

Abstract
Background
Staphylococcus aureus is an opportunistic pathogen and a leading cause of nosocomial infections. It can acquire resistance to all the antibiotics that entered the clinics to date, and the World Health Organization defined it as a high-priority pathogen for research and development of new antibiotics. A deeper understanding of the genetic variability of S. aureus in clinical settings would lead to a better comprehension of its pathogenic potential and improved strategies to contrast its virulence and resistance. However, the number of comprehensive studies addressing clinical cohorts of S. aureus infections by simultaneously looking at the epidemiology, phylogenetic reconstruction, genomic characterisation, and transmission pathways of infective clones is currently low, thus limiting global surveillance and epidemiological monitoring.
Methods
We applied whole-genome shotgun sequencing (WGS) to 184 S. aureus isolates from 135 patients treated in different operative units of an Italian paediatric hospital over a timespan of 3 years, including both methicillin-resistant S. aureus (MRSA) and methicillin-sensitive S. aureus (MSSA) from different infection types. We typed known and unknown clones from their genomes by multilocus sequence typing (MLST), Staphylococcal Cassette Chromosome mec (SCCmec), Staphylococcal protein A gene (spa), and Panton-Valentine Leukocidin (PVL), and we inferred their whole-genome phylogeny. We explored the prevalence of virulence and antibiotic resistance genes in our cohort, and the conservation of genes encoding vaccine candidates. We also performed a timed phylogenetic investigation for a potential outbreak of a newly emerging nosocomial clone.
Results
The phylogeny of the 135 single-patient S. aureus isolates showed a high level of diversity, including 80 different lineages, and co-presence of local, global, livestock-associated, and hypervirulent clones. Five of these clones do not have representative genomes in public databases. Variability in the epidemiology is mirrored by variability in the SCCmec cassettes, with some novel variants of the type IV cassette carrying extra antibiotic resistances. Virulence and resistance genes were unevenly distributed across different clones and infection types, with highly resistant and lowly virulent clones showing strong association with chronic diseases, and highly virulent strains only reported in acute infections. Antigens included in vaccine formulations undergoing clinical trials were conserved at different levels in our cohort, with only a few highly prevalent genes fully conserved, potentially explaining the difficulty of developing a vaccine against S. aureus. We also found a recently diverged ST1-SCCmecIV-t127 PVL− clone suspected to be hospital-specific, but time-resolved integrative phylogenetic analysis refuted this hypothesis and suggested that this quickly emerging lineage was acquired independently by patients.
Conclusions
Whole genome sequencing allowed us to study the epidemiology and genomic repertoire of S. aureus in a clinical setting and provided evidence of its often underestimated complexity. Some virulence factors and clones are specific of disease types, but the variability and dispensability of many antigens considered for vaccine development together with the quickly changing epidemiology of S. aureus makes it very challenging to develop full-coverage therapies and vaccines. Expanding WGS-based surveillance of S. aureus to many more hospitals would allow the identification of specific strains representing the main burden of infection and therefore reassessing the efforts for the discovery of new treatments and clinical practices.
Electronic supplementary material
The online version of this article (10.1186/s13073-018-0593-7) contains supplementary material, which is available to authorized users.
Keywords: Staphylococcus aureus, Microbial genomics, Microbial epidemiology, Bacterial pathogens
Background
Staphylococcus aureus is a bacterium commonly found on the skin (15%), in the nostrils (27%), and in the pharynx (10–20%) of healthy adults [1–3], but it is also the cause of a number of diseases, whose severity ranges from common community-associated skin infections to fatal bacteraemia [3–5]. S. aureus is a leading cause of surgical, device-related, and pleuropulmonary infections, which can result into life-threatening infective endocarditis or even sepsis [6]. The mortality of S. aureus invasive infections was extremely high (> 80%) in the pre-antibiotic era [6, 7], and only the introduction of penicillin at the beginning of the 1940s was able to contain it. However, resistant strains carrying a penicillinase/beta-lactamase quickly emerged [8–10], and more than 90% of current human-associated isolates are resistant to penicillin [6]. Similarly, the introduction of the penicillinase-resistant antibiotic methicillin was quickly followed by the emergence of methicillin-resistant S. aureus (MRSA) clones [11–13]. S. aureus is capable of acquiring resistance to virtually every antibiotic that has entered clinical use [14, 15], including recently developed agents like daptomycin and linezolid [16, 17] and the last resort antibiotic vancomycin [18, 19]. In 2017, the World Health Organization has listed vancomycin-intermediate and vancomycin-resistant MRSA among the high priority pathogens for research and development of new antibiotics [20].
S. aureus’s ability to spread worldwide and to cause outbreaks in both hospitals and the community [21, 22] has fostered the study of its global epidemiology [3, 15, 23–25]. Some lineages are very prevalent worldwide (e.g. CC5 and CC8) [24], whereas others have a more localised spreading range, like the CC5 ST612 clone, which has been found only in South Africa and Australia [24, 26]. MRSA prevalence is also highly geographically variable, ranging from < 1% in some Northern European countries to > 50% in some American and Asian countries, with livestock-associated MRSA disseminating in the last two decades [24]. Newly emerging highly pathogenic and pandemic clones have also been globally characterised [27, 28] and are often the results of recombination events as in the case of the ST239-SCCmecIII clone [25, 27, 29]. S. aureus investigations have however often underestimated the importance of non-MRSA clones, usually considering only hypervirulent or specifically relevant methicillin-sensitive S. aureus (MSSA) lineages [15], even though MSSA is the most common cause of surgical site infection [30, 31] and one of the major nosocomial pathogens [15].
Untargeted profiling of the entire S. aureus population in a given site or area is as important as its global epidemiology, and it is crucial for surveillance and prevention of local outbreaks. Some studies have for instance unbiasedly assessed the local epidemiology of nosocomial S. aureus, suggesting that this pathogen is only rarely transmitted from nurses to hospitalised patients in presence of adequate infection prevention measures [32] and that the community acts as major source of nosocomial MRSA [33]. Studies surveying the whole S. aureus population in hospitals have however focused on single aspects, like the diversity of the population, its virulence and resistance traits, and its transmission in presence of an outbreak [34–39] or in non-emergency conditions [40–42]. Despite the large body of researches on S. aureus, studies addressing a whole S. aureus infective population at a given site through whole genome sequencing to simultaneously look at the epidemiology, phylogenetic reconstruction, genomic characterisation, and transmission pathways of infective clones are currently limited [43]. Expanding these types of studies will be crucial for an in-depth global monitoring of S. aureus.
Here we report an in-depth epidemiological and genomic investigation of S. aureus infections in a paediatric hospital in Italy. With a whole-genome sequencing approach, we reconstructed the phylogenies of the clones in the cohort, characterised known clones and variants, screened for resistance and virulence genes, and tested for the presence of an outbreak. This allowed us to appreciate the high diversity of the S. aureus community, with 80 different lineages, variability of the resistance cassettes, and uneven conservation of various antigens previously clinically tested for vaccine development. We further report an increased prevalence of highly resistant and lowly virulent clones in chronic infections, and the rise of a newly emerging clone already reported in other hospitals. Overall, our results highlight the complexity of S. aureus epidemiology and advocate the need for wider genome-based analysis.
Materials and methods
Sample collection and S. aureus isolation
Samples were collected at Anna Meyer Children’s University Hospital (Florence, Italy) from 160 patients from January 2013 to December 2015. Metadata were also collected (Additional file 1: Table S1). We analysed samples obtained from the most common sites of infection for S. aureus, namely airways (bronchial aspirates, sputum or oropharyngeal and nasal swabs) or from soft-tissue and skin lesions. All samples were processed for the detection of bacteria using selective (Mannitol Salt Agar 2, bioMérieux) and chromogenic culture media for MRSA (BBL™ CHROMagar™ MRSA II, Becton Dickinson). In order to confirm species-level identification, mass spectrometry analysis was performed using matrix-assisted laser desorption/ionisation time of flight (MALDI-TOF) (VITEK® MS, bioMérieux). Antibiotic susceptibility was evaluated using the automated system VITEK®2 (bioMerieux) with the card AST-P632 (see Additional file 1: Table S1 for antibiograms). All identified strains were stored at − 80 °C for the following molecular analysis.
Molecular characterisation of S. aureus and MRSA isolates
DNA extraction was performed from pure S. aureus cultures after 24 h of incubation at 37 °C on Columbia agar + 5% sheep blood (bioMérieux) using QIAamp DNA Mini Kit (cat. num. 51306, QIAGEN, Netherlands) according to the manufacturer’s specifications. DNA was purified using Agencourt AMPure XP (Beckman Coulter, California, USA) according to the manufacturer’s specifications. Extracted DNA was stored at − 20 °C for further analyses.
In order to determine the potential virulence of SA/MRSA strains, a specific PCR assay for the presence of the gene (lukS-lukF) encoding for the Panton-Valentine Leukocidin (PVL) was set up following a previously published protocol [44]. The mecA gene and other loci of the SCCmec cassette were analysed using different multiplex PCR. The protocol suggested by Milheirico et al. [45] has been used as a screening test for most frequent SCCmec cassettes types (types I, II, III, IV, V, and VI) and then confirmed with other methods in equivocal cases [45–48].
PCR-based multilocus sequence typing (MLST) was carried out with 25 μl reaction volumes containing 2 μl of chromosomal DNA, 20 μM of each primer, 1 U of Taq DNA polymerase (Super AB Taq, AB analitica), 2.5 μl of 10× PCR buffer (supplied with the Taq polymerase), 1.5 μM MgCl2, and 250 μM each deoxynucleoside triphosphates. The PCR was performed with an initial 5-min denaturation at 95 °C, followed by 30 cycles of annealing at 55 °C for 1 min, extension at 72 °C for 1 min, and denaturation at 95 °C for 1 min, followed by a final extension step of 72 °C for 5 min. The amplified products were purified and then amplified with the BigDye® Terminator v3.1 Cycle Sequencing Kits (Applied Biosystem) with the primers used in the initial PCR amplification. The sequences of both strands were determined with an ABI Prism 310 DNA sequencer. Isolates with the same ST have identical sequences at all seven MLST loci.
Isolates sequencing and data pre-processing
DNA libraries were prepared with Nextera XT DNA Library Preparation Kit (Illumina, California, USA). Quality control was performed with Caliper LabChip GX (Perkin Elmer) prior to shotgun sequencing with MiSeq (Illumina, California, USA), with an expected sequencing depth of 260 Mb/library (expected coverage > 80×). One hundred twenty-nine million reads were generated (704 thousand reads/sample s.d. 349 thousand).
Sequences were pre-processed by removing low-quality (mean quality lower than 25) or low-complexity reads, reads mapping to human genome or to large and small ribosomal units of bacteria, fungi and human, and known contaminants (e.g. phiX174, Illumina spike-in). All genomes are available at the NCBI Sequence Read Archive (BioProject accession number PRJNA400143).
Genome assembly and annotation
Pre-processed reads were de novo assembled using SPAdes version 3.6.1 [49] and discarding contigs shorter than 1000 nt. We selected for our analysis only reconstructed genomes with an N50 > 50,000. We obtained high-quality genomes (N50 > 50,000 and less than 150 contigs) for 135 of the 160 patients enrolled. Genomes belonging to the remaining 25 patients were excluded from further analyses. Genomes were annotated with Prokka version 1.11 [50] using default parameters and adding --addgenes and --usegenus options.
Genome alignment/phylogenetic analysis
The sets of 1464 concatenated genes used as input for constructing whole cohort (Fig. 1) and strain (Fig. 2) phylogenetic trees were generated using Roary version 3.4.2 [51]. Maximum likelihood trees were inferred with RAxML version 8.0.26 [52] using a GTR replacement model with four discrete categories of Gamma. Support at nodes was estimated using 100 bootstrap pseudo-replicates (option “-f a”). The phylogenetic tree in Additional file 2: Figure S1 was inferred using the presence-absence binary matrix of the core and accessory genes computed with Roary version 3.4.2 [51] in RAxML version 8.0.26 [52] with option “-m BINGAMMA”. Phylogenetic analyses were conducted using only one single isolate per patient; when multiple isolates from different timepoints of the same patient were available, the reconstructed genome with the highest N50 and the lowest number of contigs was selected. In most cases (n = 30), patients maintained the same ST over time; in discrepant cases (n = 2), we selected the most prevalent clone.
Fig. 1.

Phylogenetic tree of the whole cohort. Phylogenetic tree based on the 1464 core genes (1,194,183 bases) of the 135 single-patient S. aureus isolates. STs are distinguished by means of numbers and background colours in the inner ring. Sample type, operative unit, PVL presence, and SCCmec type are colour-coded in the following rings. On the outermost ring, the number of virulence genes is reported as bar plot (total considered = 79)
Fig. 2.
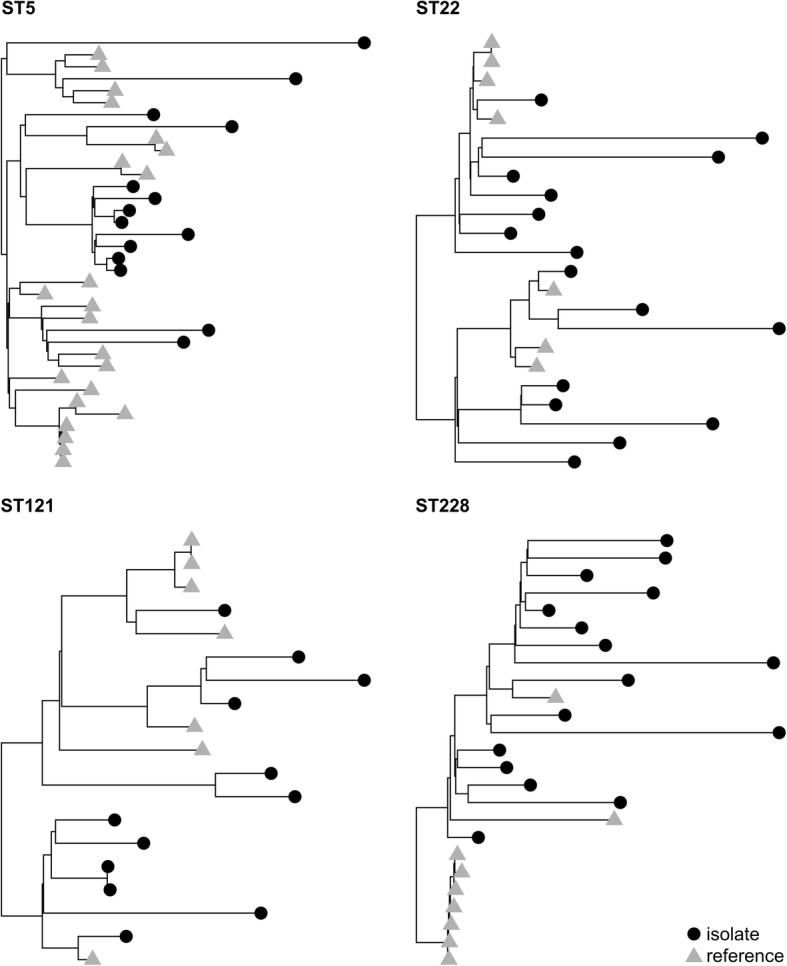
Whole-genome maximum likelihood phylogenetic trees of the four most relevant STs. All available reference genomes for ST22, ST121, and ST228 have been included. For ST5, 1478 reference genomes were available, but only 24 were included for the sake of clarity. The phylogenetic tree of ST1 and available reference genomes was also produced, but it is not reported here to avoid overlapping with Figure 5
In silico sequence type (ST), SCCmec, and spa-type identification
In order to assign SCCmec type also to equivocal cases and to confirm PCR-based SCCmec typing, the same set of primers [45] and other primer sets [53, 54] were mapped to reconstructed genomes by BLAST [55]. In most cases, the two methods were consistent. In discordant cases, PCR was repeated. Sequence typing and spa-typing were conducted using MetaMLST [56] and the DNAGear software [57] respectively.
Many isolates were not assigned a spa-type because of the limitations of short-read shotgun sequencing in repeated regions, which cause problems in genome assembly.
Virulence factors and resistance gene analysis
Selected virulence factors and resistance genes (as in [58]) were searched for by mapping reference genes (Additional file 3: Table S2) to all reconstructed genomes with BLAST [55] with the following parameters [−evalue 1e−10 -perc_identity 90 -gapopen 5 -gapextend 5] with a match > 75%. Virulence genes to be searched for were selected on the basis of a careful literature review for their clinical relevance [59–84].
Analysis of genes with available vaccine targets
Genes of interest were identified as those S. aureus vaccine candidates that had already entered clinical trials (according to http://clinicaltrials.gov as of January 2018), and those candidates that showed promising results in preclinical trials. For each genome, we extracted the reference sequences using BLAST [55] with default parameters. Extracted genes were pairwise globally aligned with the reference and evaluated for synonymous and non-synonymous single-nucleotide variation (SNVs), insertions, and/or deletions.
Bayesian divergence estimates
We estimated divergence times of ST1 SCCmecIV t127 PVL− clones using BEAST2 [85] and the core genome (core genes = 1464). We defined the best fitting model priors by testing the combination of three clock models (uncorrelated relaxed exponential, uncorrelated relaxed lognormal, and strict), three demographic models (birth-death, coalescent Bayesian skyline, and constant), and two substitution models (HKY - Hasegawa, Kishino, Yano and generalised time reversible). Bayesian Markov chain Monte Carlo were run for 500 Mio. generations and sampled every 1000 generations. We chose the combination of models that resulted in the highest Bayes factor after parameter correction using AICM in Tracer (see Additional file 4: Table S3).
Statistical tests
Associations between STs/virulence genes/antibiotic resistance markers and sample/operative unit types were found by performing Fisher’s exact test between the class of interest and the remaining set of samples.
Results and discussion
We investigated the epidemiology and the whole-genome genetics of S. aureus isolated from multiple operative units of the same paediatric hospital in Italy (Meyer’s Children Hospital, Florence). Two hundred thirty-four S. aureus isolates from 160 patients were retrieved from diverse clinical specimens, tested for antibiotic susceptibility, and subjected to whole-genome sequencing (see Materials and methods). The study produced 184 high-quality reconstructed S. aureus genomes with a N50 larger than 50,000 and less than 250 contigs (Additional file 1: Table S1). Downstream analyses are focused on the 135 high-quality strains recovered from distinct patients.
Genome sequencing highlights the presence of common clonal complexes and five newly sequenced clones
We first performed a whole-genome phylogenetic analysis to investigate the population structure of S. aureus in our cohort. The phylogeny was built using one isolate for each patient (n = 135) and using the 1464 core genes representing a core genome of > 1.19 M bases (see Materials and methods and Fig. 1). The genomic diversity of S. aureus is highlighted by the relatively large number of accessory genes even in a limited cohort of clinical isolates (n = 6909 from a pangenome of 8373 (Additional file 2: Figure S2), in concordance with a recent study based on the pangenome of 64 strains from different ecological niches [86]. The gene presence/absence phylogenetic model considering both core and genes confirmed the structure of the one built on the core genome alone, with however a slightly higher strain-diversity for isolates belonging to the same ST (Additional file 2: Figure S1). Despite this diversity, we found the presence of a reduced set of closely related strains in the cohort (Fig. 1) mostly associated with distinct multilocus sequence typing clones (STs) [87] (see Materials and methods). We identified a total of 29 different STs, with five of them—ST228, ST22, ST5, ST121, and ST1—found in at least 12 patients (Table 1 and Additional file 1: Table S1) with evidence of ST replacement in only one patient (Patient 091 switching from ST228 to ST22) of the 32 patients sampled at multiple timepoints. This longitudinal strain consistency was confirmed by whole-genome analysis (mean intra-patient variability = 56.42 SNVs), for which the replacing event in Patient 091 accounted for 6238 SNVs between the 2013 and 2016 isolates, 0.22% of the genome. The 29 identified STs belong to 14 clonal complexes (CCs), with the five most prevalent CCs (CC5, CC22, CC8, CC1, and CC121) comprising more than 60% of the isolates. Spa-typing [57] further refined the typing resolution: we found 44 distinct spa-types (Additional file 1: Table S1), with t001, t002, t008, and t127 being the most prevalent (i.e. present in > 4 isolates, Table 1). We also investigated the presence of the Panton-Valentine Leukocidin (PVL), a two-component prophage virulence factor allowing S. aureus to escape from the host immune system, that was found in 27.4% of the samples (Additional file 1: Table S1).
Table 1.
Genomic characteristics of the different STs, including SCCmec and spa-type, presence of PVL, genome length, N50 (shortest sequence length at 50% of the genome), and number of contigs, coding DNA sequences (CDS), and genes
| ST | CC | # isolates (MRSA) | Predominant SCCmec type (# isolates) | Predominant spa-type (# isolates) | # PVL+ | Avg. genome length (bp) | Avg. # contigs | Avg. N50 | Avg. # CDS | Avg. # genes |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 12 (12) | IV (11) | t127 (3) | 0 | 2,814,074.3 | 29.4 | 326,193.0 | 2601.3 | 2666.8 |
| 772 | 2 (2) | V (2) | t657 (2) | 2 | 2,768,135.0 | 46.0 | 208,282.5 | 2538.0 | 2605.0 | |
| 5 | 5 | 14 (13) | IV (10) | t002 (5) | 8 | 2,785,946.1 | 39.4 | 250,660.3 | 2580.1 | 2640.5 |
| 228 | 16 (16) | I (16) | t001 (5) | 0 | 2,837,918.4 | 81.9 | 87,688.4 | 2639.8 | 2700.7 | |
| 228 | 6 | 1 (1) | IV (1) | t5238 (1) | 0 | 2,796,820.0 | 40.0 | 150,271.0 | 2584.0 | 2648.0 |
| 7 | 7 | 3 (0) | n.a. | t1743 (1) | 0 | 2,747,478.7 | 66.0 | 147,717.0 | 2521.3 | 2588.0 |
| 8 | 8 | 11 (6) | IV (6) | t008 (6) | 5 | 2,821,267.1 | 52.7 | 259,331.5 | 2625.2 | 2681.8 |
| 293 | 2 (2) | V (1) | t037 (1) | 0 | 2,900,431.5 | 90.5 | 90,036.5 | 2697.5 | 2762.0 | |
| 241 | 1 (1) | n.d. | t030 (1) | 0 | 2,884,707.0 | 87.0 | 105,325.0 | 2707.0 | 2768.0 | |
| 247 | 1 (1) | I (1) | t197 (1) | 0 | 2,776,359.0 | 76.0 | 74,230.0 | 2567.0 | 2630.0 | |
| 10 | 10 | 1 (0) | n.a. | n.a. | 0 | 2,799,287.0 | 110.0 | 52,819.0 | 2634.0 | 2698.0 |
| 1162 | 2 (0) | n.a. | n.a. | 0 | 2,867,105.0 | 58.0 | 184,821.5 | 2702.0 | 2767.0 | |
| 15 | 15 | 5 (2) | IV (1); I (1) | t084 (1); t853 (1) | 1 | 2,719,481.8 | 45.6 | 226,394.0 | 2496.2 | 2556.6 |
| 22 | 22 | 15 (14) | IV (13) | t852 (1); t1977 (7); t223 (1); t005 (1) | 3 | 2,793,443.3 | 56.0 | 124,918.3 | 2599.5 | 2662.4 |
| 1327 | 1 (1) | IV (1) | n.a. | 0 | 2,758,892.0 | 42.0 | 167,290.0 | 2547.0 | 2612.0 | |
| 25 | 25 | 2 (0) | n.a. | t258 (1); t2242 (1) | 0 | 2,758,786.5 | 16.5 | 697,459.0 | 2554.5 | 2617.5 |
| 30 | 30 | 7 (3) | IV (3) | t019 (2) | 5 | 2,792,108.9 | 58.1 | 139,913.6 | 2603.3 | 2666.1 |
| 34 | 2 (0) | n.a. | t3905 (1) | 0 | 2,821,562.0 | 57.5 | 140,057.0 | 2665.5 | 2730.5 | |
| 45 | 45 | 8 (2) | IV (2) | t015 (2) | 0 | 2,762,203.4 | 34.4 | 390,455.1 | 2591.5 | 2654.6 |
| 59 | 59 | 3 (3) | IV (1) | t216 (1); t437 (1) | 1 | 2,799,567.0 | 53.0 | 130,817.3 | 2595.7 | 2662.0 |
| 88 | 88 | 1 (1) | IV (1) | t4701 (1) | 0 | 2,791,324.0 | 36.0 | 206,283.0 | 2575.0 | 2642.0 |
| 96 | 96 | 1 (0) | n.a. | n.a. | 1 | 2,783,146.0 | 39.0 | 141,877.0 | 2591.0 | 2652.0 |
| 97 | 97 | 2 (2) | IV (2) | t359 (1) | 0 | 2,756,222.0 | 27.0 | 401,302.0 | 2570.0 | 2635.5 |
| 121 | 121 | 12 (0) | n.a. | t3274 (1); t314 (1); t2530 (1) | 9 | 2,814,764.5 | 48.0 | 146,041.3 | 2631.3 | 2694.3 |
| 152 | 152 | 3 (2) | V (2) | t355 (1) | 2 | 2,753,826.7 | 31.0 | 267,208.7 | 2551.0 | 2608.0 |
| 395 | 395 | 1 (0) | n.a. | n.a. | 0 | 2,759,659.0 | 22.0 | 574,657.0 | 2574.0 | 2640.0 |
| 398 | 398 | 2 (1) | V (1) | t011 (1) | 0 | 27,540,120.0 | 57.5 | 20,645,930 | 2524.0 | 2589.5 |
| 942 | 942 | 1 (0) | n.a. | n.a. | 0 | 2,813,978.0 | 82.0 | 61,174.0 | 2654.0 | 2718.0 |
| – | n.a. | 3 (1) | IV (1) | n.a. | 0 | 2,739,763.7 | 46.0 | 157,754.0 | 2535.0 | 2599.3 |
The combination of the four methods (MLST, SCCmec-, and spa-typing, and PVL presence) yielded 80 different lineages. Three isolates were not assigned to any specific ST and are reported in the last row of the table
According to both antibiotic susceptibility testing (oxacillin and cefoxitin susceptibility, Additional file 1: Table S1) and genome analysis (presence of the SCCmec cassette, see Materials and methods), 63.7% of the isolates were classified as methicillin-resistant S. aureus (MRSA). Most strains (n = 54) belonged to SCCmecIV; type I cassettes were also abundant (n = 19), whereas cassettes type V (n = 8) and II (n = 1) were less represented. Methicillin resistance was unevenly distributed across the phylogenetic tree (Fig. 1) and partially independent from the STs. All CC1 isolates (n = 14, ST1 and ST772) were MRSA, and so were the isolates belonging to CC5 (n = 30, ST5 and ST228) and CC22 (n = 16, ST22 and ST1327). All CC121 (n = 12, ST121) and CC10 (n = 3, ST10 and ST1162) isolates were instead methicillin-sensitive (MSSA), and other clonal complexes (CC8, CC30, CC45) showed balanced proportions of sensitive and resistant strains. SCCmecI (n = 19) was the most CC-specific cassette, as it was found almost exclusively in CC5 isolates (ST5 and ST228), with the exception of one ST15 and one ST8 isolates, while neither SCCmecIV nor SCCmecV were associated with specific STs.
For five of the recovered STs, namely ST241, ST942, ST1162, ST1327, and ST1866, no sequenced genome is publicly available (as genomes of S. aureus in RefSeq [88] version 2017 [89]). Although a large number of S. aureus genome sequences are available in NCBI, these are biased toward a limited set of clinically relevant STs [43, 90], with many others being neglected. This underrepresentation of less-pathogenic or less-known strains may lead to a poor understanding of the host–pathogen interactions at the genomic level, and to an underestimation of emerging or re-emerging pathogenic strains [25, 43].
Co-presence of local, global, animal-associated, and hypervirulent clones
We combined the four characterisation methods (MLST, SCCmec-, spa-, and PVL typing) to identify specific known clones in the cohort, yielding 80 different lineages. The most prevalent were the South German/Italian ST228-SCCmecI clone (n = 16, 11.85%) and the E-MRSA-15 ST22-SCCmecIV clone (n = 13, 9.63%), followed by the USA400 ST1-SCCmecIV t127 (n = 11, 8.15%) clone, the USA800 paediatric clone ST5-SCCmecIV t002 (n = 10, 7.41%), and the USA500 E-MRSA-2/6 clone ST8-SCCmecIV t008 PVL− (n = 4, 2.96%) (Additional file 5: Table S4). Several other clones, including the highly virulent USA300 ST8-SCCmecIV PVL+ clone (n = 2, 1.48%), were also found, confirming a heterogeneous clone composition in Italian hospitals [91, 92]. Surprisingly, we did not isolate any ST80, the most prevalent community-associated MRSA clone in Europe [93].
We moreover identified two isolates (1.48%) belonging to the livestock-associated MRSA (LA-MRSA) ST398 clone [24, 94] (Table 1). This clone has already been reported in patients that had regular exposure to livestock in several countries [24, 95, 96] including Italy [97–99], but our results and other reports [96, 100–102] of infections in non-exposed subjects suggest that the within-subject transmission for these clones is not rare. Similar conclusions can be drawn for another LA-MRSA, namely ST97 (n = 2, 1.48%, Table 1), which is the leading cause of bovine mastitis, but is only rarely reported in humans [103–106]. This growing incidence of LA-MRSA strains (n = 4, 2.96% in our cohort) causing zoonotic infections highlights the existence of underestimated reservoirs of S. aureus strains that could become epidemic [28, 107, 108].
One isolate was assigned to ST395, which is an unusual strain unable to exchange DNA via bacteriophages with other S. aureus strains because of a modification in the wall teichoic acid (WTA) [109, 110]. The same modification, however, enables ST395 to exchange DNA with coagulase-negative staphylococci (CoNS) [110], making it particularly prone to exchange SCCmec elements and others with other commonly found staphylococci, e.g. S. epidermidis.
Genomic signatures of chronic versus acute S. aureus infections
In order to investigate the potential association of clones and antibiotic resistance with specific hospital operative units, we cross-checked the prevalence of SCCmec types, STs, and PVL+ clones with both OUs and sample types (see Materials and methods). Strains from the cystic fibrosis (CF, n = 76) unit were positively associated with the presence of SCCmecI (n = 19, ten from CF unit; p value = 0.03), a cassette known to be hospital-associated [111, 112]. Strains from the same unit were also associated with ST1 (n = 12, seven from CF unit; p value = 0.04), whereas we noted a reduced prevalence of the PVL genes (n = 37, only two from CF unit; p value = 0.0002) and of ST121 (n = 12, none from CF unit; p value = 0.02). This reflects the relatively attenuated virulence which is a well-known phenomenon in long-term S. aureus infections [113–116]. Similarly, sputum samples (n = 33; 88.7% from CF unit) were associated with ST228 (n = 16, nine from sputum; p value = 0.004) and SCCmecI (n = 19, 11 from sputum; p value = 0.0008), and negatively correlated with PVL (n = 37, only two from sputum; p value = 0.001). The high correlation of ST228 with lung isolates and specifically with CF has already been observed in Spain [111]. A similar pattern of increased resistance and lowered virulence has been observed for another sample type linked with long-term lung infections, namely broncho-aspiration material (n = 23; 78.2% from intensive care unit). Strains from this sample type were associated to SCCmecIV (n = 54, 14 from broncho-aspiration material; p value = 0.008), and with PVL− (n = 98, 23 from broncho-aspiration material; p value = 0.0005) and MRSA clones (n = 83, 21 from broncho-aspiration material; p value = 0.002), highlighting once again the loss of virulence and the acquisition of resistance in long-term lung infections [113–116].
On the contrary, patients from both emergency room (n = 5) and the infectious disease unit (n = 15) show an overrepresentation of PVL+ clones (n = 37, four from emergency room and nine from infectious diseases; p values = 0.02 and 0.005, respectively), indicative of acute rather than chronic infections. Lesion swabs (n = 31) are strongly associated with MSSA (n = 49, 31 from lesion swabs; p value = 3e−08). This sample type was also associated to the hypervirulent ST121 clone [117, 118] (n = 12, 11 from lesion swabs; p value = 2e−05) and to the presence of the PVL (n = 37, 14 from lesion swabs; p value = 3e−07), suggesting that in our cohort skin and soft tissue infections (SSTIs) are predominantly caused by hypervirulent MSSA strains. Lesion swabs from children in care at the infectious diseases unit (n = 12, 80% of the samples from this operative unit) are also characterised by high prevalence of the virulent ST45 clone [119, 120] (n = 8, three from lesion swabs; p value = 0.04) that is known to be associated with SSTIs [121–124]. The expected [125] association between PVL (n = 37) and ST121 (n = 12, nine PVL+; p value = 0.001) and ST30 (n = 7, five PVL+; p value = 0.003) supports once again the observed increased virulence of these STs [117, 118, 126, 127], which is partially in conflict with the hypothesis of lesion colonisation by commensal strains present in the skin microbiome [128, 129].
Discovery of novel variants of SCCmecIV with kanamycin, trimethoprim, and bleomycin resistance
We next investigated the specific genetic variants of the four types of SCCmec cassettes identified and discussed above. This is relevant because the epidemiology of this genetic element is disentangled by that of the rest of the genome by virtue of its high horizontal mobility [130, 131]. Moreover, the SCCmec can host genes encoding not only for resistance to beta-lactams [132, 133], but also for other antibiotic resistances or virulence factors [131].
More than a half of the MRSA isolates in our collection (n = 86) carried SCCmecIV (62.8%). This cassette type has spread widely in the last decades, often substituting the previously more prevalent nosocomial SCCmec types I and II [24, 134], and it is now common especially in European clinical isolates [24, 92]. Another cassette that has spread in recent years following a similar path is SCCmecV [134, 135], the third most prevalent cassette type in our cohort (10.5% of the MRSA isolates) after the more traditionally hospital-associated SCCmecI [24, 112] (22.1% of the MRSA isolates). We moreover isolated one MRSA carrying SCCmecII, which is widely diffused in the USA but only rarely found in Italy/Europe [25, 136]. Consistently, the SCCmecII isolate was recovered from Patient 115, which is consistent with the personal history of the patient. For two isolates, it was not possible to classify the cassette neither with PCR nor with in silico PCR using standard primers [45].
By aligning reconstructed SCCmec with reference cassettes (see Materials and methods), we observed a certain degree of variability inside the same cassette type, specifically in type IV (Fig. 3). Subtypes IVa, IVb, and IVc were identified, with some SCCmec elements showing insertions. Two cassettes in particular were not consistent with the already described subtypes: the SCCmec type IVc carried by MF062, which was enriched with genes for kanamycin [137] and bleomycin [138, 139] resistance, and the type IVa carried by MR090 that showed insertion of genes involved in resistance to trimethoprim [140, 141] (Fig. 3).
Fig. 3.

Overview of the SCCmecIV cassette variability in our cohort, compared with available reference cassettes for the recovered subtypes IVa, IVb, and IVc. Genes are marked as arrows in the direction of transcription. To avoid biases due to misassemble of the region of interest, only cassettes found on a single contig are reported. Annotated SCCmec are grouped together with the closest reference cassette subtype. Some genomes showed insertions of genes involved in resistance to trimethoprim (MR090) and to kanamycin and bleomycin (MF062)
Non-SCCmec resistance profiles show different patterns in chronic and acute infections
S. aureus can easily acquire a number of resistances, including those to the last resort antibiotics vancomycin [142, 143] and daptomycin [144]. According to results presented in previous paragraphs and elsewhere [145], resistances can occur by gene acquisition in the SCCmec cassette. Most resistances are however encoded by genes that are found in other parts of the genome or that have been horizontally transferred through different genetic elements [25]. Given the high importance of multi-drug resistance in S. aureus [20], we therefore tested the presence or absence of specific resistance genes in our cohort [146] (Fig. 4 and Additional file 3: Table S2). Consistently with previous literature [6], most of the isolates tested positive for blaZ (81.5%), responsible for penicillin resistance (96.3% concordance with antibiotic susceptibility test, as per presence of the pbp and/or mecA genes). No isolates were found positive for genes encoding resistance to vancomycin (van, 100% concordance with antibiotic susceptibility test) and to fusidic acid (fusB and far, 94.1% concordance with antibiotic susceptibility test). Antibiotic resistances were sometimes associated with specific CCs, as for the increased representation of aacA.aphD (gentamicin resistance, 92.6% concordance with antibiotic susceptibility test) and ermA (erythromycin resistance, phenotypic resistance not tested) in CC5 isolates, whose genomes tended to lack instead the blaZ gene (penicillin resistance) (Fig. 4). Overall, two isolates from acute skin infections were negative for all the resistance genes tested, while six CF and intensive care unit isolates were positive for six (33.3%) of them. This pattern of increased resistance in long-term infections, together with their observed reduced virulence, completes the scenario of reduced virulence and increased resistance that has been observed in this and previous studies [113–116].
Fig. 4.

Presence/absence profile of 79 genes encoding for virulence factors (upper part of the heatmap) and 18 genes encoding for resistance (bottom part). Some virulence and resistance factors were more represented in specific STs (only STs found in > 1 samples are specifically mentioned), as in the case of gentamicin resistance that is more prevalent in the ST228 isolates. For a more detailed overview of the single genomes’ profiles, see Additional file 3: Table S2
Emergence and disease-associated diversity of clinically relevant virulence factors
S. aureus has a large repertoire of virulence genes, and it is able to evade the host immune system through a variety of strategies. Some of the genes usually involved in immune evasion were present in almost all our isolates (Fig. 4 and Additional file 3: Table S2). These include genes encoding the phenol-soluble modulin alpha and beta and the delta-haemolysin Hld, responsible for leukocytes and erythrocyte lysis respectively [60]; the immunoglobulin-binding protein Sbi that inhibits IgG and IgA [61, 62]; and some genes part of the GIɑ genomic island (ssl6 and ssl9).
Other genes belonging to the immune evasion island IEC2 were present in many but not all isolates, for example, the one encoding for the antiplatelet extracellular fibrinogen binding protein Efb [63, 64] and those encoding various haemolysins (hla, hlg) [59, 60] (Fig. 4 and Additional file 3: Table S2). In addition to the 27.4% prevalence of the lukF and lukS PVL genes discussed above, one sample (MR029, from emergency room) was positive for the epidermal cell differentiation inhibitor Edin, which has been found to promote the translocation of S. aureus into the bloodstream [65]. One of the two USA300 isolates (MR047, from nasal swab) tested positive for the arginine catabolic mobile element (ACME), another important virulence factor (gene arcA) that has been shown to be responsible for the increased pathogenicity of S. aureus and specifically of USA300 clones [66, 67].
Many virulence genes were associated to specific STs (Fig. 4 and Additional file 3: Table S2). ST22 (n = 15), for instance, was associated with the toxic shock syndrome toxin TSST-1 (n = 8, three from ST22; p value = 0.04; present in 20% of the ST22 clones) [60, 68, 69], other pyrogenic toxin superantigens known as staphylococcal enterotoxins (SEs, mean n = 34.9 ± 28.1 s.d.; p value < 0.02 for seg, sei, sem, sen, seo, present on average in 86.7% of ST22 and 49.5% of non-ST22), and various ssl immune evasion genes (mean n = 57.4 ± 40.2 s.d.; p value < 0.01 for ssl1, ssl3, ssl4, ssl7, ssl11, ssl12, present on average in 93.3% of ST22 and 21.1% of non-ST22) [70, 71]. ST22-IV EMRSA-15 clones positive for tst1 are usually described as “Middle Eastern variant” [72–74], but a high prevalence in an Italian neonatal intensive care unit [76] and pre-school children living in Palermo, Italy [75], has been observed. Authors suggested that the Middle Eastern clone might be more widely spread than estimated and might have diffused in the Mediterranean populations as a community-acquired MRSA [75, 76], as suggested by our analysis. TSST-1 is responsible for an increased pyrogenic, emetic, and superantigen activity, together with SEs (10627489; 11544350). SEs (mean n = 34.9 ± 28.1 s.d.) were associated with all “virulent” STs, such as ST5 (n = 15, p value < 0.04 for sed, seg, sei, sej, sem, sen, seo, sep, present on average in 79.2% of ST5 and 32.3% of non-ST5), ST45 (n = 8; p value < 0.02 for sec, seg, sei, sel, sem, seo, present on average in 98.2% of ST45 and 38.9% of non-ST45), ST121 (n = 12; p value < 0.02 for seb, seg, sei, sem, sen, seo, present on average in 86.1% of ST121 and 41.7% of non-ST121), and—to a lower extent—ST30 (n = 7; p value < 0.02 for sei, sem, sen, present on average in 100% of ST30 and 50% of non-ST30).
The hypervirulent ST121 MSSA isolates obtained from lesion swabs (n = 12) were instead associated with the genes encoding for the exfoliative toxins Eta and Etb (n = 3 from ST121 swabs, and n = 0 for non-ST121, p value = 0.0006 for both genes), responsible for the skin manifestations of bullous impetigo and Staphylococcal scalded skin syndrome [77–79], the gene bbp (n = 12 from ST121, n = 7 from non-ST121; p value = 1.35e−08) that interacts with the extracellular matrix bone sialoprotein and contributes to staphylococcal arthritis and osteomyelitis [80], and the immune evasion gene ecb (n = 12 from ST121, n = 36 from non-ST121; p value = 1.51e06), which is required for the persistence of S. aureus in host tissues and the formation of abscesses [81]. The latter was also present in all and only the isolates belonging to ST1, ST7, ST10, ST15, ST30, ST34, and ST398, suggesting a strong dependence on ST (Fig. 4 and Additional file 3: Table S2).
Isolates retrieved from sputum samples of CF patients (n = 38) showed a positive association with the adhesin-encoding genes sdrD (n = 34 from CF, n = 69 from non-CF; p value = 0.03) and sdrE (n = 27 from CF, n = 48 from non-CF; p value = 0.03), and a negative association with bbp (n = 1 from CF, n = 18 from non-CF; p value = 0.01), contrary to samples from the infectious disease unit (n = 15, four positive for bbp gene). This finding is consistent with the increased need for adhesins in chronic lung infections [82, 83, 116], including in CF [84].
Conservation of genes encoding vaccine candidates
Unlike other bacterial infections, prior exposure to S. aureus does not seem to provide protective immunity [147]; therefore, vaccines are an attractive yet challenging option to prevent disease. Researchers have long attempted to produce an effective vaccine against S. aureus, but even though few have proved promising in animal models, the two vaccines so far tested in efficacy clinical trials have failed [147–153]. Since the main issue is the polymorphic expression of S. aureus surface antigens and the redundancy of its virulence proteins [147, 154, 155], we tested the prevalence and conservation of a number of genes encoding vaccine candidates described in the literature (Table 2).
Table 2.
Sequence variability of genes of interest for vaccine development. Number (and relative abundance) of isolates positive for the gene, followed by the percentage of positive isolates carrying 0 or less than 1%, 2%, 5%, or more/equal to 5% of non-synonymous SNVs or insertions-deletions (indels) with respect to reference gene. Both clinical trial IDs (ClinicalTrials.gov database identifiers, http://clinicaltrials.gov) and reference studies refer to the latest available trials
| Gene | # positive isolates (%) | Distribution of non-syn SNVs w.r.t. reference seq. | Latest trials | ClinicalTrials identifier | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | < 1% | < 2% | < 5% | ≥ 5% | indels | |||||
| clfA | 95 (70.4%) | 0% | 9.5% | 0% | 0% | 0% | 90.5% | Phases I–II | NCT01643941; NCT01364571 | [159, 160, 171] |
| csa1a | 70 (51.9%) | 41.4% | 4.3% | 0% | 0% | 0% | 54.3% | Preclinical | [168, 172] | |
| csa1b | 36 (26.7%) | 19.4% | 47.2% | 0% | 2.8% | 0% | 30.6% | |||
| esxA | 134 (99.3%) | 85.1% | 14.9% | 0% | 0% | 0% | 0% | Preclinical | [168, 172] | |
| esxB | 89 (65.9%) | 0% | 98.9% | 1.1% | 0% | 0% | 0% | Preclinical | [168, 172] | |
| esxC | 89 (65.9%) | 25.8% | 40.4% | 33.7% | 0% | 0% | 0% | |||
| esxD | 89 (65.9%) | 58.4% | 41.6% | 0% | 0% | 0% | 0% | |||
| fhuD2 | 135 (100%) | 31.1% | 68.9% | 0% | 0% | 0% | 0% | Preclinical | [168, 172] | |
| hla | 124 (91.9%) | 6.5% | 0% | 9.7% | 78.2% | 2.4% | 3.2% | Phase II | NCT02296320 | [168, 172, 173] |
| hlgA | 135 (100%) | 65.2% | 29.6% | 2.2% | 0% | 0% | 3% | |||
| hlgB | 132 (97.8%) | 11.4% | 65.2% | 22.7% | 0% | 0% | 0.8% | Preclinical | [174] | |
| hlgC | 135 (100%) | 61.5% | 8.1% | 28.1% | 2.2% | 0% | 0% | |||
| isdB | 134 (99.3%) | 11.9% | 21.6% | 0% | 0% | 0% | 66.4% | Phase III | NCT00518687 | [152, 166] |
| lukF | 37 (27.4%) | 0% | 97.3% | 0% | 0% | 0% | 2.7% | |||
| lukS | 37 (27.4%) | 56.8% | 40.5% | 0% | 0% | 0% | 2.7% | Phases I–II | NCT01011335 | [175] |
| mntC | 135 (100%) | 79.3% | 20.7% | 0% | 0% | 0% | 0% | Phases I–II | NCT01643941; NCT01364571 | [159, 160, 171] |
| tst | 8 (5.9%) | 0% | 100% | 0% | 0% | 0% | 0% | Phase I | NCT02340338 | [176] |
Among antigens that have been proposed as targets for vaccine development, the alpha haemolysin toxin gene hla [147, 156, 157] and the genes coding for capsular biosynthesis cap5 and cap8 [150, 151] are highly prevalent in our cohort (91.9% and 97.8% of the isolates respectively). Nevertheless, these genes showed a larger degree of variability compared to the others we considered, which may explain the poor results obtained in clinical trials [147, 150, 151, 156, 157]. Other genes that code for proteins used alone or in combination in vaccine formulations, such as the virulence determinant SpA [158] and the fibronectin-binding protein ClfA [159–161], are present in most of our strain collection. In some of these genes, indels are prevalent (> 90%, Table 2), but they are frequently found in repeated regions that may not critically impact the protein structure, as in the case of the spa gene.
Vaccines have also been proposed for S. aureus strains with specific characteristics. For instance, targeting the toxicity determinant TSST-1 (5.9% prevalence of tst1) [162, 163] or the PVL proteins LukF-LukS (27.4% prevalence of lukF-lukS) [164, 165] aims at selectively preventing the most virulent or lethal infections. In our cohort, despite their low prevalence, both tst1 and the PVL genes were conserved at 99%, except for a few isolates that had indels in the latter (Table 2). The gamma-haemolysins HlgAB and HlgCB genes [164, 165] were instead highly prevalent (97.8–100%) and quite conserved (69.6–94.8%). The opposite approach is targeting genes with a lower virulence profile, which may be more prevalent and conserved than those coding for highly toxic factors. Among them, the genes encoding for the manganese uptake receptor (mntC) [159–161] and for the iron acquisition factor (isdB) [152, 166], which are indeed present in all or all but one the isolates of our cohort. Non-synonymous mutations are rare in mntC (20.7% of the isolates, with only one non-synonymous SNV), and, whenever not affected by indels that may or may not affect the protein structure, also the isdB gene is highly conserved (> 99% identity, Table 2).
Finally, we also analysed the conservation of csa1A, csa1B, fhuD2, and esxA, genes recently described as being promising vaccine candidates in preclinical studies [167, 168]. The two genes encoding for the conserved antigen Csa (csa1A and csa1B) are present in 51.9% and 26.7% of the isolates respectively and are conserved in only a fraction of the cases (Table 2). By contrast, the iron uptake gene fhuD2 is present in all isolates, with a maximum of 1% non-synonymous variation in sequence (Table 2). Also the genes encoding for the ESAT-6-like secretion system (esxA, esxB, esxC, esxD) are well represented in the cohort, but only esxA is present in all but one isolate and has no non-synonymous mutations in 85.1% of the isolates (Table 2). Therefore, on the basis of their conservation, both FhuD2 and EsxA appear to be promising targets for vaccine formulations.
Phylogenetics of specific STs highlights the aggressive spread of a novel independently acquired ST1 clone
We investigated the hypothesis that some of the prevalent STs could be hospital-associated clones. We estimated the ST phylogenies using a whole-genome maximum likelihood approach (see Materials and methods). In most cases, we observed that isolates in our cohort, despite sharing the same ST, SCCmec, and spa types, were not monophyletic subtrees when considering external reference genomes for the same STs. This is the case, for example, of the ST228 and ST5 clones (Fig. 2). This suggests independent acquisition of the clones and no evidence of transmission among the selected hospitalised patients, while person-to-person transmission from healthy carriers or non-selected patients cannot be ruled out [21, 22]. Only two ST121 MSSA isolates were found to be almost identical and both were retrieved in the same time window from patients 096 and 098 (8 SNVs). For ST1, instead, all but two isolates belonged to the same sub-lineage, typed as SCCmecIV t127 PVL−.
We further estimated divergence times for all the 16 isolates belonging to the ST1 SCCmecIV t127 PVL− clone, including those obtained from earlier or later time points of the same patients. We used a Bayesian approach [85] (see Materials and methods) integrating all the reference genomes publicly available for ST1 and the two ST1 SCCmecV isolates from our cohort (Additional file 4: Table S3). These analyses were performed to test the hypothesis that all ST1 SCCmecIV t127 belong to a clone specific of Meyer’s hospital. The relaxed exponential clock model with constant coalescent prior and GTR substitution model resulted to the most appropriate model (Additional file 6: Table S5). This model estimated that Meyer’s clone has emerged approximately 6 to 28 years ago as a specific branch of the ST1 tree, which has been estimated to be 26–160 years old (Fig. 5). However, age of Meyer’s clone does not match with the time of emergence of the clone in the hospital. Moreover, an isolate obtained in a recent study investigating the spread of a ST1 SCCmecIV t127 clone in Irish hospitals [169] and carrying a virulence and resistance profile very close to the one of our cohort (differences in gene presence: 2/79 and 0/18 respectively) is phylogenetically rooted inside Meyer’s cluster (161 SNVs intra-cluster; 412 SNVs inter-cluster). These two findings suggest that ST1 SCCmecIV t127 is not specific of the Meyer Children’s hospital but might represent a newly arising community clone that is now spreading in the nosocomial environment of different countries [169, 170].
Fig. 5.

Bayesian timed tree of ST1 isolates, including reference genomes. Location and date of sample collection is reported for each isolate. For samples collected at Meyer’s Children Hospital (black circles), patient code is reported instead of location. The two North Dakota samples were collected from the same subject. “n/a” indicates that no information is available for location of sample collection. Numbers at selected nodes are posterior probabilities. Grey areas are the distributions of marginal posterior probabilities for the diversification of ST1 and the diversification of Mayer-specific clone
Conclusions
In this study, we investigated the epidemiology of S. aureus in different operative units of Anne Meyer’s Children’s University Hospital (Florence, Italy) over a timespan of 3 years by whole genome isolate sequencing. Our analyses highlighted a high diversity of STs, SCCmec, and spa-types, resulting into a wide number of clones. Some of these clones had been previously described in the literature as livestock-associated, and we described them in non-exposed children thus supporting the spreading of such clones in the non-at-risk community. We moreover described the presence of hypervirulent and geographically unusual clones, and of five STs for which no sequenced genome was available in public databases. Our refined analysis of the SCCmec cassettes highlighted the presence of further resistances and diversity within the same cassette type. On the contrary, when considering single infection types or specific STs or clones as it is usual in S. aureus epidemiological studies, the genomic diversity was limited, with an increased pattern of resistance genes in chronic patients and a larger number of virulence factors in acute infections. Altogether, these observations shed more light on the complexity of S. aureus epidemiology and on the need for a more unbiased survey of the commensal and pathogenic S. aureus community, to avoid the misrepresentation of specific genomic traits.
Whole-genome-based routine surveillance of S. aureus and other hospital-related pathogens would further allow to get a more unbiased idea of the rising clones and better informing clinical practices, which usually focused on the most dangerous or well-known strains. Performing such epidemiological studies as soon as a new putative nosocomial clone arises could allow us to conclude whether the new clone has arisen in that very hospital or it is a recent sub-clone spreading also in the non-hospitalised population and therefore more frequently isolated also in the clinics. These wider-focus studies would not only allow the assessment of the epidemiology of specific pathogens and clones in the hospital setting, but also the survey of the prevalence and conservation of their virulence and resistance traits. This could lead to the identification of antigens of interest for vaccine development and of specific sub-clones representing the main burden of infection, and therefore reassessing the efforts for the discovery of new treatments.
Whole genome sequencing studies are crucial to survey the global epidemiology of infectious agents, including S. aureus, as genome-based data are reproducible and can be easily meta-analysed without the confounding of batch effects. The meta-analysis of pathogenic, commensal, and environmental S. aureus isolates could lead to a deeper knowledge of the epidemiology of this bacterium and may help in understanding how to prevent and treat infections without boosting antibiotic resistance.
Additional files
Table S1. Characteristics of the single isolates, including collection details, genome assembly statistics, genomic features, and results of antibiotic susceptibility testing. (XLSX 69 kb)
Figure S1. Pangenome analysis statistics. Figure S2. Phylogenetic model based on gene presence/absence. (PDF 580 kb)
Table S2. Presence/absence profile of virulence and antibiotic resistance genes in the cohort. (XLSX 6063 kb)
Table S3. Characteristics of the genomes included in the ST1 analyses. (XLSX 11 kb)
Table S4. Most relevant clones represented in the cohort and their abundance. (XLSX 9 kb)
Table S5. Bayesian clock models tested and their results. (XLSX 10 kb)
Acknowledgements
The authors would like to acknowledge the laboratory of Computational Metagenomics for the valuable input. We would also like to thank Sander Wuyts and Stijn Wittouck for assistance in the plotting of the SCCmecIV graphs, and Dr. Megan Earls and Prof. David Coleman for sharing the ST1 SCCmecIV t127 genome.
Funding
This work was supported in part by the Italian Ministry of Health “Ricerca finalizzata” RF-2010-2316179 to GT. This work was also partially supported by the European Union H2020 Marie Curie grant (707345) to E.P. and by a European Union FP7 Marie Curie grant (PCIG13-618833), H2020-ERC-STG grant (MetaPG-716575), and by MIUR project FIR-RBFR13EWWI to NS.
Availability of data and materials
The datasets generated and analysed during the current study are available in the NCBI Sequence Read Archive (BioProject accession number PRJNA400143, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA400143/).
Abbreviations
- CC
Clonal complex
- CDS
Coding sequence
- CF
Cystic fibrosis
- CoNS
Coagulase-negative staphylococci
- indels
Insertions and deletions
- MLST
Multilocus sequence typing
- MRSA
Methicillin-resistant S. aureus
- MSSA
Methicillin-sensitive S. aureus
- PVL
Panton-Valentine Leukocidin
- SCCmec
Staphylococcal Cassette Chromosome mec
- SNV
Single-nucleotide variation
- Spa
Staphylococcal protein A
- ST
Sequence type
- WGS
Whole-genome shotgun sequencing
Authors’ contributions
GT, SC, and NS conceived and supervised the study. DD, NR, and FeA performed DNA extractions and library preparation. SM, FrA, and EP conducted the computational and statistical analyses. SM, DD, GG, AM, GT, and NS analysed and interpreted the results. ORS performed the phylogenetic analysis. SM, EP, DD, and NS wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The study received ethical approval from the Paediatric Ethics Committee, Autonomous Section of the Regional Ethics Committee for Clinical Trials at the Children’s Meyer Hospital, Florence on July 1, 2014. Data collection has been performed in accordance with the Declaration of Helsinki. All patients gave written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, Nouwen JL. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis. 2005;5(12):751–762. doi: 10.1016/S1473-3099(05)70295-4. [DOI] [PubMed] [Google Scholar]
- 2.Mainous AG, 3rd, Hueston WJ, Everett CJ, Diaz VA. Nasal carriage of Staphylococcus aureus and methicillin-resistant S aureus in the United States, 2001-2002. Ann Fam Med. 2006;4(2):132–137. doi: 10.1370/afm.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG., Jr Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev. 2015;28(3):603–661. doi: 10.1128/CMR.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esposito S, Noviello S, Leone S. Epidemiology and microbiology of skin and soft tissue infections. Curr Opin Infect Dis. 2016;29(2):109–115. doi: 10.1097/QCO.0000000000000239. [DOI] [PubMed] [Google Scholar]
- 5.Rhee Y, Aroutcheva A, Hota B, Weinstein RA, Popovich KJ. Evolving epidemiology of Staphylococcus aureus bacteremia. Infect Control Hosp Epidemiol. 2015;36(12):1417–1422. doi: 10.1017/ice.2015.213. [DOI] [PubMed] [Google Scholar]
- 6.Peacock SJ, Paterson GK. Mechanisms of methicillin resistance in Staphylococcus aureus. Annu Rev Biochem. 2015;84(1):577–601. doi: 10.1146/annurev-biochem-060614-034516. [DOI] [PubMed] [Google Scholar]
- 7.Skinner D. Significance of bacteremia caused by Staphylococcus aureus. Arch Intern Med. 1941;68(5):851. doi: 10.1001/archinte.1941.00200110003001. [DOI] [Google Scholar]
- 8.Rammelkamp CH, Maxon T. Resistance of Staphylococcus aureus to the action of penicillin. Exp Biol Med. 1942;51(3):386–389. doi: 10.3181/00379727-51-13986. [DOI] [Google Scholar]
- 9.Kirby WM. Extraction of a highly potent penicillin Inactivator from penicillin resistant staphylococci. Science. 1944;99(2579):452–453. doi: 10.1126/science.99.2579.452. [DOI] [PubMed] [Google Scholar]
- 10.Bondi A, Jr, Dietz CC. Penicillin resistant staphylococci. Proc Soc Exp Biol Med. 1945;60:55–58. doi: 10.3181/00379727-60-15089. [DOI] [PubMed] [Google Scholar]
- 11.Barber M. Methicillin-resistant staphylococci. J Clin Pathol. 1961;14:385–393. doi: 10.1136/jcp.14.4.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jevons MP, Coe AW, Parker MT. Methicillin resistance in staphylococci. Lancet. 1963;1(7287):904–907. doi: 10.1016/S0140-6736(63)91687-8. [DOI] [PubMed] [Google Scholar]
- 13.Parker MT, Jevons MP. A survey of methicillin resistance in Staphylococcus aureus. Postgrad Med J. 1964;40(SUPPL):170–178. doi: 10.1136/pgmj.40.Suppl.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pantosti A, Sanchini A, Monaco M. Mechanisms of antibiotic resistance in Staphylococcus aureus. Future Microbiol. 2007;2(3):323–334. doi: 10.2217/17460913.2.3.323. [DOI] [PubMed] [Google Scholar]
- 15.Monaco M, Pimentel de Araujo F, Cruciani M, Coccia EM, Pantosti A. Worldwide epidemiology and antibiotic resistance of Staphylococcus aureus. Curr Top Microbiol Immunol. 2017;409:21–56. doi: 10.1007/82_2016_3. [DOI] [PubMed] [Google Scholar]
- 16.Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, Kaplan SL, Karchmer AW, Levine DP, Murray BE, et al. Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52(3):e18–e55. doi: 10.1093/cid/ciq146. [DOI] [PubMed] [Google Scholar]
- 17.Raad I, Hanna H, Jiang Y, Dvorak T, Reitzel R, Chaiban G, Sherertz R, Hachem R. Comparative activities of daptomycin, linezolid, and tigecycline against catheter-related methicillin-resistant Staphylococcus bacteremic isolates embedded in biofilm. Antimicrob Agents Chemother. 2007;51(5):1656–1660. doi: 10.1128/AAC.00350-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kos VN, Desjardins CA, Griggs A, Cerqueira G, Van Tonder A, Holden MT, Godfrey P, Palmer KL, Bodi K, Mongodin EF, et al. Comparative genomics of vancomycin-resistant Staphylococcus aureus strains and their positions within the clade most commonly associated with Methicillin-resistant S. aureus hospital-acquired infection in the United States. MBio. 2012;3(3):e00112-12. [DOI] [PMC free article] [PubMed]
- 19.Hiramatsu K. Vancomycin-resistant Staphylococcus aureus: a new model of antibiotic resistance. Lancet Infect Dis. 2001;1(3):147–155. doi: 10.1016/S1473-3099(01)00091-3. [DOI] [PubMed] [Google Scholar]
- 20.Tacconelli E, Carrara E, Savoldi A, Harbarth S, Mendelson M, Monnet DL, Pulcini C, Kahlmeter G, Kluytmans J, Carmeli Y, et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis. 2018;18(3):318–327. doi: 10.1016/S1473-3099(17)30753-3. [DOI] [PubMed] [Google Scholar]
- 21.Tosas Auguet O, Stabler RA, Betley J, Preston MD, Dhaliwal M, Gaunt M, Ioannou A, Desai N, Karadag T, Batra R, et al. Frequent undetected ward-based methicillin-resistant Staphylococcus aureus transmission linked to patient sharing between hospitals. Clin Infect Dis. 2018;66(6):840–848. doi: 10.1093/cid/cix901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coll Francesc, Harrison Ewan M., Toleman Michelle S., Reuter Sandra, Raven Kathy E., Blane Beth, Palmer Beverley, Kappeler A. Ruth M., Brown Nicholas M., Török M. Estée, Parkhill Julian, Peacock Sharon J. Longitudinal genomic surveillance of MRSA in the UK reveals transmission patterns in hospitals and the community. Science Translational Medicine. 2017;9(413):eaak9745. doi: 10.1126/scitranslmed.aak9745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Voss A, Doebbeling BN. The worldwide prevalence of methicillin-resistant Staphylococcus aureus. Int J Antimicrob Agents. 1995;5(2):101–106. doi: 10.1016/0924-8579(94)00036-T. [DOI] [PubMed] [Google Scholar]
- 24.Stefani S, Chung DR, Lindsay JA, Friedrich AW, Kearns AM, Westh H, Mackenzie FM. Methicillin-resistant Staphylococcus aureus (MRSA): global epidemiology and harmonisation of typing methods. Int J Antimicrob Agents. 2012;39(4):273–282. doi: 10.1016/j.ijantimicag.2011.09.030. [DOI] [PubMed] [Google Scholar]
- 25.Chambers HF, Deleo FR. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol. 2009;7(9):629–641. doi: 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jansen van Rensburg MJ, Eliya Madikane V, Whitelaw A, Chachage M, Haffejee S, Gay Elisha B. The dominant methicillin-resistant Staphylococcus aureus clone from hospitals in Cape Town has an unusual genotype: ST612. Clin Microbiol Infection. 2011;17(5):785–792. doi: 10.1111/j.1469-0691.2010.03373.x. [DOI] [PubMed] [Google Scholar]
- 27.Harris SR, Feil EJ, Holden MT, Quail MA, Nickerson EK, Chantratita N, Gardete S, Tavares A, Day N, Lindsay JA, et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science. 2010;327(5964):469–474. doi: 10.1126/science.1182395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mediavilla JR, Chen L, Mathema B, Kreiswirth BN. Global epidemiology of community-associated methicillin resistant Staphylococcus aureus (CA-MRSA) Curr Opin Microbiol. 2012;15(5):588–595. doi: 10.1016/j.mib.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 29.Deurenberg RH, Stobberingh EE. The evolution of Staphylococcus aureus. Infect Genet Evol. 2008;8(6):747–763. doi: 10.1016/j.meegid.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 30.Point prevalence survey of healthcare-associated infections and antimicrobial use in European long-term care facilities, May–September 2010 [https://publications.europa.eu/en/publication-detail/-/publication/d6355bbd-f651-4770-bcad-0456b1dc2515 Accessed 11 Apr 2018]. [DOI] [PMC free article] [PubMed]
- 31.Sievert DM, Ricks P, Edwards JR, Schneider A, Patel J, Srinivasan A, Kallen A, Limbago B, Fridkin S, National Healthcare Safety Network T et al. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009-2010. Infect Control Hosp Epidemiol. 2013;34(1):1–14. doi: 10.1086/668770. [DOI] [PubMed] [Google Scholar]
- 32.Price JR, Cole K, Bexley A, Kostiou V, Eyre DW, Golubchik T, Wilson DJ, Crook DW, Walker AS, Peto TEA, et al. Transmission of Staphylococcus aureus between health-care workers, the environment, and patients in an intensive care unit: a longitudinal cohort study based on whole-genome sequencing. Lancet Infect Dis. 2017;17(2):207–214. doi: 10.1016/S1473-3099(16)30413-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prosperi M, Veras N, Azarian T, Rathore M, Nolan D, Rand K, Cook RL, Johnson J, Morris JG, Jr, Salemi M. Molecular epidemiology of community-associated methicillin-resistant Staphylococcus aureus in the genomic era: a cross-sectional study. Sci Rep. 2013;3:1902. doi: 10.1038/srep01902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harris SR, Cartwright EJ, Torok ME, Holden MT, Brown NM, Ogilvy-Stuart AL, Ellington MJ, Quail MA, Bentley SD, Parkhill J, et al. Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect Dis. 2013;13(2):130–136. doi: 10.1016/S1473-3099(12)70268-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stein M, Navon-Venezia S, Chmelnitsky I, Kohelet D, Schwartz O, Agmon O, Somekh E. An outbreak of new, nonmultidrug-resistant, methicillin-resistant Staphylococcus aureus strain (sccmec type iiia variant-1) in the neonatal intensive care unit transmitted by a staff member. Pediatr Infect Dis J. 2006;25(6):557–559. doi: 10.1097/01.inf.0000219407.31195.44. [DOI] [PubMed] [Google Scholar]
- 36.Bertin ML, Vinski J, Schmitt S, Sabella C, Danziger-Isakov L, McHugh M, Procop GW, Hall G, Gordon SM, Goldfarb J. Outbreak of methicillin-resistant Staphylococcus aureus colonization and infection in a neonatal intensive care unit epidemiologically linked to a healthcare worker with chronic otitis. Infect Control Hosp Epidemiol. 2006;27(6):581–585. doi: 10.1086/504933. [DOI] [PubMed] [Google Scholar]
- 37.Coombs GW, Van Gessel H, Pearson JC, Godsell MR, O'Brien FG, Christiansen KJ. Controlling a multicenter outbreak involving the New York/Japan methicillin-resistant Staphylococcus aureus clone. Infect Control Hosp Epidemiol. 2007;28(7):845–852. doi: 10.1086/518726. [DOI] [PubMed] [Google Scholar]
- 38.Saiman L, Cronquist A, Wu F, Zhou J, Rubenstein D, Eisner W, Kreiswirth BN, Della-Latta P. An outbreak of methicillin-resistant Staphylococcus aureus in a neonatal intensive care unit. Infect Control Hosp Epidemiol. 2003;24(5):317–321. doi: 10.1086/502217. [DOI] [PubMed] [Google Scholar]
- 39.Wang JT, Chang SC, Ko WJ, Chang YY, Chen ML, Pan HJ, Luh KT. A hospital-acquired outbreak of methicillin-resistant Staphylococcus aureus infection initiated by a surgeon carrier. J Hosp Infect. 2001;47(2):104–109. doi: 10.1053/jhin.2000.0878. [DOI] [PubMed] [Google Scholar]
- 40.Givney R, Vickery A, Holliday A, Pegler M, Benn R. Methicillin-resistant Staphylococcus aureus in a cystic fibrosis unit. J Hosp Infect. 1997;35(1):27–36. doi: 10.1016/S0195-6701(97)90165-1. [DOI] [PubMed] [Google Scholar]
- 41.Blok HE, Troelstra A, Kamp-Hopmans TE, Gigengack-Baars AC, Vandenbroucke-Grauls CM, Weersink AJ, Verhoef J, Mascini EM. Role of healthcare workers in outbreaks of methicillin-resistant Staphylococcus aureus: a 10-year evaluation from a Dutch university hospital. Infect Control Hosp Epidemiol. 2003;24(9):679–685. doi: 10.1086/502275. [DOI] [PubMed] [Google Scholar]
- 42.Tong SY, Holden MT, Nickerson EK, Cooper BS, Koser CU, Cori A, Jombart T, Cauchemez S, Fraser C, Wuthiekanun V, et al. Genome sequencing defines phylogeny and spread of methicillin-resistant Staphylococcus aureus in a high transmission setting. Genome Res. 2015;25(1):111–118. doi: 10.1101/gr.174730.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Copin R, Shopsin B, Torres VJ. After the deluge: mining Staphylococcus aureus genomic data for clinical associations and host-pathogen interactions. Curr Opin Microbiol. 2017;41:43–50. doi: 10.1016/j.mib.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lina G, Piemont Y, Godail-Gamot F, Bes M, Peter MO, Gauduchon V, Vandenesch F, Etienne J. Involvement of Panton-Valentine leukocidin-producing Staphylococcus aureus in primary skin infections and pneumonia. Clin Infect Dis. 1999;29(5):1128–1132. doi: 10.1086/313461. [DOI] [PubMed] [Google Scholar]
- 45.Milheirico C, Oliveira DC, de Lencastre H. Update to the multiplex PCR strategy for assignment of mec element types in Staphylococcus aureus. Antimicrob Agents Chemother. 2007;51(9):3374–3377. doi: 10.1128/AAC.00275-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oliveira DC, de Lencastre H. Multiplex PCR strategy for rapid identification of structural types and variants of the mec element in methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2002;46(7):2155–2161. doi: 10.1128/AAC.46.7.2155-2161.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oliveira DC, Milheirico C, de Lencastre H. Redefining a structural variant of staphylococcal cassette chromosome mec, SCCmec type VI. Antimicrob Agents Chemother. 2006;50(10):3457–3459. doi: 10.1128/AAC.00629-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Milheirico C, Oliveira DC, de Lencastre H. Multiplex PCR strategy for subtyping the staphylococcal cassette chromosome mec type IV in methicillin-resistant Staphylococcus aureus: 'SCCmec IV multiplex. J Antimicrob Chemother. 2007;60(1):42–48. doi: 10.1093/jac/dkm112. [DOI] [PubMed] [Google Scholar]
- 49.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 51.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MT, Fookes M, Falush D, Keane JA, Parkhill J. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31(22):3691–3693. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang K, McClure JA, Elsayed S, Louie T, Conly JM. Novel multiplex PCR assay for characterization and concomitant subtyping of staphylococcal cassette chromosome mec types I to V in methicillin-resistant Staphylococcus aureus. J Clin Microbiol. 2005;43(10):5026–5033. doi: 10.1128/JCM.43.10.5026-5033.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boye K, Bartels MD, Andersen IS, Moller JA, Westh H. A new multiplex PCR for easy screening of methicillin-resistant Staphylococcus aureus SCCmec types I-V. Clin Microbiol Infect. 2007;13(7):725–727. doi: 10.1111/j.1469-0691.2007.01720.x. [DOI] [PubMed] [Google Scholar]
- 55.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 56.Zolfo M, Tett A, Jousson O, Donati C, Segata N. MetaMLST: multi-locus strain-level bacterial typing from metagenomic samples. Nucleic Acids Res. 2017;45(2):e7. doi: 10.1093/nar/gkw837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.AL-T F, Brunel AS, Bouzinbi N, Corne P, Banuls AL, Shahbazkia HR. DNAGear—a free software for spa type identification in Staphylococcus aureus. BMC Res Notes. 2012;5:642. doi: 10.1186/1756-0500-5-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gordon NC, Price JR, Cole K, Everitt R, Morgan M, Finney J, Kearns AM, Pichon B, Young B, Wilson DJ, et al. Prediction of Staphylococcus aureus antimicrobial resistance by whole-genome sequencing. J Clin Microbiol. 2014;52(4):1182–1191. doi: 10.1128/JCM.03117-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Inoshima I, Inoshima N, Wilke GA, Powers ME, Frank KM, Wang Y, Bubeck Wardenburg J. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat Med. 2011;17(10):1310–1314. doi: 10.1038/nm.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dinges MM, Orwin PM, Schlievert PM. Exotoxins of Staphylococcus aureus. Clin Microbiol Rev. 2000;13(1):16–34. doi: 10.1128/CMR.13.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith EJ, Visai L, Kerrigan SW, Speziale P, Foster TJ. The Sbi protein is a multifunctional immune evasion factor of Staphylococcus aureus. Infect Immun. 2011;79(9):3801–3809. doi: 10.1128/IAI.05075-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haupt K, Reuter M, van den Elsen J, Burman J, Halbich S, Richter J, Skerka C, Zipfel PF. The Staphylococcus aureus protein Sbi acts as a complement inhibitor and forms a tripartite complex with host complement factor H and C3b. PLoS Pathog. 2008;4(12):e1000250. doi: 10.1371/journal.ppat.1000250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shannon O, Flock JI. Extracellular fibrinogen binding protein, Efb, from Staphylococcus aureus binds to platelets and inhibits platelet aggregation. Thromb Haemost. 2004;91(4):779–789. doi: 10.1160/TH03-05-0287. [DOI] [PubMed] [Google Scholar]
- 64.Shannon O, Uekotter A, Flock JI. Extracellular fibrinogen binding protein, Efb, from Staphylococcus aureus as an antiplatelet agent in vivo. Thromb Haemost. 2005;93(5):927–931. doi: 10.1160/TH04-08-0501. [DOI] [PubMed] [Google Scholar]
- 65.Courjon J, Munro P, Benito Y, Visvikis O, Bouchiat C, Boyer L, Doye A, Lepidi H, Ghigo E, Lavigne JP, et al. EDIN-B promotes the translocation of Staphylococcus aureus to the bloodstream in the course of pneumonia. Toxins (Basel) 2015;7(10):4131–4142. doi: 10.3390/toxins7104131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Diep BA, Stone GG, Basuino L, Graber CJ, Miller A, des Etages SA, Jones A, Palazzolo-Ballance AM, Perdreau-Remington F, Sensabaugh GF, et al. The arginine catabolic mobile element and staphylococcal chromosomal cassette mec linkage: convergence of virulence and resistance in the USA300 clone of methicillin-resistant Staphylococcus aureus. J Infect Dis. 2008;197(11):1523–1530. doi: 10.1086/587907. [DOI] [PubMed] [Google Scholar]
- 67.Ellington MJ, Yearwood L, Ganner M, East C, Kearns AM. Distribution of the ACME-arcA gene among methicillin-resistant Staphylococcus aureus from England and Wales. J Antimicrob Chemother. 2008;61(1):73–77. doi: 10.1093/jac/dkm422. [DOI] [PubMed] [Google Scholar]
- 68.Kim J, Urban RG, Strominger JL, Wiley DC. Toxic shock syndrome toxin-1 complexed with a class II major histocompatibility molecule HLA-DR1. Science. 1994;266(5192):1870–1874. doi: 10.1126/science.7997880. [DOI] [PubMed] [Google Scholar]
- 69.Miethke T, Duschek K, Wahl C, Heeg K, Wagner H. Pathogenesis of the toxic shock syndrome: T cell mediated lethal shock caused by the superantigen TSST-1. Eur J Immunol. 1993;23(7):1494–1500. doi: 10.1002/eji.1830230715. [DOI] [PubMed] [Google Scholar]
- 70.Rooijakkers SH, van Strijp JA. Bacterial complement evasion. Mol Immunol. 2007;44(1-3):23–32. doi: 10.1016/j.molimm.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 71.Thammavongsa V, Kim HK, Missiakas D, Schneewind O. Staphylococcal manipulation of host immune responses. Nat Rev Microbiol. 2015;13(9):529–543. doi: 10.1038/nrmicro3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Udo EE, Boswihi SS, Al-Sweih N. High prevalence of toxic shock syndrome toxin-producing epidemic methicillin-resistant Staphylococcus aureus 15 (EMRSA-15) strains in Kuwait hospitals. New Microbes New Infect. 2016;12:24–30. doi: 10.1016/j.nmni.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Al Laham N, Mediavilla JR, Chen L, Abdelateef N, Elamreen FA, Ginocchio CC, Pierard D, Becker K, Kreiswirth BN. MRSA clonal complex 22 strains harboring toxic shock syndrome toxin (TSST-1) are endemic in the primary hospital in Gaza, Palestine. PLoS One. 2015;10(3):e0120008. doi: 10.1371/journal.pone.0120008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Biber A, Abuelaish I, Rahav G, Raz M, Cohen L, Valinsky L, Taran D, Goral A, Elhamdany A, Regev-Yochay G, et al. A typical hospital-acquired methicillin-resistant Staphylococcus aureus clone is widespread in the community in the Gaza strip. PLoS One. 2012;7(8):e42864. doi: 10.1371/journal.pone.0042864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Geraci DM, Bonura C, Giuffre M, Aleo A, Saporito L, Graziano G, Valenti RM, Mammina C. tst1-positive ST22-MRSA-IVa in healthy Italian preschool children. Infection. 2014;42(3):535–538. doi: 10.1007/s15010-013-0583-z. [DOI] [PubMed] [Google Scholar]
- 76.Geraci DM, Giuffre M, Bonura C, Matranga D, Aleo A, Saporito L, Corsello G, Larsen AR, Mammina C. Methicillin-resistant Staphylococcus aureus colonization: a three-year prospective study in a neonatal intensive care unit in Italy. PLoS One. 2014;9(2):e87760. doi: 10.1371/journal.pone.0087760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Amagai M, Matsuyoshi N, Wang ZH, Andl C, Stanley JR. Toxin in bullous impetigo and staphylococcal scalded-skin syndrome targets desmoglein 1. Nat Med. 2000;6(11):1275–1277. doi: 10.1038/81385. [DOI] [PubMed] [Google Scholar]
- 78.Hanakawa Y, Schechter NM, Lin C, Garza L, Li H, Yamaguchi T, Fudaba Y, Nishifuji K, Sugai M, Amagai M, et al. Molecular mechanisms of blister formation in bullous impetigo and staphylococcal scalded skin syndrome. J Clin Invest. 2002;110(1):53–60. doi: 10.1172/JCI0215766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ladhani S. Understanding the mechanism of action of the exfoliative toxins of Staphylococcus aureus. FEMS Immunol Med Microbiol. 2003;39(2):181–189. doi: 10.1016/S0928-8244(03)00225-6. [DOI] [PubMed] [Google Scholar]
- 80.Tung H, Guss B, Hellman U, Persson L, Rubin K, Ryden C. A bone sialoprotein-binding protein from Staphylococcus aureus: a member of the staphylococcal Sdr family. Biochem J. 2000;345(Pt 3):611–619. doi: 10.1042/bj3450611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jongerius I, von Kockritz-Blickwede M, Horsburgh MJ, Ruyken M, Nizet V, Rooijakkers SH. Staphylococcus aureus virulence is enhanced by secreted factors that block innate immune defenses. J Innate Immun. 2012;4(3):301–311. doi: 10.1159/000334604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sanchez CJ, Jr, Mende K, Beckius ML, Akers KS, Romano DR, Wenke JC, Murray CK. Biofilm formation by clinical isolates and the implications in chronic infections. BMC Infect Dis. 2013;13:47. doi: 10.1186/1471-2334-13-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Paharik AE, Horswill AR. The staphylococcal biofilm: adhesins, regulation, and host response. Microbiol Spectr. 2016;4(2):VMBF-0022-2015. [DOI] [PMC free article] [PubMed]
- 84.Schwab UE, Wold AE, Carson JL, Leigh MW, Cheng PW, Gilligan PH, Boat TF. Increased adherence of Staphylococcus aureus from cystic fibrosis lungs to airway epithelial cells. Am Rev Respir Dis. 1993;148(2):365–369. doi: 10.1164/ajrccm/148.2.365. [DOI] [PubMed] [Google Scholar]
- 85.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29(8):1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bosi E, Monk JM, Aziz RK, Fondi M, Nizet V, Palsson BO. Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc Natl Acad Sci U S A. 2016;113(26):E3801–E3809. doi: 10.1073/pnas.1523199113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant DA, et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A. 1998;95(6):3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35(Database issue):D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Haft DH, DiCuccio M, Badretdin A, Brover V, Chetvernin V, O'Neill K, Li W, Chitsaz F, Derbyshire MK, Gonzales NR, et al. RefSeq: an update on prokaryotic genome annotation and curation. Nucleic Acids Res. 2018;46(D1):D851–D860. doi: 10.1093/nar/gkx1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Planet PJ, Narechania A, Chen L, Mathema B, Boundy S, Archer G, Kreiswirth B. Architecture of a species: Phylogenomics of Staphylococcus aureus. Trends Microbiol. 2017;25(2):153–166. doi: 10.1016/j.tim.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 91.Mato R, Campanile F, Stefani S, Crisostomo MI, Santagati M, Sanches SI, de Lencastre H. Clonal types and multidrug resistance patterns of methicillin-resistant Staphylococcus aureus (MRSA) recovered in Italy during the 1990s. Microb Drug Resist. 2004;10(2):106–113. doi: 10.1089/1076629041310109. [DOI] [PubMed] [Google Scholar]
- 92.Campanile F, Bongiorno D, Perez M, Mongelli G, Sessa L, Benvenuto S, Gona F, Participants ASS, Varaldo PE, Stefani S. Epidemiology of Staphylococcus aureus in Italy: first nationwide survey, 2012. J Glob Antimicrob Resist. 2015;3(4):247–254. doi: 10.1016/j.jgar.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 93.Vandenesch F, Naimi T, Enright MC, Lina G, Nimmo GR, Heffernan H, Liassine N, Bes M, Greenland T, Reverdy ME, et al. Community-acquired methicillin-resistant Staphylococcus aureus carrying Panton-Valentine leukocidin genes: worldwide emergence. Emerg Infect Dis. 2003;9(8):978–984. doi: 10.3201/eid0908.030089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Witte W, Strommenger B, Stanek C, Cuny C. Methicillin-resistant Staphylococcus aureus ST398 in humans and animals, Central Europe. Emerg Infect Dis. 2007;13(2):255–258. doi: 10.3201/eid1302.060924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.van Cleef BA, Graveland H, Haenen AP, van de Giessen AW, Heederik D, Wagenaar JA, Kluytmans JA. Persistence of livestock-associated methicillin-resistant Staphylococcus aureus in field workers after short-term occupational exposure to pigs and veal calves. J Clin Microbiol. 2011;49(3):1030–1033. doi: 10.1128/JCM.00493-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cuny C, Kock R, Witte W. Livestock associated MRSA (LA-MRSA) and its relevance for humans in Germany. Int J Med Microbiol. 2013;303(6-7):331–337. doi: 10.1016/j.ijmm.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 97.Monaco M, Pedroni P, Sanchini A, Bonomini A, Indelicato A, Pantosti A. Livestock-associated methicillin-resistant Staphylococcus aureus responsible for human colonization and infection in an area of Italy with high density of pig farming. BMC Infect Dis. 2013;13:258. doi: 10.1186/1471-2334-13-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pan A, Battisti A, Zoncada A, Bernieri F, Boldini M, Franco A, Giorgi M, Iurescia M, Lorenzotti S, Martinotti M, et al. Community-acquired methicillin-resistant Staphylococcus aureus ST398 infection, Italy. Emerg Infect Dis. 2009;15(5):845–847. doi: 10.3201/eid1505.081417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Soavi L, Stellini R, Signorini L, Antonini B, Pedroni P, Zanetti L, Milanesi B, Pantosti A, Matteelli A, Pan A, et al. Methicillin-resistant Staphylococcus aureus ST398, Italy. Emerg Infect Dis. 2010;16(2):346–348. doi: 10.3201/eid1602.091478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mammina C, Cala C, Plano MR, Bonura C, Vella A, Monastero R, Palma DM. Ventilator-associated pneumonia and MRSA ST398, Italy. Emerg Infect Dis. 2010;16(4):730–731. doi: 10.3201/eid1604.091584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.van Cleef BA, Monnet DL, Voss A, Krziwanek K, Allerberger F, Struelens M, Zemlickova H, Skov RL, Vuopio-Varkila J, Cuny C, et al. Livestock-associated methicillin-resistant Staphylococcus aureus in humans, Europe. Emerg Infect Dis. 2011;17(3):502–505. doi: 10.3201/eid1703.101036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Verkade E, Bergmans AM, Budding AE, van Belkum A, Savelkoul P, Buiting AG, Kluytmans J. Recent emergence of Staphylococcus aureus clonal complex 398 in human blood cultures. PLoS One. 2012;7(10):e41855. doi: 10.1371/journal.pone.0041855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sung JM, Lloyd DH, Lindsay JA. Staphylococcus aureus host specificity: comparative genomics of human versus animal isolates by multi-strain microarray. Microbiology. 2008;154(Pt 7):1949–1959. doi: 10.1099/mic.0.2007/015289-0. [DOI] [PubMed] [Google Scholar]
- 104.Spoor LE, McAdam PR, Weinert LA, Rambaut A, Hasman H, Aarestrup FM, Kearns AM, Larsen AR, Skov RL, Fitzgerald JR. Livestock origin for a human pandemic clone of community-associated methicillin-resistant Staphylococcus aureus. MBio. 2013;4(4):e00356–13. [DOI] [PMC free article] [PubMed]
- 105.Cuny C, Wieler LH, Witte W. Livestock-associated MRSA: the impact on humans. Antibiotics (Basel) 2015;4(4):521–543. doi: 10.3390/antibiotics4040521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Feltrin F, Alba P, Kraushaar B, Ianzano A, Argudin MA, Di Matteo P, Porrero MC, Aarestrup FM, Butaye P, Franco A, et al. A livestock-associated, multidrug-resistant, methicillin-resistant Staphylococcus aureus clonal complex 97 lineage spreading in dairy cattle and pigs in Italy. Appl Environ Microbiol. 2015;82(3):816–821. doi: 10.1128/AEM.02854-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fitzgerald JR. Livestock-associated Staphylococcus aureus: origin, evolution and public health threat. Trends Microbiol. 2012;20(4):192–198. doi: 10.1016/j.tim.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 108.Harrison EM, Coll F, Toleman MS, Blane B, Brown NM, Torok ME, Parkhill J, Peacock SJ. Genomic surveillance reveals low prevalence of livestock-associated methicillin-resistant Staphylococcus aureus in the East of England. Sci Rep. 2017;7(1):7406. doi: 10.1038/s41598-017-07662-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Winstel V, Liang C, Sanchez-Carballo P, Steglich M, Munar M, Broker BM, Penades JR, Nubel U, Holst O, Dandekar T, et al. Wall teichoic acid structure governs horizontal gene transfer between major bacterial pathogens. Nat Commun. 2013;4:2345. doi: 10.1038/ncomms3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Larsen J, Andersen PS, Winstel V, Peschel A. Staphylococcus aureus CC395 harbours a novel composite staphylococcal cassette chromosome mec element. J Antimicrob Chemother. 2017;72(4):1002–1005. doi: 10.1093/jac/dkw544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Molina A, Del Campo R, Maiz L, Morosini MI, Lamas A, Baquero F, Canton R. High prevalence in cystic fibrosis patients of multiresistant hospital-acquired methicillin-resistant Staphylococcus aureus ST228-SCCmecI capable of biofilm formation. J Antimicrob Chemother. 2008;62(5):961–967. doi: 10.1093/jac/dkn302. [DOI] [PubMed] [Google Scholar]
- 112.Asghar AH. Molecular characterization of methicillin-resistant Staphylococcus aureus isolated from tertiary care hospitals. Pak J Med Sci. 2014;30(4):698–702. doi: 10.12669/pjms.304.4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.McAdam PR, Holmes A, Templeton KE, Fitzgerald JR. Adaptive evolution of Staphylococcus aureus during chronic endobronchial infection of a cystic fibrosis patient. PLoS One. 2011;6(9):e24301. doi: 10.1371/journal.pone.0024301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Goerke C, Wolz C. Adaptation of Staphylococcus aureus to the cystic fibrosis lung. Int J Med Microbiol. 2010;300(8):520–525. doi: 10.1016/j.ijmm.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 115.Kahl BC. Impact of Staphylococcus aureus on the pathogenesis of chronic cystic fibrosis lung disease. Int J Med Microbiol. 2010;300(8):514–519. doi: 10.1016/j.ijmm.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 116.Cullen L, McClean S. Bacterial adaptation during chronic respiratory infections. Pathogens. 2015;4(1):66–89. doi: 10.3390/pathogens4010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rao Q, Shang W, Hu X, Rao X. Staphylococcus aureus ST121: a globally disseminated hypervirulent clone. J Med Microbiol. 2015;64(12):1462–1473. doi: 10.1099/jmm.0.000185. [DOI] [PubMed] [Google Scholar]
- 118.Goering RV, Shawar RM, Scangarella NE, O'Hara FP, Amrine-Madsen H, West JM, Dalessandro M, Becker JA, Walsh SL, Miller LA, et al. Molecular epidemiology of methicillin-resistant and methicillin-susceptible Staphylococcus aureus isolates from global clinical trials. J Clin Microbiol. 2008;46(9):2842–2847. doi: 10.1128/JCM.00521-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Moore CL, Osaki-Kiyan P, Perri M, Donabedian S, Haque NZ, Chen A, Zervos MJ. USA600 (ST45) methicillin-resistant Staphylococcus aureus bloodstream infections in urban Detroit. J Clin Microbiol. 2010;48(6):2307–2310. doi: 10.1128/JCM.00409-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Roberts JC. Classification of epidemic community-acquired methicillin-resistant Staphylococcus aureus by anatomical site of isolation. Biomed Res Int. 2014;2014:904283. doi: 10.1155/2014/904283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.David MZ, Boyle-Vavra S, Zychowski DL, Daum RS. Methicillin-susceptible Staphylococcus aureus as a predominantly healthcare-associated pathogen: a possible reversal of roles? PLoS One. 2011;6(4):e18217. doi: 10.1371/journal.pone.0018217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tinelli M, Monaco M, Vimercati M, Ceraminiello A, Pantosti A. Methicillin-susceptible Staphylococcus aureus in skin and soft tissue infections, Northern Italy. Emerg Infect Dis. 2009;15(2):250–257. doi: 10.3201/eid1502.080010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Baranovich T, Zaraket H, Shabana II, Nevzorova V, Turcutyuicov V, Suzuki H. Molecular characterization and susceptibility of methicillin-resistant and methicillin-susceptible Staphylococcus aureus isolates from hospitals and the community in Vladivostok, Russia. Clin Microbiol Infect. 2010;16(6):575–582. doi: 10.1111/j.1469-0691.2009.02891.x. [DOI] [PubMed] [Google Scholar]
- 124.Wu D, Wang Q, Yang Y, Geng W, Wang Q, Yu S, Yao K, Yuan L, Shen X. Epidemiology and molecular characteristics of community-associated methicillin-resistant and methicillin-susceptible Staphylococcus aureus from skin/soft tissue infections in a children's hospital in Beijing, China. Diagn Microbiol Infect Dis. 2010;67(1):1–8. doi: 10.1016/j.diagmicrobio.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 125.Rasigade JP, Laurent F, Lina G, Meugnier H, Bes M, Vandenesch F, Etienne J, Tristan A. Global distribution and evolution of Panton-Valentine leukocidin-positive methicillin-susceptible Staphylococcus aureus, 1981-2007. J Infect Dis. 2010;201(10):1589–1597. doi: 10.1086/652008. [DOI] [PubMed] [Google Scholar]
- 126.Isobe H, Takano T, Nishiyama A, Hung WC, Kuniyuki S, Shibuya Y, Reva I, Yabe S, Iwao Y, Higuchi W, et al. Evolution and virulence of Panton-Valentine leukocidin-positive ST30 methicillin-resistant Staphylococcus aureus in the past 30 years in Japan. Biomed Res. 2012;33(2):97–109. doi: 10.2220/biomedres.33.97. [DOI] [PubMed] [Google Scholar]
- 127.Fernandez S, Ledo C, Lattar S, Noto Llana M, Bertelli AM, Di Gregorio S, Sordelli DO, Gomez MI, Mollerach ME. High virulence of methicillin resistant Staphylococcus aureus ST30-SCCmecIVc-spat019, the dominant community-associated clone in Argentina. Int J Med Microbiol. 2017;307(4-5):191–199. doi: 10.1016/j.ijmm.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 128.Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat Rev Microbiol. 2018;16(3):143–155. doi: 10.1038/nrmicro.2017.157. [DOI] [PubMed] [Google Scholar]
- 129.Tett A, Pasolli E, Farina S, Truong DT, Asnicar F, Zolfo M, Beghini F, Armanini F, Jousson O, De Sanctis V, et al. Unexplored diversity and strain-level structure of the skin microbiome associated with psoriasis. NPJ Biofilms Microbiomes. 2017;3:14. doi: 10.1038/s41522-017-0022-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hiramatsu K, Cui L, Kuroda M, Ito T. The emergence and evolution of methicillin-resistant Staphylococcus aureus. Trends Microbiol. 2001;9(10):486–493. doi: 10.1016/S0966-842X(01)02175-8. [DOI] [PubMed] [Google Scholar]
- 131.Hanssen AM, Ericson Sollid JU. SCCmec in staphylococci: genes on the move. FEMS Immunol Med Microbiol. 2006;46(1):8–20. doi: 10.1111/j.1574-695X.2005.00009.x. [DOI] [PubMed] [Google Scholar]
- 132.Hartman B, Tomasz A. Altered penicillin-binding proteins in methicillin-resistant strains of Staphylococcus aureus. Antimicrob Agents Chemother. 1981;19(5):726–735. doi: 10.1128/AAC.19.5.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Archer GL, Niemeyer DM, Thanassi JA, Pucci MJ. Dissemination among staphylococci of DNA sequences associated with methicillin resistance. Antimicrob Agents Chemother. 1994;38(3):447–454. doi: 10.1128/AAC.38.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Campanile F, Bongiorno D, Falcone M, Vailati F, Pasticci MB, Perez M, Raglio A, Rumpianesi F, Scuderi C, Suter F, et al. Changing Italian nosocomial-community trends and heteroresistance in Staphylococcus aureus from bacteremia and endocarditis. Eur J Clin Microbiol Infect Dis. 2012;31(5):739–745. doi: 10.1007/s10096-011-1367-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Valsesia G, Rossi M, Bertschy S, Pfyffer GE. Emergence of SCCmec type IV and SCCmec type V methicillin-resistant Staphylococcus aureus containing the Panton-Valentine leukocidin genes in a large academic teaching hospital in Central Switzerland: external invaders or persisting circulators? J Clin Microbiol. 2010;48(3):720–727. doi: 10.1128/JCM.01890-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tenover FC, Goering RV. Methicillin-resistant Staphylococcus aureus strain USA300: origin and epidemiology. J Antimicrob Chemother. 2009;64(3):441–446. doi: 10.1093/jac/dkp241. [DOI] [PubMed] [Google Scholar]
- 137.Pedersen LC, Benning MM, Holden HM. Structural investigation of the antibiotic and ATP-binding sites in kanamycin nucleotidyltransferase. Biochemistry. 1995;34(41):13305–13311. doi: 10.1021/bi00041a005. [DOI] [PubMed] [Google Scholar]
- 138.Gennimata D, Davies J, Tsiftsoglou AS. Bleomycin resistance in Staphylococcus aureus clinical isolates. J Antimicrob Chemother. 1996;37(1):65–75. doi: 10.1093/jac/37.1.65. [DOI] [PubMed] [Google Scholar]
- 139.Dortet L, Girlich D, Virlouvet AL, Poirel L, Nordmann P, Iorga BI, Naas T. Characterization of BRPMBL, the bleomycin resistance protein associated with the Carbapenemase NDM. Antimicrob Agents Chemother. 2017;61(3):AAC–02413. [DOI] [PMC free article] [PubMed]
- 140.Burdeska A, Ott M, Bannwarth W, Then RL. Identical genes for trimethoprim-resistant dihydrofolate reductase from Staphylococcus aureus in Australia and Central Europe. FEBS Lett. 1990;266(1-2):159–162. doi: 10.1016/0014-5793(90)81529-W. [DOI] [PubMed] [Google Scholar]
- 141.Rouch DA, Messerotti LJ, Loo LS, Jackson CA, Skurray RA. Trimethoprim resistance transposon Tn4003 from Staphylococcus aureus encodes genes for a dihydrofolate reductase and thymidylate synthetase flanked by three copies of IS257. Mol Microbiol. 1989;3(2):161–175. doi: 10.1111/j.1365-2958.1989.tb01805.x. [DOI] [PubMed] [Google Scholar]
- 142.Whitener CJ, Park SY, Browne FA, Parent LJ, Julian K, Bozdogan B, Appelbaum PC, Chaitram J, Weigel LM, Jernigan J, et al. Vancomycin-resistant Staphylococcus aureus in the absence of vancomycin exposure. Clin Infect Dis. 2004;38(8):1049–1055. doi: 10.1086/382357. [DOI] [PubMed] [Google Scholar]
- 143.Tenover FC, Weigel LM, Appelbaum PC, McDougal LK, Chaitram J, McAllister S, Clark N, Killgore G, O'Hara CM, Jevitt L, et al. Vancomycin-resistant Staphylococcus aureus isolate from a patient in Pennsylvania. Antimicrob Agents Chemother. 2004;48(1):275–280. doi: 10.1128/AAC.48.1.275-280.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Stefani S, Campanile F, Santagati M, Mezzatesta ML, Cafiso V, Pacini G. Insights and clinical perspectives of daptomycin resistance in Staphylococcus aureus: a review of the available evidence. Int J Antimicrob Agents. 2015;46(3):278–289. doi: 10.1016/j.ijantimicag.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 145.Noto MJ, Kreiswirth BN, Monk AB, Archer GL. Gene acquisition at the insertion site for SCCmec, the genomic island conferring methicillin resistance in Staphylococcus aureus. J Bacteriol. 2008;190(4):1276–1283. doi: 10.1128/JB.01128-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wright GD. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol. 2007;5(3):175–186. doi: 10.1038/nrmicro1614. [DOI] [PubMed] [Google Scholar]
- 147.Giersing BK, Dastgheyb SS, Modjarrad K, Moorthy V. Status of vaccine research and development of vaccines for Staphylococcus aureus. Vaccine. 2016;34(26):2962–2966. doi: 10.1016/j.vaccine.2016.03.110. [DOI] [PubMed] [Google Scholar]
- 148.Verkaik NJ, van Wamel WJ, van Belkum A. Immunotherapeutic approaches against Staphylococcus aureus. Immunotherapy. 2011;3(9):1063–1073. doi: 10.2217/imt.11.84. [DOI] [PubMed] [Google Scholar]
- 149.Salgado-Pabon W, Schlievert PM. Models matter: the search for an effective Staphylococcus aureus vaccine. Nat Rev Microbiol. 2014;12(8):585–591. doi: 10.1038/nrmicro3308. [DOI] [PubMed] [Google Scholar]
- 150.Fattom AI, Horwith G, Fuller S, Propst M, Naso R. Development of StaphVAX, a polysaccharide conjugate vaccine against S. aureus infection: from the lab bench to phase III clinical trials. Vaccine. 2004;22(7):880–887. doi: 10.1016/j.vaccine.2003.11.034. [DOI] [PubMed] [Google Scholar]
- 151.Fattom A, Matalon A, Buerkert J, Taylor K, Damaso S, Boutriau D. Efficacy profile of a bivalent Staphylococcus aureus glycoconjugated vaccine in adults on hemodialysis: phase III randomized study. Hum Vaccin Immunother. 2015;11(3):632–641. doi: 10.4161/hv.34414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Fowler VG, Allen KB, Moreira ED, Moustafa M, Isgro F, Boucher HW, Corey GR, Carmeli Y, Betts R, Hartzel JS, et al. Effect of an investigational vaccine for preventing Staphylococcus aureus infections after cardiothoracic surgery: a randomized trial. JAMA. 2013;309(13):1368–1378. doi: 10.1001/jama.2013.3010. [DOI] [PubMed] [Google Scholar]
- 153.Fowler VG, Jr, Proctor RA. Where does a Staphylococcus aureus vaccine stand? Clin Microbiol Infect. 2014;20(Suppl 5):66–75. doi: 10.1111/1469-0691.12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Golubchik T, Batty EM, Miller RR, Farr H, Young BC, Larner-Svensson H, Fung R, Godwin H, Knox K, Votintseva A, et al. Within-host evolution of Staphylococcus aureus during asymptomatic carriage. PLoS One. 2013;8(5):e61319. doi: 10.1371/journal.pone.0061319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dreisbach A, van der Kooi-Pol MM, Otto A, Gronau K, Bonarius HP, Westra H, Groen H, Becher D, Hecker M, van Dijl JM. Surface shaving as a versatile tool to profile global interactions between human serum proteins and the Staphylococcus aureus cell surface. Proteomics. 2011;11(14):2921–2930. doi: 10.1002/pmic.201100134. [DOI] [PubMed] [Google Scholar]
- 156.Hua L, Cohen TS, Shi Y, Datta V, Hilliard JJ, Tkaczyk C, Suzich J, Stover CK, Sellman BR. MEDI4893* promotes survival and extends the antibiotic treatment window in a Staphylococcus aureus immunocompromised pneumonia model. Antimicrob Agents Chemother. 2015;59(8):4526–4532. doi: 10.1128/AAC.00510-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bagnoli F. Staphylococcus aureus toxin antibodies: good companions of antibiotics and vaccines. Virulence. 2017;8(7):1037–1042. doi: 10.1080/21505594.2017.1295205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yang L, Zhou H, Cheng P, Yang Y, Tong Y, Zuo Q, Feng Q, Zou Q, Zeng H. A novel bivalent fusion vaccine induces broad immunoprotection against Staphylococcus aureus infection in different murine models. Clin Immunol. 2018;188:85–93. doi: 10.1016/j.clim.2017.12.012. [DOI] [PubMed] [Google Scholar]
- 159.Frenck RW, Jr, Creech CB, Sheldon EA, Seiden DJ, Kankam MK, Baber J, Zito E, Hubler R, Eiden J, Severs JM, et al. Safety, tolerability, and immunogenicity of a 4-antigen Staphylococcus aureus vaccine (SA4Ag): results from a first-in-human randomised, placebo-controlled phase 1/2 study. Vaccine. 2017;35(2):375–384. doi: 10.1016/j.vaccine.2016.11.010. [DOI] [PubMed] [Google Scholar]
- 160.Begier E, Seiden DJ, Patton M, Zito E, Severs J, Cooper D, Eiden J, Gruber WC, Jansen KU, Anderson AS, et al. SA4Ag, a 4-antigen Staphylococcus aureus vaccine, rapidly induces high levels of bacteria-killing antibodies. Vaccine. 2017;35(8):1132–1139. doi: 10.1016/j.vaccine.2017.01.024. [DOI] [PubMed] [Google Scholar]
- 161.Anderson AS, Miller AA, Donald RG, Scully IL, Nanra JS, Cooper D, Jansen KU. Development of a multicomponent Staphylococcus aureus vaccine designed to counter multiple bacterial virulence factors. Hum Vaccin Immunother. 2012;8(11):1585–1594. doi: 10.4161/hv.21872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Roetzer A, Jilma B, Eibl MM. Vaccine against toxic shock syndrome in a first-in-man clinical trial. Expert Rev Vaccines. 2017;16(2):81–83. doi: 10.1080/14760584.2017.1268921. [DOI] [PubMed] [Google Scholar]
- 163.Narita K, Hu DL, Asano K, Nakane A. Vaccination with non-toxic mutant toxic shock syndrome toxin-1 induces IL-17-dependent protection against Staphylococcus aureus infection. Pathog Dis. 2015;73(4):ftv023. [DOI] [PubMed]
- 164.Rouha H, Badarau A, Visram ZC, Battles MB, Prinz B, Magyarics Z, Nagy G, Mirkina I, Stulik L, Zerbs M, et al. Five birds, one stone: neutralization of alpha-hemolysin and 4 bi-component leukocidins of Staphylococcus aureus with a single human monoclonal antibody. MAbs. 2015;7(1):243–254. doi: 10.4161/19420862.2014.985132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Badarau A, Rouha H, Malafa S, Battles MB, Walker L, Nielson N, Dolezilkova I, Teubenbacher A, Banerjee S, Maierhofer B, et al. Context matters: the importance of dimerization-induced conformation of the LukGH leukocidin of Staphylococcus aureus for the generation of neutralizing antibodies. MAbs. 2016;8(7):1347–1360. doi: 10.1080/19420862.2016.1215791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Moustafa M, Aronoff GR, Chandran C, Hartzel JS, Smugar SS, Galphin CM, Mailloux LU, Brown E, Dinubile MJ, Kartsonis NA, et al. Phase IIa study of the immunogenicity and safety of the novel Staphylococcus aureus vaccine V710 in adults with end-stage renal disease receiving hemodialysis. Clin Vaccine Immunol. 2012;19(9):1509–1516. doi: 10.1128/CVI.00034-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Schluepen C, Malito E, Marongiu A, Schirle M, McWhinnie E, Lo Surdo P, Biancucci M, Falugi F, Nardi-Dei V, Marchi S, et al. Mining the bacterial unknown proteome: identification and characterization of a novel family of highly conserved protective antigens in Staphylococcus aureus. Biochem J. 2013;455(3):273–284. doi: 10.1042/BJ20130540. [DOI] [PubMed] [Google Scholar]
- 168.Bagnoli F, Fontana MR, Soldaini E, Mishra RP, Fiaschi L, Cartocci E, Nardi-Dei V, Ruggiero P, Nosari S, De Falco MG, et al. Vaccine composition formulated with a novel TLR7-dependent adjuvant induces high and broad protection against Staphylococcus aureus. Proc Natl Acad Sci U S A. 2015;112(12):3680–3685. doi: 10.1073/pnas.1424924112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Earls MR, Kinnevey PM, Brennan GI, Lazaris A, Skally M, O'Connell B, Humphreys H, Shore AC, Coleman DC. The recent emergence in hospitals of multidrug-resistant community-associated sequence type 1 and spa type t127 methicillin-resistant Staphylococcus aureus investigated by whole-genome sequencing: implications for screening. PLoS One. 2017;12(4):e0175542. doi: 10.1371/journal.pone.0175542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.David MD, Kearns AM, Gossain S, Ganner M, Holmes A. Community-associated meticillin-resistant Staphylococcus aureus: nosocomial transmission in a neonatal unit. J Hosp Infect. 2006;64(3):244–250. doi: 10.1016/j.jhin.2006.06.022. [DOI] [PubMed] [Google Scholar]
- 171.Creech CB, Frenck RW, Jr, Sheldon EA, Seiden DJ, Kankam MK, Zito ET, Girgenti D, Severs JM, Immermann FW, McNeil LK, et al. Safety, tolerability, and immunogenicity of a single dose 4-antigen or 3-antigen Staphylococcus aureus vaccine in healthy older adults: results of a randomised trial. Vaccine. 2017;35(2):385–394. doi: 10.1016/j.vaccine.2016.11.032. [DOI] [PubMed] [Google Scholar]
- 172.Torre A, Bacconi M, Sammicheli C, Galletti B, Laera D, Fontana MR, Grandi G, De Gregorio E, Bagnoli F, Nuti S, et al. Four-component Staphylococcus aureus vaccine 4C-staph enhances Fcgamma receptor expression in neutrophils and monocytes and mitigates S. aureus infection in neutropenic mice. Infect Immun. 2015;83(8):3157–3163. doi: 10.1128/IAI.00258-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yu XQ, Robbie GJ, Wu Y, Esser MT, Jensen K, Schwartz HI, Bellamy T, Hernandez-Illas M, Jafri HS. Safety, tolerability, and pharmacokinetics of MEDI4893, an investigational, extended-half-life, anti-Staphylococcus aureus alpha-toxin human monoclonal antibody, in healthy adults. Antimicrob Agents Chemother. 2017;61(1):e01020–16. [DOI] [PMC free article] [PubMed]
- 174.Delfani S, Mohabati Mobarez A, Imani Fooladi AA, Amani J, Emaneini M. Protection of mice against Staphylococcus aureus infection by a recombinant protein ClfA-IsdB-Hlg as a vaccine candidate. Med Microbiol Immunol. 2016;205(1):47–55. doi: 10.1007/s00430-015-0425-y. [DOI] [PubMed] [Google Scholar]
- 175.Landrum ML, Lalani T, Niknian M, Maguire JD, Hospenthal DR, Fattom A, Taylor K, Fraser J, Wilkins K, Ellis MW, et al. Safety and immunogenicity of a recombinant Staphylococcus aureus alpha-toxoid and a recombinant Panton-Valentine leukocidin subunit, in healthy adults. Hum Vaccin Immunother. 2017;13(4):791–801. doi: 10.1080/21645515.2016.1248326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Schwameis M, Roppenser B, Firbas C, Gruener CS, Model N, Stich N, Roetzer A, Buchtele N, Jilma B, Eibl MM. Safety, tolerability, and immunogenicity of a recombinant toxic shock syndrome toxin (rTSST)-1 variant vaccine: a randomised, double-blind, adjuvant-controlled, dose escalation first-in-man trial. Lancet Infect Dis. 2016;16(9):1036–1044. doi: 10.1016/S1473-3099(16)30115-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Characteristics of the single isolates, including collection details, genome assembly statistics, genomic features, and results of antibiotic susceptibility testing. (XLSX 69 kb)
Figure S1. Pangenome analysis statistics. Figure S2. Phylogenetic model based on gene presence/absence. (PDF 580 kb)
Table S2. Presence/absence profile of virulence and antibiotic resistance genes in the cohort. (XLSX 6063 kb)
Table S3. Characteristics of the genomes included in the ST1 analyses. (XLSX 11 kb)
Table S4. Most relevant clones represented in the cohort and their abundance. (XLSX 9 kb)
Table S5. Bayesian clock models tested and their results. (XLSX 10 kb)
Data Availability Statement
The datasets generated and analysed during the current study are available in the NCBI Sequence Read Archive (BioProject accession number PRJNA400143, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA400143/).