

Abstract
BACKGROUND & AIMS: Steatohepatitis (SH) and SH-associated hepatocellular carcinoma (HCC) are of considerable clinical significance. SH is morphologically characterized by steatosis, liver cell ballooning, cytoplasmic aggregates termed Mallory-Denk bodies (MDBs), inflammation, and fibrosis at late stage. Disturbance of the keratin cytoskeleton and aggregation of keratins (KRTs) are essential for MDB formation. METHODS: We analyzed livers of aged Krt18−/− mice that spontaneously developed in the majority of cases SH-associated HCC independent of sex. Interestingly, the hepatic lipid profile in Krt18−/− mice, which accumulate KRT8, closely resembles human SH lipid profiles and shows that the excess of KRT8 over KRT18 determines the likelihood to develop SH-associated HCC linked with enhanced lipogenesis. RESULTS: Our analysis of the genetic profile of Krt18−/− mice with 26 human hepatoma cell lines and with data sets of >300 patients with HCC, where Krt18−/− gene signatures matched human HCC. Interestingly, a high KRT8/18 ratio is associated with an aggressive HCC phenotype. CONCLUSIONS: We can prove that intermediate filaments and their binding partners are tightly linked to hepatic lipid metabolism and to hepatocarcinogenesis. We suggest KRT8/18 ratio as a novel HCC biomarker for HCC.
Introduction
Hepatocellular carcinoma (HCC) is the most common obesity-related cancer, ranking as the second cause of cancer-related death [1], [2], [3]. The epidemiology of HCC is characterized through geographic trends and different risk factors [4]. Chronic hepatitis B and C are the most frequent etiologic risk factors for HCC [5]. In sub-Saharan Africa and eastern Asia, the main risk factors are aflatoxin B1 and chronic hepatitis B. On the contrary, in the USA, Japan, and Europe, the main risk factors are chronic hepatitis C and alcohol abuse [4].
Various factors common to the Western lifestyle like intake of diets rich in saturated fats, central obesity, and sedentary behavior are risk factors of nonalcoholic fatty liver disease (NAFLD) [6], [7]. Previously, NAFLD was described as the hepatic manifestation of the metabolic syndrome (MetS) [8], but NAFLD is not only a hepatic manifestation of MetS but may inception of the development of MetS [9], [10]. NAFLD is becoming a major cause of HCC, and as many as 50% of NAFLD-HCCs occur in patients without cirrhosis and are often detected at a late tumor stage [11], [12].
Nonalcoholic steatohepatitis (NASH) is a well-characterized cause of cirrhosis and is associated with the development of HCC [13].
HCC is a highly heterogeneous disease; Hoshida and colleagues have tried to develop genomics-based classification for HCC and observed three subclassifications for HCC (S1–3 group) [14], [15], [16]. The signatures of S1 display aberrant activation of the WNT signaling pathway, S2 was characterized through proliferation as well as activation of MYC and AKT, and S3 was linked with differentiation of hepatocytes [14], [15].
Depending on the etiology, alcoholic steatohepatitis (ASH) and NASH can be distinguished. ASH and NASH are characterized by ballooned hepatocytes; the correct identification of ballooned hepatocytes at routine hematoxylin and eosin–stained liver sections is challenging. For accurate characterization, specialized stains such as cytoplasmic keratin 8/18 immunohistochemistry may allow a more consistent detection of ballooned hepatocytes. Other histological features, such as microgranulomas, Mallory-Denk body (MDB), lipogranulomas, megamitochondria, acidophil bodies, iron, and glycogenated nuclei, may occur but do not contribute to the diagnosis of NASH [12], [17], [18], [19], [20], [21], [22]. MDBs are mainly composed of KRT8, KRT18, attached p62/SEQUESTOSOME 1 (p62), and ubiquitin. Under physiological conditions, KRT8 and 18 are present in a 1:1 ratio and assembled as intermediate filaments.
The role of KRTs in liver diseases is underlined by the fact that expression of a dominant-negative Krt18 mutant in mouse liver resulted in chronic hepatitis with increased hepatocyte fragility and higher susceptibility to acute drug-induced liver injury [18]. Lack of Krt8 or Krt18 in mice predisposes to liver injury and FAS- but not TNFα-mediated apoptosis [23]. The relevance of KRT sequence variants for human liver disease was substantiated by detection of KRT mutations in patients of different ethnic backgrounds and with acute and chronic liver diseases [24], [25].
The sequence of events leading to NASH and HCC is still poorly understood, although it is widely accepted that inflammation, oxidative stress, and fibrosis-promoting stimuli are critical for NASH development [26], [27]. It has been demonstrated that FAS expression, activation of caspases-3 and -7, and hepatocyte apoptosis are enhanced in the liver of NASH patients, which positively correlated with biochemical and histopathological markers of liver injury [26], [27].
In the present study, we aimed to answer some crucial questions related to development of SH and liver tumorigenesis in an HCC setting by analyzing and comparing molecular events in mouse models, human hepatoma cell lines, and human liver tissue.
Materials and Methods
Human Liver Tissue
Human HCC samples were obtained from the HCC Genomic Consortium [1]. Human liver biopsies were obtained from the Biobank of the Medical University of Graz and the Department of Pathology, University of Heidelberg. Biopsies were registered in the respective biobanks and kept anonymous. The research project was authorized by the ethical committees of the Medical University of Graz (ref. no. 1.0 24/11/2008) and the University of Heidelberg [6]. The study protocol was in accordance with the ethical guidelines of the Helsinki Declaration. Patients were enrolled after given written informed consent.
Mice
During breeding and experiments, the animals were housed in a rodent facility with climate-controlled rooms (20°C-24°C temperature and 50%-60% humidity) and a 12-hour light–dark rhythm and fed a standard diet (sniff Spezialitäten GmbH, Soest, Germany) and tap water ad libitum. The specific pathogen-free quality of the animals was confirmed by a sentinel program according to FELASA recommendations. Experimental protocols were in accordance with the Austrian Animal Protection Law, Veterinary office, Vienna. The study was approved by the institutional ethics committee of the Medical University of Graz, and an experimental license was granted under BMWF-535233 (Austrian Federal Ministry of Science and Research).
After having performed embryo transfers for rederivation of the mutant line, heterozygous FVB/N-Krt18+/− mice were backcrossed to 129P2/OlaHsd wild-type (wt) mice over more than 10 generations [7], [13], [28] to generate a congenic strain. Genotypes were determined by PCR analysis of tail DNA. The following primers were used: Krt8-868 (5′-GGC CCT GCC CTC TAG TGT-3′) and Krt8-1377 (5′-AGG GGT ATC ACC TTG TCA AT-3′) for amplification of the wt allele; Krt8-868 and neo-249 (5′-CCT TCC CGC TTC AGT TAC-3′) for amplification of the mutated allele (Krt8−/−). Breeding and genotyping of 129P2.FVB/N-Krt18 mutant and 129P2/ OlaHsd wt mice were performed as described [7], [13], [28]. Krt18-BN1 (5′-GTC CTC GGC ACC CTG TAA CCT G-3′) and Krt18-BN2 (5′-CGG TCT GGA TTC CAC CCA TTC-3′) for amplification of the wt allele, Krt18-HPRT1 (5′-GCA GTC CCA GCG TCG TGA TTA-3′) and Krt18-HPRT2 (5′-AGT TTG CAT TGT TTT ACC AGT GTC-3′) for amplification of the mutated allele (Krt18−/−). PCR products were separated on a 1% agarose gel in 1× TBE buffer. The expected product from Krt8-868 and Krt8-1377 is 529 bp, and that from Krt8-868 and neo-249 is 700 bp. The expected product from Krt18-BN1and Krt18-BN2 is 1200 bp, and that from Krt18-HPRT1 and Krt18-HPRT1 is 430 bp. Mice were sacrificed by cervical dislocation; livers were removed and analyzed as indicated next.
Colocalization of hTAA with p62 and K8/18 in MDBs (Triple-Label Immunofluorescence)
Mouse liver tissues were fixed in 4% neutral buffered formaldehyde solution and embedded in paraffin. Seven-micrometer–thick sections were deparaffinized and washed with PBS for 10 minutes. H-HTAA, synthesis of a library of oligothiophenes and their utilization as fluorescent ligands for spectral assignment of protein aregates (1:500 in PBS) [17], was applied, and sections were incubated for 20 minutes. The sections were then washed with PBS (3 × 10 minutes) and microwaved in target retrieval solution (Dako), pH 9, for 40 minutes at 150 W. Thereafter, the sections were immunostained with antibodies to KRT8 (Novocastra)/KRT18 (Neomarker) (1:100) and to anti-p62/SQSTM1 [17] (1:200, Progen); Alexa Fluor 633 anti-mouse (1:200, Invitrogen) and Rhodamine Red-X conjugated goat anti-guinea pig immunoglobulin (1:200, Jackson Immune Research) were used as secondary antibodies.
Gene Expression Profiling of wt, Krt18+/− and Krt18−/− Mouse Livers (SuperArray)
Total RNA was isolated from cryopreserved non-neoplastic liver tissue and tumors using TRIzol reagent (Invitrogen). Genomic DNA contamination was eliminated by DNase treatment following the protocol of the RNeasy Mini kit from Qiagen. The cDNA was applied to the Mouse RT2 Profiler PCR array Mouse liver cancer (PAMM-133Z array, SABiosciences) following the manufacturer's instructions. Real-Time SYBR Green/ROX PCR mix was purchased from SA Bioscience. The arrays were run on the ABI PRISM 7900HT Sequence Detection System using the Sequence Detector Software Version 2.3 from Applied Biosystems. Data were analyzed using the online SABiosience RT2 Profiler PCR array data analysis tool based on the ΔΔCt method (RT2 profiler PCR array data analysis Version 3.5). Gene expression was normalized to five housekeeping genes: β-actin (Actb), β-2 microglobulin (B2m), glyceraldehyde-3-phosphate dehydrogenase (Gapdh), Glucuronidase, beta (Gusb), heat shock protein 90 alpha (cytosolic), and class B member1 (Hsp90ab1) expressed as fold change over the control group (wt tumors compared to Krt18+/− and Krt18−/− livers).
KRT8/KRT18 Expression Ratio and HCC Early Recurrence
To evaluate prognostic relevance of KRT8/KRT18 ratio, we analyzed mRNA expression profiles of two cohorts of HCC patients previously described [1] (Lonzano et al., 2012) (the NCBI Gene Expression Omnibus accession number for the gene expression profiling of 91 HCCs with HVC virus etiology reported in this paper is GSE9843) for early HCC recurrence within 2 years after surgery, assumed to reflect primary tumor cell dissemination. KRT8/KRT18 mRNA expression ratios greater than 1 standard deviation above mean were categorized into the high–KRT8/KRT18 ratio group. Association with outcome was evaluated with Kaplan-Meier curve and log-rank test.
Quantitative Real-Time PCR
Routine protocols were performed. Primers and probes (synthesized by MWG-Biotech AG, Germany) are listed in Table S1.
Fatty Acid Measurement by Gas Chromatography–Mass Spectrometry (GC-MS)
Cryopreserved murine liver samples were lyophilized, dispersed in 500 μl methanol/toluene/sulfuric acid (50:50:2) [v/v/v], incubated at 55°C overnight, and neutralized by 400 μl of a 0.5-M NH4CO3, 2-M KCl solution. After centrifugation, the supernatant was derivatized with 25 μl N-methyl-N-(trimethylsilyl)trifluoroacetamide at 37°C for 1 hour and subsequently analyzed on a Thermo Fisher Focus gas chromatograph coupled to an DSQII mass selective detector (Fisher Scientific, Schwerte, Germany) equipped with a nonpolar J&WDB-5HT capillary column.
Prediction Analysis
As expression of those keratins' mRNA and related information is visualized correlating KRT expression levels with molecular HCC S1/S2 subclasses in a panel of HCC cell lines. The prediction of S1/S2 subclass was made by Nearest Template Prediction module of Gene pattern genomic analysis toolkit (www.broadinstitute.org/genepattern). The prediction was made based on proximity to the expression pattern of S1/S2 subclass signature genes measured by cosine distance. Significance of the proximity was evaluated as prediction confidence P value based on empirical null distribution of the distance determined by randomly picking the same number of genes from the whole-genome microarray data and recalculating the distance 1000 times.
MR Imaging
MR imaging was performed on a 3-T whole-body scanner with a 3.5-mm–diameter volume coil (RAPID Biomedical GmbH, Würzburg, Germany) in order to gain high SNR within the liver.
All images were acquired in a nontriggered manner in the axial plane. T2*-weighted 2D-gradient-recalled echo in-phase images (repetition time/echo time = 177/7.1 milliseconds, al = 35°, matrix = 184 × 256, field of view = 32 × 45 mm, 10 slices at 1.5 mm, averages = 4) were used for contrast-enhanced imaging. Superparamagnetic iron oxide (Resovist; Schering AG, Berlin, Germany) in a dose of 15 μmol Fe/kg BW was used as a T2* agent for focal liver lesion detection. The dose was kept as clinically recommended to ensure longitudinal studies. After a precontrast T2*-weighted gradient-recalled echo image was acquired, the bolus of 100 μl Resovist was applied intravenously into tail vein followed by a 75-μl saline flush. Immediately after injection, the T2* postcontrast scan was performed.
Array Comparative Genome Hybridization (aCGH) Analysis
Paraffin-embedded liver tumors were microdissected and hybridized against age-matched non-neoplastic wt liver tissue as described previously [21]. Labeling was performed following the protocol of BioPrime Array CGH Genomic Labeling Module by Invitrogen, and the samples were hybridized on an 8 × 60k CGH Array under the conditions of the Agilent protocol (Version 7.2). Hybridization buffer (Agilent in situ hybridization kit plus) was added, and the samples were applied to microarrays enclosed in Agilent SureHyb-enabled hybridization chambers. The array was analyzed with the Agilent DNA Microarray Scanner G2505C and the feature extraction software Agilent Feature Extraction 11.0.1.1. The data were analyzed by the statistical software R. Amplifications (indicated in red) and deletions (indicated in green) of chromosomal regions ranged from ≤1 megabase (MB) to 160 MB (the NCBI Gene Expression Omnibus accession number for the murine HCC data reported in this paper is GSE47212).
Statistical Analysis
A chi-square test with a significance level of P = .05 was used for P value calculation. Data are reported as median values with 95% confidence interval limits. These estimates were computed by nonparametric bootstrap using DATAPLOT (National Institute of Standards and Technology, Statistical Engineering Division, Gaithersburg, MD) and GraphPadPrism software.
Morphometric data present a hierarchical structure, where portal fields or fields of view (level-1 sampling units) are nested within mice (level-2 sampling units). A two-level linear regression model by means of MLwiN 2.02 software (Centre for Multilevel Modelling, University of Bristol, UK) was fitted to calculate means and confidence intervals. Finally, a two-way ANOVA with Bonferroni correction was performed to test the significance of differences between groups.
aCGH was performed by MSMAD [29], [30], a novel median smoothing median absolute deviation method which is more sensitive than conventional aCGH techniques. It is based on the assumption of rank order dependence of copy number changes and the jump character of these changes in the sequence of log2 ratios. In addition to MSMAD, the standard Gain and Loss Analysis of DNA (GLAD) method of Bioconductor was applied throughout as a reference [31]. The majority of gains and losses are equally supported by both techniques.
Results
Liver Phenotype of Aged Krt18−/− Mice: a Novel Steatohepatitis Model
We have recently reported Krt18−/− mice as a novel spontaneous SH-associated HCC mouse model [21].
To further address the contribution of KRT8 and KRT18 with respect to liver pathology in livers of wt, Krt8+/−, Krt8−/−, Krt18+/−, and Krt18−/− mice aged 3, 6, 12, and 17-20 months, we performed, in addition to light microscopy, double immunofluorescence microscopy using antibodies against K8/18 and p62. Moreover, heptameric oligothiophene, h-HTAA [17], was used to confirm β-sheet conformation (Figure 1, Figure S1).
Figure 1.

Immunofluorescent characterization of wt, Krt8−/−, and Krt18−/− mice. (A) Double immunofluorescence staining of representative wt, Krt8−/−, and Krt18−/− mouse liver paraffin sections stained with KRT8/18 (green/Alexa Fluor 488) and p62CT (red/Rhodhamine) antibodies for colocalization of p62 with KRT8/K18. In wt mice, mild steatosis without MDBs is observed. The livers of wt, Krt8−/−, and Krt18−/− mice show no MDB formation at the age of 3 months. (B) Yellow color indicates MDB formation with colocalization of p62 and KRT8/18 (white arrowheads) in 17- to 20-month-old Krt18−/− mice, whereas 17- to 20-month-old wt livers lack MDBs. (C) K8, p62, and the conjugated polyelectrolyte probe h-HTAA colocalize in MDBs of 17- to 20-month-old Krt18−/− livers (white arrowheads). Scale bars: 20 μm and 50 μm. Inserts present higher magnifications.
Figure S1.
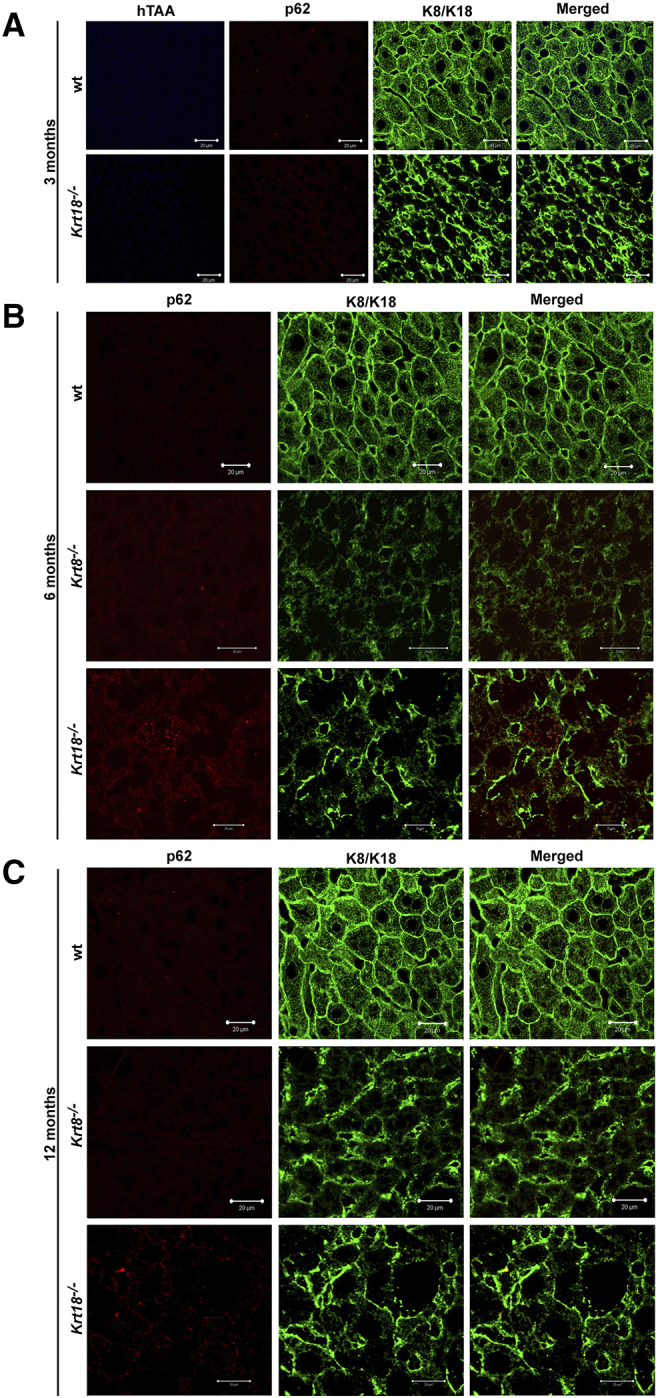
Immunofluorescent characterization of wt, Krt8-/-, and Krt18-/-mice.
(A) Double immunofluorescence staining of representative 3-month-old wt and Krt18-/- mouse liver paraffin sections stained with K8/18 (green/Alexa Fluor 488), p62CT (red/Rhodhamine) and the conjugated polyelectrolyte probe hTAA. No colocalization is seen. (B) Double immunofluorescence staining of representative 6-month-old wt, Krt8-/-, and Krt18-/- mouse liver paraffin sections stained with K8/18 (green/Alexa Fluor 488) and p62CT (red/Rhodhamine). No colocalization is observed. (C) Double immunofluorescence staining of representative 12-month-old wt, Krt8-/-, and Krt18-/- mouse liver paraffin sections stained with K8/18 (green/Alexa Fluor 488) and p62CT (red/Rhodhamine). No colocalization is observed. Scale bar: 20 μm.
In 17- to 20-month-old Krt18−/− mice, we observed the entire morphologic spectrum of characteristics of SH in humans (Figure 1, B and C). MDBs were exclusively present in aged Krt18−/− mice, whereas aged Krt8−/−, Krt8+/−, and wt mice lacked MDBs (Figure 1, Figure S1).
Changes in Hepatic Fatty Acid Composition of Krt18−/− Mice
In order to characterize the hepatic lipid profile of aged mice, we performed a fatty acid (FA) analysis by GC-MS. In livers of 6-month-old Krt18+/− and Krt18−/− mice, total FA amounts were significantly increased compared to wt mice (Figure 2, Figure S2; Table S1). Moreover, the composition of FAs palmitic acid (16:0) and eicosadienoic acid (20:2) were significantly elevated in Krt18+/− and Krt18−/− livers (Figure 2). Analysis of total FAs showed a significant increase in 17- to 20-month-old Krt18+/− and Krt18−/− animals compared to 3-month-old mice (Figure 2 and Table S1). In contrast, the livers of the differently aged wt mice revealed no significant changes in their total FA content, except for 17- to 20-month-old wt animals, which displayed significantly higher degree of total FAs compared to 3-month-old mice (Figure 2 and Table S1).
Figure 2.

GC-MS analysis of livers of 17- to 20-month-old wt, Krt18+/−, and Krt18−/− mice. (A) GC-MS analysis shows significantly increased amounts of FAs in livers of 6-month-old Krt18+/− and Krt18−/− mice compared to age-matched wt animals. Data are presented as μg/mg liver tissue, results are shown as mean, and error bars indicate standard error of the mean (SEM). *, **, *** indicate statistical significance (Student's t test): *P < .05; **P < .001; ***P < .0001. (B) GC-MS analysis illustrates elevation of palmitic acid (16:0), palmitoleic acid (16:1), stearic acid (18:0), oleic acid (18:1), linoleic acid (18:2), arachidonic acid (20:4), and docosahexaenoic acid (22:6) when comparing wt against Krt18+/− and Krt18−/− livers. Data are presented as μg/mg liver tissue, results are shown as mean, and error bars indicate SEM. *, **, *** indicate statistical significance (Student's t test): *P < .05; **P < .001; ***P < .0001.
Figure S2.

Total hepatic fatty acids and hepatic fatty acid profile of Krt8-/-and p62-/-livers.
Total hepatic FAs were measured by GC-MS. (A) Analysis of total FAs shows a marked tendency of reduction in 6- and 12-month-old Krt8-/- animals compared to age-matched wt and Krt8+/- mice. Estimation of the total FA content in 3- and 6-month-old p62-/- mice and their respective littermates reveals no significant changes (see also Table S2). (B) The livers of the different aged wt, Krt8+/-, and Krt8-/- mice as well as the livers of the p62-/- and their respective wt controls show no significant changes in their profile (see also Table S2). Data are presented as μg/mg liver tissue, results are shown as mean, and error bars indicate SEM. *, **, *** indicate statistical significance (Student’s t test): *P<0.05; **P<0.001; ***P<0.0001.
The hepatic FA profile showed significant differences in 3-month-old livers (wt, Krt18+/−, and Krt18−/−) with respect to palmitic acid (16:0) and oleic acid (18:1) compared to livers of 12- and 17- to 20-month-old mice (Figure 2 and Table S1). High KRT8/ 18 ratio leads to alterations in the hepatic fatty acid profile. Beside an increased overall hight of hepatic fatty acids levels of myristic acid, palmitic acid, and eicosadienoic acid were significantly elevated.
Steatohepatitis-Related Transcript Changes
In order to understand the molecular basis of the changes listed above, the expression of genes involved in SH, including sterol regulatory element binding transcription factor 1 (Srebp1c), forkhead box O1 (FoxO1), and FoxO3 (Figure 3A), inflammation drivers like mitogen-activated protein kinase 8 (JNK1), tumor necrosis factor α (TNFα), and nuclear factor of kappa light polypeptide gene enhancer in B cells 1 (NFκb1) (Figure 3B) were analyzed by qRT-PCR. We also investigated autophagy-inducing genes, including Map1lc3α, Map1lc3β, and Atg5, at four time points (3, 6, 12, and 17-20 months) in wt, Krt18+/−, and Krt18−/− mice (Figure 3C). We measured mRNA levels of p62 and Ubiquitin, encoding for essential MDB protein components and the oxidative stress–related gene nuclear factor, erythroid derived 2, like 2 (Nrf2) (Figure 3D).
Figure 3.

Transcriptional profile of wt, Krt18+/−, and Krt18−/− livers for inflammation and autophagy-related genes at various time points. qRT-PCR analysis for mRNA expression of candidate genes in livers at 3, 6, 12, and 17-20 months. (A) Expression levels of Srebp1, FoxO1, and FoxO3 as SH-related mRNAs. (B) Jnk1, Tnfα, and Nfκb1 as inflammation markers. (C) Sequestosome 1/p62 and Ubiquitin as indicators of protein aggregation and Nrf2 as oxidative stress related marker. (D) Map1lc3α, Map1lc3β, and Atg5 mRNA as markers for autophagy are presented as fold change log10. qRT-PCR analysis of 3-, 6-, 12-, and 17- to 20-month-old wt, Krt18+/−, and Krt18−/− murine livers indicated by symbols (circles for wt, squares for Krt18+/−, and triangles for Krt18−/− mice). Each data point reflects the expression of a particular gene resulting from three to five different mice normalized to the mean expression value of the respective gene in wt livers. Significance values are described by asterisks and hash above horizontal bars. Data are presented as fold change log 10, results are shown as mean, and error bars indicate SD. *, **, *** indicate statistical significance (two-way ANOVA with Bonferroni correction): *P < .05; **P < .001; ***P < .0001.
Before the onset of SH, Srebp1c was significantly increased from the age of 3 to 6 months in Krt18−/− mice compared to wt livers at the age of 6 months. After 6 months, Srebp1c decreased to baseline levels. Nfκb1 was significantly elevated in Krt18−/− compared to wt livers at 3 months of age. Significantly increased expression levels of p62 and Ubiquitin were observed in Krt18−/− livers compared to age-matched wt controls at 6 months before showing morphologically visible SH features. Both p62 and Ubiquitin significantly differed between 3-, 6-, and 12-month-old mice. Over time, the two mRNAs attenuated to (normal) basal levels. Nrf2 was significantly elevated in Krt18−/− compared to wt livers at 6 months of age and significantly decreased in 6- to 12-month-old animals (Figure 3B). The following NAFLD-related mRNAs remained largely unchanged: malonyl-CoA decarboxylase (Mlycd), microsomal triglyceride transfer protein (Mttp), ATP-binding cassette, sub-family C, member 2 (Cftr/Mrp) (Figure S3A), insulin receptor substrate 1 (Isr1), glutamate-cysteine ligase, modifier subunit (Gclm), fatty acid synthase (Fasn) (Figure S3B), diacylglycerol O-acyltransferase 2 (Dgat2), angiotensin II receptor 1a (Agtr1α), adiponectin receptor 2 (Adipor2) (Figure S3C), and peroxisome proliferator activated receptor alpha (Pparα) (Figure S3D). Thus, relevant molecular changes as similarly seen in human HCC associated with SH were observed in Krt18−/− mice already at an initial phase of SH before the full-blown morphological features were present/could be detected.
Figure S3.

Transcriptional profile of wt, Krt18+/-, and Krt18-/-livers for NAFLD-related genes at various time points.
qRT-PCR analysis for mRNA expression of candidate genes in murine livers at 3, 6, 12, and 18 month of age. (A) Expression levels of Mlycd, Mttp, Cftr/Mrp, (B) Isr1, Gclm, Fasn, (C) Dgat2, Agtr1α, Adipor2, and (D) Pparα are indicated by symbols (circles for wt, squares for Krt18+/-, and triangles for Krt18-/- mice) and presented as fold change log10. Each data point reflects the expression of a particular gene resulting from three to six different mice, normalized to the mean expression value of the respective gene in wt livers. Results are shown as mean, and error bars indicate SD. Significance values are described by asterisks above horizontal bars. *, **, *** indicate statistical significance (two-way ANOVA with Bonferroni posttest): *P≤0.05; **P<0.001; ***P<0.0001.
Steatohepatitis-Associated Liver Tumors
Resovist-enhanced MRI scanning of male and female wt, Krt18+/−, and Krt18−/− mice exhibited tumor formation at 17-20 months of age (Figure 4D). Moreover, at 17-20 months of age, higher HCC incidences were seen in males compared to females of each genotype. At the age of 17-20 months, however, 43% of wt (18/42), 38% of Krt18+/− (31/81), and 57% of Krt18−/− (40/70) mice had developed liver tumors which in some cases displayed signs of neovascularization comparable to vascular patterns reminiscent of human HCC (Figure 4D) [21].
Figure 4.
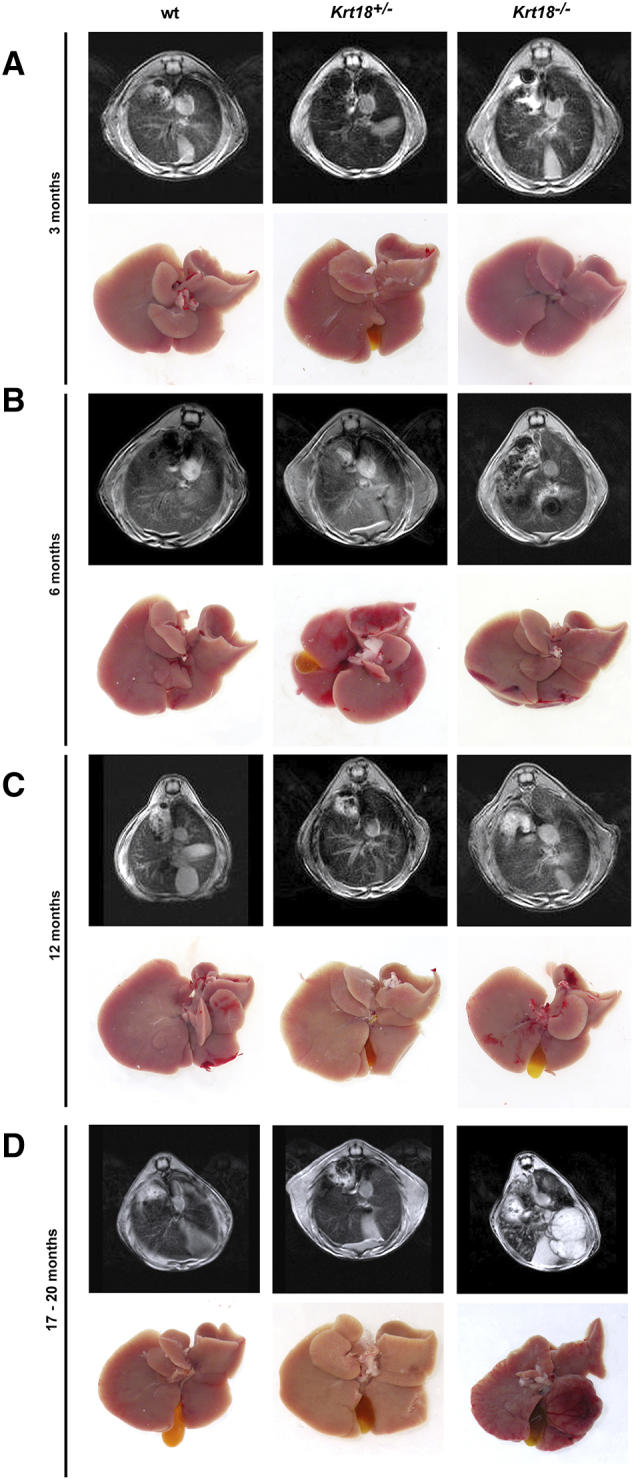
Chronic liver injury and HCC development in Krt18−/− mice. (A) Representative MRI after administration of Resovist (upper panel) and gross pictures of 3-month-old wt, Krt18+/−, and Krt18−/− mice (lower panel). (B) Representative MRI after administration of Resovist (upper panel) and gross pictures of 6-month-old wt, Krt18+/−, and Krt18−/− mice (lower panel). (C) Representative MRI after administration of Resovist (upper panel) and gross pictures of 12-month-old wt, Krt18+/−, and Krt18−/− mice (lower panel). (D) Representative MRI after administration of Resovist (upper panel) and gross pictures of 17- to 20-month-old wt, Krt18+/−, and Krt18−/− mice (lower panel).
To investigate whether 129P2/OlaHsd wt mice are more susceptible to develop liver tumors, an independent cohort of mice was analyzed, revealing the occurrence of liver tumors at the age of 17-20 months with a similar frequency as our wt mice (data not shown).
In 17- to 20-month-old Krt18−/− mice, we often encountered multiple HCCs but rarely detected lung metastases. Microdissected Krt18−/− HCC and lung metastases derived from the same Krt18−/− mouse were analyzed for chromosomal aberrations. The lung metastases and the corresponding HCC displayed significantly overlapping chromosomal aberrations throughout the entire genome (P < .0001), suggesting a clonal relationship between the primary and metastatic lesion (Figure 5A).
Figure 5.

Genetic profile of murine HCCs and primary HCC with lung metastases. (A) Comparison of primary HCC with lung metastases reveals a clonal evolution process. (B) Association of KRT8/18 ratio with early HCC recurrence after surgical resection. Kaplan-Meier curves for a subset of previously reported cohorts of HCC patients with updated clinical follow-up data (A: n = 69, B: n = 221) are shown [28], [35]. High KRT8/18 ratio indicates >1 standard deviation above mean. Difference between the high and low groups is evaluated by log-rank test. (C) Hepatoma cell lines are ordered based on KRT8/18 expression levels. HCC cell lines with higher KRT8/18 ratio (above median) are characterized by an S1 signature (eight cell lines) and suppression of S2 signature (nine cell lines) with statistical significance. Expression of those keratin mRNAs and related information is visualized correlating KRT expression levels with molecular HCC S1/S2/S3 subclasses. The prediction of distinguishing S1 versus S2 subclass is made by nearest template prediction [16]. Gray color indicates prediction confidence P > .25 (i.e., nonsignificant prediction) as the prediction confidence was calculated as ALPHA-1-FETOPROTEIN (AFP)–expressing cell lines were predicted as presenting S2 signature consistent with a previous publication (http://www.ncbi.nlm.nih.gov/pubmed/19723656).
No chromosomal aberrations were detected in 3-month-old wt mouse livers which served as controls. As previously reported, aCGH analysis using the MSMAD method [29] revealed many chromosomal aberrations in all investigated Krt18−/− HCCs. Amplifications and deletions of chromosomal regions ranged from ≤1 MB to 160 MB and were detected in most autosomes.
KRT8/18 Ratio as a Novel Biomarker in Recurrence Prediction in Patients with HCC
Several studies suggested various tissue biomarkers as potentially useful in predicting outcome of HCC patients upon treatment, particularly in order to improve the stratification of candidates for liver transplantation or more aggressive chemotherapy [15], [32]. Various types of markers including histological, immunohistochemical, and molecular markers have been identified [33]. However, conclusive studies defining molecular subclasses are lacking, and defining new biomarkers with predictive power for disease progression (survival or recurrence) is needed [33], [34]. Thus, we assessed the clinical relevance of the KRT8/18 ratio signature in predicting early human HCC recurrence. We identified that a high KRT8/18 ratio signature is associated with early recurrence in HCC patients in two independent cohorts: the HBV-related HCC cohort showed a strong tendency towards significance (KRT8/18 high: n = 28, KRT8/18 low: 193, P = .16) [35]. What is more, the HCV-related HCC cohort showed a statistical significance (KRT8/18 high: n = 9, KRT8/18 low: 60, P = .02) [28] (NCBI Gene Expression Omnibus, accession # GSE14520) (Figure 5B).
KRT8/18 Ratio in Established Hepatoma Cell Lines
Next, we tested the KRT8/KRT18 ratio and its correlation to the genomewide transcriptome landscape in a comprehensive panel of 26 human hepatoma cell lines being part of a data subset (Cancer Cell Line Encyclopedia; http://www.ncbi.nlm.nih.gov/pubmed/22460905). It has been repeatedly reported that human hepatoma cell lines are clustered into two distinct groups based on transcriptional profiling [36], [37]. We previously showed that these two subclasses represent transcriptional profiles of aggressive human HCC subclasses (subclasses S1 and S2) as determined by a meta-analysis of multiple human cohorts. S1 tumors are characterized by TGF-β/WNT and nuclear β-CATENIN activation, molecular features of epithelial-mesenchymal transition, and a more disseminative phenotype as evidenced by higher incidence of early tumor recurrence after surgery [14]. Interestingly, HCC cell lines with higher KRT8/18 ratio were characterized by the presence of the S1 signature with statistical significance (KRT8/18 ratio above median: S1/S2 = 7/2 cell lines, below median: 1/7 cell lines) (Figure 5C).
We further confirmed that the S1 signature was enriched in KRT8/18 high cell lines by using Gene Set Enrichment Analysis with statistical significance (P = .04) [38] (Figure 5C).
Discussion
NASH represents a progressive form of NAFLD and is characterized by three elementary lesions: steatosis, lobular inflammation, and hepatocellular ballooning. The lobular inflammation and hepatocellular ballooning are major factors for the diagnosis of NASH. NASH can be diagnosed in the absence of fibrosis [12], [39]. It has been reported that in approximately 11% of NASH patients, cirrhosis arises in a time frame of 15 years, and nearly 7% develop HCC within 6.5 years [40], [41]. Several studies revealed that an imbalance of lipid metabolism has a strong impact on the development of NASH, hepatic fibrosis, cirrhosis, and HCC [42]. Some studies provide evidence that a disturbed cytoskeleton is involved in triggering SH [18], [43].
Generation of a hepatic fatty acid profile showed that, at 3 and 6 months of age, significant differences were detectable when comparing wt and Krt18−/− mice, giving evidence that, in this time frame, essential steps occur which drive the development of SH. Lipid accumulation is probably due to an increased expression of Srebp1c, a key regulator of lipogenic gene expression. Furthermore, Mttp, which is a component of lipoproteins, tended to be downregulated. In line with that finding, mice with a liver specific knockout of Mttp develop steatosis [44]. This result is in accordance with other metabolomic studies which reported changes in the lipid and glucose metabolism as critical events in NAFLD [42].
Srebpb1 is the main regulator of lipogenesis, and its counterpart and master regulator of ß-oxidation is Pparα [45], [46], [47]. qRT-PCR analyses revealed a significant increase of Srebpb1 mRNA level in Krt18−/− mice aged 3 and 6 months, whereas no increased mRNA expression of Pparα was identified (Figure 3, Figure S3). During the same time period, enhanced mRNA expression levels of main components of MDBs and an important indicator of oxidative stress were detected. These important indicators underline our theory of an early event which promotes the generation of hepatotoxic particles.
Importantly, Krt18−/− liver tumors bear a high potential for metastasis formation (Figure 5A). This assumption could be verified by CGH analyses of a lung metastasis found in a 17- to 20-month-old Krt18−/− mouse (Figure 5A).
To investigate the relevance of our model for human disease, we compared Krt18−/− mice with human hepatoma cell lines (n = 26) and HCC patient data (n = 300), revealing that Krt18−/− mice mimic a particular human HCC gene signature (Figure 5, B and C).
We were able to show that Krt18−/−-associated liver tumors resemble human HCCs of subclass S1 according to an established molecular HCC classification and, are therefore, most probably WNT/β-CATENIN driven [14].
Despite having generated a preclinical HCC model that is SH-triggered, we report here for the first time about significant clinical implication of KRT8/18 ratio with respect to recurrence rates of human HCC (Figure 5B) and mice. Thus, the obesity-associated HCC development in patients could resemble a chaotropic protein folding leading to lipid metabolism disturbance, probably associated due to carcinoma formation with elevated ROS causing DNA damage and lipid peroxidation, inactivation of phosphatases, and progression of cancer. Interestingly, the lipid profile of hepatocytes of Krt18−/− mice changed at the age of 6 months where transcript levels of NRF2, p62, and Ubiquitin were elevated (Figure 3). Histopathological examination of aged Krt8−/− liver sections revealed neither MDB formation nor enhanced steatosis and tumor development which were detectable in Krt18−/− mice (Figure 1A). This suggests that K8 and the Krt8/18 ratio have an important functional implication on the homeostasis of lipid metabolism and on tumor formation.
Thus, our study elucidates the impact of an excess of Krt8 and its influence on the hepatic lipid profile and cancer phenotype in a novel mouse model.
Our findings about relative protein excess–driven disease could have high impact in the identification and treatment of SH and SH-associated HCC in the future.
In summary, we discovered that a high KRT8/18 ratio leads to a changed hepatic lipid profile finally resulting in ballooning with MDB formation. During an early preaggregation phase, the starting point for SH development is laid and consequently followed by liver tumor formation. Taken together, we can prove that intermediate filaments and their binding partners are tightly linked to hepatic lipid metabolism and to hepatocarcinogenesis (Figure 6).
Figure 6.

Scheme of SH-induced HCC development. Schematic overview of SH induced hepatocarcinogenesis: KRT8/18 (1:1 ratio, wt condition, left column); KRT8< 18 (Krt8−/− condition, middle column); KRT8>18 (Krt18−/− condition, right column): upon oxidative stress over 3, 6, 12, or 17-20 months, macro- and mediovesicular steatosis (large and medium yellow dots) is induced in both wt and Krt8−/− hepatocytes (hexagons) without MDB formation (left and middle column), whereas in livers of Krt18−/− mice, upon oxidative stress in the same time periods, next to macro- and mediovesicular steatosis, a microvesicular steatosis (large, medium, and small yellow dots) is induced.
mRNA levels of p62 and Ubiquitin, components of MDBs (red transparent aggregates), are elevated in 6-month-old Krt18−/− mice and after 17-20 months, MDBs are formed (MDBs: irregular red aggregates, right column). At the age of 17-20 months, liver tumors with SH features and pleomorphic tumor cells with enlarged, hyperchromatic nuclei are formed in Krt18−/− mice (right column, right down).
The following are the supplementary data related to this article.
Immunofluorescent characterisation of wt, Krt8-/- and Krt18-/- mice.
qRT-Primer Sequences Mus musculus
Tabularization of the different qRT-Primer and their sequence which have been used for qRT-PCR experiments.
Authors’ contributions
N. G. S., K. B., A. K. M., and J. H. made the concept and design of the study. N. G. S., K. B., A. K. M., S. M. K., J. U., S. K., K. K., X. C., Y. H., V. S., M. G. S., V. M., C. S., A. T., L. F. F., T. S., T. K., and T. R. performed experiments and procedures. N. G. S., K. B., A. K. M., J. U., N. M. B., H. M., M. G. S., C. D., A. L., K. P. R. N., T. M. M., P. S., A. K., R. M., and J. H. performed the writing of article.
Acknowledgements
We are thankful to Gerda and Hans Heid for providing K8 and K18 antibodies. We thank Peter Bannasch for providing human tissues. We thank Monika Artl for her help with the CGH analysis. This study was supported by grants of the “Kurt und Senta Herrmann Stiftung” (to M.G.S. and J.H.) and the Austrian Genome Programme GEN-AU (to K.Z. and to T.R.), and R. M. was supported by a private donation from Lichtenstein. Work in the Magin lab was partially supported by the DFG (MA1316-15, MA1316-17, MA1316-19, MA1316-21, INST 268/230-1). This work was supported by the German Research Foundation grant STR 1095/4-2 and Else Kröner Exzellenzstipendium (to P.S.). The research leading to these results has received support from the Innovative Medicines Initiative Joint Undertaking under grant agreement no. 115234, resources of which are composed of financial contribution from the European Union's Seventh Framework Programme (FP7/2007-2013) and EFPIA companies (to JH.).
Footnotes
Financial support statement: The authors disclose no competing financial interests.
Conflicts of interest statement: All authors have no conflicts of interest.
References
- 1.Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245–1255. doi: 10.1016/S0140-6736(11)61347-0. [DOI] [PubMed] [Google Scholar]
- 2.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zucman-Rossi J. Molecular classification of hepatocellular carcinoma. Dig Liver Dis. 2010;42(Suppl. 3):S235–S241. doi: 10.1016/S1590-8658(10)60511-7. [DOI] [PubMed] [Google Scholar]
- 4.Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–1314. doi: 10.1016/S0140-6736(18)30010-2. [DOI] [PubMed] [Google Scholar]
- 5.Massarweh NN, El-Serag HB. Epidemiology of hepatocellular carcinoma and intrahepatic cholangiocarcinoma. Cancer Control. 2017;24(3) doi: 10.1177/1073274817729245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moore JB. Non-alcoholic fatty liver disease: the hepatic consequence of obesity and the metabolic syndrome. Proc Nutr Soc. 2010;69:211–220. doi: 10.1017/S0029665110000030. [DOI] [PubMed] [Google Scholar]
- 8.Ballestri S, Zona S, Targher G, Romagnoli D, Baldelli E, Nascimbeni F, Roverato A, Guaraldi G, Lonardo A. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J Gastroenterol Hepatol. 2016;31(5):936–944. doi: 10.1111/jgh.13264. [DOI] [PubMed] [Google Scholar]
- 9.Lonardo A, Nascimbeni F, Maurantonio M, Marrazzo A, Rinaldi L, Adinolfi LE. Nonalcoholic fatty liver disease: evolving paradigms. World J Gastroenterol. 2017;23(36):6571–6592. doi: 10.3748/wjg.v23.i36.6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lonardo A, Nascimbeni F, Targher G, Bernardi M, Bonino F, Bugianesi E, Casini, Gastaldelli AA, Marchesini G, Marra F. AISF position paper on nonalcoholic fatty liver disease (NAFLD): updates and future directions. Dig Liver Dis. 2017;49:471–483. doi: 10.1016/j.dld.2017.01.147. [DOI] [PubMed] [Google Scholar]
- 11.Piscaglia F, Svegliati-Baroni G, Barchetti A, Pecorelli A, Marinelli S, Tiribelli C, Bellentani S. Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: a multicenter prospective study. Hepatology. 2016;63(3):827–838. doi: 10.1002/hep.28368. [DOI] [PubMed] [Google Scholar]
- 12.Nascimbeni F, Ballestri S, Machado MV, Mantovani A, Cortez-Pinto H, Targher G, Lonardo A. Clinical relevance of liver histopathology and different histological classifications of NASH in adults. Expert Rev Gastroenterol Hepatol. 2017;12(4):351–367. doi: 10.1080/17474124.2018.1415756. [DOI] [PubMed] [Google Scholar]
- 13.Ascha MS, Hanouneh IA, Lopez R, Tamimi TA, Feldstein AF, Zein NN. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology. 2010;51:1972–1978. doi: 10.1002/hep.23527. [DOI] [PubMed] [Google Scholar]
- 14.Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, Villanueva A, Newell P, Ikeda K, Hashimoto M. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69:7385–7392. doi: 10.1158/0008-5472.CAN-09-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoshida Y, Toffanin S, Lachenmayer A, Villanueva A, Minguez B, Llovet JM. Molecular classification and novel targets in hepatocellular carcinoma: recent advancements. Semin Liver Dis. 2010;30:35–51. doi: 10.1055/s-0030-1247131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Villanueva A, Minguez B, Forner A, Reig M, Llovet JM. Hepatocellular carcinoma: novel molecular approaches for diagnosis, prognosis, and therapy. Annu Rev Med. 2010;61:317–328. doi: 10.1146/annurev.med.080608.100623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahajan V, Klingstedt T, Simon R, Nilsson KP, Thueringer A, Kashofer K, Haybaeck J, Denk H, Abuja PM, Zatloukal K. Cross beta-sheet conformation of keratin 8 is a specific feature of Mallory-Denk bodies compared to other hepatocyte inclusions. Gastroenterology. 2011;141(3):1080–1090. doi: 10.1053/j.gastro.2011.05.039. [DOI] [PubMed] [Google Scholar]
- 18.Strnad P, Stumptner C, Zatloukal K, Denk H. Intermediate filament cytoskeleton of the liver in health and disease. Histochem Cell Biol. 2008;129:735–749. doi: 10.1007/s00418-008-0431-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zatloukal K, French SW, Stumptner C, Strnad P, Harada M, Toivola DM, Cadrin M, Omary MB. From Mallory to Mallory-Denk bodies: what, how and why? Exp Cell Res. 2007;313:2033–2049. doi: 10.1016/j.yexcr.2007.04.024. [DOI] [PubMed] [Google Scholar]
- 20.Toivola DM, Boor P, Alam C, Strnad P. Keratins in health and disease. Curr Opin Cell Biol. 2015;32:73–81. doi: 10.1016/j.ceb.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 21.Bettermann K, Mehta AK, Hofer EM, Wohlrab C, Golob-Schwarzl N, Svendova V, Schimek MG, Stumptner C, Thüringer A, Speicher MR. Keratin 18-deficiency results in steatohepatitis and liver tumors in old mice: a model of steatohepatitis-associated liver carcinogenesis. Oncotarget. 2016;7(45):73309–73322. doi: 10.18632/oncotarget.12325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haybaeck J, Stumptner C, Thueringer A, Kolbe T, Magin TM, Hesse M, Fickert P, Tsybrovskyy O, Müller H, Trauner M. Genetic background effects of keratin 8 and 18 in a DDC-induced hepatotoxicity and Mallory-Denk body formation mouse model. Lab Invest. 2012;92(6):857–867. doi: 10.1038/labinvest.2012.49. [DOI] [PubMed] [Google Scholar]
- 23.Tamai Y, Ishikawa T, Bösl MR, Mori M, Nozaki M, Baribault H, Oshima RG, Taketo MM. Cytokeratins 8 and 19 in the mouse placental development. J Cell Biol. 2000;151:563–572. doi: 10.1083/jcb.151.3.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ku NO, Lim JK, Krams SM, Esquivel CO, Keeffe EB, Wright TL, Parry DA, Omary MB. Keratins as susceptibility genes for end-stage liver disease. Gastroenterology. 2005;129:885–893. doi: 10.1053/j.gastro.2005.06.065. [DOI] [PubMed] [Google Scholar]
- 25.Strnad P, Lienau TC, Tao GZ, Lazzeroni LC, Stickel F, Schuppan D, Omary MB. Keratin variants associate with progression of fibrosis during chronic hepatitis C infection. Hepatology. 2006;43:1354–1363. doi: 10.1002/hep.21211. [DOI] [PubMed] [Google Scholar]
- 26.Adams LA, Lindor KD. Nonalcoholic fatty liver disease. Ann Epidemiol. 2007;17:863–869. doi: 10.1016/j.annepidem.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 27.Angulo P. Obesity and nonalcoholic fatty liver disease. Nutr Rev. 2007;65:S57–S63. doi: 10.1111/j.1753-4887.2007.tb00329.x. [DOI] [PubMed] [Google Scholar]
- 28.Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, LeBlanc AC, Donovan DJ, Thung SN, Solé M. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008;68:6779–6788. doi: 10.1158/0008-5472.CAN-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Budinska E, Gelnarova E, Schimek MG. MSMAD: a computationally efficient method for the analysis of noisy array CGH data. Bioinformatics. 2009;25(6):703–713. doi: 10.1093/bioinformatics/btp022. [DOI] [PubMed] [Google Scholar]
- 30.Denk H, Stumptner C, Zatloukal K. Mallory bodies revisited. J Hepatol. 2000;32:689–702. doi: 10.1016/s0168-8278(00)80233-0. [DOI] [PubMed] [Google Scholar]
- 31.Hupé P, Stransky N, Thiery JP, Radvanyi F, Barillot E. Analysis of array CGH data: from signal ratio to gain and loss of DNA regions. Bioinformatics. 2004;20:3413–3422. doi: 10.1093/bioinformatics/bth418. [DOI] [PubMed] [Google Scholar]
- 32.Lachenmayer A, Alsinet C, Chang CY, Llovet JM. Molecular approaches to treatment of hepatocellular carcinoma. Dig Liver Dis. 2010;42(Suppl. 3):S264–S272. doi: 10.1016/S1590-8658(10)60515-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Villanueva A, Hoshida Y, Battiston C, Tovar V, Sia D, Alsinet C, Cornella H, Liberzon A, Kobayashi M, Kumada H. Combining clinical, pathology, and gene expression data to predict recurrence of hepatocellular carcinoma. Gastroenterology. 2011;140:1501–1512. doi: 10.1053/j.gastro.2011.02.006. e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nault JC, De Reyniès A, Villanueva A, Calderaro J, Rebouissou S, Couchy G, Decaens T, Franco D, Imbeaud S, Rousseau F. A Hepatocellular carcinoma 5-gene score associated with survival of patients after liver resection. Gastroenterology. 2013;145(1):176–187. doi: 10.1053/j.gastro.2013.03.051. [DOI] [PubMed] [Google Scholar]
- 35.Roessler S, Jia HL, Budhu A, Forgues M, Ye QH, Lee JS, Thorgeirsson SS, Sun Z, Tang ZY, Qin LX. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 2010;70:10202–10212. doi: 10.1158/0008-5472.CAN-10-2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawai HF, Kaneko S, Honda M, Shirota Y, Kobayashi K. alpha-fetoprotein-producing hepatoma cell lines share common expression profiles of genes in various categories demonstrated by cDNA microarray analysis. Hepatology. 2001;33:676–691. doi: 10.1053/jhep.2001.22500. [DOI] [PubMed] [Google Scholar]
- 37.Lee JS, Thorgeirsson SS. Functional and genomic implications of global gene expression profiles in cell lines from human hepatocellular cancer. Hepatology. 2002;35:1134–1143. doi: 10.1053/jhep.2002.33165. [DOI] [PubMed] [Google Scholar]
- 38.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brunt EM. Nonalcoholic fatty liver disease: pros and cons of histologic systems of evaluation. Int J Mol Sci. 2016;17(1) doi: 10.3390/ijms17010097. [pii: E97] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farrell GC, van Rooyen D, Gan L, Chitturi S. NASH is an inflammatory disorder: pathogenic, prognostic and therapeutic implications. Gut Liver. 2012;6:149–171. doi: 10.5009/gnl.2012.6.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Torres DM, Williams CD, Harrison SA. Features, diagnosis, and treatment of nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2012;10:837–858. doi: 10.1016/j.cgh.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 42.Beyoglu DaI JR. The metabolomic window into hepatobiliary disease. J Hepatol. 2013;59(4):842–858. doi: 10.1016/j.jhep.2013.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lackner C, Gogg-Kamerer M, Zatloukal K, Stumptner C, Brunt EM, Denk H. Ballooned hepatocytes in steatohepatitis: the value of keratin immunohistochemistry for diagnosis. J Hepatol. 2008;48:821–828. doi: 10.1016/j.jhep.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 44.Newberry EP, Xie Y, Kennedy SM, Graham MJ, Crooke RM, Jiang H, Chen A, Ory DS, Davidson NO. Prevention of hepatic fibrosis with liver microsomal triglyceride transfer protein deletion in liver fatty acid binding protein null mice. Hepatology. 2017;65(3):836–852. doi: 10.1002/hep.28941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matherly SC, Puri P. Mechanisms of simple hepatic steatosis: not so simple after all. Clin Liver Dis. 2012;16:505–524. doi: 10.1016/j.cld.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 47.Varga T, Czimmerer Z, Nagy L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim Biophys Acta. 2011;1812:1007–1022. doi: 10.1016/j.bbadis.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Immunofluorescent characterisation of wt, Krt8-/- and Krt18-/- mice.
qRT-Primer Sequences Mus musculus
Tabularization of the different qRT-Primer and their sequence which have been used for qRT-PCR experiments.