

Abstract
Since the first microRNA (miRNA) was discovered, a lot of studies have confirmed the associations between miRNAs and human complex diseases. Besides, obtaining and taking advantage of association information between miRNAs and diseases play an increasingly important role in improving the treatment level for complex diseases. However, due to the high cost of traditional experimental methods, many researchers have proposed different computational methods to predict potential associations between miRNAs and diseases. In this work, we developed a computational model of Random Forest for miRNA-disease association (RFMDA) prediction based on machine learning. The training sample set for RFMDA was constructed according to the human microRNA disease database (HMDD) version (v.)2.0, and the feature vectors to represent miRNA-disease samples were defined by integrating miRNA functional similarity, disease semantic similarity, and Gaussian interaction profile kernel similarity. The Random Forest algorithm was first employed to infer miRNA-disease associations. In addition, a filter-based method was implemented to select robust features from the miRNA-disease feature set, which could efficiently distinguish related miRNA-disease pairs from unrelated miRNA-disease pairs. RFMDA achieved areas under the curve (AUCs) of 0.8891, 0.8323, and 0.8818 ± 0.0014 under global leave-one-out cross-validation, local leave-one-out cross-validation, and 5-fold cross-validation, respectively, which were higher than many previous computational models. To further evaluate the accuracy of RFMDA, we carried out three types of case studies for four human complex diseases. As a result, 43 (esophageal neoplasms), 46 (lymphoma), 47 (lung neoplasms), and 48 (breast neoplasms) of the top 50 predicted disease-related miRNAs were verified by experiments in different kinds of case studies. The results of cross-validation and case studies indicated that RFMDA is a reliable model for predicting miRNA-disease associations.
Keywords: microRNA, disease, association prediction, Random Forest
Graphical Abstract
Introduction
MicroRNAs (miRNAs) are a series of endogenous small non-coding RNAs (about 22 nt), which can suppress the expression of target genes by inducing the cutting degradation of mRNA, translation inhibition, or other modality regulation mechanism.1, 2, 3, 4 Line-4 and let-7, the first two miRNAs that were discovered more than 20 years ago,5, 6, 7 act as positive regulators coincidently. Since then, as many studies on miRNAs have been carried out, a mass of miRNAs was found in viruses, green algae plants, and animals.8 Furthermore, several studies have demonstrated that miRNAs are in connection with many important biological processes, such as cell growth,9 cell death,10 cell proliferation,11 immune reaction,12 signal transduction,13 tumor invasion,14 and viral infection.15 Hence, it is no surprise that miRNAs could be associated with different kinds of diseases.16
With the development of biotechnology and accumulation of theories, more and more associations between miRNAs and diseases have been discovered. Yao et al.17 found that miRNA-103 and miRNA-107 inhibit the translation of cofilin. Moreover, the decrease of miRNA-103 or miRNA-107 levels and the increase of cofilin protein levels happened at the same time in a transgenic mouse model of Alzheimer’s disease. Gao et al.18 discovered the phenomenon that deregulation of miRNA-145 and miRNA-199 expression happened in previous stages of hepatitis B virus (HBV)-associated multi-step hepatocarcinogenesis. In addition, miRNA-155 plays an important role in the induction of chronic gastritis and colitis and the T cell-mediated control of Helicobacter pylori infection.19 A recent study also showed that miRNA-23, miRNA-24, and miRNA-27 contained underlying therapeutic factors in ischemic heart and vascular disorders disease.20 Hence, there is no doubt that obtaining and taking advantage of association information between miRNAs and diseases could improve the treatment level for complex diseases. However, since experimental methods may consume plenty of time, numerous materials, and a lot of labor to find associations, proposing efficient computational methods based on existing databases is expected to significantly reduce the workload. Indeed, a growing number of computational models have been developed to predict potential associations between miRNAs and diseases in recent years.21, 22, 23, 24, 25, 26
On the basis of a reasonable conjecture that functionally similar miRNAs tend to link with diseases that have similar phenotypes, lots of computational methods are proposed for predicting associations between miRNAs and diseases.27, 28, 29 Jiang et al.30 created a hypergeometric distribution-based computational method by integrating miRNA functional similarity network, disease phenotype similarity network, and known human miRNA-disease association network. However, only using the information of miRNA neighbors resulted in an unsatisfactory prediction performance of this model. Later, Shi et al.31 built a computational model to identify unknown miRNA-disease associations by using the algorithm of random walk on the protein-protein interaction (PPI) network. Under the conjecture that a miRNA may be associated with a certain disease when target genes of the miRNA have connection with this disease, they predicted novel miRNA-disease associations by integrating PPI network, miRNA-target interaction network, and gene-disease interaction network. Mørk et al.32 proposed a miRNA-protein-disease (miRPD) association prediction model to judge whether miRNAs link with diseases via considering the underlying proteins. After combining the known miRNA-disease associations, text-mined disease-protein associations, and predicted miRNA-protein associations into a scoring framework, they finally ranked the miRNA-disease pairs to infer miRNA-disease associations. Xu et al.33 developed a comprehensive prioritization method for prioritizing miRNAs associated with disease, without using any known miRNA-disease associations, by integrating a few diseases’ phenotypes with suited mRNAs and miRNA expression profiles. However, all of the above computational models have a common shortcoming because they relied much on miRNA-target interactions with high false-positive and false-negative ratios.
There were also some classical calculation models without depending on miRNA-target interactions. A prediction method named “human disease-related miRNA prediction” (HDMP) was presented.34 To get a good performance, they computed the functional similarity of two miRNAs by integrating information of phenotype similarity between diseases and description of disease terms and distributing higher weights to miRNAs that were members of a miRNA cluster and/or family. Nevertheless, HDMP could not work for novel diseases without known associated miRNAs because the prediction process was mainly based on miRNAs’ neighbors. Chen et al.35 proposed a method named “random walk with restart for miRNA-disease association” (RWRMDA) to predict miRNA-disease associations by applying random walk with restart to find candidate miRNAs for the concerned disease. RWRMDA achieved a satisfactory performance, but it still failed to seek potential associated miRNAs for new diseases without any known related miRNAs. Later, Xuan et al.36 presented a prediction method named “miRNAs associated with disease prediction” (MIDP), still based on random walk, which utilized different kinds of topologies and features of nodes. They extended the work on a miRNA-disease bilayer network so that their model could be used to predict candidate diseases without any known associated miRNAs. Chen et al.37 further proposed a model named “within and between score for miRNA-disease association” (WBSMDA) prediction, which could avoid the above limitation as well. They calculated within scores, based on the information of known associated miRNA-disease pairs, and between scores, according to the information of unlabeled miRNA-disease pairs. WBSMDA could infer not only potential miRNAs for novel disease but also potential diseases for novel miRNA. Another computational method named “heterogeneous graph inference for miRNA-disease association” (HGIMDA) prediction was built by Chen et al.38 to predict miRNA-disease associations through an iterative process. They obtained a convergent association probability matrix after some steps, since the two similarity matrices for miRNAs and diseases were respectively normalized properly. Similarly, HGIMDA could also work for new diseases as well as new miRNAs.
Li et al.39 proposed a matrix completion algorithm named “matrix completion for miRNA-disease association” (MCMDA) prediction by updating the adjacency matrix more efficiently based on the known miRNA-disease association information only. Yu et al.40 applied the maximizing information flow approach (MaxFlow) for the first time to predict miRNA-disease associations through integrating disease phenotypic and semantic similarity network, miRNA functional similarity network, and known miRNA-disease association network into a phenome-microRNAome network and maximizing network information flow. Chen et al.41 presented a computational model of ranking-based K-nearest neighbor (KNN) for miRNA-disease association prediction (RKNNMDA). In the model, the K-nearest neighbor algorithm was used to search for k-nearest neighbors by combining known similarity networks. This model could also work for diseases (or miRNAs) without any known associated miRNAs (or diseases).
In addition, prediction models based on machine learning have also been frequently utilized to search for potential miRNA-disease associations. Xu et al.42 proposed a model named “miRNA target-dysregulated network” (MTDN) based on support vector machine (SVM) to prioritize candidate disease-related miRNAs for prostate cancer. The algorithm MTDN was utilized to define four features of a miRNA, and then the SVM classifier divided miRNAs into two categories, positive and negative, as the result of prediction. Later, Chen and Yan43 developed a semi-supervised learning method named “regularized least-squares for miRNA-disease association” (RLSMDA) prediction to infer miRNA-disease associations. Negative samples are not necessary in the model, but it’s still hard to select optimal parameter values. Next Chen et al.44 proposed a computational model called “restricted Boltzmann machine for multiple types of miRNA-disease association” (RBMMMDA) to predict different types of associations between miRNAs and diseases. Compared with former methods, which could only predict binary miRNA-disease associations, RBMMMDA could obtain both new miRNA-disease associations and corresponding association types.
As we know, Random Forest has been successfully applied to a wide range of bioinformatics problems, including protein or peptide identification,45 in vivo transcription factor-binding prediction,46 enhancer identification,47 and functional annotation of non-coding SNPs.48, 49 In this study, we developed a novel efficient computational model of Random Forest for miRNA-disease association (RFMDA) prediction (motivated by the study of Cheng et al.50). First, we constructed training samples for data preparation. Second, RFMDA made full use of biological information of miRNAs and diseases by integrating the miRNA functional similarity network or disease semantic similarity network with the Gaussian interaction profile kernel similarity network for miRNAs or diseases. Each miRNA-disease pair (m(i), d(j)) was represented by a feature vector based on the above similarity networks. Then, a feature selection method was used to cut down the dimensionality of feature vectors for efficiently distinguishing associated miRNA-disease pairs from unassociated miRNA-disease pairs and decreasing computational cost. Finally, a Random Forest prediction model could be obtained by training the samples mentioned in the first step.
To evaluate the performance of RFMDA, local and global leave-one-out cross-validations (LOOCVs) as well as 5-fold cross-validation were implemented. As a result, the areas under the curve (AUCs) of global and local LOOCVs were 0.8891 and 0.8323, respectively; and, the AUC obtained from 5-fold cross-validation was 0.8818 ± 0.0014. Besides, we implemented three types of case studies on esophageal neoplasms, lymphoma, lung neoplasms, and breast neoplasms. The top 10 and top 50 candidate miRNAs associated with these four diseases obtained from RFMDA were verified by experimental reports in some representative databases. As a result, 86% (esophageal neoplasms), 92% (lymphoma), 94% (lung neoplasms), and 96% (breast neoplasms) of the top 50 predicted miRNAs were respectively verified by recent experimental results. The data demonstrated that RFMDA is an excellent method to predict potential miRNA-disease associations.
Results
Performance Evaluation
To evaluate the performance of RFMDA, LOOCVs and 5-fold cross-validation were utilized based on the known miRNA-disease associations in the human microRNA disease database (HMDD) version (v.)2.0. The dataset contains 5,430 known miRNA-disease associations between 495 miRNAs and 383 diseases. In our model, all known miRNA-disease associations were treated as positive samples, while the unknown miRNA-disease pairs were treated as unlabeled samples. Global LOOCV and local LOOCV are two categories of LOOCV. For global LOOCV, each positive sample would be left out in turn as a test sample, and other positive samples were used to train the model. RFMDA would give a predicted score to the test sample and each unlabeled sample. After sorting all scores in decreasing order, we could obtain the ranking of the test sample. Finally, 5,430 rankings could be obtained by this way.
Then we drew the Receiver Operating Characteristic (ROC) curves by plotting the true positive rate (TPR, sensitivity) against the false positive rate (FPR, 1-specificity) with different thresholds. Sensitivity shows the percentage of test samples that was ranked in front of the given threshold, while specificity demonstrates the percentage of negative miRNA-disease associations whose ranks were lower than the given threshold. The ROC AUC was regarded as a standard for performance evaluation. A higher AUC indicates more excellent prediction performance of a prediction model. What makes a difference in local LOOCV is that the test sample was ranked with miRNAs that have no known association with the investigated disease according to the prediction scores.
As shown in Figures 1 and 2, RFMDA achieved an AUC of 0.8891, which is higher than AUCs of 0.8781 (HGIMDA), 0.8749 (MCMDA), 0.8624 (MaxFlow), 0.8426 (RLSMDA), 0.8366 (HDMP), and 0.8030 (WBSMDA) in global LOOCV. Besides, in local LOOCV, HGIMDA, MCMDA, MaxFlow, RLSMDA, HDMP, WBSMDA, MIDP, MiRAI, and RWRMDA obtained AUCs of 0.8077, 0.7718, 0.7774, 0.6953, 0.7702, 0.8031, 0.8196, 0.6299, and 0.7891, respectively. The AUCs of all the nine models were lower than RFMDA’s AUC of 0.8323. As we can see, MIDP, RWRMDA, and MiRAI only appeared in local LOOCV comparison. On the one hand, MIDP and RWRMDA were based on random walk, which is a local method so that these two methods could not be used to predict for all diseases simultaneously. On the other hand, the association score between a disease (miRNA) and its candidate miRNAs (diseases) computed by MiRAI was extremely correlated with how many known miRNAs (diseases) associated with the disease (miRNAs). For a disease (miRNA) with more known associated miRNAs (diseases), the prediction scores between the disease (miRNA) and its candidate miRNAs (diseases) would be higher. Therefore, it is unfair to compare the prediction scores obtained from different diseases. MiRAI obtained a lower AUC after being implemented on our training dataset because the model suffered from the data sparsity problem. In our training dataset, the majority of 383 diseases (495 miRNAs) were associated with only a few miRNAs (diseases). However, MiRAI was implemented on the dataset that included 83 diseases with at least 20 known associated miRNAs for each in the original literature.51 It is obvious that AUCs of RFMDA were higher than all of the previous methods mentioned above both in local and global LOOCVs. Both the global and local LOOCVs showed the excellent prediction performance of our model.
Figure 1.

AUCs of RFMDA and HGIMDA, RLSMDA, HDMP, WBSMDA, MaxFlow, and MCMDA under Global LOOCV
As one can see, RFMDA achieved AUCs of 0.8891 under global LOOCV, which were higher than those of previous models.
Figure 2.

AUCs of RFMDA and HGIMDA, RLSMDA, HDMP, WBSMDA, RWRMDA, MaxFlow, MCMDA, MIDP, and MiRAI under Local LOOCV
As one can see, RFMDA achieved AUCs of 0.8323 under local LOOCV, which were higher than those of previous models.
As for 5-fold cross-validation, we evenly divided positive samples into 5 parts, and each part would be treated as test samples in turn; and, each miRNA-disease pair in the test sample would be ranked with all unlabeled samples based on their prediction scores. The whole process was repeated 100 times to avoid evaluation bias. RFMDA achieved an AUC of 0.8818 ± 0.0014 in 5-fold cross-validation. The average AUC of 0.8818 under 100 cross-validation is still higher than the average AUCs of 0.8767 (MCMDA), 0.8579 (MaxFlow), 0.8569 (RLSMDA), 0.8342 (HDMP), and 0.8185 (WBSMDA). The average AUC revealed the superiority of our model, and the SD of 0.0014 demonstrated the stability of RFMDA.
Case Studies
To further evaluate the prediction performance of our model, we carried out three types of case studies on four diseases. The first type was implemented on esophageal neoplasms and lymphoma. Here, all known miRNA-disease associations in the HMDD v.2.0 were put into the training set of RFMDA. We selected the top 50 predicted miRNAs associated with the investigated disease based on their prediction scores, and then we validated them in another two databases, namely, dbDEMC52 and miR2Disease.53
Esophageal neoplasms ranked eighth in the most common cancers and sixth in cancer mortality all over the world, according to the literature.54 In the United States, about 10 in 100,000 people die of esophageal neoplasms every year, and the number of male patients was about four times as many as female patients.55 Diagnosing the disease in the early stages would enhance the survival rate of patients.56 Many experiments have confirmed that there are many miRNAs related to esophageal neoplasms. For example, the methylation ratios of miRNA-34a, miRNA-34b/c, and miRNA-129-2 are 66.67%, 40.74%, and 96.30%, respectively, in esophageal squamous cell carcinoma, which are obviously higher than those in non-tumor tissues.57 We selected esophageal neoplasm as an example in the first type of case study, then RFMDA was implemented to predict miRNAs potentially associated with the disease. As a result, 10 of the top 10 and 43 of the top 50 predicted miRNAs were verified by experimental data in dbDEMC or miR2Disease (see Table 1).
Table 1.
Top 50 miRNAs Associated with Esophageal Neoplasms Were Predicted by RFMDA Based on Known Associations in the HMDD v.2.0
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa-mir-127 | dbDEMC | hsa-mir-30a | dbDEMC |
| hsa-let-7g | dbDEMC | hsa-mir-125b | dbDEMC |
| hsa-mir-222 | dbDEMC | hsa-mir-7 | dbDEMC |
| hsa-mir-221 | dbDEMC | hsa-mir-18a | dbDEMC |
| hsa-mir-30c | dbDEMC | hsa-mir-95 | dbDEMC |
| hsa-mir-146b | dbDEMC | hsa-let-7i | dbDEMC |
| hsa-mir-372 | dbDEMC | hsa-mir-181a | dbDEMC |
| hsa-mir-181b | dbDEMC | hsa-mir-204 | dbDEMC |
| hsa-mir-10b | dbDEMC | hsa-mir-107 | unconfirmed |
| hsa-mir-93 | dbDEMC | hsa-mir-451 | dbDEMC and miR2Disease |
| hsa-mir-16 | dbDEMC | hsa-mir-122 | dbDEMC |
| hsa-mir-200b | dbDEMC | hsa-mir-335 | unconfirmed |
| hsa-mir-142 | dbDEMC | hsa-let-7f | dbDEMC |
| hsa-mir-191 | dbDEMC | hsa-mir-29a | unconfirmed |
| hsa-mir-9 | dbDEMC | hsa-mir-139 | dbDEMC |
| hsa-mir-199b | dbDEMC | hsa-mir-140 | dbDEMC |
| hsa-mir-137 | dbDEMC | hsa-mir-218 | dbDEMC |
| hsa-mir-20b | dbDEMC | hsa-mir-135a | unconfirmed |
| hsa-mir-132 | dbDEMC | hsa-mir-125a | dbDEMC |
| hsa-mir-18b | dbDEMC | hsa-mir-194 | dbDEMC |
| hsa-mir-449a | unconfirmed | hsa-mir-29b | dbDEMC and miR2Disease |
| hsa-mir-449b | unconfirmed | hsa-mir-30e | dbDEMC |
| hsa-mir-106a | dbDEMC | hsa-mir-27b | unconfirmed |
| hsa-mir-373 | dbDEMC and miR2Disease | hsa-mir-193b | dbDEMC |
| hsa-mir-224 | dbDEMC | hsa-mir-195 | dbDEMC |
The top 1–25 related miRNAs are recorded in the first column, and the top 26–50 related miRNAs are recorded in the third column. As we can see 10, 19, and 43 of the top 10, top 20, and top 50 were verified by databases.
Lymphoma is a cancer that starts in the lymphocytes or white blood cells.58 These cells play a critical role in the immune system, which can help us fight against various diseases in the human body.59 Hodgkin lymphoma and non-Hodgkin lymphoma are two main kinds of lymphoma that can happen in both children and adults.60 Recently, many miRNAs have been verified to be associated with lymphoma in different mechanisms. For example, plasma miRNA-92a values in non-Hodgkin lymphoma were about 5%, which were far less than those in heathy subjects.61 Besides, several miRNAs were discovered significantly overexpressed in splenic marginal zone lymphoma, including miRNA-21, miRNA-155, and miRNA-146a.62 We took lymphoma as another example in the first type of case study, and we utilized RFMDA to predict lymphoma-associated miRNAs. Finally, 9 of the top 10 and 46 of the top 50 predicted miRNAs were confirmed by experimental data in dbDEMC or miR2Disease (see Table 2).
Table 2.
Top 50 miRNAs Associated with Lymphoma Were Predicted by RFMDA Based on Known Associations in the HMDD v.2.0
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa-let-7b | dbDEMC | hsa-mir-199a | dbDEMC |
| hsa-mir-125b | unconfirmed | hsa-mir-106a | dbDEMC and miR2Disease |
| hsa-let-7a | dbDEMC | hsa-mir-29a | dbDEMC |
| hsa-let-7c | dbDEMC | hsa-mir-182 | dbDEMC |
| hsa-mir-34a | dbDEMC | hsa-mir-23b | dbDEMC |
| hsa-let-7e | dbDEMC and miR2Disease | hsa-mir-15b | dbDEMC |
| hsa-mir-106b | dbDEMC | hsa-mir-29b | dbDEMC |
| hsa-let-7d | dbDEMC | hsa-mir-27a | dbDEMC |
| hsa-mir-145 | dbDEMC and miR2Disease | hsa-mir-141 | dbDEMC |
| hsa-let-7i | dbDEMC | hsa-mir-22 | dbDEMC |
| hsa-mir-143 | dbDEMC and miR2Disease | hsa-mir-195 | dbDEMC |
| hsa-mir-222 | dbDEMC | hsa-mir-30a | dbDEMC |
| hsa-mir-221 | dbDEMC and miR2Disease | hsa-mir-196a | dbDEMC |
| hsa-mir-223 | dbDEMC | hsa-mir-1 | dbDEMC |
| hsa-mir-127 | dbDEMC and miR2Disease | hsa-mir-32 | dbDEMC |
| hsa-mir-25 | dbDEMC | hsa-mir-95 | dbDEMC and miR2Disease |
| hsa-mir-30c | dbDEMC | hsa-mir-34b | dbDEMC |
| hsa-mir-146b | unconfirmed | hsa-mir-148a | dbDEMC |
| hsa-let-7f | dbDEMC | hsa-mir-183 | dbDEMC |
| hsa-mir-181b | dbDEMC | hsa-mir-10b | dbDEMC |
| hsa-mir-214 | dbDEMC | hsa-mir-132 | dbDEMC |
| hsa-mir-191 | dbDEMC | hsa-mir-133a | dbDEMC |
| hsa-let-7g | dbDEMC | hsa-mir-199b | dbDEMC |
| hsa-mir-34c | unconfirmed | hsa-mir-335 | dbDEMC |
| hsa-mir-100 | dbDEMC | hsa-mir-372 | unconfirmed |
The top 1–25 related miRNAs are recorded in the first column, and the top 26–50 related miRNAs are recorded in the third column. As we can see 9, 18, and 46 of the top 10, top 20, and top 50 were verified by databases.
RFMDA was also implemented to predict potential miRNAs for 381 other diseases in the HMDD v.2.0 apart from esophageal neoplasms and lymphoma. The whole prediction list is shown in Table S1. The table includes three kinds of information: the disease, the miRNA, and the predicted association score.
To prove the ability of our model in predicting new diseases without known associated miRNAs, we selected lung neoplasm as an example in the second type of case study. Here, before training the model, we removed all known associations of lung neoplasms. Then, we ranked all the 495 miRNAs based on their predicted association scores, and we validated the top 50 miRNAs in the HMDD v.2.0, dbDEMC, and miR2Disease. As a result, 10 of the top 10 and 47 of the top 50 miRNAs were confirmed by these databases (see Table 3).
Table 3.
Top 50 miRNAs Associated with Lung Neoplasms Were Predicted by RFMDA after Hiding All Known Associations about Lung Neoplasms Based in the HMDD v.2.0
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa-mir-133a | dbDEMC and HMDD | hsa-mir-192 | dbDEMC, miR2Disease, and HMDD |
| hsa-mir-150 | dbDEMC, miR2Disease, and HMDD | hsa-mir-130a | dbDEMC and miR2Disease |
| hsa-mir-196a | dbDEMC and HMDD | hsa-mir-10a | dbDEMC |
| hsa-mir-210 | dbDEMC, miR2Disease, and HMDD | hsa-mir-200c | dbDEMC, miR2Disease, and HMDD |
| hsa-mir-182 | dbDEMC, miR2Disease, and HMDD | hsa-mir-148a | dbDEMC and HMDD |
| hsa-mir-204 | miR2Disease | hsa-mir-17 | miR2Disease and HMDD |
| hsa-mir-100 | dbDEMC and HMDD | hsa-mir-146a | dbDEMC, miR2Disease, and HMDD |
| hsa-mir-199b | dbDEMC, miR2Disease, and HMDD | hsa-mir-206 | HMDD |
| hsa-mir-196b | dbDEMC | hsa-mir-203 | dbDEMC, miR2Disease, and HMDD |
| hsa-mir-31 | dbDEMC, miR2Disease, and HMDD | hsa-mir-20a | dbDEMC, miR2Disease, and HMDD |
| hsa-mir-335 | miR2Disease and HMDD | hsa-mir-26a | dbDEMC, miR2Disease, and HMDD |
| hsa-mir-30a | miR2Disease and HMDD | hsa-mir-302b | dbDEMC |
| hsa-mir-1 | dbDEMC, miR2Disease, and HMDD | hsa-mir-224 | dbDEMC, miR2Disease, and HMDD |
| hsa-mir-296 | dbDEMC | hsa-mir-302c | dbDEMC |
| hsa-mir-205 | dbDEMC, miR2Disease, and HMDD | hsa-mir-181a | dbDEMC and HMDD |
| hsa-mir-27a | dbDEMC and HMDD | hsa-mir-221 | dbDEMC and HMDD |
| hsa-mir-21 | dbDEMC, miR2Disease, and HMDD | hsa-mir-95 | miR2Disease and HMDD |
| hsa-mir-183 | dbDEMC, miR2Disease, and HMDD | hsa-mir-143 | dbDEMC, miR2Disease, and HMDD |
| hsa-mir-22 | miR2Disease and HMDD | hsa-mir-32 | miR2Disease and HMDD |
| hsa-mir-200a | dbDEMC, miR2Disease, and HMDD | hsa-mir-135b | dbDEMC and HMDD |
| hsa-mir-181b | dbDEMC and HMDD | hsa-mir-135a | dbDEMC and HMDD |
| hsa-mir-146b | miR2Disease and HMDD | hsa-mir-302a | unconfirmed |
| hsa-mir-107 | dbDEMC and HMDD | hsa-mir-7 | miR2Disease and HMDD |
| hsa-mir-34c | dbDEMC and HMDD | hsa-mir-218 | dbDEMC, miR2Disease, and HMDD |
| hsa-mir-372 | unconfirmed | hsa-mir-491 | unconfirmed |
The top 1–25 related miRNAs are recorded in the first column, and the top 26–50 related miRNAs are recorded in the third column. As we can see 10, 20, and 47 of the top 10, top 20, and top 50 were verified by databases.
We took breast neoplasms (BNs) as an example in the third type of case study to evaluate the performance of RFMDA using another miRNA-disease association database. Here, we used the associations in the HMDD v.1.0 as our training set. The HMDD v.1.0 contains 1,395 known associations between 271 miRNAs and 137 diseases. The whole prediction process was similar to the first type of case study. Finally, the data showed that 10 of the top 10 and 48 of the top 50 predicted miRNAs were confirmed by experimental data recorded in the HMDD v.2.0, dbDEMC, and miR2Disease (see Table 4).
Table 4.
Top 50 miRNAs Associated with Breast Neoplasms Were Predicted by RFMDA Based on Known Associations in the HMDD v.1.0
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa-mir-223 | dbDEMC and HMDD | hsa-mir-520b | dbDEMC and HMDD |
| hsa-mir-24 | dbDEMC and HMDD | hsa-mir-23b | dbDEMC and HMDD |
| hsa-let-7b | dbDEMC and HMDD | hsa-mir-148a | dbDEMC, miR2Disease, and HMDD |
| hsa-mir-126 | dbDEMC, miR2Disease, and HMDD | hsa-mir-135a | dbDEMC and HMDD |
| hsa-mir-373 | dbDEMC, miR2Disease, and HMDD | hsa-mir-182 | dbDEMC, miR2Disease, and HMDD |
| hsa-mir-32 | dbDEMC | hsa-mir-142 | unconfirmed |
| hsa-mir-16 | dbDEMC and HMDD | hsa-let-7i | dbDEMC, miR2Disease, and HMDD |
| hsa-let-7c | dbDEMC and HMDD | hsa-mir-128b | miR2Disease |
| hsa-mir-150 | dbDEMC | hsa-mir-335 | dbDEMC, miR2Disease, and HMDD |
| hsa-mir-29c | dbDEMC, miR2Disease, and HMDD | hsa-mir-15b | dbDEMC |
| hsa-mir-372 | dbDEMC | hsa-mir-98 | dbDEMC and miR2Disease |
| hsa-mir-101 | dbDEMC, miR2Disease, and HMDD | hsa-mir-181a | dbDEMC, miR2Disease, and HMDD |
| hsa-let-7e | dbDEMC and HMDD | hsa-mir-183 | dbDEMC and HMDD |
| hsa-mir-106a | dbDEMC | hsa-mir-26a | dbDEMC, miR2Disease, and HMDD |
| hsa-let-7g | dbDEMC and HMDD | hsa-mir-100 | dbDEMC and HMDD |
| hsa-mir-99b | dbDEMC | hsa-mir-107 | dbDEMC and HMDD |
| hsa-mir-192 | dbDEMC | hsa-mir-224 | dbDEMC and HMDD |
| hsa-mir-30e | unconfirmed | hsa-mir-92b | dbDEMC |
| hsa-mir-199b | dbDEMC and HMDD | hsa-mir-95 | dbDEMC |
| hsa-mir-27a | dbDEMC, miR2Disease, and HMDD | hsa-mir-22 | dbDEMC, miR2Disease, and HMDD |
| hsa-mir-130a | dbDEMC | hsa-mir-196b | dbDEMC |
| hsa-mir-195 | dbDEMC, miR2Disease, and HMDD | hsa-mir-191 | dbDEMC, miR2Disease, and HMDD |
| hsa-mir-30a | miR2Disease and HMDD | hsa-mir-18b | dbDEMC and HMDD |
| hsa-mir-203 | dbDEMC, miR2Disease, and HMDD | hsa-mir-186 | dbDEMC |
| hsa-mir-92a | HMDD | hsa-mir-424 | dbDEMC |
The top 1–25 related miRNAs are recorded in the first column, and the top 26–50 related miRNAs are recorded in the third column. As we can see 10, 19, and 48 of the top 10, top 20, and top 50 were verified by databases.
Discussion
With the development of experimental technologies and computational tools, more and more miRNAs have been discovered in recent years. Various associations between miRNAs and diseases have attracted the attention of researchers. These associations play an important role in the prevention, diagnosis, and treatment of complex human diseases. However, using traditional experimental methods to find miRNA-disease associations may be expensive and inefficient. Thus, we proposed an efficient computational model of RFMDA based on machine learning to predict potential miRNA-disease associations.
Positive samples and negative samples were selected from known miRNA-disease associations and unlabeled miRNA-disease pairs, respectively, according to the HMDD v.2.0. We represented each miRNA-disease pair as a feature vector by integrating information of miRNA functional similarity, disease semantic similarity, and Gaussian interaction profile kernel similarity of miRNA and disease. RFMDA predicted unknown miRNA-disease associations by labeling them with scores after implementing the Random Forest algorithm. Based on LOOCV and 5-fold cross-validation, RFMDA obtained more excellent performance than lots of previous reliable computational models, such as MiRAI, MIDP, RWRMDA, MCMDA, WBSMDA, HDMP, RLSMDA, MaxFlow, and HGIMDA. In addition, the results of three types of case studies for four complex human diseases (esophageal neoplasms, lymphoma, lung neoplasms, and breast neoplasms) further demonstrated that RFMDA was a reliable prediction model.
There were several important factors that contributed to the satisfying performance of RFMDA. First, RFMDA made full use of biological information, including known miRNA-disease associations, miRNA functional similarity, disease semantic similarity, and Gaussian interaction profile kernel similarity of miRNA and disease when we constructed feature vectors for miRNA-disease pairs. Second, each miRNA-disease pair was represented by a feature vector, and then a feature selection method was used to cut down the dimension of feature vector. The method could select robust features for each miRNA-disease pair. In this way, RFMDA could efficiently distinguish related miRNA-disease pairs from unrelated miRNA-disease pairs. Finally, the model of RFMDA had a good generalization ability, which benefitted from utilizing an unbiased estimator for generalization error in the Random Forest algorithm and that the parameters of Random Forest were easy to select.
However, some limitations still exist in the model of RFMDA. RFMDA requires training samples, including both positive samples and negative samples. As we know, it is difficult or even impossible to obtain reliable negative samples. We utilized a random selection method to select negative samples based on unknown miRNA-disease associations. That may influence the final result of prediction. Besides, in the HMDD v.2.0, there are only 5,430 known miRNA-disease associations, which are far less than unknown associations between 383 diseases and 495 miRNAs. Finally, not just similarity information can be used to the constructed feature vector, with a deeper understanding of mechanisms both for miRNAs and diseases. Thus, we believe that the performance of RFMDA will be much better in the future.
Materials and Methods
Human miRNA-Disease Associations
The information of 5,430 known human miRNA-disease associations between 383 diseases and 495 miRNAs was obtained from the HMDD v.2.0.63 We constructed an adjacency matrix A with 383 (nd) rows and 495 (nm) columns to briefly store the information of known and unknown miRNA-disease associations between 383 diseases and 495 miRNAs. The element A(d(i), m(j)) is equal to 1 when miRNA m(j) had been verified to be associated with disease d(i), otherwise 0.
miRNA Functional Similarity
Under the assumption that functionally similar miRNAs tend to link with phenotypically similar diseases, Wang et al.28 developed a method to compute the miRNA functional similarity score of two miRNAs. We downloaded the miRNA functional similarity scores from http://www.cuilab.cn/. Then, we constructed the miRNA functional similarity matrix FS with 495 rows and 495 columns, where the element FS(m(i), m(j)) denotes the functional similarity score between miRNA m(i) and miRNA m(j).
Disease Semantic Similarity Model 1
Medical subject headings (MeSH) disease descriptors were downloaded from the National Library of Medicine (https://www.nlm.nih.gov/), which furnished a rigorous system for disease classification. In the system, each disease could be described by a directed acyclic graph (DAG), in which the nodes represent diseases and each of the direct edges connects two nodes from parent node to child node. A disease D can be described as DAGD = (D, TD, ED), where TD is a node set containing disease D and its ancestor diseases and ED is an edge set containing the corresponding edges.28 Actually, Xiang et al.64 utilized MeSH gene descriptors to calculate dissimilarity between genes by the GenoMeSH algorithm. Here, we computed disease semantic similarity based on MeSH disease descriptors in another method according to previous study.34 Specifically, we defined the contribution of disease t to the semantic value of disease D as follows.
| (Equation 1) |
where is the semantic contribution decay factor. It will reduce the contribution of disease t if t is different from D. Besides, the contribution of disease D to its own semantic value is equal to 1.
Moreover, the semantic value DV1(D) of disease D was defined as follows.
| (Equation 2) |
The semantic similarity value between disease d(i) and d(j) could be computed based on a conjecture that two diseases will be more similar if they share a larger part of their DAGs,
| (Equation 3) |
where SS1 is a disease semantic similarity matrix with 383 rows and 383 columns and the element SS1(d(i),d(j)) represents the semantic similarity of d(i) and d(j) based on disease semantic similarity model 1.
Disease Semantic Similarity Model 2
Each disease can be described as a hierarchical DAG in which the parent node represents a more general disease and the child node represents a more specific disease. According to disease semantic similarity model 1, the contributions of different diseases in the same layer of DAGD to the semantic value of D are at a same level. However, these diseases may appear in other DAGs, and the number of DAGs in which they appear may be different. Thus, we believe that the contributions of these diseases should be distinguished. The contributions of diseases appearing in other DAGs more frequently should be less than specific diseases that appear in fewer DAGs. According to previous study,34 the contribution of disease t to the semantic value of disease D can be calculated as follows.
| (Equation 4) |
The semantic similarity value between disease d(i) and d(j) was calculated similarly to the disease semantic similarity model 1 as follows,
| (Equation 5) |
where DV2(d(i)) and DV2(d(j)) are semantic values of d(i) and d(j), respectively, which can be calculated similarly to Equation 2. SS2 is another disease semantic similarity matrix with 383 rows and 383 columns, and the element SS2(d(i), d(j)) represents the semantic similarity of d(i) and d(j) based on disease semantic similarity model 2.
Gaussian Interaction Profile Kernel Similarity for Diseases
Under the assumption that similar diseases are more likely to be related with functionally similar miRNAs and vice versa, the Gaussian interaction profile kernel similarity for diseases can be computed.65 We defined binary vector IP(d(u)) to represent the interaction profiles of disease d(u) by observing whether d(u) is associated with each of the 495 miRNAs. The binary vector IP(d(u)) is equivalent to the u-th row vector of adjacency matrix A. Then the Gaussian interaction profile kernel similarity between d(u) and d(v) was defined as follows,
| (Equation 6) |
where parameter was implemented to tune the kernel bandwidth, which was calculated via normalizing the original parameter as follows.
| (Equation 7) |
Gaussian Interaction Profile Kernel Similarity for miRNAs
The Gaussian profile kernel similarity between miRNAs was calculated similarly to the method of disease Gaussian interaction profile kernel similarity computation:
| (Equation 8) |
| (Equation 9) |
where binary vector IP(m(u)) (or IP(m(v))) represents the interaction profiles of miRNA m(u) (or m(v)) by observing whether m(u) (or m(v)) is associated with each of the 383 diseases and is equivalent to the u-th (or v-th) column vector of adjacency matrix A.
Integrated Similarity for Diseases
To make full use of disease semantic similarity 1, disease semantic similarity 2, and disease Gaussian interaction profile kernel similarity, an integrated disease similarity matrix SD was constructed by integrating the above similarities. According to previous study,37 the element SD(d(u), d(v)) represented integrated similarity between disease d(u) and d(v) and was defined as follows,
| (Equation 10) |
where d(u) and d(v) have semantic similarity if both d(u) and d(v) have their own DAGs.
Integrated Similarity for miRNAs
We integrated miRNA functional similarity and miRNA Gaussian interaction profile kernel similarity in a similar way into the integrated miRNA similarity. Thus, the integrated similarity between miRNA m(i) and m(j) was calculated as follows.
| (Equation 11) |
RFMDA
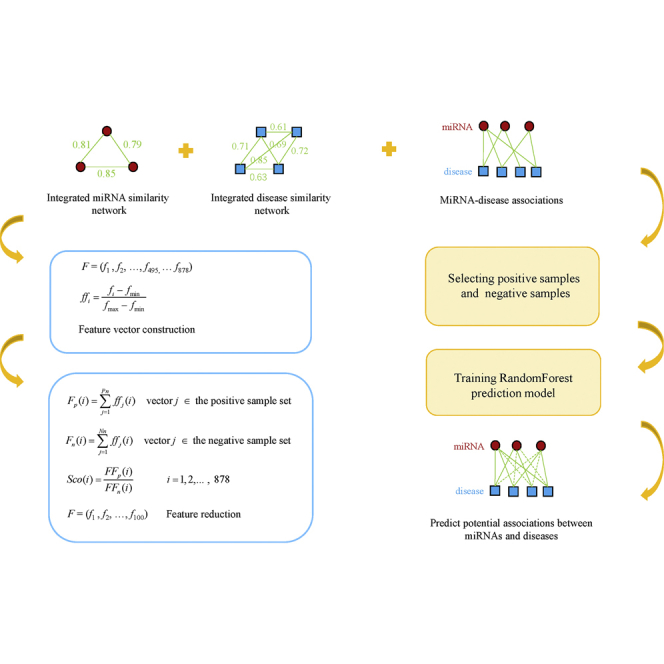
The RFMDA model was constructed based on Random Forest for predicting miRNA-disease associations, which can be divided into four steps (see Figure 3; motivated by the study of Cheng et al.50): (1) selecting positive samples and negative samples; (2) constructing feature vectors to represent samples; (3) reducing the dimension of feature; (4) constructing the final prediction model and predicting miRNA-disease associations. The details of each step are described below.
Figure 3.

Flowchart of RFMDA Model to Predict Potential Associations between miRNAs and Diseases
First, we constructed a training sample set by selecting both positive samples and negative samples according to the ratio of 1:1. The 5,430 known associated miRNA-disease pairs were extracted from the HMDD v.2.0 to compose the positive sample set. Based on the assumption that if there is no confirmed association between a miRNA and a disease, then the miRNA and the disease constitute a negative sample, we randomly selected 5,430 negative samples to compose a negative sample set. The process of randomly selecting negative samples can be roughly divided into three steps. Above all, we randomly selected one of the 383 diseases; then we randomly selected a miRNA from the 495 miRNAs; finally, the disease and the miRNA constitute a negative sample if the miRNA-disease pair isn’t a member of the 5,430 known miRNA-disease associations. We repeated this process until 5,430 negative samples were obtained. Our training sample set consisted of positive and negative sample sets.
Second, we represented each miRNA-disease pair by a feature vector. The semantic similarity 1, semantic similarity 2, and Gaussian interaction profile kernel similarity between each disease can be calculated. For each disease, there were 383 integrated similarity values. We used integrated semantic similarity values as features to represent each disease by a 383-dimensional feature vector. For example, we represented disease d(u) by a feature vector,
| (Equation 12) |
where SD(d(u)) is the u-th row vector of matrix SD, and av is the integrated similarity value between disease d(u) and d(v).
For each miRNA, we could obtain 495 integrated similarity values through integrating miRNA functional similarity and Gaussian interaction kernel profile similarity between the miRNA and all 495 miRNAs, including itself. A miRNA m(i) could be represented by a 495-dimensional feature vector in a similar way to disease,
| (Equation 13) |
where SM(m(i)) is the i-th column vector of matrix SM, and bj is the integrated similarity value between miRNA m(i) and m(j).
Therefore, each miRNA-disease sample could be described by an 878-dimensional vector based on integrated similarity for the miRNA and integrated similarity for the disease,
| (Equation 14) |
F = (f1, f2, …, f495, …, f878), where (f1, f2, …, f495) represents the 495 integrated similarity values of the miRNA, and (f496, f497, …, f878) represents the 383 integrated similarity values of the disease. Then we normalized fi to ffi as follows,
| (Equation 15) |
where fmax and fmin are the maximum and the minimum of all fi (i = 1, 2, …, 878).
Third, we reduced the dimension of feature vectors to reduce the computational cost and obtain more effective features. Our purpose was to select those discriminative features that either frequently appear in the positive sample set but seldom appear in the negative sample set or frequently appear in the negative sample set but seldom appear in the positive sample set. We used ffj(i) to denote the i-th feature of j-th vector and let Fp(i) and Fn(i) respectively represent the feature occurrence frequency in the positive sample set and negative sample set. Fp(i) and Fn(i) can be computed as follows,
| (Equation 16) |
| (Equation 17) |
where Pn and Nn are the number of positive samples and negative samples, respectively.
Fp(i) and Fn(i) are futher normalized to FFp(i) and FFn(i) in a similar way to Equation 15. Then the final score of every feature can be calculated by
| (Equation 18) |
To achieve our purpose, Sco(i) can be utilized to judge whether the i-th feature is effective, as Sco(i) measures the relative enrichment of the i-th feature in the positive samples over the negative samples. For a feature i, when FFp(i) in P is large but FFn(i) in N is small, Sco(i) will be large. On the contrary, Sco(i) will be small when FFp(i) in P is small but FFn(i) in N is large. The most effective feature has the largest or smallest score. In this study, we cut down the dimension of feature vector from 878 to 100. Thus, 50 features with the largest scores and 50 features with the smallest scores were selected to represent each sample by a 100-dimensional vector, which could improve the ability of our model to distinguish positive miRNA-disease associations from negative associations.
Finally, RandomForestRegressor, an algorithm package of Random Forest, was implemented to train the prediction model by training sample set. More specifically, each of the samples in the training set was represented by a 100-dimensional vector according to step 2 and step 3. Each sample in the positive sample set was given a label of 1, and each sample in the negative sample set was given a label of 0. Then we put these training samples’ data into the package of Random Forest. After training, we obtained a prediction model that could infer potential miRNA-disease associations by scoring miRNA-disease samples. The higher score of a miRNA-disease sample indicates that the miRNA is more likely to be associated with the disease. It’s worth noting that the max_features, n_estimators, and min_samples_leaf, main parameters of RandomForestRegressor, were set to 0.2, 100, and 10, respectively, according to empirical data.
Author Contributions
All authors contributed important elements to the work presented herein. X.C. conceived the project, developed the prediction method, designed the experiments, analyzed the results, and wrote the paper. C.-C.W. implemented the experiments, analyzed the results, and wrote the paper. J.Y. and Z.-H.Y. analyzed the results and revised the paper. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
X.C. was supported by the National Natural Science Foundation of China under grant 61772531.
Footnotes
Supplemental Information includes one table and can be found with this article online at https://doi.org/10.1016/j.omtn.2018.10.005.
Contributor Information
Xing Chen, Email: xingchen@amss.ac.cn.
Zhu-Hong You, Email: zhuhongyou@ms.xjb.ac.cn.
Supplemental Information
References
- 1.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 3.Meister G., Tuschl T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004;431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- 4.Ambros V. microRNAs: tiny regulators with great potential. Cell. 2001;107:823–826. doi: 10.1016/s0092-8674(01)00616-x. [DOI] [PubMed] [Google Scholar]
- 5.Lee R.C., Feinbaum R.L., Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 6.Reinhart B.J., Slack F.J., Basson M., Pasquinelli A.E., Bettinger J.C., Rougvie A.E., Horvitz H.R., Ruvkun G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 7.Wightman B., Ha I., Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- 8.Griffiths-Jones S., Saini H.K., van Dongen S., Enright A.J. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ambros V. MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell. 2003;113:673–676. doi: 10.1016/s0092-8674(03)00428-8. [DOI] [PubMed] [Google Scholar]
- 10.Xu P., Guo M., Hay B.A. MicroRNAs and the regulation of cell death. Trends Genet. 2004;20:617–624. doi: 10.1016/j.tig.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 11.Zhang K., Guo L. MiR-767 promoted cell proliferation in human melanoma by suppressing CYLD expression. Gene. 2018;641:272–278. doi: 10.1016/j.gene.2017.10.055. [DOI] [PubMed] [Google Scholar]
- 12.Taganov K.D., Boldin M.P., Chang K.J., Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui Q., Yu Z., Purisima E.O., Wang E. Principles of microRNA regulation of a human cellular signaling network. Mol. Syst. Biol. 2006;2:46. doi: 10.1038/msb4100089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma L., Teruya-Feldstein J., Weinberg R.A. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–688. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 15.Miska E.A. How microRNAs control cell division, differentiation and death. Curr. Opin. Genet. Dev. 2005;15:563–568. doi: 10.1016/j.gde.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 16.Calin G.A., Dumitru C.D., Shimizu M., Bichi R., Zupo S., Noch E., Aldler H., Rattan S., Keating M., Rai K. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA. 2002;99:15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao J., Hennessey T., Flynt A., Lai E., Beal M.F., Lin M.T. MicroRNA-related cofilin abnormality in Alzheimer’s disease. PLoS ONE. 2010;5:e15546. doi: 10.1371/journal.pone.0015546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao P., Wong C.C., Tung E.K., Lee J.M., Wong C.M., Ng I.O. Deregulation of microRNA expression occurs early and accumulates in early stages of HBV-associated multistep hepatocarcinogenesis. J. Hepatol. 2011;54:1177–1184. doi: 10.1016/j.jhep.2010.09.023. [DOI] [PubMed] [Google Scholar]
- 19.Oertli M., Engler D.B., Kohler E., Koch M., Meyer T.F., Müller A. MicroRNA-155 is essential for the T cell-mediated control of Helicobacter pylori infection and for the induction of chronic Gastritis and Colitis. J. Immunol. 2011;187:3578–3586. doi: 10.4049/jimmunol.1101772. [DOI] [PubMed] [Google Scholar]
- 20.Bang C., Fiedler J., Thum T. Cardiovascular importance of the microRNA-23/27/24 family. Microcirculation. 2012;19:208–214. doi: 10.1111/j.1549-8719.2011.00153.x. [DOI] [PubMed] [Google Scholar]
- 21.Chen X., Wang L., Qu J., Guan N.N., Li J.Q. Predicting miRNA-disease association based on inductive matrix completion. Bioinformatics. 2018 doi: 10.1093/bioinformatics/bty503. Published online June 22, 2018. [DOI] [PubMed] [Google Scholar]
- 22.Chen X., Xie D., Wang L., Zhao Q., You Z.H., Liu H. BNPMDA: Bipartite Network Projection for MiRNA-Disease Association prediction. Bioinformatics. 2018;34:3178–3186. doi: 10.1093/bioinformatics/bty333. [DOI] [PubMed] [Google Scholar]
- 23.Chen X., Huang L. LRSSLMDA: Laplacian Regularized Sparse Subspace Learning for MiRNA-Disease Association prediction. PLoS Comput. Biol. 2017;13:e1005912. doi: 10.1371/journal.pcbi.1005912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.You Z.H., Huang Z.A., Zhu Z., Yan G.Y., Li Z.W., Wen Z., Chen X. PBMDA: A novel and effective path-based computational model for miRNA-disease association prediction. 2017;13:e1005455. doi: 10.1371/journal.pcbi.1005455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen X., Huang L., Xie D., Zhao Q. EGBMMDA: Extreme Gradient Boosting Machine for MiRNA-Disease Association prediction. Cell Death Dis. 2018;9:3. doi: 10.1038/s41419-017-0003-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X., Zhou Z., Zhao Y. ELLPMDA: Ensemble learning and link prediction for miRNA-disease association prediction. 2018;15:807–818. doi: 10.1080/15476286.2018.1460016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goh K.I., Cusick M.E., Valle D., Childs B., Vidal M., Barabási A.L. The human disease network. Proc. Natl. Acad. Sci. USA. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang D., Wang J., Lu M., Song F., Cui Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics. 2010;26:1644–1650. doi: 10.1093/bioinformatics/btq241. [DOI] [PubMed] [Google Scholar]
- 29.Chen X., Xie D., Zhao Q., You Z.H. MicroRNAs and complex diseases: from experimental results to computational models. Brief. Bioinform. 2017 doi: 10.1093/bib/bbx130. Published online October 17, 2017. [DOI] [PubMed] [Google Scholar]
- 30.Jiang Q., Hao Y., Wang G., Juan L., Zhang T., Teng M., Liu Y., Wang Y. Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC Syst. Biol. 2010;4(Suppl 1):S2. doi: 10.1186/1752-0509-4-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi H., Xu J., Zhang G., Xu L., Li C., Wang L., Zhao Z., Jiang W., Guo Z., Li X. Walking the interactome to identify human miRNA-disease associations through the functional link between miRNA targets and disease genes. BMC Syst. Biol. 2013;7:101. doi: 10.1186/1752-0509-7-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mørk S., Pletscher-Frankild S., Palleja Caro A., Gorodkin J., Jensen L.J. Protein-driven inference of miRNA-disease associations. Bioinformatics. 2014;30:392–397. doi: 10.1093/bioinformatics/btt677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu C., Ping Y., Li X., Zhao H., Wang L., Fan H., Xiao Y., Li X. Prioritizing candidate disease miRNAs by integrating phenotype associations of multiple diseases with matched miRNA and mRNA expression profiles. Mol. Biosyst. 2014;10:2800–2809. doi: 10.1039/c4mb00353e. [DOI] [PubMed] [Google Scholar]
- 34.Xuan P., Han K., Guo M., Guo Y., Li J., Ding J., Liu Y., Dai Q., Li J., Teng Z., Huang Y. Prediction of microRNAs associated with human diseases based on weighted k most similar neighbors. PLoS ONE. 2013;8:e70204. doi: 10.1371/journal.pone.0070204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen X., Liu M.X., Yan G.Y. RWRMDA: predicting novel human microRNA-disease associations. Mol. Biosyst. 2012;8:2792–2798. doi: 10.1039/c2mb25180a. [DOI] [PubMed] [Google Scholar]
- 36.Xuan P., Han K., Guo Y., Li J., Li X., Zhong Y., Zhang Z., Ding J. Prediction of potential disease-associated microRNAs based on random walk. Bioinformatics. 2015;31:1805–1815. doi: 10.1093/bioinformatics/btv039. [DOI] [PubMed] [Google Scholar]
- 37.Chen X., Yan C.C., Zhang X., You Z.H., Deng L., Liu Y., Zhang Y., Dai Q. WBSMDA: Within and Between Score for MiRNA-Disease Association prediction. Sci. Rep. 2016;6:21106. doi: 10.1038/srep21106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen X., Yan C.C., Zhang X., You Z.H., Huang Y.A., Yan G.Y. HGIMDA: Heterogeneous graph inference for miRNA-disease association prediction. Oncotarget. 2016;7:65257–65269. doi: 10.18632/oncotarget.11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J.Q., Rong Z.H., Chen X., Yan G.Y., You Z.H. MCMDA: Matrix completion for MiRNA-disease association prediction. Oncotarget. 2017;8:21187–21199. doi: 10.18632/oncotarget.15061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu H., Chen X., Lu L. Large-scale prediction of microRNA-disease associations by combinatorial prioritization algorithm. Sci. Rep. 2017;7:43792. doi: 10.1038/srep43792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen X., Wu Q.F., Yan G.Y. RKNNMDA: Ranking-based KNN for MiRNA-Disease Association prediction. RNA Biol. 2017;14:952–962. doi: 10.1080/15476286.2017.1312226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu J., Li C.X., Lv J.Y., Li Y.S., Xiao Y., Shao T.T., Huo X., Li X., Zou Y., Han Q.L. Prioritizing candidate disease miRNAs by topological features in the miRNA target-dysregulated network: case study of prostate cancer. Mol. Cancer Ther. 2011;10:1857–1866. doi: 10.1158/1535-7163.MCT-11-0055. [DOI] [PubMed] [Google Scholar]
- 43.Chen X., Yan G.Y. Semi-supervised learning for potential human microRNA-disease associations inference. Sci. Rep. 2014;4:5501. doi: 10.1038/srep05501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen X., Yan C.C., Zhang X., Li Z., Deng L., Zhang Y., Dai Q. RBMMMDA: predicting multiple types of disease-microRNA associations. Sci. Rep. 2015;5:13877. doi: 10.1038/srep13877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ulintz P.J., Zhu J., Qin Z.S., Andrews P.C. Improved classification of mass spectrometry database search results using newer machine learning approaches. Mol. Cell. Proteomics. 2006;5:497–509. doi: 10.1074/mcp.M500233-MCP200. [DOI] [PubMed] [Google Scholar]
- 46.Xu T., Li B., Zhao M., Szulwach K.E., Street R.C., Lin L., Yao B., Zhang F., Jin P., Wu H., Qin Z.S. Base-resolution methylation patterns accurately predict transcription factor bindings in vivo. Nucleic Acids Res. 2015;43:2757–2766. doi: 10.1093/nar/gkv151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rajagopal N., Xie W., Li Y., Wagner U., Wang W., Stamatoyannopoulos J., Ernst J., Kellis M., Ren B. RFECS: a random-forest based algorithm for enhancer identification from chromatin state. PLoS Comput. Biol. 2013;9:e1002968. doi: 10.1371/journal.pcbi.1002968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ritchie G.R., Dunham I., Zeggini E., Flicek P. Functional annotation of noncoding sequence variants. Nat. Methods. 2014;11:294–296. doi: 10.1038/nmeth.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen L., Jin P., Qin Z.S. DIVAN: accurate identification of non-coding disease-specific risk variants using multi-omics profiles. Genome Biol. 2016;17:252. doi: 10.1186/s13059-016-1112-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng Z., Huang K., Wang Y., Liu H., Guan J., Zhou S. Selecting high-quality negative samples for effectively predicting protein-RNA interactions. BMC Syst. Biol. 2017;11(Suppl 2):9. doi: 10.1186/s12918-017-0390-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pasquier C., Gardès J. Prediction of miRNA-disease associations with a vector space model. Sci. Rep. 2016;6:27036. doi: 10.1038/srep27036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang Z., Ren F., Liu C., He S., Sun G., Gao Q., Yao L., Zhang Y., Miao R., Cao Y. dbDEMC: a database of differentially expressed miRNAs in human cancers. BMC Genomics. 2010;11(Suppl 4):S5. doi: 10.1186/1471-2164-11-S4-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jiang Q., Wang Y., Hao Y., Juan L., Teng M., Zhang X., Li M., Wang G., Liu Y. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37:D98–D104. doi: 10.1093/nar/gkn714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Parkin D.M., Bray F., Ferlay J., Pisani P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 55.Bosetti C., Levi F., Ferlay J., Garavello W., Lucchini F., Bertuccio P., Negri E., La Vecchia C. Trends in oesophageal cancer incidence and mortality in Europe. Int. J. Cancer. 2008;122:1118–1129. doi: 10.1002/ijc.23232. [DOI] [PubMed] [Google Scholar]
- 56.Daly J.M., Fry W.A., Little A.G., Winchester D.P., McKee R.F., Stewart A.K., Fremgen A.M. Esophageal cancer: results of an American College of Surgeons Patient Care Evaluation Study. J. Am. Coll. Surg. 2000;190:562–572. doi: 10.1016/s1072-7515(00)00238-6. [DOI] [PubMed] [Google Scholar]
- 57.Chen X., Hu H., Guan X., Xiong G., Wang Y., Wang K., Li J., Xu X., Yang K., Bai Y. CpG island methylation status of miRNAs in esophageal squamous cell carcinoma. Int. J. Cancer. 2012;130:1607–1613. doi: 10.1002/ijc.26171. [DOI] [PubMed] [Google Scholar]
- 58.Nayak L.M., Deschler D.G. Lymphomas. Otolaryngol. Clin. North Am. 2003;36:625–646. doi: 10.1016/s0030-6665(03)00033-1. [DOI] [PubMed] [Google Scholar]
- 59.Barajas Torres R.L., Domínguez Cruz M.D., Borjas Gutiérrez C., Ramírez Dueñas Mde.L., Magaña Torres M.T., González García J.R. 1,2:3,4-Diepoxybutane Induces Multipolar Mitosis in Cultured Human Lymphocytes. Cytogenet. Genome Res. 2016;148:179–184. doi: 10.1159/000445858. [DOI] [PubMed] [Google Scholar]
- 60.Intlekofer A.M., Younes A. Precision therapy for lymphoma--current state and future directions. Nat. Rev. Clin. Oncol. 2014;11:585–596. doi: 10.1038/nrclinonc.2014.137. [DOI] [PubMed] [Google Scholar]
- 61.Ohyashiki K., Umezu T., Yoshizawa S., Ito Y., Ohyashiki M., Kawashima H., Tanaka M., Kuroda M., Ohyashiki J.H. Clinical impact of down-regulated plasma miR-92a levels in non-Hodgkin’s lymphoma. PLoS ONE. 2011;6:e16408. doi: 10.1371/journal.pone.0016408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peveling-Oberhag J., Crisman G., Schmidt A., Döring C., Lucioni M., Arcaini L., Rattotti S., Hartmann S., Piiper A., Hofmann W.P. Dysregulation of global microRNA expression in splenic marginal zone lymphoma and influence of chronic hepatitis C virus infection. Leukemia. 2012;26:1654–1662. doi: 10.1038/leu.2012.29. [DOI] [PubMed] [Google Scholar]
- 63.Li Y., Qiu C., Tu J., Geng B., Yang J., Jiang T., Cui Q. HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014;42:D1070–D1074. doi: 10.1093/nar/gkt1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xiang Z., Qin T., Qin Z.S., He Y. A genome-wide MeSH-based literature mining system predicts implicit gene-to-gene relationships and networks. BMC Syst. Biol. 2013;7(Suppl 3):S9. doi: 10.1186/1752-0509-7-S3-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Laarhoven T., Nabuurs S.B., Marchiori E. Gaussian interaction profile kernels for predicting drug-target interaction. Bioinformatics. 2011;27:3036–3043. doi: 10.1093/bioinformatics/btr500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.