and Seidenberg MS (2009). Genotype differences in cognitive functioning in Noonan syndrome. Genes Brain Behav 8, 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S, and Wenthold RJ (2005). The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron 47, 845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauner C, and Köhr G (2011). Triheteromeric NR1/NR2A/NR2B receptors constitute the major N-methyl-D-aspartate receptor population in adult hippocampal synapses. J. Biol. Chem 286, 7558–7566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves SA, Ueki K, Sinha B, Difiglia M, and Louis DN (1996). Regional expression and subcellular localization of the tyrosine-specific phosphatase SH-PTP2 in the adult human nervous system. Neuroscience 71, 1037–1042. [DOI] [PubMed] [Google Scholar]
- Robert A, and Howe JR (2003). How AMPA receptor desensitization depends on receptor occupancy. J. Neurosci 23, 847–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R, Nollau P, Ho HY, Kirschner MW, and Mayer BJ (2001). Nck and phosphatidylinositol 4,5-bisphosphate synergistically activate actin polymerization through the N-WASP-Arp2/3 pathway. J. Biol. Chem 276, 26448–26452. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, and Westbrook GL (1993). Calcium-induced actin depolymerization reduces NMDA channel activity. Neuron 10, 805–814. [DOI] [PubMed] [Google Scholar]
- Sarkozy A, Digilio MC, and Dallapiccola B (2008). Leopard syndrome. Orphanet J. Rare Dis 3, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumilina EV, Negulyaev YA, Morachevskaya EA, Hinssen H, and Khaitlina SY (2003). Regulation of sodium channel activity by capping of actin filaments. Mol. Biol. Cell 14, 1709–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DB, and Johnson KS (1988). Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67, 31–40. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MWW, Lombroso PJ, Gouras GK, and Greengard P (2005). Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci 8, 1051–1058. [DOI] [PubMed] [Google Scholar]
- Stern P, Béhé P, Schoepfer R, and Colquhoun D (1992). Single-channel conductances of NMDA receptors expressed from cloned cDNAs: comparison with native receptors. Proc. Biol. Sci 250, 271–277. [DOI] [PubMed] [Google Scholar]
- Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A, Jeffery S, et al. (2001). Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet 29, 465–468. [DOI] [PubMed] [Google Scholar]
- Tartaglia M, Gelb BD, and Zenker M (2011). Noonan syndrome and clinically related disorders. Best Pract. Res. Clin. Endocrinol. Metab 25, 161–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenot E, Moreau AW, Rousseau V, Combeau G, Domenichini F, Jacquet C, Goupille O, Amar M, Kreis P, Fossier P, and Barnier JV (2011). p21-Activated kinase 3 (PAK3) protein regulates synaptic transmission through its interaction with the Nck2/Grb4 protein adaptor. J. Biol. Chem 286, 40044–40059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, McGinley MJ, and Westbrook GL (2013). Triheteromeric NMDA receptors at hippocampal synapses. J. Neurosci 33, 9150–9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, and Dingledine R (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven W, Wingbermühle E, Egger J, Van der Burgt I, and Tuinier S (2008). Noonan syndrome: psychological and psychiatric aspects. Am. J. Med. Genet. A 146A, 191–196. [DOI] [PubMed] [Google Scholar]
- Woolley CS, Gould E, Frankfurt M, and McEwen BS (1990). Naturally occurring fluctuation in dendritic spine density on adult hippocampal pyramidal neurons. J. Neurosci 10, 4035–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Levy AD, Rosenberg BJ, Higley MJ, and Koleske AJ (2016). Disruption of coordinated presynaptic and postsynaptic maturation underlies the defects in hippocampal synapse stability and plasticity in Abl2/Arg-deficient mice. J. Neurosci 36, 6778–6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Zhang B, Lu W, Peng L, Yang Q, Cao W, Lin S, Yu W, Li X, Ke Y, et al. (2017). Increased Src family kinase activity disrupts excitatory synaptic transmission and impairs remote fear memory in forebrain Shp2-deficient mice. Mol. Neurobiol 54, 7235–7250. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang X-F, Fleming MR, Amiri A, El-Hassar L, Surguchev AA, Hyland C, Jenkins DP, Desai R, Brown MR, et al. (2016). Kv3.3 channels bind Hax-1 and Arp2/3 to assemble a stable local actin network that regulates channel gating. Cell 165, 434–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.