

Visual Abstract

Key Words: atherosclerosis, inflammasome, methylation, miRNA, TNFRSF9, VSMC
Abbreviations and Acronyms: 5-aza, 5-aza-2ʹ-deoxycytidine; 5-hmC, 5-hydroxymethylcytosine; ApoE, apolipoprotein E; BSP, bisulfite genomic sequencing; cDNA, complementary deoxyribonucleic acid; ChIP, chromatin immunoprecipitation; CVD, cardiovascular disease; DNA, deoxyribonucleic acid; DNMT, deoxyribonucleic acid methyltransferase; IL, interleukin; miR-145, microRNA 145; mRNA, messenger ribonucleic acid; MSP, methylation-specific polymerase chain reaction; NFATc1, nuclear factor of activated T cells 1; RNA, ribonucleic acid; SMA, smooth muscle actin; TET, ten-eleven translocation; TNF, tumor necrosis factor; UTR, untranslated region; VSMC, vascular smooth muscle cell; WT, wild type
Highlights
-
•
miR-145 in vessels decreases with plaque progression.
-
•
DNMT1 and TET2 dynamic imbalance leads to miR-145 promoter hypermethylation.
-
•
Reduction of miR-145 activates NLRP3 inflammasome through CD137/NFATc1 signaling.
-
•
DNMT1 and TET2 could be promising therapeutic candidates for atherosclerosis in the future.
Summary
Two major issues are involved in clinical atherosclerosis treatment. First, there are no significant clinical markers for early diagnosis of atherosclerosis. Second, the plaque will not regress once it initiates even if the risk factors are removed. In this paper, the research shows that the hypermethylation level of the microRNA 145 (miR-145) promoter is related to a DNMT1 and TET2 dynamic imbalance. The reduction of miR-145 causes NLRP3 (nucleotide-binding oligomerization domain-like receptor protein 3) inflammasome activation through CD137/NFATc1 signaling. These findings could be a potential target for plaque regression in the future.
Atherosclerosis is a chronic inflammatory disease that involves multiple cell types such as macrophages, endothelial cells, and vascular smooth muscle cells (VSMCs). Although many drugs or treatments have been identified to prevent atherosclerosis progression, none have been found capable of completely reversing plaque formation (1). Atherosclerosis has a memory effect and progresses at its own pace once atherosclerotic plaques are formed (2).
The memory effects are mediated by epigenetic modifications including noncoding ribonucleic acid (RNA), deoxyribonucleic acid (DNA) methylation, and histone acetylation. A recent study showed that epigenetic modification can be heritable and dynamic in cells even after the stress has been removed (3). MicroRNAs (miRNAs) are small noncoding RNAs that bind to the 3ʹ untranslated region (UTR) of messenger RNA (mRNA) to depress target protein translation, which has been associated with many diseases such as tumors, immune dysfunction, hypertension, diabetes mellitus, and cardiovascular disease (CVD). miR-145 is one of the most important miRNAs in CVD, especially in atherosclerosis and pulmonary hypertension, because of its abundant expression in VSMCs and its role in VSMC phenotype transformation 4, 5, 6. miR-145 maintains VSMCs in the contraction phenotype, which is more resistant to atherogenic factors. miR-145 inhibits the abnormal proliferation of VSMCs during the initiation of plaques by directly stimulating the translation of myocardin or suppressing Krüppel-like factor 5 and Krüppel-like factor 4 (7). miR-145−/− mice develop spontaneous atherosclerotic lesions without hypercholesterolemia (8). Overexpression of miR-145 in VSMCs alleviates atherosclerosis in apolipoprotein E (ApoE)–deficient mice (ApoE−/−mice) and increases the stability of lesions by elevating the VSMCs and collagen content 9, 10. Although these previous studies demonstrated that miR-145 plays a protective role in atherosclerosis progression, the reason for the consistent down-regulation of miR-145 in plaques remains unknown.
DNA methylation is one of the most understood epigenetic modifications. In brief, the cytosines in CpG islands (CG-dinucleotide–rich regions) can be converted to 5-methylcytosine (5-mC) catalyzed by DNA methyltransferases (DNMTs) and further combined by methyl-CpG–binding proteins (11). DNA methylation is a negative regulatory mechanism in cells through gene silencing in eukaryotic genomes. The methylation status is a dynamic process mediated by the methylation system of DNMTs or by a demethylation system such as TET2 (ten-eleven translocation 2) (12). Although still controversial, the demethylation agent 5-aza-2ʹ-deoxycytidine (5-aza) was shown to inhibit atherosclerosis formation 13, 14. Recent studies have shown that the expression of miR-145 is also regulated by methylation. Abnormally high methylation of the miR-145 promoter was reported to cause a low expression level of miR-145 15, 16, 17. However, whether the silence of miR-145 in CVD (especially in atherosclerosis) is also mediated by methylation and which enzyme might be involved in miR-145 methylation in plaques require further investigation.
In this paper, we sought to study the methylation status of the miR-145 promoter in atherosclerosis. We found that the hypermethylation of miR-145 might account for its low expression and subsequent activation of downstream inflammation signaling in vivo and in vitro.
Methods
Ethics statement and tissue samples
Male 6- to 8-week-old ApoE−/− mice were purchased from Vital River Laboratories (a distributor of Jackson Laboratory, Beijing, China) and housed in pathogen-free standard conditions. Mice were fed a Western diet (0.15% cholesterol, 20% fat) and euthanized through CO2 anesthesia at different weeks on the basis of experimental demand. Aortas were removed, and the portion from the root to the ascending aorta was cross-sectioned (5 μm) for staining; the other parts were used for DNA, RNA, or protein extraction. All animal experimental procedures were conducted in conformity with institutional guidelines for the care and use of laboratory animals at Jiangsu University, Zhenjiang, China, and conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (publication No. 85-23, revised 1996).
Cell culture and treatment
Mouse VSMCs were isolated from the aortas of male 8-week-old C57BL/6J mice as reported previously 18, 19. Cells were cultured in DF12 (Dulbecco’s modified Eagle’s medium and Ham’s F-12 nutrient mixture) containing 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C with 5% CO2. Cells at <5 passages were used in all cell experiments. For related experiments, cells were treated with 10 ng/ml tumor necrosis factor (TNF)-α (PeproTech, Rocky Hill, New Jersey) for 24 h or TNF-α plus 5 μmol/ml of 5-aza (MilliporeSigma, St. Louis, Missouri) for 48 h. In a CD137-associated assay, VSMCs were pre-treated with TNF-α (10 ng/ml) for 24 h to elevate the CD137 levels on the membrane and further treated with 10 μg/ml recombinant CD137L (Sangon Biotech, Shanghai, China) to activate CD137 signaling (20).
5-aza treatment of ApoE−/− mice
Eight-week-old ApoE−/− mice were fed a Western diet for 12 weeks to initiate plaque formation and then divided into a 5-aza group and a saline group. C57BL/6 mice were fed a Western diet for 24 weeks, as was the wild-type (WT) group. Mice were administered saline or 5-aza injection IP (0.25 mg/kg body weight, 3 times per week) for another 12 weeks 21, 22 and euthanized through CO2 anesthesia, as described previously. The serum was stored overnight and collected at 1,000 g for 20 min for enzyme-linked immunosorbent assay, and aortas were dissected for immune staining or further assay.
Bisulfite genomic sequencing and methylation-specific polymerase chain reaction
DNA was extracted from cells or tissues with a DNeasy Blood & Tissue Kit (QIAGEN, Germantown, Maryland), and bisulfite conversion was performed through an EpiTect Fast DNA bisulfite kit (QIAGEN). Generally, aortas were cut into small pieces, supplemented with 20 μl of proteinase K in buffers, and incubated at 56°C until completely lysed. DNA was extracted, and 1 μg of DNA was used for bisulfite conversion. The subsequent assays were performed according to the protocol. After conversion, the DNA was used in a methylation-specific polymerase chain reaction (MSP) for the +129 CpG site or sent to Sangon Biotech for bisulfite genomic sequencing (BSP) detection. The primer sequence is shown in Supplemental Table 1.
Histology, immunohistochemistry, and immunofluorescence
Aortic paraffin sections were subjected to dewaxing and hydration. Masson staining was used to observe the fibrous cap in plaques. The plaque size and necrotic core were measured using 3 sections per mouse through Image-Pro Plus (Media Cybernetics, Inc., Rockville, Maryland). After antigen retrieval and blocking, immunostaining was performed using the following primary antibodies: Mac3 (BD Pharmingen, San Diego, California), nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) (Abcam, Cambridge, United Kingdom; 1:200), and α-smooth muscle actin (α-SMA) (MilliporeSigma; 1:500). After they were washed in phosphate-buffered saline, sections were further incubated with goat biotinylated anti-rabbit IgG (Boster, Pleasanton, California; 1:200) for immunohistochemistry or DyLight 594 goat anti-rabbit (Abbkine, Wuhan, China) for immunofluorescence. Images were obtained with an Olympus microscope.
Western blot
Proteins were extracted using radioimmunoprecipitation assay, separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, and then transferred to polyvinylidene fluoride membranes. The membranes were blocked and incubated with antibodies against interleukin (IL)-β, NLRP3, CD137, DNMT3a, and DNMT3b (Abcam); TET2 (Proteintech, Wuhan, China); and DNMT1 and nuclear factor of activated T cells 1 (NFATc1, Cell Signaling Technology, Inc., Danvers, Massachusetts). Membranes were washed 3 times and incubated with a horseradish peroxidase–labeled secondary antibody. Bands were detected by enzyme-linked chemiluminescence according to the manufacturer’s protocol (ECL, Bio-Rad, Hercules, California).
Chromatin immunoprecipitation assay at the miR-145 promoter
A chromatin immunoprecipitation (ChIP) assay was performed according to the instructions of the SimpleChIP enzymatic chromatin IP kit (Cell Signaling Technology). Cells (1.2 × 107) were collected and cross-linked with formaldehyde at a final concentration of 37%. The nuclei in the cell lysate were treated with 0.5 μl of micrococcal nuclease per 4 × 106 cells to digest chromatin, and nuclei were broken with an ultrasonic sonicator (25 kw/20 s) 3 times. Anti-DNMT1 (Abcam) or anti-TET2 (Proteintech) was coated overnight to pull down the chromatin. Rabbit anti-H3 and rabbit IgG served as the positive and negative control, respectively. Relative data quantification was performed using the 2−ΔΔCt method, and the result was calculated in the form of 2% input, as follows: Enrichment = 2% × 2(CtInput−CtChIP).
miRNA extraction and real-time quantitative polymerase chain reaction
Total RNA or miRNAs were extracted from cells or tissue samples with TRIzol (Invitrogen/Thermo Fisher Scientific, Waltham, Massachusetts) according to the manufacturer's protocol. Complementary DNA (cDNA) synthesis of mRNA or miRNA was performed with Thermo Fisher reverse transcription reagents or an miRNA First Strand cDNA Synthesis kit (Sangon Biotech), respectively, as instructed by each product manual. GAPDH or U6 was used as a control. The primers were synthesized by Sangon Biotech.
Vectors, cell culture, and treatment
The eukaryotic expression vectors p3×FLAG-CMV-24 and PLKO.1-puro were obtained from MilliporeSigma. The NFATc1 and CD137 overexpression vectors with and without the 3ʹUTR were constructed by amplifying mouse spleen cDNA with Phanta Super-Fidelity DNA Polymerase (Vazyme Biotech, Nanjing, China) and cloning the amplified products at the Kpn I/BamH I sites of the p3×FLAG-CMV-24 vector to obtain the constructs p3×FLAG-NFATc1-3ʹUTR, p3×FLAG-NFATc1, p3×FLAG-CD137-3ʹUTR, and p3×FLAG-CD137. The TET2 or NFATc1 overexpression and small hairpin RNA targeting NFATc1 (sh-NFATc1) or DNMT1 (sh-DNMT1) knockdown sequences were inserted into the pLenti-puro or PLKO.1 vector, respectively, to create an overexpression or knockdown plasmid. pLenti-NFATc1 and pLenti-TET2 plasmids were constructed using ECOR I/Xba I sites. 293T cells were obtained from ATCC (Manassas, Virginia). The miR-145 mimic/inhibitor and mimic control/inhibitor control were purchased from Biomics (Nantong, China). Cells were transfected accordingly through Lipofectamine 2000 (Thermo Fisher).
Lentivirus package and infection
The lentivirus plasmids constructed above were packaged in 293T cells by cotransfecting a lentiviral vector and packaging plasmids PMD2G (0.5 μg/well) and PSPAX2 (1 μg/well) with Lipofectamine 2000. Cell supernatants were collected to obtain viruses at 24 h and 48 h. Acquired lentiviruses were used to infect VSMCs cultured in 60-mm dishes, which reached 60% confluence in the presence of polybrene (8 μg/ml), and were screened with puromycin (3.5 μg/ml) to establish the corresponding stable cell lines.
Luciferase reporter assay
PsiCHECK2 reporter vectors containing the 3ʹUTRs of NFATc1 or CD137 and their mutants were cotransfected with mimic/inhibitors or mimic control/inhibitor control into 293T cells after the procedure. At 48 h after transfection, luciferase activity was determined (Promega, Madison, Wisconsin).
Enzyme-linked immunosorbent assay
Cell supernatants and mouse sera, which were stored at 4°C overnight and acquired after being centrifuged at 1,000 g for 20 min, were sent to Multi Sciences Biotech Co. (Hangzhou, China) to detect the level of mature IL-1β in vivo or vitro.
DNMT1 activity assay and 5-hydroxymethylcytosine detection
DNMT1 activity and 5-hydroxymethylcytosine (5-hmC) level were detected by an EpiQuik DNA methyltransferase 1 activity assay kit (EpiGentek, Farmingdale, New York) and a Quest 5-hmC detection kit (Zymo Research, Irvine, California) to observe DNMT1 or TET2 activity, respectively. The assays were performed according to the manufacturers’ instructions. The results were detected by a microplate reader at 450 nm or by real-time polymerase chain reaction. DNMT1 activity (OD/h/μg) was calculated as follows, where OD indicates optimal density: (no inhibitor OD – blank OD) × 1,000/DNMT1 amount (ng) added in the reaction × h.
miRNA mimic or inhibitor transfection
Mimics and inhibitors, which increase or decrease miR-145, respectively, were purchased from RiboBio (Guangzhou, Guangdong, China). VSMCs were seeded in a 6-well plate and were transfected when they reached 50% confluence. Appropriate concentration mimics or inhibitors were transfected to cells through Lipofectamine 2000, following the manufacturer’s instructions. For the in vivo study, 50 mg/kg miR-145 antagomir and the relative antagomir control were injected into ApoE−/− mice via tail veins every 3 days for 6 weeks.
Statistical analysis
Histology and immunohistochemistry were analyzed with Image-Pro Plus. Triplicate wells were used for each treatment within an experiment, and at least 3 independent experimental studies were performed for each type of experiment. Data are expressed as the mean ± SEM of at least 3 independent experiments and were compared by a Student’s t-test or ANOVA with Newman-Keuls post hoc tests (multiple comparisons) using SPSS version 12.0 (SPSS, Chicago, Illinois). Two-tailed p < 0.05 was considered significant.
Results
Depression of miR-145 associated with plaque progression in ApoE−/− mice
Although a reduced miR-145 level is found in plaques, it is not known whether reduced miR-145 is associated with plaque progression. Thus, we analyzed the miR-145 level in the aortas of ApoE−/− mice fed a Western diet (0, 12, and 24 weeks). Masson staining revealed a gradual increase in plaque content with a necrotic core (Figures 1A and 1B), and Mac3 staining was used as a marker for inflammation (Figures 1C and 1D). As expected, the level of miR-145 gradually decreased with the progression of plaques and inflammation (Figure 1E).
Figure 1.

Decreased Level of miR-145 With Plaque Progression
(AandB) Six-week-old ApoE−/− mice were fed a Western diet for 0, 12, and 24 weeks, and sections of ascending aortas were stained with Masson to observe the fibers. Lesion size and necrotic core were measured by Image-Pro Plus. Values are mean ± SEM from 6 animals in each group; *p < 0.05 vs. the 0-week group. (CandD) Immunohistochemistry of Mac3, which represents macrophage infiltration, with staining to show the inflammatory content of plaques. Mean density and IOD were calculated for each group. Values are mean ± SEM from 6 animals in each group; *p < 0.05 vs. the 0-week group. (E) Total RNAs were extracted from aortas and reverse transcribed to complementary deoxyribonucleic acid, and the level of miR-145 was measured. Values are mean ± SEM from 6 animals in each group; *p < 0.05 vs. the 0-week group. ApoE−/− = apolipoprotein E–deficient; IOD = integrated optimal density; miR-145 = microRNA 145; RNA = ribonucleic acid.
Hypermethylation of the miR-145 promoter observed in vivo and in vitro
Through NCBI (National Center for Biotechnology Information) and MethPrimer database analysis, we found that there was 1 CpG island with 7 CpG sites in the miR-145 promoter (Supplemental Figure I). BSP suggested that the total methylation degree of the CpG island increased with plaque progression. In addition, the +129 CpG site in the island was altered significantly (Figures 2A to 2C). Given that miR-145 was abundant and played an important role in VSMC function, we analyzed the miR-145 methylation status under inflammatory conditions in vitro. Different concentrations of TNF-α were used to stimulate VSMCs. Although 20 and 40 ng/ml of TNF-α induced down-regulation of miR-145, the 10 ng/ml concentration showed a significant effect and was used for subsequent experiments (Supplemental Figure II). To further investigate whether the depression of miR-145 induced by TNF-α was mediated by methylation, 5-aza was used to inhibit methylation. Interestingly, 5-aza treatment did not affect miR-145 expression in normal VSMCs but rescued the miR-145 reduction in VSMCs treated with TNF-α (Figure 2D). We also performed BSP to define the miR-145 CpG island methylation status in cells. In accordance with animal studies, TNF-α induced CpG island hypermethylation compared with that of the control group. Interestingly, the +129 CpG site also seemed to be a significant site, as was found for plaques. This finding implies that the +129 CpG site might be the major point regulating miR-145 expression and might be the downstream target of inflammatory cytokines (Figures 2E and 2F). To validate this result, we designed a pair of primers containing the +129 CpG site for MSP under treatment with several inflammatory cytokines. As shown in Supplemental Figure III, TNF-α, IL-1β, and interferon-γ all induced methylation of the +129 CpG site.
Figure 2.



Methylation Status of the miR-145 Promoter In Vivo and In Vitro
(A) The CpG island of the miR-145 promoter was predicted, and primers were designed using MethPrimer (23). (BandC) BSP and the relative methylation ratio (20 clones) of each CpG site of the miR-145 promoter in aortas of ApoE−/− mice fed a Western diet (0, 12, and 24 weeks) are shown. Values are mean ± SEM from 6 animals in each group; *p < 0.05. (D) Level of miR-145 in vascular smooth muscle cells treated with TNF-α (10 ng/ml) or 5-aza (0, 2.5, or 5 nmol). Values are mean ± SEM of 3 samples in each group; *p < 0.05. (EandF) BSP and methylation rate of the miR-145 promoter in vascular smooth muscle cells treated with TNF-α or 5-aza. Values are mean ± SEM of 3 samples in each group; *p < 0.05, ***p < 0.01. 5-aza = 5-aza-2ʹ-deoxycytidine; BSP = bisulfite genomic sequencing; Con = control; TNF = tumor necrosis factor.
DNMT1 and TET2 altered dynamically in plaques and VSMCs
To clarify the underlying mechanisms of miR-145 methylation, we analyzed the expression of DNMT proteins and TET2 both in vivo and in vitro. We found increased expression of DNMT1 and decreased expression of TET2 during plaque progression (at 0, 6, 12, 18, and 24 weeks), whereas the expression of DNMT3a and DNMT3b was not altered (Figures 3A and 3B). The results were also confirmed in VSMCs treated with TNF-α (Figures 3C and 3D). Therefore, DNMT1 and TET2 were investigated further in VSMCs. We also found that TNF-α increased both the expression and activity of DNMT1 while decreasing the levels of TET2 and 5-hmC, which could be alleviated by 5-aza (Figures 3E to 3H).
Figure 3.

Expression and Activity of Methylation-Associated Proteins In Vivo and In Vitro
(AandB) The protein level of methylation-associated proteins (DNMT1, DNMT3a, DNMT3b, and Tet2) in aortas of ApoE−/− mice fed a Western diet (0, 6, 12, 18, and 24 weeks). Values are mean ± SEM from 6 animals in each group; *p < 0.05 vs. the 0-week group. (CandD) Protein level of methylation-associated proteins in vascular smooth muscle cells treated with TNF-α (5 or 10 ng/ml). Values are mean ± SEM of 3 samples in each group; *p < 0.05. (EandF) Protein-level alterations in DNMT1 and Tet2 induced by TNF-α in vascular smooth muscle cells were rescued with 5-aza treatment. Values are mean ± SEM of 3 samples in each group; *p < 0.05. (G) DNMT1 enzyme activity was measured in vascular smooth muscle cells treated with TNF-α or TNF-α + 5-aza. Values are mean ± SEM of 3 samples in each group; *p < 0.05. H, 5-hmC content, which represents Tet2 activity in vascular smooth muscle cells treated with TNF-α or TNF-α + 5-aza (n = 3). Values are mean ± SEM from 3 samples in each group; *p < 0.05. 5-hmC = 5-hydroxymethylcytosine; DNMT = deoxyribonucleic acid methyltransferase; Tet2 = ten-eleven translocation 2; other abbreviations as in Figure 2.
Methylation of miR-145 mediated by DNMT1 and TET2
Given that methylation dynamic alteration can be regulated by inflammation through DNMT1 and TET2, these 2 enzymes were either knocked down or overexpressed in subsequent studies. Real-time polymerase chain reaction results showed that miR-145 expression was increased in the sh-DNMT1 group or pLenti-TET2 group compared with the control group (Figures 4A and 4B). MSP was performed to determine the +129 CpG site methylation status of miR-145, as described previously. The bands of the methylated specific primer were obvious in the sh-con + TNF-α group or the pLenti-con + TNF-α group, whereas DNMT1 knockdown or TET2 overexpression led to the appearance of unmethylated specific bands instead of methylated specific bands, which meant demethylation of the +129 CpG site (Figure 4C). The ChIP assay verified that binding of DNMT1 to the miR-145 promoter was increased, whereas the binding ability to TET2 was decreased in the TNF-α group (Figures 4D and 4E).
Figure 4.


Methylation of the miR-145 Promoter Was Mediated by DNMT1 and Tet2 Dynamic Alteration
(AandB) DNMT1 knockdown or Tet2 overexpression efficiency and the effect on miR-145 expression in vascular smooth muscle cells treated with TNF-α. Values are mean ± SEM of 3 samples in each group; *p < 0.05. (C) Methylation-specific polymerase chain reaction of the +129 CpG site in vascular smooth muscle cells treated with TNF-α with knockdown of DNMT1 or overexpression of Tet2. The methylated specific bands were obvious in the sh-con or Plenti-con groups treated with TNF-α, whereas the unmethylated specific bands were obvious in the sh-DNMT1 + TNF-α group and Plenti-Tet2 + TNF-α group. (D) Chromatin immunoprecipitation assay identified the miR-145 promoter sequence binding with DNMT1 and Tet2. (E) The relative enrichment rate of the chromatin immunoprecipitation assay was determined by real time polymerase chain reaction. Values are mean ± SEM of 3 samples in each group; *p < 0.05 vs. the con group. IgG = immunoglobulin G; mRNA = messenger ribonucleic acid; sh- = small hairpin RNA targeting; other abbreviations as in Figures 2 and 3.
Demethylation treatment inhibited NLRP3 inflammasome activation
In Figure 1, we demonstrated that the degree of inflammation is associated with miR-145 expression and methylation. Given that the NLRP3 inflammasome is one of the most important molecules in inflammatory reactions, we examined whether NLRP3 activation was regulated by methylation. Immunofluorescence was performed to determine the location of NLRP3. In aortic sections of ApoE−/− mice fed a Western diet, we found that the expression of NLRP3 increased with plaque progression. In aortic sections from severe plaques, NLRP3 was detected in VSMCs in the fibrous cap (Figure 5A). Western blotting also showed that the protein levels of NLRP3 and IL-1β were increased with plaque progression (Figure 5B). These results imply that NLRP3 can contribute to plaque progression and rupture. We then administered 5-aza (0.25 mg/kg) to ApoE−/− mice to confirm the effect of DNA demethylation on NLRP3 inflammasomes. 5-Aza treatment alleviated plaque formation and NLRP3 expression in ApoE−/− mice (Figures 5C to 5E). Both the total methylation rate and the +129 CpG site methylation rate were increased in the saline group compared with the 5-aza group and WT mouse group (Figure 5F). Enzyme-linked immunosorbent assay also revealed a lower level of IL-1β in the serum of mice treated with 5-aza (Figure 5G). We then explored the association between methylation and NLRP3 in vitro and found that 5-aza treatment, DNMT1 knockdown, or overexpression of TET2 in VSMCs attenuated the NLRP3 activation induced by TNF-α, which implied that demethylation treatment can attenuate inflammasomes through the modulation of DNMT1 or TET2 (Figure 5H).
Figure 5.




Demethylation Treatment Ameliorated NLRP3 Inflammasome Activation
(A) Immunofluorescence of NLRP3 and α-SMA in ascending aortas of ApoE−/− mice fed a Western diet (0, 12, and 24 weeks). (B) Western blot and quantitative results of NLRP3, pro-IL-1β, and mature IL-1β in aortas of ApoE−/− mice fed a Western diet (0, 6, 12, 18, and 24 weeks). Values are mean ± SEM from 6 animals in each group; *p < 0.05 vs. the 0-week group. (C) Immunofluorescence of NLRP3 and α-SMA in aortas of WT mice fed a 24-week Western diet or ApoE−/− mice fed a 12-week Western diet and treated with 5-aza or phosphate-buffered saline for another 12 weeks (0.25 mg/kg body weight, 3 times per week). Values are mean ± SEM from 6 animals in each group; *p < 0.05. (DandE) Masson and Mac3 immunohistochemistry in aortas of WT mice or ApoE−/− mice treated with 5-aza to analyze plaque components. Values are mean ± SEM from 6 animals in each group; *p < 0.05. (F) BSP and relative methylation rate (20 clones) of the +129 CpG site of the miR-145 promoter in aortas of ApoE−/− mice treated with 5-aza. Values are mean ± SEM from 6 animals in each group; *p < 0.05. (G) Concentration of IL-1β in the serum of ApoE−/− mice treated with 5-aza. Values are mean ± SEM from 6 animals in each group; *p < 0.05. (H) Western blot of NLRP3, pro-IL-1β, and mature IL-1β in vascular smooth muscle cells with demethylation treatment (5-aza, knockdown of DNMT1, or overexpression of Tet2). Values are mean ± SEM of 3 samples in each group; *p < 0.05. α-SMA = α-smooth muscle actin; IL = interleukin; NLRP3 = nucleotide-binding oligomerization domain-like receptor protein 3; WT = wild-type; other abbreviations as in Figures 1, 2, and 3.
miR-145 mediates the inhibitory effect of 5-aza on NLRP3 inflammasomes
miR-145 is one of the most important protective miRNAs in atherosclerosis, and its anti-inflammatory effect has been reported. We demonstrated that 5-aza treatment enhanced the expression of miR-145; however, whether miR-145 could inhibit NLRP3 inflammasomes remained unknown. An miRNA mimic and inhibitor were used to up-regulate or down-regulate miR-145, respectively. Protein levels of NLRP3 or IL-1β were elevated under TNF stimulation in VSMCs but were decreased in the presence of the miR-145 mimic (Figures 6A and 6B). Importantly, the anti-inflammasome effect of 5-aza was inhibited by blocking miR-145 with the miR-145 inhibitor (Figures 6C and 6D). IL-1β levels were elevated in both the TNF-α group and the miR-145 inhibitor group, which indicates activation of the NLRP3 inflammasome (Figures 6E and 6F). In ApoE−/− mice fed a Western diet, the miR-145 antagomir eliminated the benefits of 5-aza treatment on NLRP3 activation and plaque stability. Compared with mice in the 5-aza group, mice in the miR-145 antagomir plus 5-aza group showed more NLRP3 expression in VSMCs and displayed a larger necrotic core in lesions (Supplemental Figure IV).
Figure 6.


MiR-145 Inhibited NLRP3 Inflammasome Activation Induced by TNF-α in Vascular Smooth Muscle Cells
(AandB) Western blot and quantitative results of NLRP3, pro-IL-1β, and mature IL-1β in vascular smooth muscle cells treated with TNF-α and the miR-145 mimic (100 nmol/l). Values are mean ± SEM of 3 samples in each group; *p < 0.05. (CandD) Western blot and quantitative results of NLRP3, pro-IL-1β, and mature IL-1β in vascular smooth muscle cells treated with 5-aza and the miR-145 inhibitor (100 nmol/l). Values are mean ± SEM of 3 samples in each group; *p < 0.05. (E) Concentration of IL-1β in supernatants of vascular smooth muscle cells with different treatments. Values are mean ± SEM of 3 samples in each group; *p < 0.05. (F) Immunofluorescence of IL-1β accumulation in vascular smooth muscle cells with different treatments. INC = inhibitor control; NC = mimic control; other abbreviations as in Figures 1, 2, and 3 and 5.
CD137 and NFATc1 are the downstream target genes of miR-145
To clarify how miR-145 regulates NLRP3, we investigated the potential target gene of miR-145. CD137, also called TNFRSF9, belongs to the TNF receptor superfamily. Several studies have demonstrated that CD137 plays an important role in immune reactions and inflammation. Our previous study discovered that CD137/NFATc1 signaling accelerates plaque formation and induces IL-1β secretion in VSMCs (data not shown). TargetScan and PicTar revealed that both CD137 and NFATc1 are direct target genes of miR-145 (Supplemental Figure V). Thus, we hypothesized that the effect of miR-145 on NLRP3 could be associated with the CD137/NFATc1 pathway. mRNA levels of CD137 and NFATc1 were decreased or increased gradually when cells were transfected with the miR-145 mimic or inhibitor, respectively (Figure 7A). Moreover, the miR-145 mimic inhibited the CD137 and NFATc1 proteins induced by TNF-α, whereas the miR-145 inhibitor inhibited the effect of 5-aza (Figure 7B). To define the target site in the 3ʹUTRs (noncoding regions), we cloned the CD137 or NFATc1 gene, which included the 3ʹUTRs, into a p3×FLAG-CMV-24 eukaryotic expression plasmid. The mimic inhibited the plasmid in the WT group, whereas no changes were observed if the 3ʹUTR was deleted (Figure 6C). The dual-luciferase assay showed a similar tendency (Figure 6D). Together, these data suggest that CD137 and NFATc1 are the target genes of miR-145.
Figure 7.

CD137/NFATc1 Axis Was Directly Targeted by miR-145
(A) Relative mRNA expression levels of NFATc1 and CD137 in the miR-145 mimic- or inhibitor-treated group were determined by quantitative polymerase chain reaction. Values are mean ± SEM of 3 samples in each group; *p < 0.05. (B) Protein levels of CD137 and NFATc1 altered by TNF-α or 5-aza could be rescued by an miR-145 mimic or inhibitor. (C) Expression of eukaryotic vectors with or without CD137 or NFATc1 3ʹUTR regulated by the miR-145 mimic. (D) Luciferase assay of the CD137 and NFATc1 3ʹUTR or the corresponding mutation plasmid treated with the mimic. Values are mean ± SEM of 3 samples in each group; *p < 0.05. MUT-UTR = mutation plasmid; NFATc1 = nuclear factor of activated T cells 1; n.s. = nonsignificant; other abbreviations as in Figures 2, 4, 5, and 6.
CD137/NFATc1 signaling contributed to NLRP3 activation
Lastly, we investigated the effect of CD137/NFATc1 signaling on NLRP3 inflammasome activity. A recombinant CD137L protein was used to activate CD137 as described previously. Western blotting showed that NLRP3 expression was enhanced by activation of the CD137 signaling pathway. Levels of pre-IL-1β and mature IL-1β were increased in the CD137L group compared with the control group and could be prevented by knocking down NFATc1 (Figures 8A to 8C).
Figure 8.

CD137/NFATc1 Axis Activated NLRP3 Inflammasome in Vascular Smooth Muscle Cells
(A) Western blot and quantitative results of NLRP3, pro-IL-1β, and mature IL-1β in vascular smooth muscle cells treated with TNF-α or recombinant CD137L. Values are mean ± SEM of 3 samples in each group; *p < 0.05. (B) Western blot and quantitative results of NFATc1, NLRP3, pro-IL-1β, and mature IL-1β in vascular smooth muscle cells with knocked-down NFATc1. Values are mean ± SEM of 3 samples in each group; *p < 0.05. (C) Concentration of IL-1β in supernatants of vascular smooth muscle cells with different treatments. Values are mean ± SEM of 3 samples in each group; *p < 0.05. Abbreviations as in Figures 2, 5, and 7.
Discussion
In this study, we investigated the mechanism involved in abnormal depression of miR-145, which is regarded as a protective factor in atherosclerosis formation. Through both in vitro and in vivo studies, we found that inflammation could induce aberrant hypermethylation of the miR-145 promoter, which was mediated by DNMT1 and TET2 dynamic cooperation. Knockdown of DNMT1, overexpression of TET2, or 5-aza treatment demethylated miR-145 and elevated its expression. The elevation of miR-145 led to repression of the target genes CD137/NFATc1 and reduced the activation of NLRP3 inflammasomes in VSMCs.
Atherosclerosis is a chronic inflammatory disease (24). Unlike tumors, atherosclerosis is not caused by gene mutations. However, the plaques do not regress even after risk factors are removed 25, 26, which can be explained by epigenetic alterations (27). Both DNA methylation and miRNA are involved in the development of atherosclerosis. miR-145 is generally considered a protective factor in VSMC phenotype and viability (28). In the present study, we found that the expression of miR-145 gradually decreased with plaque progression. Genome-wide DNA methylation sequencing revealed that the miR-145 promoter was hypermethylated following plaque aggregation or TNF-α stimulation in VSMCs. Through bioinformatics analysis, we found that there was one CpG island within the miR-145 promoter that contained 7 CpG sites. Interestingly, the +29, +118, and +140 CpG sites could be involved in early plaques, whereas the other 4 CpG sites were methylated in advanced plaques. Among these, the +129 CpG site was methylated with plaque progression and might play a critical role in miR-145 depression. Consequently, we further investigated the relationship between the +129 CpG site and inflammation. We treated the VSMCs with 3 inflammatory cytokines (TNF-α, IL-1β, and interferon-γ). MSP revealed that the methylation status of the +129 CpG site can be regulated by inflammatory cytokines.
A positive correlation between DNA methylation and atherosclerotic lesion grade has been established through genome-wide DNA methylation sequencing in human aortas (29). In mammalian cells, the methylation is catalyzed by DNMTs, which consist of DNMT1, DNMT3a, and DNMT3b. Among these, DNMT3a and DNMT3b mediate de novo DNA methylation, whereas DNMT1 maintains newly synthesized DNA methylation (30). Unlike the methylation system, the demethylation system was poorly defined. Despite this finding, TET2 was regarded as the most known demethyltransferase, which could oxidize 5-methylcytosine to 5-hmC and finally convert it to unmethylated cytosine 31, 32. To explain the mechanism of miR-145 methylation, we explored the role of these 4 enzymes in plaques and VSMCs. Consistent with the findings by Peng et al., an increased expression of DNMT1 and decreased expression of TET2 were observed in plaque formation, whereas the other 2 enzymes, DNMT3a and 3b, were not altered significantly. Furthermore, a similar tendency existed in VSMCs treated with TNF-α, which implies that DNMT1 and TET2 might be the principal molecules mediating the inflammation-associated methylation in atherosclerosis. Both knockdown of DNMT1 and overexpression of TET2 led to miR-145 elevation and demethylation of the +129 CpG site in VSMCs.
It is known that inflammation is a key initiator and driver of atherosclerosis, but the initial factors linking inflammation to atherosclerosis remain unknown. The NLRP3 inflammasome, the most well-known inflammasome, had been identified as the key regulator, which can intensify inflammation in response to a wide range of harmful signals 33, 34. Increasing evidence suggests that the NLRP3 inflammasome and downstream cytokine IL-1β, which is activated by NLRP3/caspase 1, could be associated with atherogenesis. Furthermore, it has been reported that IL-1β is involved in VSMC proliferation, migration, calcification, and senescence 35, 36, 37. Inhibition of NLRP3 or IL-1β led to plaque reduction and stabilization 38, 39, 40; however, the association between NLRP3 and epigenetics, especially in atherosclerosis formation, has not been confirmed. Therefore, we analyzed NLRP3 expression and activation in vivo and in vitro. We showed that 5-aza treatment inhibited NLRP3 expression and reduced IL-1β levels in ApoE−/− mice. In VSMCs, 5-aza treatment, knockdown of DNMT1, or overexpression of TET2 rescued the NLRP3 activation induced by TNF-α. To explore whether this phenomenon was mediated by miR-145, the inhibitor of miR-145 was used simultaneously with 5-aza to block miRNA expression. Inhibition of miR-145 elevated the NLRP3 and IL-1β levels, which were reduced by 5-aza. These data suggest that miR-145 could be the key molecule between methylation and NLRP3. miRNAs repress mRNA through binding to its 3ʹUTR. Because NLRP3 and IL-1β were not the direct targets of miR-145, as evidenced by the bioinformatics analysis, we hypothesized that miR-145 represses NLRP3 activity through targeting another inflammation pathway. Recent studies have demonstrated that CD137/NFATc1 is an important signaling pathway in VSMC inflammation and plaque formation 18, 41, 42. We previously observed elevated IL-1β levels in VSMC supernatants when CD137 was activated (data not shown). Therefore, NLRP3 might be associated with CD137 signaling. TargetScan and PicTar revealed that both CD137 and the downstream NFATc1 could be the target genes of miR-145. Thus, we hypothesized that CD137/NFATc1 signaling might be the mediator between miR-145 and NLRP3. We proved that miR-145 represses the mRNA of both CD137 and NFATc1 and decreases their protein levels directly. Knockdown of NFATc1 alleviates NLRP3 inflammasome activation induced by CD137 signaling.
Conclusions
In this study, we found that miR-145 expression in plaques was regulated by DNMT1-mediated or TET2-mediated promoter hypermethylation. The decreased expression of miR-145 led to CD137/NFATc1 up-regulation, which induced NLRP3 activation, inflammation, and atherosclerosis progression. Our findings provide new insights linking inflammation to atherosclerosis and open the door to the development of new therapies (Figure 9).
Figure 9.
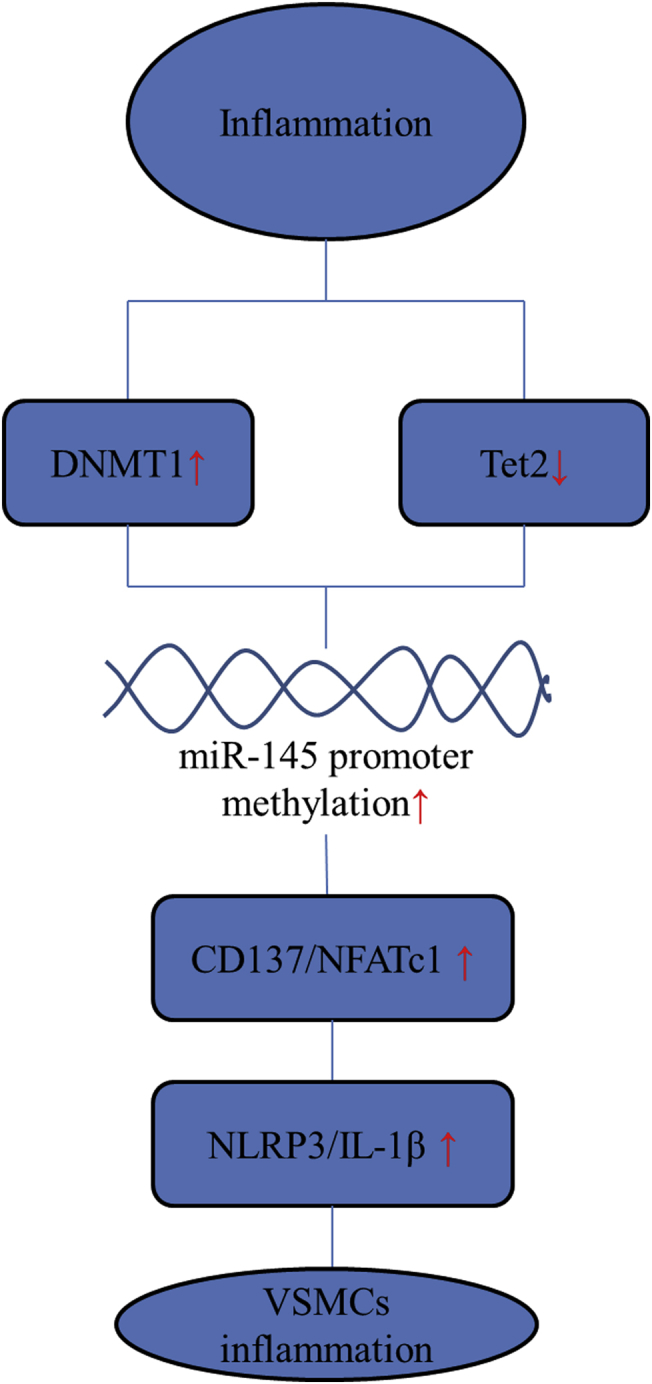
Hypermethylation of the miR-145 Promoter Is the Key Regulator of NLRP3 Inflammasome Activation and Plaque Formation
Inflammation-induced DNMT1 elevation or Tet2 reduction in vascular smooth muscle cells (VSMCs) of the aortic wall, which led to miR-145 promoter hypermethylation and its low expression. Low levels of miR-145 weakened the inhibitory effect of miR-145 on the CD137/NFATc1 axis, which further activated the NLRP3 inflammasome to amplify the inflammatory reaction and induce plaque formation or rupture. Abbreviations as in Figures 3, 5, and 7.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: This paper demonstrates that DNMT1 and TET2 dynamic balance contributes to methylation status changes under inflammation. It is implied that other genes may also be controlled by these 2 enzymes.
TRANSLATIONAL OUTLOOK: The abnormal hypermethylation of the miR-145 promoter could be the initiating step of plaque and contributes to plaque progression. Therapeutic reagents that alleviate the methylation level might be used to treat plaque. More importantly, the methylation status of miR-145 could also be used in determining the prognosis for atherosclerosis.
Footnotes
This project was supported by the National Natural Science Foundation of China (81670405, 81370409), the Natural and Social Development Foundation of Jiangsu Province (BK20161355, BE2017699, CXTDA2017010), and the Social Development Foundation of Zhenjiang (SH2017015, SH2016037). The authors have reported that they have no relationships relevant to the contents of this paper to disclose.
All authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the JACC: Basic to Translational Scienceauthor instructions page.
Appendix
References
- 1.Yao L., Heuser-Baker J., Herlea-Pana O. Bone marrow endothelial progenitors augment atherosclerotic plaque regression in a mouse model of plasma lipid lowering. Stem Cells. 2012;30:2720–2731. doi: 10.1002/stem.1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhuang J., Luan P., Li H. The Yin–Yang dynamics of DNA methylation is the key regulator for smooth muscle cell phenotype switch and vascular remodeling. Arterioscler Thromb Vasc Biol. 2017;37:84–97. doi: 10.1161/ATVBAHA.116.307923. [DOI] [PubMed] [Google Scholar]
- 3.Muka T., Koromani F., Portilla E. The role of epigenetic modifications in cardiovascular disease: a systematic review. Int J Cardiol. 2016;212:174–183. doi: 10.1016/j.ijcard.2016.03.062. [DOI] [PubMed] [Google Scholar]
- 4.Guo X., Yu L., Chen M. miR-145 mediated the role of aspirin in resisting VSMCs proliferation and anti-inflammation through CD40. J Transl Med. 2016;14:211. doi: 10.1186/s12967-016-0961-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sala F., Aranda J.F., Rotllan N. MiR-143/145 deficiency attenuates the progression of atherosclerosis in Ldlr-/-mice. Thromb Haemost. 2014;112:796–802. doi: 10.1160/TH13-11-0905. [published correction appears in Thromb Haemost 2014;112:796–802] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vengrenyuk Y., Nishi H., Long X. Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler Thromb Vasc Biol. 2015;35:535–546. doi: 10.1161/ATVBAHA.114.304029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wei Y., Nazari-Jahantigh M., Neth P., Weber C., Schober A. MicroRNA-126, -145, and -155: a therapeutic triad in atherosclerosis? Arterioscler Thromb Vasc Biol. 2013;33:449–454. doi: 10.1161/ATVBAHA.112.300279. [DOI] [PubMed] [Google Scholar]
- 8.Boettger T., Beetz N., Kostin S. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest. 2009;119:2634–2647. doi: 10.1172/JCI38864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lovren F., Pan Y., Quan A. MicroRNA-145 targeted therapy reduces atherosclerosis. Circulation. 2012;126:S81–S90. doi: 10.1161/CIRCULATIONAHA.111.084186. [DOI] [PubMed] [Google Scholar]
- 10.Cordes K.R., Sheehy N.T., White M.P. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705–710. doi: 10.1038/nature08195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ambrosi C., Manzo M., Baubec T. Dynamics and context-dependent roles of DNA methylation. J Mol Biol. 2017;429:1459–1475. doi: 10.1016/j.jmb.2017.02.008. [DOI] [PubMed] [Google Scholar]
- 12.Kohli R.M., Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502:472–479. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hai Z., Zuo W. Aberrant DNA methylation in the pathogenesis of atherosclerosis. Clin Chim Acta. 2016;456:69–74. doi: 10.1016/j.cca.2016.02.026. [DOI] [PubMed] [Google Scholar]
- 14.Fernandez-Sanles A., Sayols-Baixeras S., Subirana I., Degano I.R., Elosua R. Association between DNA methylation and coronary heart disease or other atherosclerotic events: a systematic review. Atherosclerosis. 2017;263:325–333. doi: 10.1016/j.atherosclerosis.2017.05.022. [DOI] [PubMed] [Google Scholar]
- 15.Donzelli S., Mori F., Bellissimo T. Epigenetic silencing of miR-145-5p contributes to brain metastasis. Oncotarget. 2015;6:35183–35201. doi: 10.18632/oncotarget.5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xia W., Chen Q., Wang J. DNA methylation mediated silencing of microRNA-145 is a potential prognostic marker in patients with lung adenocarcinoma. Sci Rep. 2015;5:16901. doi: 10.1038/srep16901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang W., Ji G., Xiao X. Epigenetically regulated miR-145 suppresses colon cancer invasion and metastasis by targeting LASP1. Oncotarget. 2016;7:68674–68687. doi: 10.18632/oncotarget.11919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhong W., Li B., Li X.Y. Amelioration of inflammatory cytokines mix stimulation: a pretreatment of CD137 signaling study on VSMC. Mediators Inflamm. 2017;2017:1382805. doi: 10.1155/2017/1382805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rong J.X., Shapiro M., Trogan E., Fisher E.A. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A. 2003;100:13531–13536. doi: 10.1073/pnas.1735526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olofsson P.S., Soderstrom L.A., Wagsater D. CD137 is expressed in human atherosclerosis and promotes development of plaque inflammation in hypercholesterolemic mice. Circulation. 2008;117:1292–1301. doi: 10.1161/CIRCULATIONAHA.107.699173. [DOI] [PubMed] [Google Scholar]
- 21.Cao Q., Wang X., Jia L. Inhibiting DNA methylation by 5-aza-2'-deoxycytidine ameliorates atherosclerosis through suppressing macrophage inflammation. Endocrinology. 2014;155:4925–4938. doi: 10.1210/en.2014-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang C., Yi J., Gong X. Anti-oxidative and anti-inflammatory benefits of the ribonucleoside analogue 5-azacitidine in mice with acetaminophen-induced toxic hepatitis. Int Immunopharmacol. 2017;48:91–95. doi: 10.1016/j.intimp.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 23.Li L.C., Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 24.Gistera A., Hansson G.K. The immunology of atherosclerosis. Nat Rev Nephrol. 2017;13:368–380. doi: 10.1038/nrneph.2017.51. [DOI] [PubMed] [Google Scholar]
- 25.Puri R., Nissen S.E., Ballantyne C.M. Factors underlying regression of coronary atheroma with potent statin therapy. Eur Heart J. 2013;34:1818–1825. doi: 10.1093/eurheartj/eht084. [DOI] [PubMed] [Google Scholar]
- 26.Puri R., Nissen S., Shao M. Coronary atheroma volume and cardiovascular events during maximally intensive statin therapy. Eur Heart J. 2013;34:3182–3190. doi: 10.1093/eurheartj/eht260. [DOI] [PubMed] [Google Scholar]
- 27.Khyzha N., Alizada A., Wilson M.D., Fish J.E. Epigenetics of atherosclerosis: emerging mechanisms and methods. Trends Mol Med. 2017;23:332–347. doi: 10.1016/j.molmed.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 28.Hutcheson R., Terry R., Chaplin J. MicroRNA-145 restores contractile vascular smooth muscle phenotype and coronary collateral growth in the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2013;33:727–736. doi: 10.1161/ATVBAHA.112.301116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valencia-Morales Mdel P., Zaina S., Heyn H. The DNA methylation drift of the atherosclerotic aorta increases with lesion progression. BMC Med Genomics. 2015;8:7. doi: 10.1186/s12920-015-0085-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li E., Zhang Y. DNA methylation in mammals. Cold Spring Harb Perspect Biol. 2014;6:a019133. doi: 10.1101/cshperspect.a019133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ito S., Shen L., Dai Q. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He Y.F., Li B.Z., Li Z. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bakker P.J., Butter L.M., Kors L. Nlrp3 is a key modulator of diet-induced nephropathy and renal cholesterol accumulation. Kidney Int. 2014;85:1112–1122. doi: 10.1038/ki.2013.503. [DOI] [PubMed] [Google Scholar]
- 34.Martinon F., Agostini L., Meylan E., Tschopp J. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol. 2004;14:1929–1934. doi: 10.1016/j.cub.2004.10.027. [DOI] [PubMed] [Google Scholar]
- 35.Bessueille L., Fakhry M., Hamade E., Badran B., Magne D. Glucose stimulates chondrocyte differentiation of vascular smooth muscle cells and calcification: a possible role for IL-1β. FEBS Lett. 2015;589:2797–2804. doi: 10.1016/j.febslet.2015.07.045. [DOI] [PubMed] [Google Scholar]
- 36.Yoon J., Ryoo S. Arginase inhibition reduces interleukin-1beta-stimulated vascular smooth muscle cell proliferation by increasing nitric oxide synthase-dependent nitric oxide production. Biochem Biophys Res Commun. 2013;435:428–433. doi: 10.1016/j.bbrc.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 37.Gardner S.E., Humphry M., Bennett M.R., Clarke M.C. Senescent vascular smooth muscle cells drive inflammation through an interleukin-1α-dependent senescence-associated secretory phenotype. Arterioscler Thromb Vasc Biol. 2015;35:1963–1974. doi: 10.1161/ATVBAHA.115.305896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Heijden T., Kritikou E., Venema W. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoproteinE–deficient mice. Arterioscler Thromb Vasc Biol. 2017;37:1457–1461. doi: 10.1161/ATVBAHA.117.309575. [DOI] [PubMed] [Google Scholar]
- 39.Zheng F., Xing S., Gong Z., Mu W., Xing Q. Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein E-deficient mice. Mediators Inflamm. 2014;2014:1–8. doi: 10.1155/2014/507208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peiro C., Lorenzo O., Carraro R., Sanchez-Ferrer C.F. IL-1beta inhibition in cardiovascular complications associated to diabetes mellitus. Front Pharmacol. 2017;8:363. doi: 10.3389/fphar.2017.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhong W., Li B., Yang P. CD137-CD137L interaction modulates neointima formation and the phenotype transformation of vascular smooth muscle cells via NFATc1 signaling. Mol Cell Biochem. 2017;439:65–74. doi: 10.1007/s11010-017-3136-4. [DOI] [PubMed] [Google Scholar]
- 42.Soderstrom L.A., Jin H., Caravaca A.S. Increased carotid artery lesion inflammation upon treatment with the CD137 agonistic antibody 2A. Circ J. 2017;81:1945–1952. doi: 10.1253/circj.CJ-17-0230. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.