

Abstract
N6-Methyladenosine (m6A) modification of messenger RNA (mRNA) is emerging as an important regulator of gene expression that impacts different developmental and biological processes, and altered m6A homeostasis is linked to cancer1-5. m6A is catalyzed by METTL3 and enriched in the 3’ untranslated region (3’ UTR) of a large subset of mRNAs at sites close to the stop codon5. METTL3 can promote translation but the mechanism and widespread relevance remain unknown1. Here we show that METTL3 enhances translation only when tethered to reporter mRNA at sites close to the stop codon supporting a mRNA looping mechanism for ribosome recycling and translational control. Electron microscopy reveals the topology of individual polyribosomes with single METTL3 foci found in close proximity to 5’ cap-binding proteins. We identify a direct physical and functional interaction between METTL3 and the eukaryotic translation initiation factor 3 subunit h (eIF3h). METTL3 promotes translation of a large subset of oncogenic mRNAs, including Bromodomain-containing protein 4 (BRD4) that are also m6A-modified in human primary lung tumors. The METTL3-eIF3h interaction is required for enhanced translation, formation of densely packed polyribosomes, and oncogenic transformation. METTL3 depletion inhibits tumorigenicity and sensitizes lung cancer cells to BRD4 inhibition. These findings uncover a mRNA looping mechanism of translation control and identify METTL3-eIF3h as a potential cancer therapeutic target.
Keywords: N6-methyladenosine, m6A, METTL3, polysome, translation, closed loop, eIF3h, lung adenocarcinoma, oncogene, BRD4, JQ1
Despite awareness of the biological importance of m6A in various organisms, the mechanisms for how m6A regulates gene expression remain poorly understood. YTH-domain containing proteins can specifically bind m6A-modified RNA to regulate mRNA splicing, export, stability, and translation5-8. In addition, we found that tethering METTL3 to mRNA reporters promotes translation and METTL3 enhances translation of oncogenes including EGFR in human lung cancer cells1.
The initiation step of translation is rate limiting and a closed-loop model was proposed to facilitate multiple rounds of mRNA translation. This is supported by functional and physical interaction between the capped 5’ terminus and the polyadenylated 3’ terminus of mRNA mediated by translation initiation factor eIF4G and PABPC1 (poly(A) binding protein cytoplasmic 1)9-12. Our data support an alternative closed loop model, where circularization of the mRNA is mediated by association between the eIF3h subunit at the 5’-end of the mRNA and METTL3 bound to specific sites near the translation stop codon. We find that METTL3 promotes translation of a large subset of oncogenic mRNAs and that METTL3-eIF3h interaction is required for oncogenic transformation. These findings provide new insights into the mechanism of translation control and suggest that METTL3-eIF3h and downstream oncogenes could be therapeutic targets for cancer.
To test a mRNA looping model we first examined whether the METTL3 binding position on mRNA is important for its ability to enhance translation. Using reporters with MS2 binding sites located at different positions (Extended Data Fig. 1a) we found that direct METTL3 tethering can promote translation only when bound to the 3’ UTR at a position near the stop codon. In vitro translation assays performed with cell lystates, recombinant METTL3 protein, and in vitro transcribed mRNAs confirmed that full-length METTL3 or an amino-terminal fragment (1-200 aa) that is sufficient to promote reporter translation in cells (Extended Data Fig. 2), enhanced translation of a luciferase reporter mRNA (Extended Data Fig. 1e-g). METTL3 tethering had a stronger affect on mRNAs without a poly (A) tail (Extended Data Fig. 1g), which is consistent with some redundancy between METTL3 and eIF4G-PABPC1-mediated looping for poly (A)-containing mRNAs in these in vitro assays.
To provide evidence that METTL3 is bound to polyribosomes FLAG-METTL3-containing mRNA ribonucleoprotein complexes (mRNPs) were affinity purified, incubated with α-METTL3 gold-labeled antibodies, subjected to sucrose gradient fractionation, and then analyzed by electron microscopy (EM) (Fig. 1a). This revealed gold-labeled METTL3 in the individual polyribosomes (Extended Data Fig. 4a, b). We performed similar experiments using either α-CBP80 or α-eIF4E gold-labeled antibodies together with the α-METTL3 particles. Since the α-CBP80 and α-eIF4E gold particles were larger they could be distinguished from the α-METTL3 particles. Individual polyribosomes containing double-labeled gold particles showed that each METTL3 signal is in close proximity (<20 nm) to a cap-binding protein (Fig. 1b and Extended Data Fig. 4c, d). This reveals the topology of individual endogenous METTL3-bound polyribosomes and support that METTL3 mediates the looping of mRNA to promote efficient translation.
Fig. 1 |. METTL3 enhances translation of target mRNAs by interacting with eIF3h.
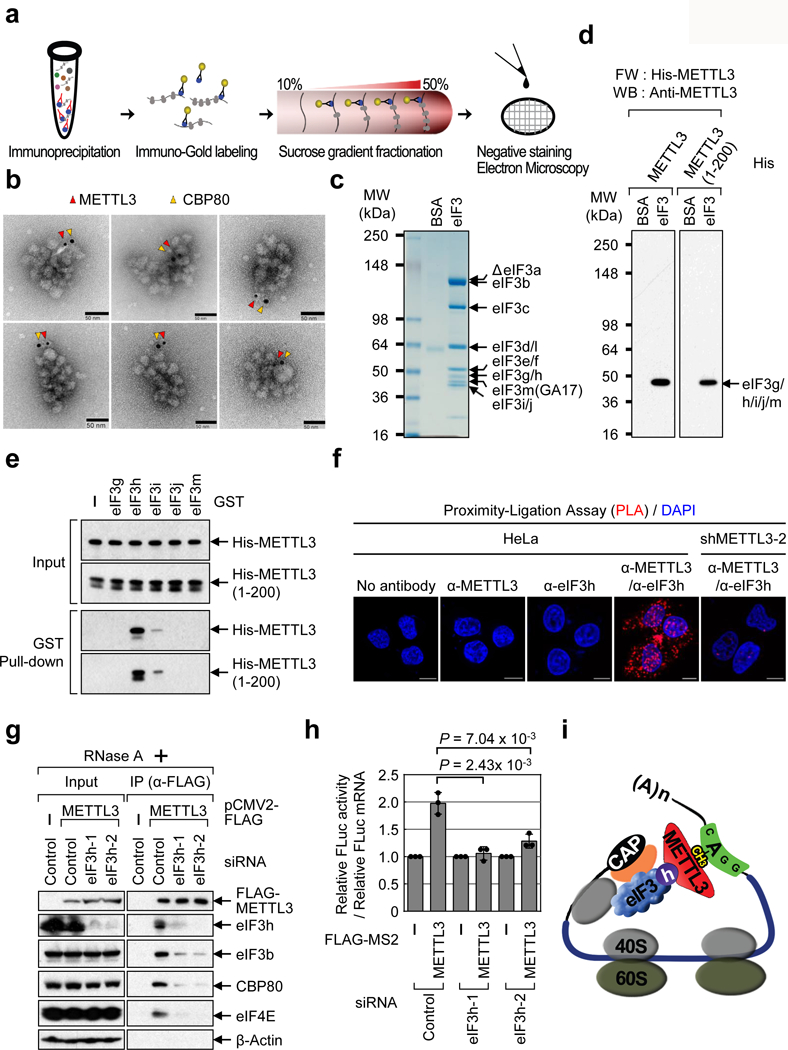
a, Electron microscopy (EM) procedure. b, EM images of polyribosomes. Red arrows; METTL3 with immuno-gold particle (6 nm), yellow arrows; CBP80 with immuno-gold particle (10 nm). Three independently performed experiments show similar results. c-d, Far Western (FW). c, Staining of eIF3 complex. A breakdown product is denoted (ΔeIF3a). Two independently performed experiments show similar results. d, FW of purified eIF3 complex. Two independently performed experiments show similar results. e, GST-tagged eIF3 subunits and co-purified His-METTL3 or 1-200 aa analyzed by Western blotting. Two independently performed experiments show similar results. f, Proximity ligation assay (PLA). Two independently performed experiments show similar results. g, Co-IPs from control or eIF3h knockdown cells. Two independently performed experiments show similar results. h, Tethering assays. Error bars = mean ± SD; n = 3 biologically independent samples, two-sided t-test. i, Model.
Full length METTL3 as well as the 1-200 aa, and 1-350 aa fragments were found to associate with m7GTP-Agarose in cap-binding assays (Extended Data Fig. 3a). This result is highly consistent with tethering assays (Extended Data Fig. 2) and support that the 1-200 aa fragment of METTL3 interacts with translation initiation factor(s) to promote translation. Knockdown of METTL3 had no effect on the association of cap-binding proteins or translation initiation factors (Extended Data Fig. 3b). Thus translation initiation complex formation does not require METTL3. Conversely, the association of METTL3 with m7GTP-Agarose was dramatically diminished using lysates depleted for CTIF, eIF4GI or eIF3b, supporting that the association of METTL3 with m7GTP-Agarose is mediated through an interaction with general translation initiation factor(s) (Extended Data Fig. 3c).
A large-scale purification and mass spectroscopy characterization of FLAG-METTL3-containing complexes identified numerous translation factors (Extended Data Fig. 3d, and data not shown). Gene ontology (GO) analysis of the METTL3-interacting proteins identified ‘mRNA metabolic processes’, ‘RNA processing’, and ‘Translation’ as the most significantly enriched categories (Extended Data Fig. 3e, f). Considering this and our previous observation that METTL3 knockdown diminishes the association of eIF3 with cap-binding proteins in co-IPs, we hypothesized that METTL3 might interact directly with certain component(s) of the multi-subunit eIF3 complex.
To test whether METTL3 interacts with any of the 13 subunit(s) of eIF3. Recombinant METTL3 and 1-200 aa were used for Far-Western blotting with a purified human eIF3 complex (Extended Data Fig. 4e and Fig. 1c). METTL3 and 1-200 aa both specifically bound to a single band that most likely corresponds to eIF3g, -h, -i, -j, or -m (Fig. 1d). To further confirm this interaction and to define the particular subunit(s) that interacts with METTL3, we individually expressed and purified the GST-tagged eIF3 subunits from bacteria (Extended Data Fig. 4f) and tested them for binding to His-METTL3 using in vitro binding assays with either His-METTL3 or 1-200 aa. METTL3 (and 1-200 aa) were found to specifically interact with eIF3h (Fig. 1e). The interaction between eIF3h with METTL3 was further confirmed using a α-METTL3 antibody (that recognizes a 1-250 aa METTL3 epitope) to specifically disrupt this eIF3h-METTL3 interaction (Extended Data Fig. 4g). Additional GST pull-down experiments identified the Mpr1p/Pad1p N-terminal (MPN) domain13 as necessary and sufficient to interact with METTL3 (Extended Data Fig. 4h-j). Notably, the MPN domain faces the solvent side of the ribosome and is likely accessible for interaction with METTL3 without impairing 80S assembly13.
An in situ proximity ligation assay (PLA) confirmed METTL3-eIF3h proximity in cells (Fig. 1f). co-IP experiments revealed that METTL3 association with translation initiation factors is dependent on eIF3h (Fig. 1g). We next tested the functional interaction between METTL3 and eIF3h, and found depletion of eIF3h abrogated the enhanced translation by METTL3 (Extended Data Fig. 4k-m and Fig. 1h). Overall, our results support a model whereby mRNA circularization and translation control is mediated by a specific METTL3-eIF3h interaction (Fig. 1i).
We next examined the widespread impact of METTL3 on mRNA translation (Fig. 2, and Extended Data Fig. 5a). METTL3 depletion caused an increase in the 80S ribosome peak and a corresponding reduction of polyribosome peak (Fig. 2a). METTL3 depletion had a negligible effect on steady state mRNA abundance whereas translation efficiency of a large subset (4,267) of mRNAs was reduced by at least 2-fold in METTL3-depleted cells (Fig. 2b-d). Comparison of these genes with previously reported METTL3 PAR-CLIP data14 identified 809 mRNAs that are both bound and translationally regulated by METTL3 (Fig. 2d). mRNAs on this list of METTL3 target mRNAs have on average longer 3’ UTRs compared to all mRNAs (Fig. 2e). Gene ontology (GO) showed that these mRNAs are involved in tumor progression and apoptosis (Extended Data Fig. 5b). qRT-PCR analysis confirmed that METTL3 depletion had a modest effect on mRNA abundance (Extended Data Fig. 5c), but strongly decreased translation of target mRNAs (Fig. 2f). Moreover, neither global mRNA stability analysis by RNA-seq (Fig. 2g) nor qRT-PCR analysis of individual genes (Extended Data Fig. 5d, e) showed any differences in mRNA stability upon METTL3 depletion. We isolated endogenous METTL3 mRNPs and confirmed the association with target mRNAs by qRT-PCR (Fig. 2h and Extended Data Fig. 6a). Western blotting showed decreased protein expression from these target mRNAs in the METTL3 knockdown samples (Fig. 2i), whereas analysis of mRNA sequencing data revealed that the splicing patterns of these target mRNAs was unaltered (Extended Data Fig. 6b). Depletion of YTHDF1, a m6A-reader protein implicated in translational control had no effect on the expression of these METTL3 targets (Extended Data Fig. 6c)7. Western blotting showed strongly reduced endogenous BRD4 protein expression upon knockdown of eIF3h without affecting BRD4 mRNA abundance or the levels of METTL3 protein (Fig. 2j and Extended Data Fig. 6d). Expression of a shRNA-resistant FLAG-METTL3 rescued the expression of BRD4 and CD9 proteins. However, expression of a catalytic mutant of METTL3 failed to recover target gene expression (Fig. 2k). METTL3 knockdown in A549 lung cancer cells similarly led to decreased expression of BRD4 and other targets (Fig. 2l), and treatment of METTL3-depleted and control- A549 cells with the BRD4 inhibitor JQ1 revealed that the METTL3 depleted cells are more sensitive to pharmacological BRD4 inhibition (Fig. 2m, n and Extended Data Fig. 6e).
Fig. 2 |. METTL3 promotes translation of a large subset of mRNAs.
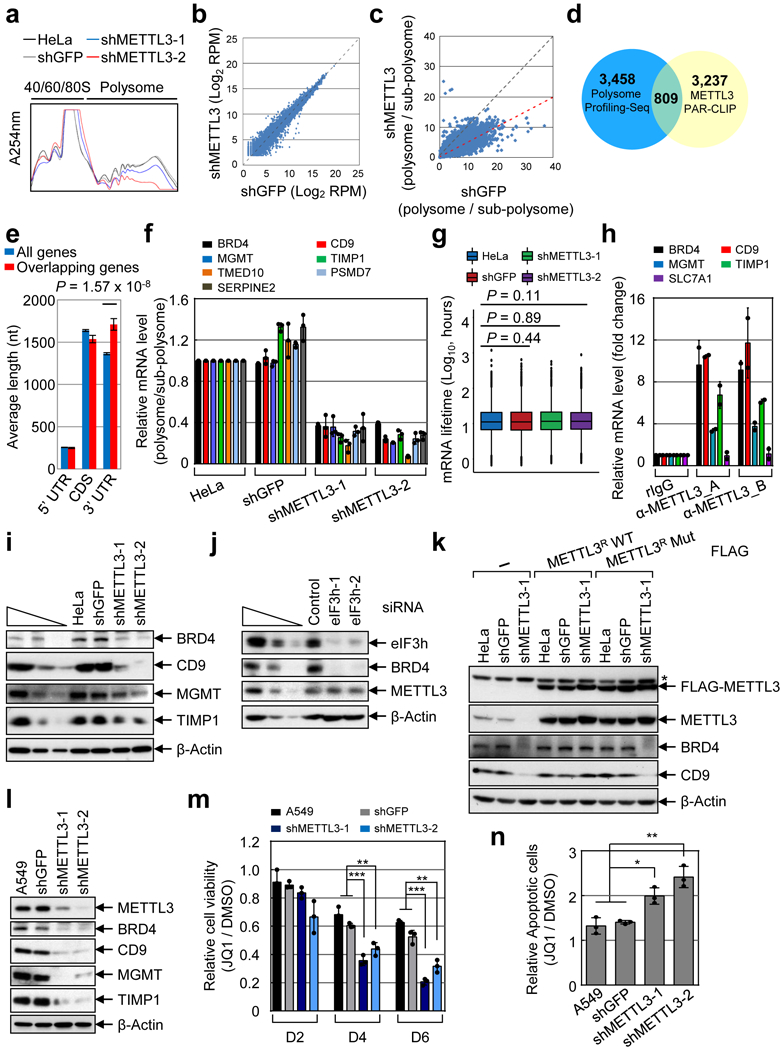
a, Polysome profile. Two independently performed experiments show similar results. b, Scatter plot of RNA-Seq data. Average read number from two METTL3 knockdowns is plotted. c, Scatter plot of translation efficiency (TE). Average read number from two shMETTL3 samples. d, Venn diagram showing mRNAs with >2-fold change in TE and METTL3 PAR-CLIP data. e, Features of overlapping mRNAs (n=809) from (d) was compared with all (18,115) expressed genes. Error bars = mean ± SEM; unpaired t-test (two-sided). f, qRT-PCR analysis. Error bars = mean ± SD; n = 3 technical replicates. g, Box plot represents global mRNA (12,479 mRNAs) stability profiling from two biological replicates. Two-sided Wilcoxon rank sum test; the results are statistically not significant. h, qRT-PCR analysis of METTL3-associated mRNAs using two different α-METTL3 antibodies. Error bars =mean ± SD; n = 2 independent experiments. i-l, Western blot. At least two independently performed experiments show similar results. m, MTS assay of A549 cellular proliferation upon JQ1 treatment. Error bars =mean ± SD; n = 3 independent experiments. Two-sided t-test, multiple comparison for the p-values; *** P < 0.001, ** P < 0.01. n, Quantification of (sum of early and late) apoptotic cells. Error bars +mean ± SD; n = 3 independent experiments. Two-sided t-test, multiple comparison for the p-values; * p<0.05, **p<0.01.
Considering that 1-200 aa is sufficient to directly interact with eIF3h (Fig. 1 and Extended Data Fig. 4) and that 1-200 aa can promote translation in tethering experiments whereas 1-150 aa does not (Extended Data Fig. 2), we reasoned that a region between 150-200 aa must be important for the physical and functional METTL3-eIF3h interaction. Secondary structure predictions identified a putative alpha helix (150-161 aa) that is highly conserved in mammals (Extended Data Fig. 7a, b)15,16. Moreover, 3D modeling identified a putative structured module (Extended Data Fig. 7c)17. We therefore generated a mutant version of METTL3 with a single aa substitution of a highly conserved Alanine (A155P) to disrupt this putative helical structure. co-IP confirmed substantially impaired interaction of A155P with translation initiation factors specifically in RNase-treated samples (Fig. 3a). Importantly, the A155P mutation did not disrupt METTL3 interaction with METTL14 (Fig. 3a). Tethering experiments demonstrated that the A155P mutant is strongly impaired in promoting mRNA translation (Fig. 3b and Extended Data Fig. 7d, e). Moreover, expression of METTL3 A155P in METTL3-depleted cells failed to rescue the expression of endogenous target proteins (Fig. 3c). This effect was not due to altered mRNA association of A155P with endogenous target mRNAs (Fig. 3d and Extended Data Fig. 7f). In summary, while METTL3 A155P mutant can associate with METTL14 and be loaded on mRNAs, its ability to interact with initiation factors and promote mRNA translation is severely compromised.
Fig. 3 |. METTL3-eIF3h interaction is crucial for enhanced mRNA translation and polysome conformation.
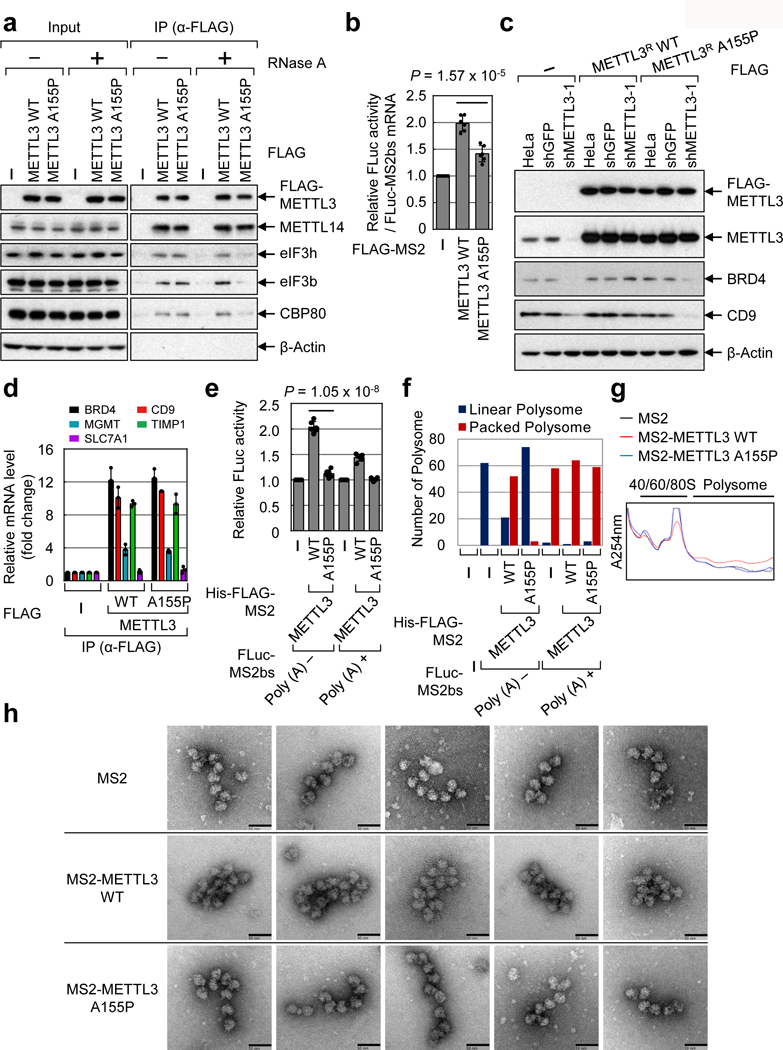
a, Co-IPs. Two independently performed experiments show similar results. b, Tethering assay. Error bars =mean ± SD; n = 6 independent experiments; two-sided t-test. c, Western blot. Two independently performed experiments show similar results. d, qRT-PCR. Error bars =mean ± SD; n = 2 independent experiments. e, In vitro translation with Rabbit reticulocyte lysate. Error bars =mean ± SD; n = 6 independent experiments; two-sided t-test. f, Analysis of 20 images from each sample in (e). g, Peak analysis of polysome profiling coupled with in vitro translation. Two independently performed experiments show similar results. h, EM images of polyribosomes. Images were taken from the samples in (g). Scale bar, 50 nm. Two independently performed experiments show similar results.
We next used in vitro reconstituted translation assays to explore the effect of 3’ UTR-bound METTL3 on translation efficiency and polysome conformation (Fig. 3e-f, Extended Data Fig. 7g and Extended Data Fig. 9a). Consistent with Extended Data Fig. 1g, tethering of recombinant METTL3 increased translation efficiency with a stronger effect on mRNA without a poly (A) tail (Fig. 3e). In contrast, tethered A155P had no significant effect on translation (Fig. 3e). EM analysis of samples from the in vitro translation reactions showed mostly densely packed polysome structures formed when METTL3 was tethered to reporter mRNAs without a poly (A) tail (Extended Data Fig. 9a). In contrast, almost all the polysomes observed in the A155P sample appeared as linear polysomes (as in the control sample). Only dispersed ribosomes were observed in reactions without mRNA (Fig. 3f and Extended Data Fig. 9a). In all samples with poly (A)+ containing mRNAs mostly packed polysomes were observed irrespective of the protein that was tethered. Since the reporter mRNA contains a very short 3’UTR it expected that the known eIF4GI-PABP interaction is likely responsible for observed packed polysomes in poly (A)+ samples (Fig. 3f and Extended Data Fig. 9a). We further analyzed in vitro translation assays by sucrose gradient fractionation. Tethering of WT METTL3 tethering resulted in a larger polysome peak compared to the A155P mutant (Fig. 3g). Individual polysomes were analyzed by EM (Fig. 3g, h). Polysomes formed with WT METTL3 appeared more densely packed compared to the more linear polysomes that formed with the A155P mutant or MS2 control (Fig. 3h). Taken together, these results strongly support our model that METTL3 through its interaction with eIF3h promotes translation through its effects on polysome conformation.
Considering that METTL3 regulates the translation of a large subset of genes that are involved in tumor progression and apoptosis (Extended Data Fig. 5b), we examined the role of METTL3 in cancer. Immunohistochemistry (IHC) staining of primary human lung adenocarcinoma samples and adjacent normal control tissue revealed that METTL3 expression is significantly increased in lung tumors and correlates with tumor stage (Fig. 4a, b and Extended Data Fig. 8a). METTL3 depletion in A549 lung cancer cells resulted in significantly smaller tumors in mouse xenografts (Fig. 4c and Extended Data Fig. 8b-d).
Fig. 4 |. Role of METTL3 and m6A in lung cancer cells and primary human tumors.
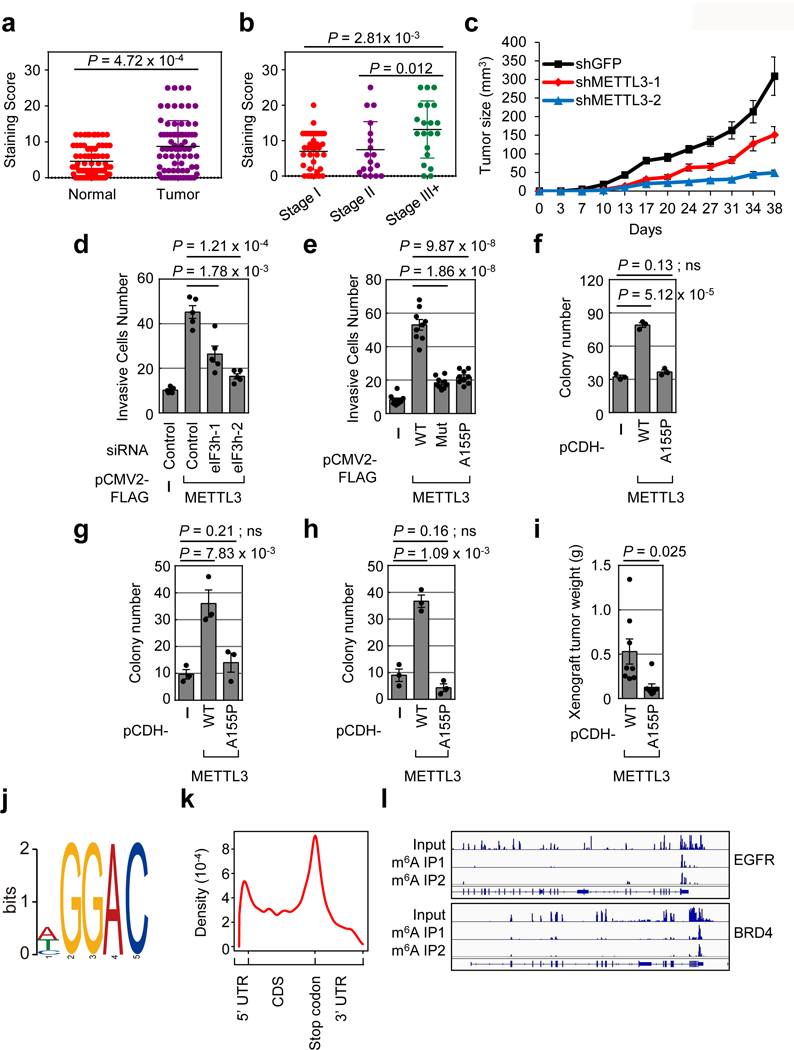
a-b, METTL3 IHC in primary lung adenocarcinoma and adjacent normal tissue. a, Error bars =mean ± SD; n = 75; two-sided Wilcoxon signed-rank test. b, . Error bars =mean ± SD; Stage I, n =37; Stage II, n =18; Stage III, n =20; two-sided Wilcoxon signed-rank test. c, Tumor-growth of xenografts from A549 cells stably expressing indicated shRNAs. Error bars =mean ± SEM; n=5 independent mice. d-e, Quantification of invasive BJ cells. Cells were transiently transfected with indicated siRNAs (d) or plasmids (e). Error bars =mean ± SEM; n = 5 for (d), n = 9 for (e) independent experiments; two-sided t-test. f, Quantification of NIH-3T3 cells colony formation. Error bars =mean ± SEM; n = 3 independent experiments; two-sided t-test; ns, not significant. g-h, Quantification of MEFs (g) or MB352 (h) cells colony formation. Error bars =mean ± SEM; n = 3 independent experiments; two-sided t-test; ns, not significant. i, Tumor weight of xenografts derived from NIH-3T3 cells stably expressing indicated proteins. There was no tumor formation in empty vector group during the observed period. Error bars =mean ± SEM; n = 8 independent mice; two-sided t-test. j-l, Global profiling of m6A targets in primary lung cancer samples. j, Sequence motif identified in m6A MeRIP-seq. k, Metagene analysis of m6A peaks. l, Integrative genomics viewer plots of representative m6A containing genes. Four lung tumors show similar results.
We next examined the relevance of the METTL3-eIF3h interaction and mRNA looping in the context of cancer cell biology. Knockdown of eIF3h suppressed the ability of METTL3 to promote cellular invasion (Fig. 4d and Extended Data Fig. 8e). Moreover, unlike WT METTL3 protein, expression of either a catalytically inactive mutant, or A155P mutant was unable to promote the invasive capability of lung fibroblasts (Fig. 4e and Extended Data Fig. 8f). Furthermore, METTL3 overexpression was sufficient to promote the oncogenic transformation of NIH-3T3 cells, mouse embryonic fibroblasts (MEFs) or MB352 (p53 null MEFs) cells, whereas METTL3 A155P had no significant affect in these 3D soft agar colony formation assays (Fig. 4f-h and Extended Data Fig. 8g-h). The oncogenic function of METTL3 was also studied in the mouse xenografts. NIH-3T3 cells with ectopic expression of METTL3 WT, METTL3 A155P, or the empty vector control were injected into nude mice to determine their in vivo tumorigenic capacities. METTL3 WT overexpression promoted in vivo tumor growth, whereas METTL3 A155P showed an impaired ability to promote tumor growth. As control, no tumors were detected in mice injected with control NIH-3T3 cells (Fig. 4i and Extended Data Fig. 8i). Overall, these results support that the METTL3-eIF3h mediated mRNA looping is critical for the oncogenic function of METTL3.
meRIP-seq performed on four primary human lung tumors identified patient specific and commonly m6A-modified mRNAs in lung cancer (Extended Data Fig. 8j and Table S3). An expected “GGAC” motif was identified and m6A peaks are predominantly localized near the translation stop codon (Fig. 4j-k and Extended Data Fig. 8k). Gene ontology analysis revealed that the common methylated genes are enriched in the signature genes of neoplasms and cancer, including the known oncogenes EGFR and BRD4 (Fig. 4l and Extended Data Fig. 9b). These data were consistent with m6A features identified in cell lines, with >50% of the peaks found in both tumors and cancer cell lines (Extended Data Fig. 9c).
We propose that METTL3 promotes oncogene translation and tumorigenesis through a mRNA looping mechanism. This is supported by; 1) the position-dependent effects of METTL3 tethering on mRNA translation, 2) EM visualization of METTL3 bound to endogenous polyribosomes and its proximity to cap-binding proteins, 3) METTL3 interaction with eIF3h, and 4) Disruption of METTL3-eIF3h interaction abolishes the ability of METTL3 to promote translation, affect polysome conformation, or promote oncogenic transformation. This METTL3-eIF3h loop presumably promotes translation through ribosome recycling in a way similar to that proposed for eIF4G-PABPC1-mediated mRNA looping. While there is likely some redundancy between these mRNA circularization mechanisms, looping between the stop codon and the 5’ end might represent a more productive way to recycle ribosomes rather than via the 3’ end, especially for mRNAs with long 3’ UTRs since the ribosomes will dissociate from the mRNP once released at the stop codon.
The complexity of eIF3 and the specialized roles of individual subunits is only beginning to be appreciated18,19. Imbalanced expression of eIF3 subunits is found in various tumors20 and expression in fibroblasts can promote malignant transformation21. Increased eIF3h expression is found in different tumor types and in many cases is due to amplification of a chromosomal region that includes eIF3h at 8q23.3 and the nearby MYC oncogene22,23. METTL3 is overexpressed in many different cancer types (Extended Data Fig. 10a), and the expression of METTL3 and eIF3h is often positively correlated (Extended Data Fig. 10b, c). Our findings could pave the way for the development of new cancer therapeutic strategies.
Methods
Cell Culture and transfection
Human lung cancer cell lines (A549 and H1299), HEK293T, BJ, NIH-3T3, HeLa, MEFs were cultured with DMEM supplemented with 10% fetal bovine serum (FBS) and antibiotics. Cells were grown in a 5% CO2 cell culture incubator at 37°C. Cell lines were authenticated with morphology, karyotyping, and PCR based approaches by ATCC. The cell lines were tested for potential mycoplasma contamination and confirmed that they are mycoplasma negative. Transfection of plasmids was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Down regulation of target genes by siRNA was performed using Lipofectamine RNAi Max (Invitrogen). The following siRNA sequences were used in this study: 5′-r(GAUAGAUGGCCUUGUGGUA)(UU)-3′ for eIF3h-1; 5′-r(GCGGAGCCUUCGCCAUGUA)(UU)-3′ for eIF3h-2; 5′-r(UGAGAAAGGAGGAGAGGAA)d(TT)-3′ for eIF4GI; 5′-r(UCAACCUCUUUACGGAUUU)d(TT)-3′ for eIF3b; and 5′-r(GCAUCAACCUGAAUGACAU)(UU)-3′ for CTIF.
Virus Production and generation of stable knockdown and over-expression cells
Virus mediated generation of stable knockdown and over-expression cells were performed as described previously1. Briefly, shRNA containing pLKO.1 vector was co-transfected with pLP1, pLP2, and VSVG into 293T cells. For over-expression, pCDH vectors containing the METTL3 WT and A155P cDNA were co-transfected with Delta 8.9 and VSVG plasmids into 293T cells. Viruses were collected at 48 hr and 72 hr after transfection and then used to infect cells with Polybrene (8 mg/ml, Sigma); 48 hr after infection, puromycin was added to the culture medium to select the infected cells.
Plasmid construction
pFLAG-METTL3 WT, pFLAG-MS2-METTL3 and pFLAG-MS2-METTL3 Mut, were described previously1. pFLAG-METTL3 A155P plasmids was generated by inducing point mutation in pFLAG-METTL3 WT using Q5® Site-Directed Mutagenesis Kit (NEB E0554). Plasmids pFLAG-MS2-METTL3 (1-100), pFLAG-MS2-METTL3 (1-150), pFLAG-MS2-METTL3 (1-200), pFLAG-MS2-METTL3 (1-350) and pFLAG-MS2-METTL3 (101-580) was constructed by substitution of PCR amplified each METTL3 fragment into NotI-METTL3 WT-BglII site of pFLAG-MS2-METTL3. For stable METTL3 over-expression, METTL3 WT and A155P sequence were PCR amplified and cloned into the NheI and NotI sites of pCDH-CMV-MCS-EF1-Puro plasmid. For expression of recombinant METTL3 proteins, METTL3 full length and N-terminal amino acids 1-200 cDNA were cloned into the pETDuet-1 and pET His6 GST TEV LIC cloning vector individually. For expression of recombinant METTL3 proteins for in vitro translation, FLAG-MS2, FLAG-MS2-METTL3 (1-200), FLAG-MS2-METTL3, FLAG-MS2-METTL3 A155P were cloned into pETDuet-1. For bacteria protein expression of human eIF3h, eIF3j and eIF3m that express N-terminal GST-fused proteins, BamHI/EcoRI fragment of pGEX2TK vector was ligated to the PCR amplified BamHI/EcoRI fragment that contained eIF3h, eIF3j, or eIF3m. In addition, for the pGEX2TK-eIF3g or -eIF3i, BglII/EcoRI fragment of pGEX2TK vector was ligated to the PCR amplified BglII/EcoRI fragment that contained either eIF3g or eIF3i, respectively. For bacteria expression of eIF3h deletion mutants were generated by ligation of BamHI/EcoRI fragment of pGEX2TK vector with BamHI/EcoRI fragment of either PCR amplified eIF3h (1-222) or eIF3h (29-222). The pGL3c_TK luciferase reporter (FLuc) and pGL3c_TK luciferase reporter containing 2X MS2 binding sites near the stop codon (FLuc-MS2bs) were described previously1. The 2X MS2 binding site sequence was PCR amplified from FLuc-MS2bs and inserted into the NcoI site of pGL3c_TK luciferase reporter to make the FLuc-5’UTR-MS2bs reporter that the MS2 binding sites are located in the 5’UTR region of luciferase gene. The 2X MS2 binding site sequence and GFP sequence (from CAG-GFP, Addgene Plasmid #16664) were cloned into the XbaI site of pGL3c_TK luciferase reporter (FLuc) to make the FLuc-MS2bs-GFP and FLuc-GFP-MS2bs reporters. For pFLAG-tethering effector plasmids, METTL3 shRNA resistance plasmids were generated by introducing synonymous mutations in the shRNA targeting sequence using the Q5® Site-Directed Mutagenesis Kit (NEB E0554). All cloning primers are listed in Table S1.
In vitro translation assay
H1299 cells were harvested and resuspended in hypotonic buffer [10 mM Hepes (pH 7.4), 10 mM potassium acetate, 1.5 mM magnesium acetate, and 2.5 mM DTT]. Cells were then incubated on ice for 30min and ruptured by passing 10 times through a 25-gauge needle attached to a 3-mL syringe. The cell homogenate was centrifuged at 13,000 × g for 15 min at 4 °C. The supernatant was collected and used for in vitro translation. In vitro translation reactions were performed for 1 hour at 30°C using either H1299 cytoplasmic cell extracts or rabbit reticulocyte lysate (RRL) (Thermo FisherScientific, AM1200) in 20 μL reaction mixtures containing 100 ng of in vitro transcribed reporter mRNAs and 500 ng of either purified recombinant His-FLAG-MS2, His-FLAG-MS2-METTL3 or His-FLAG-MS2-METTL3 (1-200) protein. The activity of in vitro-translated luciferase was measured by Luciferase assay kit (Promega, E1960) according to the manufacturer’s instructions. Reporter mRNAs either presence or absence of poly (A) tails were in vitro transcribed using PCR amplified FLuc-MS2bs fragment with following primers; 5′-GACTAGTAATACGACTCACTATAGGGGCCACCATGGAAGACGCCAAAAACATAAAG-3′ (sense) and 5′-TCTAGACCCCGGGAGCATGGGTGAT-3′ (antisense) for the FLuc-MS2bs Poly (A)- mRNA, and 5′-GACTAGTAATACGACTCACTATAGGGGCCACCATGGAAGACGCCAAAAACATAAAG-3′ (sense) and 5′-TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTCTAGACCCCGGGAGCATGGGTGAT-3′ (antisense) for the FLuc-MS2bs Poly (A)+ mRNAs.
RNA Isolation and qRT-PCR
The details of RNA isolation and qRT-PCR assays are as described previously1. In brief, RNA was extracted from cells, co-immunoprecipitation or sucrose gradient fractionation samples using Trizol (Invitrogen) following the manufacturer’s instructions. qRT-PCR analyses were performed using SYBR Green PCR Master Mix with the Step One Real-Time PCR System (AppliedBiosystems). All primers used in this study are listed in Table S2. For the analysis of global or individual mRNA lifetime, METTL3-depleted or control HeLa cells (60-mm culture dishes) were treated with Actinomycin D (5 μg/ml), then harvested at 0h, 2h, 4h, and 6h.
Luciferase assay and translation efficiency
Dual luciferase assays were performed according to the manufacturer’s protocol (Promega). Firefly luciferase (FLuc) activity was normalized to the Renilla luciferase (RLuc) activity. Relative FLuc activity was normalized to the relative FLuc mRNAs. The normalized FLuc activity (translation efficiency) in the presence of FLAG-MS2 was set to 1.
Polysome Fractionation and RNA-seq
METTL3-depleted or control HeLa cells (four 150-mm culture dishes) were treated with 100 μg/ml cycloheximide (Sigma) for 10 min at 37°C. Cells were then lysed and layered onto 10%-50% sucrose gradient tube and centrifuged at 36,000 rpm in a Beckman SW-41Ti rotor for 2.5 hr at 4°C. Gradients were fractionated and monitored at absorbance 254 nm (Brandel). Collected fractions were pulled into sub-polysome fraction and polysome fraction. Then, total RNA, sub-polysome and polysome samples were subjected to RNA-seq. Poly-A selected mRNAs were purified and used for library construction using TruSeq Stranded mRNA Sample Prep Kits (Illumina RS-122-2101) and sequenced with Illumina NextSeq 500. ERCC RNA Spike-In Control Mixes (Ambion) were added into each sample before constructing the library to normalize the reads.
Co-immunoprecipitation, Mass spectrometry, and Western Blot
Co-immunoprecipitation (co-IP) and western blot were performed as previously1. Briefly, FLAG-METTL3 expressing HeLa or H1299 cells was harvested and lysed using NET-2 buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM phenylmethanesulfonylfluoride, 2 mM benzamidine, 1% NP-40] then the supernatant was subjected to IP using Anti-Flag M2 Affinity Gel (Sigma-Aldrich). Where indicated, the affinity elute was subjected to SDS-PAGE followed by either Colloidal Coomassie blue staining or Western blotting. Bands were excised and subjected to Mass spectrometric sequencing as previously1. The following antibodies were used for Western blotting: METTL3 (Proteintech, 15073-1-AP; Abcam, ab195352), β-actin (Abcam, ab8227), eIF3h (Abcam, ab60942) CBP80 (Gift from Dr. Yoon Ki Kim, Korea University), CTIF (Gift from Dr. Yoon Ki Kim, Korea University), eIF4E (Cell Signaling Technology, #2067), eIF3b (Santa Cruz Biotechnology,sc-16377), eIF4GI (Cell Signaling Technology, #2498), FLAG (Sigma, A8592), BRD4 (Abcam, ab128874), CD9 (Cell Signaling Technology, #13174), MGMT (Cell Signaling Technology, #2739), TIMP1 (Cell Signaling Technology, #8946) and FTO (Phosphosolution, 597-FTO).
Protein Expression, Purification, and GST Pull-Down Assay.
Plasmids expressing the recombinant proteins were transformed into BL21 Escherichia coli then the recombinant proteins were induced by IPTG at 20 °C overnight. The bacteria were pelleted and resuspended in protease inhibitor containing PBST buffer and then lysed by sonication. His-tag recombinant proteins were purified using Ni-NTA agarose (Qiagen 30210). The GST-tagged proteins were purified using the glutathione sepharose (BioVision 6655) following the manufacturer’s protocol. For GST pull down assay, equal amount of GST fusion proteins or GST control bound to glutathione sepharose were incubated with purified recombinant His-tagged METTL3 full length or N-terminal (1-200) fragment for 1 hour at 4 °C, after extensive washing, the proteins bound to the sepharose were resolved on SDS-polyacrylamide gels and detected by western blot analysis.
Far-Western Blotting
Far western blotting was performed with biochemically purified human eIF3 protein complex that was resolved by SDS-PAGE and then transferred to Hybond ECL nitrocellulose membrane. The membrane was first incubated in blocking buffer (100 mM Tris (pH 7.5), 100 mM potassium acetate, 2 mM magnesium acetate, 0.1 mM EDTA, 10% glycerol, 1 mM PMSF, 1 mM benzamidine, and 0.05% Tween 20, 5% non-fat milk) at 4 °C overnight, then the membrane was incubated with blocking buffer containing 5 μg of purified recombinant METTL3 full-length or METTL3 (1-200) proteins at 4 °C for another 24 hours. After that, the membrane was incubated with METTL3 antibody for Western blotting analysis.
Electron Microscopy
FLAG-METTL3 expressing H1299 cells were harvested and lysed using NET-2 buffer, then the supernatant was subjected to IP using Anti-Flag M2 Affinity Gel (Sigma-Aldrich). Resin-bound mRNP complexes were eluted using 3XFLAG peptides (Sigma, F3290). Where indicated, during the elution, α-METTL3 antibody (Proteintech, 15073-1-AP) and gold nanoparticle (6nm) conjugated α-rabbit IgG were added with/without either α-CBP80 antibody or α-eIF4E antibody that was gold nanoparticle (10nm) conjugated using GOLD conjugation kit (Abcam, ab201808) according to the manufacturer’s instructions. The elutes were then fractionated using 10%−50% sucrose gradients. Each fraction was applied to an EM grid (EMS, G400-Cu) covered with a thin layer of carbon and after 1 min the excess suspension was sucked up with a filter paper. The grid was washed twice with water and 0.7% uranyl formate, and then negatively stained for 20 seconds with 0.7% uranyl formate. The specimens were examined in Tecnai G2 Spirit BioTWIN Transmission Electron Microscope (FEI company) with AMT 2k CCD camera equipped. Direct magnification of 68,000x was used to detect images from Extended Data Fig. 10a, whereas magnification of 98,000x was used to detect images from Fig. 1b, 3h, Extended Data Fig. 4a and Extended Data Fig. 4c. Polysome numbers were counted using 20 individual pictures for each sample with direct magnification of 30,000x in Fig. 3f. All the images shown in the Figures were cropped sections. The average distance between immuno-gold particles in Extended Data Fig. 4d were measured from the images in Fig. 1b and Extended Data Fig. 4c using the electron microscope software (AMT Capture engine). In vitro translation reactions were performed for 1 hour at 30°C using RRL, and then the total reaction mixture was subjected to illustra™ MicroSpin™ S-400 HR Columns (Figure 3f and Extended Data Fig. 10a) or sucrose gradient fractionation (Fig. 3g and 3h). The elutes were then applied to an EM grid and analyzed by electron microscopy.
Cap-association assay using m7GTP-Agarose
To analyze the interaction of METTL3 to cap-binding protein complex, cells were lysed using NET2 buffer and total cell extracts were incubated with m7GTP-Agarose (Jena Bioscience, AC-155S) for 2 hours at 4°C. Then, the beads were washed for five times and suspended in SDS sample buffer. The eluted samples were analyzed by Western blot. Where indicated, 75 μM of m7G(5’)ppp(5’)G Cap Analog (Ambion, AM8048) was added into the sample and incubated with m7GTP-Agarose.
Cell Proliferation, Apoptosis and Invasion Assays
Cell proliferation, apoptosis and invasion assays were performed as described1. Briefly, for cell proliferation, 700 cells were seeded in a 96-well plate on day 0 with the pertinent treatment. 500 nM of JQ1 were used. Absorbances at 490 nm were measured using CellTiter 96 AQueous One Solution Cell Proliferation Assay kit (Promega) on day 2, day 4, and day 6 to measure the cellular proliferation. The numbers of apoptotic cells were quantified by flow cytometric assays using Annexin V-FITC Apoptosis Detection Kit (BioVision) five days after 500 nM of JQ1 treatment and cell seeding. Cell invasion was measured using BioCoat Matrigel Invasion Chamber (Corning) following the manufacturer’s instructions.
Soft Agar Colony Formation Assays
NIH-3T3, MEFs and MB352 cells at 30% confluence were infected with the lentivirus expressing indicated protein for 48 h in the presence of 8 μg/ml polybrene (Sigma). Two days after infection, puromycin was added to the media at 2.5 μg/ml, and cells were selected for 1 week. Selected 50,000 live NIH-3T3 cells, 100,000 live MEFs, or 100,000 live MB352 (p53 null MEFs) cells were mixed with 0.35% top-agar and were plated onto 0.6% base-agar in six-well plates. 25 days (NIH-3T3) or 30 days (MEFS or MB352) after plating the cells into soft agar, colony numbers were counted. The colony numbers were counted by openCFU.
In situ Proximity ligation assay (PLA)
HeLa Cells were incubated with primary antibodies (rabbit α-METTL3 antibody and mouse α-eIF3h antibody) in blocking solution at 4°C for 2 h. Cells were then washed for five times for 5min in PBS plus 0.1% Tween 20. Then, cells were incubated with secondary proximity probes (α-Rabbit-PLUS and α-Mouse-MINUS) (Sigma, DUO92101) for 90 min at 37 °C. Cells were washed five times for 5 min in 10 mM Tris-HCl (pH 7.5) plus 0.1% Tween 20 at 37 °C, then twice for 5 min in PBS plus 0.1% Tween 20. All subsequent steps were performed according to the manufacturer’s instruction. Cells were observed with a Zeiss LSM 710 Multiphoton Laser Scanning Confocal.
Immunohistochemistry (IHC) staining
The human lung cancer tumor array was purchased from Biomax (HLug-Ade150CS-01). Slide was baked for 60 minutes in an oven set to 60°C and then loaded into the Bond III staining platform with appropriate labels. Antigen was retrieved by Bond Epitope Retrieval 2 for 20 minutes. Then the slide was incubated with METLL3 antibody (Abcam, ab195352) at 1:500 for 30 minutes at RT. Primary antibody was detected using Bond Polymer Refine Detection kit. Slides were developed in DAB then dehydrated and coverslipped. Each sample was score by the percentage of positive stained cells (percentage score: 1-5) and the staining intensity (intensity score: 1-5). Then the sample staining score was calculated by multiplying the percentage score and the intensity score.
In vivo tumor xenograft
All research involving animals was complied with protocols approved by the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee. 4-6 weeks old female NU/J (Nude) immunodeficient mice (Jackson Laboratory #002019) were used for subcutaneous injections. Randomly divided 5 mice (A549 cells) or 8 mice (NIH-3T3 cells) were used for each group. 100,000 A549 cells or 1,500,000 NIH-3T3 cells in serum-free medium and growth factor reduced Matrigel (Corning #354230) (1:1) were inoculated into the flank of nude mice. The xenograft tumor formation was monitored by calipers twice a week. The recipient mice were monitored and euthanized when the tumors reached 1 cm in diameter. The tumor volume was calculated by use of a formula 1/2 (length X width2). The investigator was blinded to group allocation.
m6A MeRIP-Seq and Data Analysis
Performed as previously described1. All the studies involving human patient samples were complied with protocols approved by Institutional Review Board (IRB). Informed consent was obtained from all participants. Portions of fresh tumor tissue approximately 0.5 × 0.5 × 0.5cm were snap frozen and preserved at −80C, pulverized in liquid nitrogen and stabilized in Trizol for total RNA isolation. Then the mRNA purification from total RNA was performed using PolyATtract mRNA Isolation Systems (Promega). 2μg of the purified mRNA was fragmented and immunoprecipitated with α-m6A antibody (Synaptic Systems, 202003). The purified RNA fragments from m6A MeRIP were used for library construction using the TruSeq Stranded mRNA Sample Prep Kits (Illumina RS-122-2101) and sequenced with Illumina NextSeq 500. Reads mapping, peak calling, metagene analysis and motif search were performed as previously1. To identify the alternative splicing events, all the clean RNA-Seq reads of control and METTL3 knockdown samples were firstly trimmed to same length with 72 bp, which were then aligned against the human hg19 (GRCh37) reference genome using Tophat224. rMATS v3.2.525 was used to detect the splicing events and significant splicing differences between METTL3 knockdown and control samples. To analyze the global profiling of mRNA lifetime, the clean reads were aligned to human reference genome (hg19) using Tophat224 after trimming the adapters and filtering low quality sequences from the raw data. The reads mapped to each gene were counted using HTSeq26 based on the GENCODE gene model (v19)27. The raw counts were then normalized as Reads per kilobase per million mapped reads (RPKM). ERCC RNA Spike-In Control Mixes (Ambion) were added into each sample before constructing the library to normalize the reads. mRNA lifetime was calculated according to the method in the previous study8. To analyze METTL3 or eIF3h expression level among TCGA tumors, RNA-Seq data for 33 TCGA tumor types were downloaded from Genomic Data Commons Data Portal (GDC) of TCGA (http://cancergenome.nih.gov/) using R package TCGAbiolinks28 The expression matrix was then constructed by merging the TPM (Transcripts Per Million) values of all downloaded RNA-seq samples. The tumor types without corresponding normal tissue samples were excluded and the retained 24 tumor types with normal tissues were used to draw the boxplot of gene expression. p-values are calculated by Wilcoxon rank-sum test, with asterisks indicating statistical significance.
Statistics and Reproducibility
Data are presented as the mean ± SEM or mean ± SD. Statistical significance was determined by a Student’s two-tailed t-test for qRT-PCRs and Luciferase assays. Where it is applicable, Shapiro-Wilk test and Q-Q plotting (quantile-quantile plot,) were performed prior to Student’s two-tailed t-test to assess if the data plausibly came from a normal distribution. Where indicated, Shapiro-Wilk signed-rank test was used. P<0.05 was considered statistically significant. Sample size for Extended Data Fig. 10a-b; n = 408, n = 1,095, n = 304, n = 36, n = 285, n = 184, n = 155, n = 520, n = 66, n = 533, n = 290, n = 371, n = 515, n = 501, n = 178, n = 179, n = 497, n = 94, n =259, n = 103, n = 415, n = 505, n = 120 and n = 176, from the order of left to right for TP; n = 19, n = 133, n = 3, n = 9, n = 41, n = 11, n = 5, n = 44, n = 25, n = 72, n = 32, n = 50, n = 59, n = 51, n = 4, n = 3, n = 52, n = 10, n = 2, n =1, n = 35, n = 59, n = 2 and n = 24, from the order of left to right for NT. Sample size for Extended Data Fig. 10c; n = 285, n = 184, n = 155, n = 371, n = 515, n = 501, n = 497 and n = 94, from the order of left to right.
Extended Data
Extended Data Fig. 1 |. METTL3 binding close to the stop codon enhances translation.
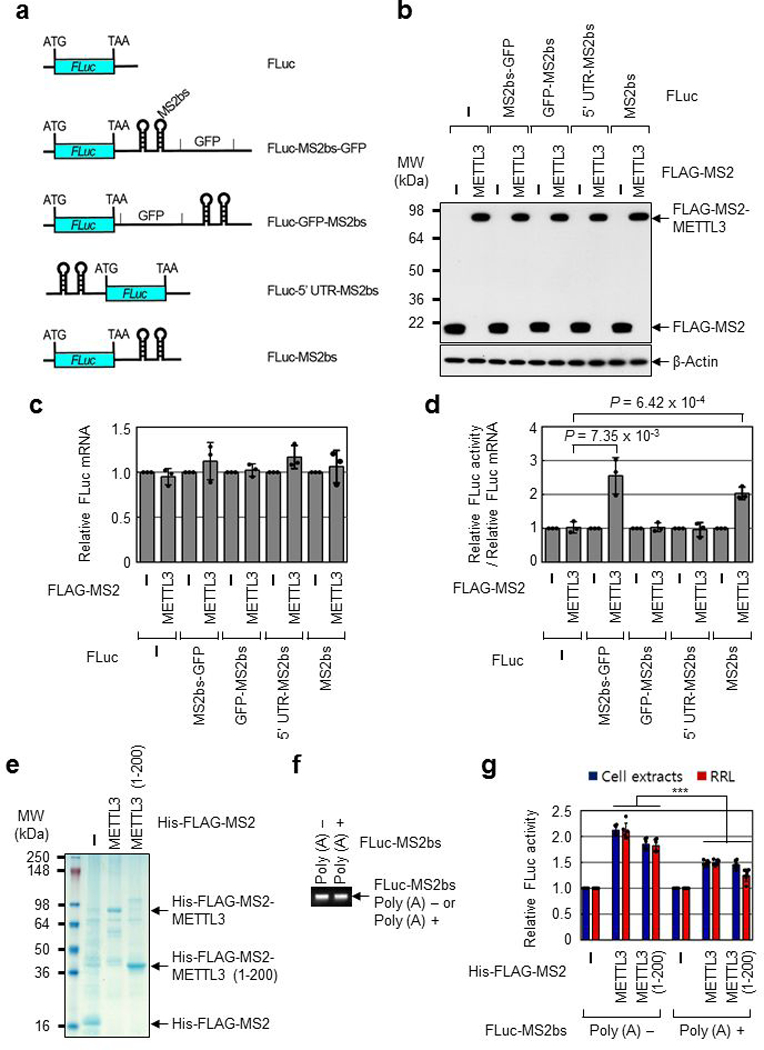
a, Schematic diagram of reporter plasmids containing Firefly luciferase cDNA and different positions of MS2 binding sites. b, Western blotting with indicated antibodies. Two independently performed experiments show similar results. c, qRT-PCR analysis of reporter mRNAs. Each tested reporter mRNAs were normalized to RLuc mRNAs. The FLuc:RLuc ratio for each construct with FLAG-MS2 expression was set to 1. Error bars represent mean ± SD; n = 3 biologically independent samples. d, Tethering assay to measure translation efficiency as described in (Fig. 1h). Error bars represent mean ± SD; n = 3 biologically independent samples; two-sided t-test. e, Colloidal Coomassie blue staining of recombinant protein His-FLAG-MS2, His-FLAG-MS2-METTL3, or His-FLAG-MS2-METTL3 (1-200). Two independently performed experiments show similar results. f, Ethidium bromide-stained agarose gel electrophoresis of the indicated in vitro transcribed reporter mRNAs; FLuc-MS2bs without poly (A) tail (Poly (A) -) or FLuc-MS2bs with 30nt poly (A) tail (Poly (A) +). Two independently performed experiments show similar results. g, In vitro translation of reporter mRNAs using either H1299 cell extracts or Rabbit reticulocyte lysate (RRL). The levels of in vitro-translated FLuc protein were analyzed using luciferase assays. Value of FLuc activity in the presence of His-FLAG-MS2 recombinant protein was set to 1.0. Error bars represent mean ± SD; n = 6 independent experiments. Two-sided t-test, *** denotes multiple comparison for the p-values showing P < 0.001.
Extended Data Fig. 2 |. N-terminal region of METTL3 promotes translation.
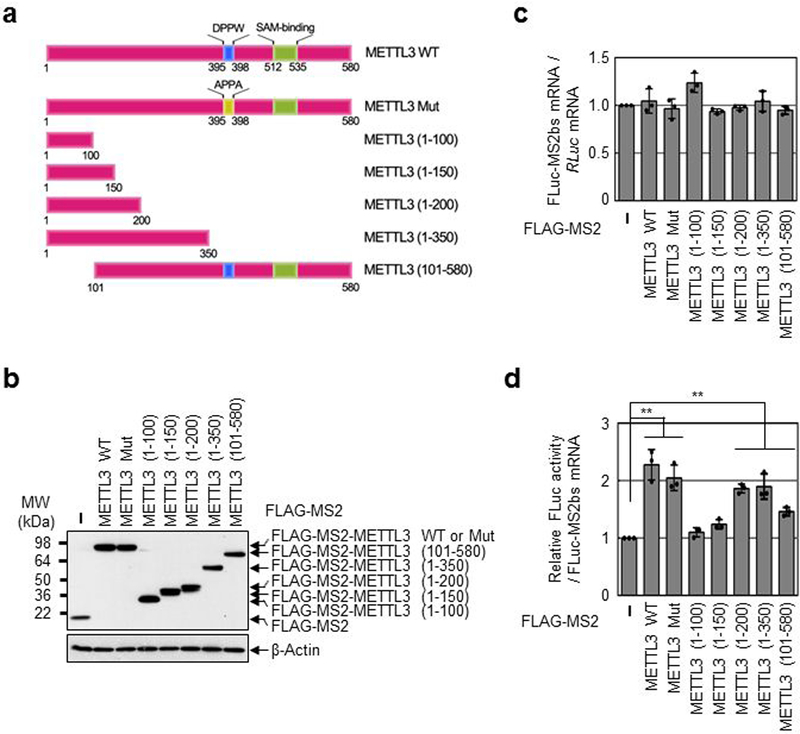
a, Schematic diagram of METTL3 deletion mutants or mutation in METTL3 catalytic domain. b, Western blotting with indicated antibodies. Two independently performed experiments show similar results. c, qRT-PCR analysis of reporter mRNAs. FLuc-MS2bs mRNA levels were normalized to RLuc mRNAs. The FLuc:RLuc ratio obtained in FLAG-MS2 (control) was set to 1. Error bars represent mean ± SD; n = 3 biologically independent samples. d, Tethering assay to measure translation efficiency of reporter mRNAs as described in (Fig. 1 h). Error bars represent mean ± SD; n = 3 biologically independent samples. Two-sided t-test, ** denotes multiple comparison for the p-values showing P < 0.01.
Extended Data Fig. 3 |. METTL3 associates with translation initiation factors.
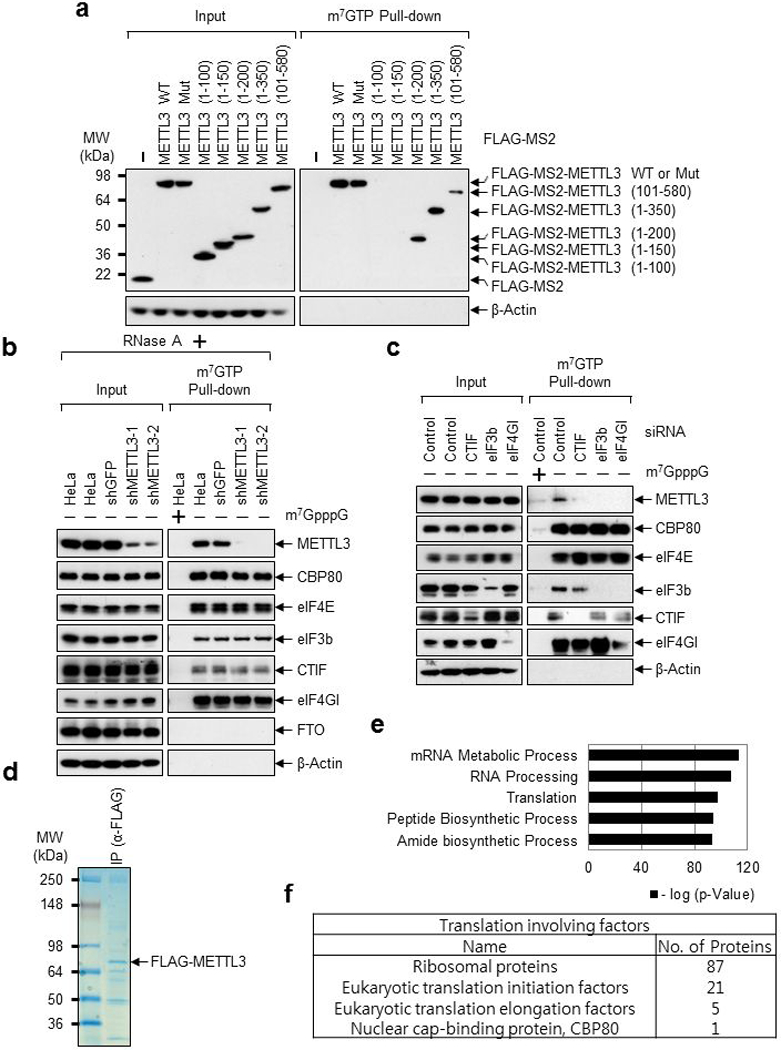
a, Deletion mutants of METTL3 were expressed in HeLa cell. The total-cell extracts (Input) and the cap-associated protein samples were analyzed by Western blotting using the indicated antibodies. Two independently performed experiments show similar results. b, Cap-association assay with METTL3 depletion. The total-cell extracts (Input) and the cap-bound protein samples were analyzed by Western blotting using the indicated antibodies. m7GpppG cap analogue was used for antagonizing cap-associating proteins binding to m7GTP-Agarose. Two independently performed experiments show similar results. c, Same as (b) except HeLa cells were transfected with CTIF, eIF3b or eIF4GI siRNA. Two independently performed experiments show similar results. d-f, Mass spectrometry of FLAG-METTL3 interacting proteins. d, Proteins that were co-immunopurified with FLAG-METTL3 subjected to 4-12% Tris-Glycine SDS-PAGE. Colloidal Coomassie blue staining was performed. n=1 independent experiment. e, Gene ontology analysis of the identified proteins from Mass spectrometry. n=1 independent experiment. Hypergeometric distribution (one-tail) with Bonferroni adjustment was used to determine enrichment statistical significance. f, Table showing the translation involving factors identified from Mass spectrometry.
Extended Data Fig. 4 |. N-terminal region of METTL3 directly interacts with MPN domain of eIF3h.
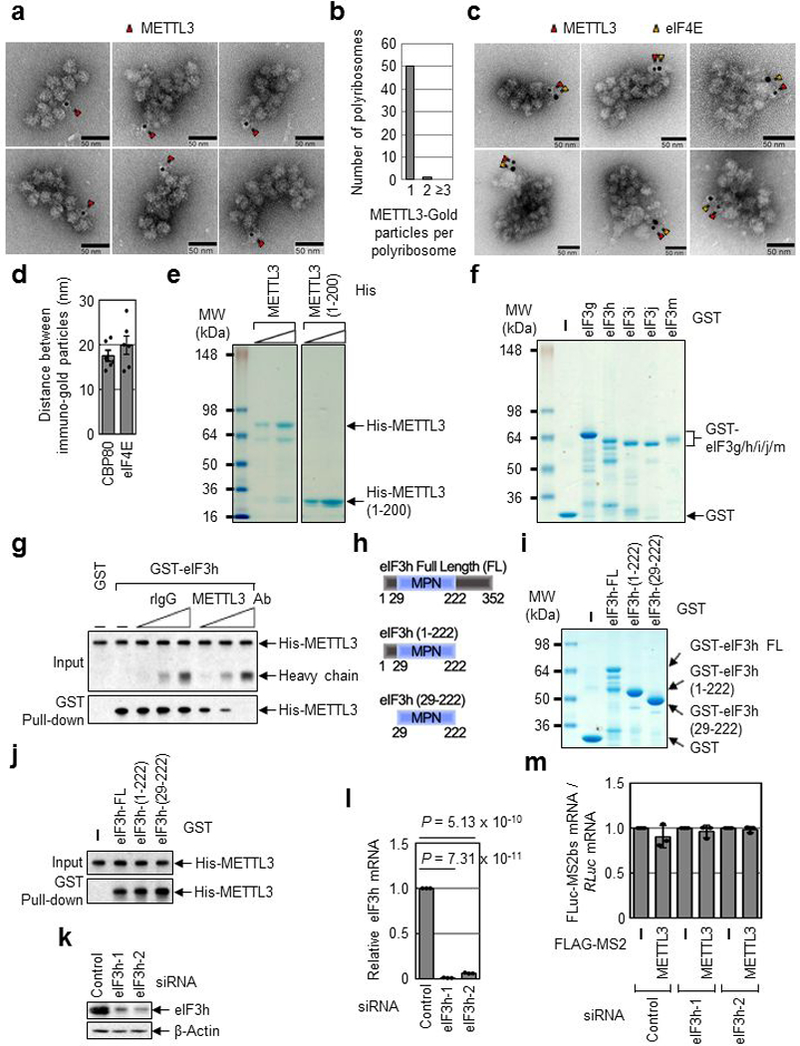
a, EM images of polyribosome with METTL3-gold particle labeling. Red arrows indicate METTL3 with immuno-gold particle (6 nm). Three independently performed experiments show similar results. b, Counting of METTL3 with gold particle labeling in each polyribosome. c, EM images of polyribosome with METTL3 and eIF4E. Red arrows indicate METTL3 with immuno-gold particle (6 nm) and yellow arrows indicate eIF4E with immuno-gold particle (10 nm). Four independently performed experiments show similar results. d, Average distance between immuno-gold particles was measured. n = 6 biologically independent samples from at least three independent experiments. Error bars represent mean ± SD. e, Colloidal Coomassie blue staining of recombinant protein His-METTL3 or His-METTL3 1-200 amino acid fragments (1-200). Two independently performed experiments show similar results. f, Colloidal Coomassie blue staining of recombinant GST-tagged protein eIF3g, eIF3h, eIF3i, eIF3j or eIF3m. Two independently performed experiments show similar results. g, GST-eIF3h was co-purified with His-METTL3 in the presence of either rabbit IgG (rIgG) or α-METTL3 antibody. Levels of co-purified His-METTL3 were analyzed by Western blotting. Two independently performed experiments show similar results. h, Schematic diagram of human eIF3h deletion mutants. i, Colloidal Coomassie blue staining of recombinant GST-eIF3h, -eIF3h (1-222) or -eIF3h (29-222). n = 1 independent experiments. j, GST pull-down of indicated eIF3h deletion mutants. Co-purified His-METTL3 was analyzed by Western blotting. n = 1 independent experiments. k, Western blotting demonstrates efficient knockdown of eIF3h protein. Three independently performed experiments show similar results. l, qRT-PCR analysis demonstrates efficient down regulation of eIF3h mRNA. Error bars represent mean ± SD; n = 3 biologically independent samples; two-sided t-test. m, qRT-PCR analysis of reporter mRNAs. FLuc-MS2bs reporter mRNAs were normalized to RLuc mRNAs. The FLuc:RLuc ratio obtained in FLAG-MS2 was set to 1. Error bars represent mean ± SD; n = 3 biologically independent samples.
Extended Data Fig. 5 |. METTL3 has no significant effect on mRNA stability.
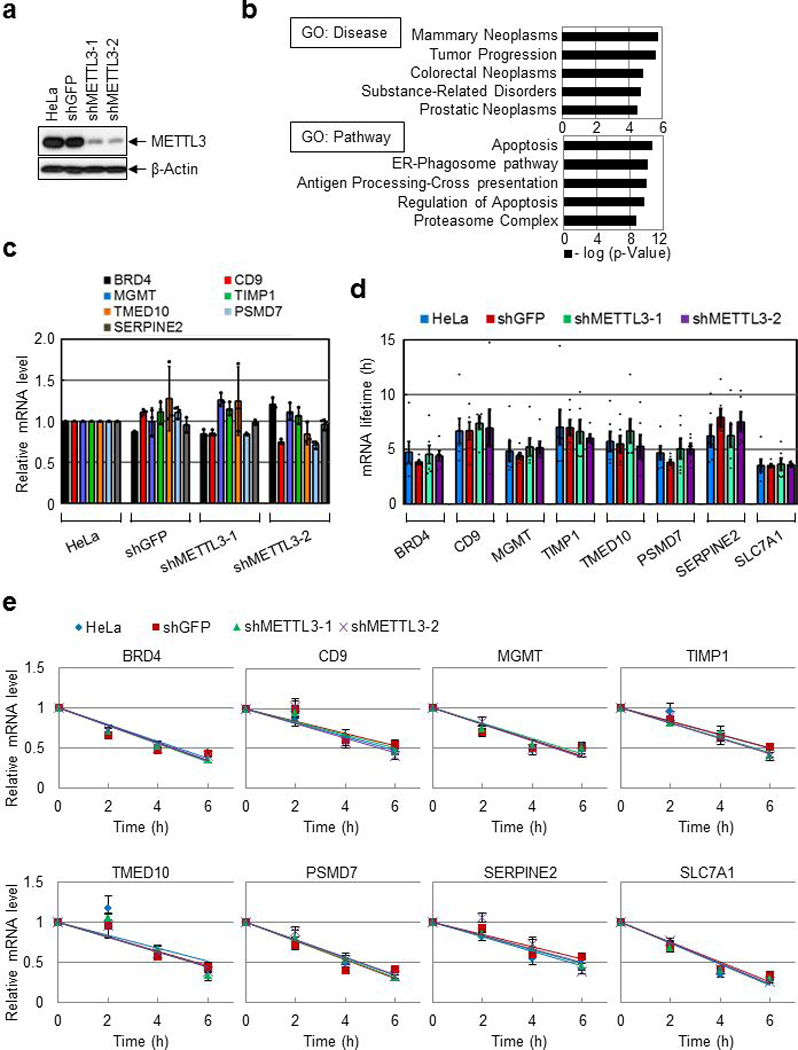
a, Western blotting with indicated antibodies. Three independently performed experiments show similar results. b, Gene ontology analysis of the overlapping mRNAs (n=809) in (Fig. 2d). Hypergeometric distribution (one-tail) with Bonferroni adjustment was used to determine enrichment statistical significance. c, qRT-PCR analysis using indicated primers. Error bars represent mean ± SD; n = 3 technical replicates. d-e, Half-life of endogenous mRNAs was analyzed by qRT-PCR using indicated primers. Error bars represent mean ± SEM; n = 6 independent experiments. For (d), two-sided t-test; multiple comparison for the p-values showed that there were no significant differences between the samples for all the tested mRNAs, P > 0.05.
Extended Data Fig. 6 |. Widespread role of METTL3 in oncogene translation.
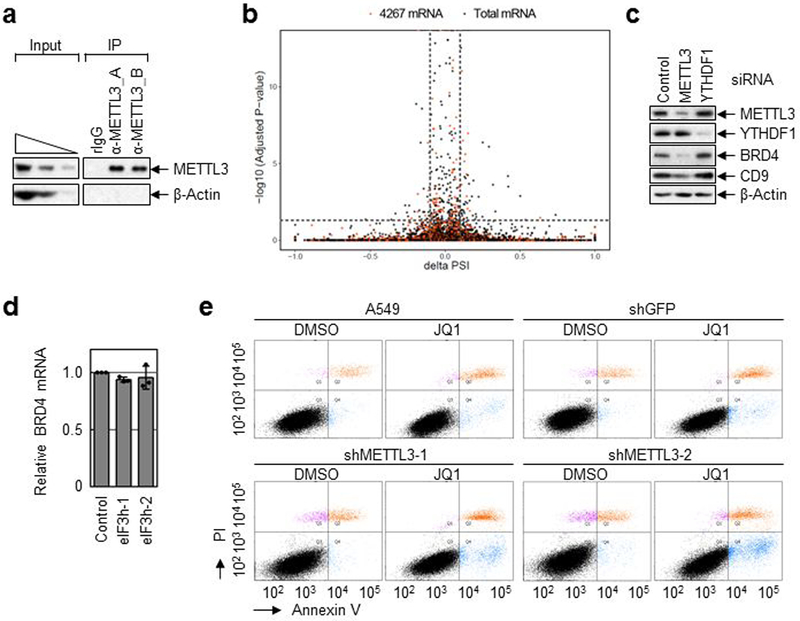
a, IP of endogenous METTL3 and Western blotting analysis using indicated antibodies. Two independently performed experiments show similar results. b, Density plot reflects the distribution of changes in percent spliced In (ΔPSI) values and according p-values for alternative splicing events detected by rMATs v3.2.5 (rMATs is developed based on a hierarchical framework and likelihood-ratio test was used to detect differential splicing). Splicing events at a FDR < 5% and deltaPSI > 0.1 are considered as significant. Black dots indicate total mRNAs. Red dots (4,276 mRNAs) indicate more than 2-fold less translating mRNAs in METTL3 depleted cells. c, Western blot using indicated antibodies in control-, METTL3- or YTHDF1-knockdwon cells. Two independently performed experiments show similar results. d, qRT-PCR analysis of endogenous BRD4 mRNAs. Error bars represent mean ± SEM; n = 3 biologically independent samples. e, Annexin V/PI staining of METTL3 knockdown and control A549 cells upon JQ1 treatment that was analyzed by FACS. n = 3 independent experiments.
Extended Data Fig. 7 |. Identification of a conserved Alanine residue in the N-terminal region of METTL3 required for its interaction with eIF3h.
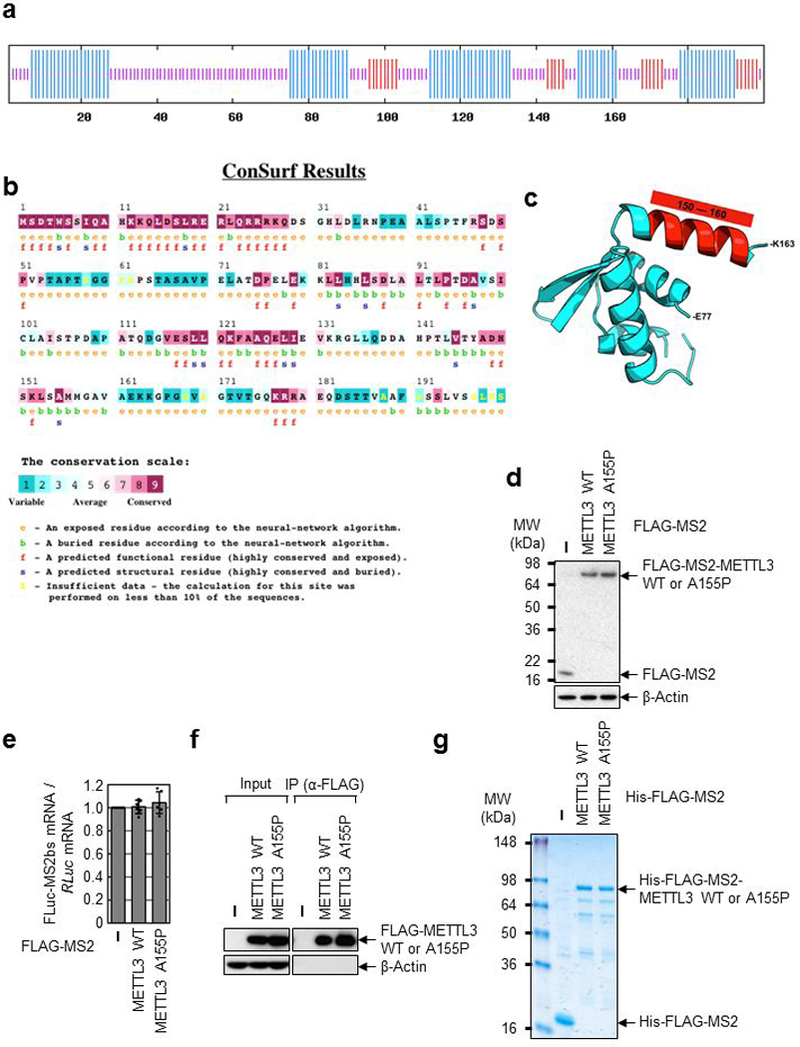
a, Secondary structure prediction of the N-terminal (1-200) region of METTL3 protein showing putative alpha helices (blue lines). b, Evolutionary conservation of the N-terminal (1-200) region METTL3 protein. c, Computational modeling of the 3D structure of the N-terminal (77-163) region METTL3 protein, based on the coordinates of PDB: 3HHH. d, Western blotting analysis using indicated antibodies. Two independently performed experiments show similar results. e, qRT-PCR analysis of reporter mRNAs. FLuc-MS2bs mRNA levels were normalized to RLuc mRNAs. The FLuc:RLuc ratio obtained in FLAG-MS2 (control) was set to 1. Error bars represent mean ± SD; n = 6 independent experiments. f, IP of FLAG-METTL3 WT or A155P and Western blotting analysis using indicated antibodies. Two independently performed experiments show similar results. g, Staining of recombinant protein His-FLAG-MS2-METTL3 WT or His-FLAG-MS2-METTL3 A155P. Two independently performed experiments show similar results.
Extended Data Fig. 8 |. METTL3 expression correlates with lung tumor stage and promotes tumorigenicity.
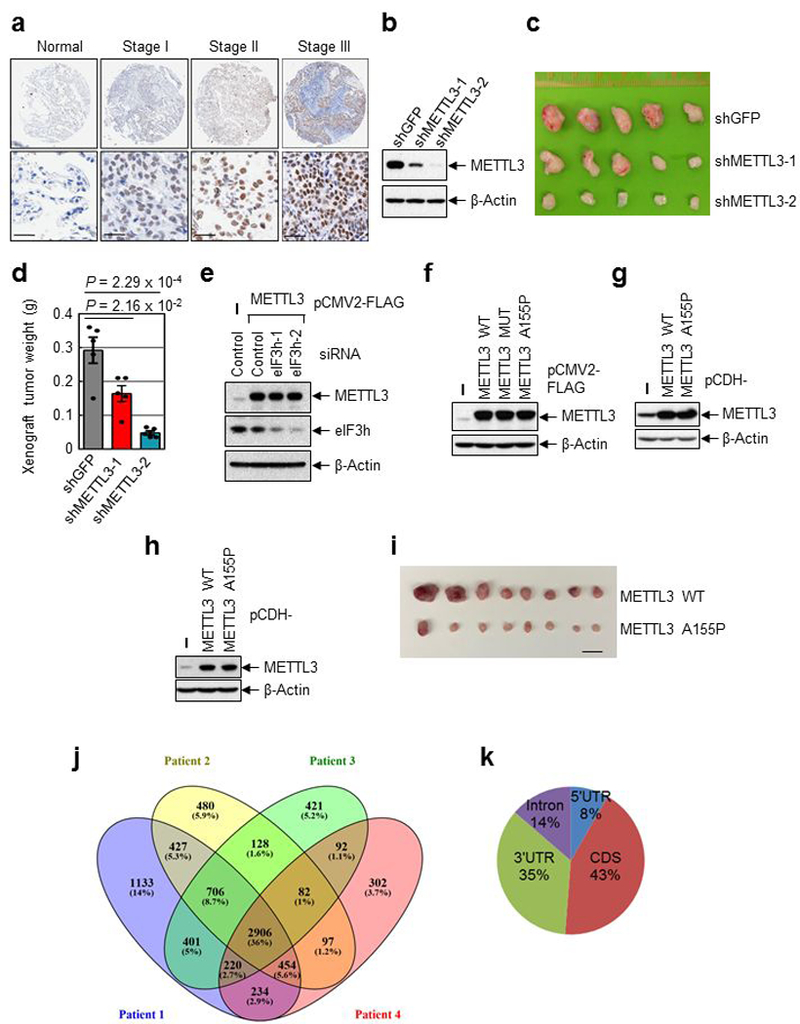
a, Representative staining image of control and different stages lung cancer samples (n=75). Lower panels show the enlarged sections of the upper ones. Scale bar=30μM. b, Western blotting analysis using indicated antibodies. n = 1 independent experiments. c-d, Tumor images (c) and plot of tumor weight (d) at the endpoint in the xenograft experiment. Error bars represent mean ± SEM; n = 5 independent mice; two-sided t-test. e-h, Western blotting analysis using indicated antibodies. For (e), two independently performed experiments show similar results. For (f-h), n = 1 independent experiments. i, Tumor images at the endpoint in the xenograft experiment. Scale bar, 20mm. j, Overlapping of m6A containing genes identified in four lung cancer patient samples. k, Distribution of m6A sites.
Extended Data Fig. 9 |. Polysome conformation is affected by METTL3 and m6A modification in primary human lung tumors.
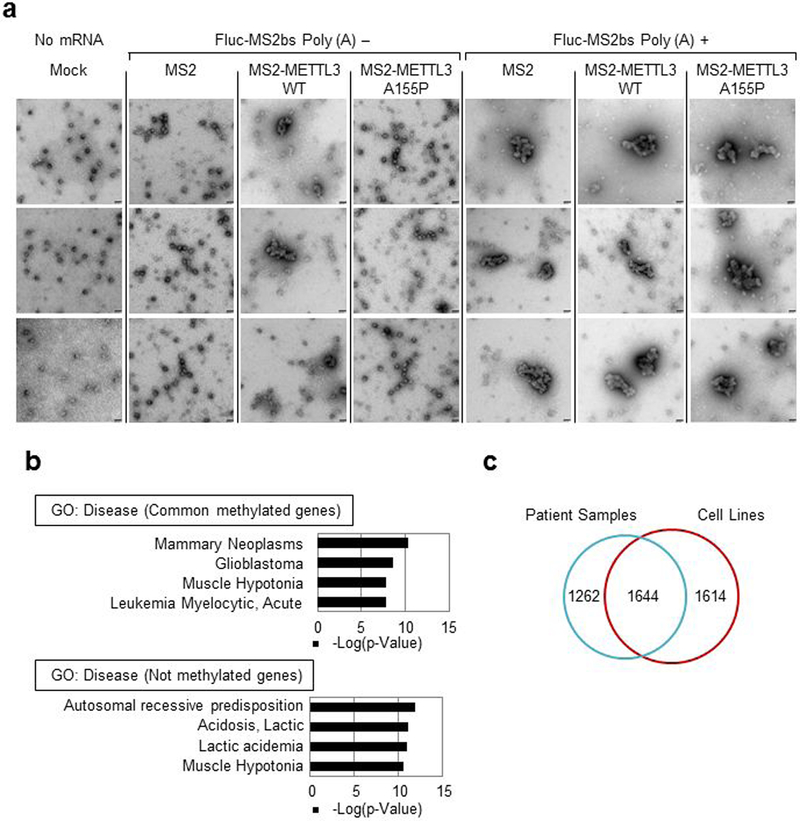
a, EM images of polyribosomes. Images were taken from the samples in (Fig. 3e). Scale bar, 50 nm. Six independently performed experiments show similar results. b, Gene ontology analysis. Common methylated genes refers to the methylated genes in all four patient samples. Not methylated genes refers to the genes not methylated in any of the four patient samples. Hypergeometric distribution (one-tail) with Bonferroni adjustment was used to determine enrichment statistical significance. c, Venn Diagram showing m6A peak overlap between patient tumor samples and cells (H1299 and A549).
Extended Data Fig. 10 |. Expression of METTL3 and eIF3h is positively correlated in many tumor types.
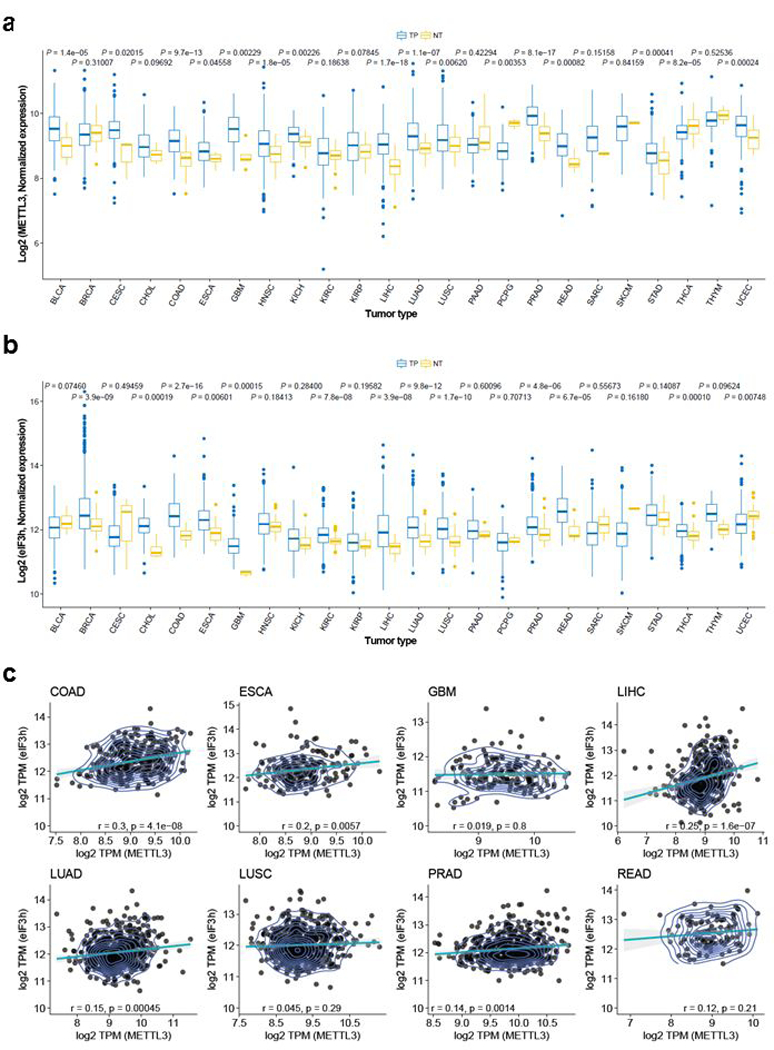
a, METTL3 gene expression among TCGA tumors. Box plots display the full range of variation based on the five number summaries (minimum, first quartile, median, third quartile, and maximum). TP= primary solid tumor, NT= solid tissue normal. Two-sided Wilcoxon signed-rank test was used for statistical significance. b, eIF3h gene expression among TCGA tumors. Box plots display the full range of variation based on the five number summaries (minimum, first quartile, median, third quartile, and maximum). TP= primary solid tumor, NT= solid tissue normal. Two-sided Wilcoxon signed-rank test was used for statistical significance. c, Plot illustrating the Pearson’s correlations of expression level between METTL3 and eIF3h in eight TCGA tumors, in which both METTL3 and eIF3h are significantly changed compared with normal tissues.
Supplementary Material
Acknowledgements
Thanks to Dr. Yoon Ki Kim for CBP80 and CTIF antibodies. SL was supported by a Damon Runyon-Sohn Pediatric Fellowship (DRSG-7–13) and a grant from Alex’s Lemonade Stand Foundation. R.I.G was supported by grants from the US National Institute of General Medical Sciences (NIGMS) (R01GM086386) and National Cancer Institute (NCI) (R01CA211328).
Footnotes
Code availability
Script and code used for data analysis can be found at [https://github.com/rnabioinfor/rnamethy].
Data availability
The m6A MeRIP-Seq and RNA-seq data have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE117299. All other data are available from the authors on request.
References
- 1.Lin S, Choe J, Du P, Triboulet R & Gregory RI The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol Cell 62, 335–345, doi: 10.1016/j.molcel.2016.03.021 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barbieri I et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 552, 126–131, doi: 10.1038/nature24678 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vu LP et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med 23, 1369–1376, doi: 10.1038/nm.4416 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen M et al. RNA N6-methyladenosine methyltransferase METTL3 promotes liver cancer progression through YTHDF2 dependent post-transcriptional silencing of SOCS2. Hepatology, doi: 10.1002/hep.29683 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Meyer KD & Jaffrey SR The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol 15, 313–326, doi: 10.1038/nrm3785 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X et al. N-methyladenosine-dependent regulation of messenger RNA stability. Nature, doi:nature12730[pii]10.1038/nature12730 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X et al. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 161, 1388–1399, doi: 10.1016/j.cell.2015.05.014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu C et al. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol 10, 927–929, doi: 10.1038/nchembio.1654 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Imataka H, Gradi A & Sonenberg N A newly identified N-terminal amino acid sequence of human eIF4G binds poly(A)-binding protein and functions in poly(A)-dependent translation. Embo J 17, 7480–7489, doi: 10.1093/emboj/17.24.7480 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wells SE, Hillner PE, Vale RD & Sachs AB Circularization of mRNA by eukaryotic translation initiation factors. Molecular cell 2, 135–140 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Borman AM, Michel YM, Malnou CE & Kean KM Free poly(A) stimulates capped mRNA translation in vitro through the eIF4G-poly(A)-binding protein interaction. J Biol Chem 277, 36818–36824, doi: 10.1074/jbc.M205065200 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Tarun SZ Jr. & Sachs AB Association of the yeast poly(A) tail binding protein with translation initiation factor eIF-4G. The EMBO journal 15, 7168–7177 (1996). [PMC free article] [PubMed] [Google Scholar]
- 13.des Georges A et al. Structure of mammalian eIF3 in the context of the 43S preinitiation complex. Nature 525, 491–495, doi: 10.1038/nature14891 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu J et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 10, 93–95, doi: 10.1038/nchembio.1432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ashkenazy H et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44, W344–350, doi: 10.1093/nar/gkw408 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kouza M, Faraggi E, Kolinski A & Kloczkowski A The GOR Method of Protein Secondary Structure Prediction and Its Application as a Protein Aggregation Prediction Tool. Methods Mol Biol 1484, 7–24, doi: 10.1007/978-1-4939-6406-2_2 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Kelley LA, Mezulis S, Yates CM, Wass MN & Sternberg MJ The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10, 845–858, doi: 10.1038/nprot.2015.053 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee AS, Kranzusch PJ & Cate JH eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature 522, 111–114, doi: 10.1038/nature14267 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee AS, Kranzusch PJ, Doudna JA & Cate JH eIF3d is an mRNA cap-binding protein that is required for specialized translation initiation. Nature 536, 96–99, doi: 10.1038/nature18954 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hershey JW The role of eIF3 and its individual subunits in cancer. Biochim Biophys Acta 1849, 792–800, doi: 10.1016/j.bbagrm.2014.10.005 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Zhang L, Pan X & Hershey JW Individual overexpression of five subunits of human translation initiation factor eIF3 promotes malignant transformation of immortal fibroblast cells. J Biol Chem 282, 5790–5800, doi: 10.1074/jbc.M606284200 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Mahmood SF et al. A siRNA screen identifies RAD21, EIF3H, CHRAC1 and TANC2 as driver genes within the 8q23, 8q24.3 and 17q23 amplicons in breast cancer with effects on cell growth, survival and transformation. Carcinogenesis 35, 670–682, doi: 10.1093/carcin/bgt351 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Zhang L, Smit-McBride Z, Pan X, Rheinhardt J & Hershey JW An oncogenic role for the phosphorylated h-subunit of human translation initiation factor eIF3. J Biol Chem 283, 24047–24060, doi: 10.1074/jbc.M800956200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim D et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36, doi: 10.1186/gb-2013-14-4-r36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen S et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci U S A 111, E5593–5601, doi: 10.1073/pnas.1419161111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harrow J et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22, 1760–1774, doi: 10.1101/gr.135350.111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anders S, Pyl PT & Huber W HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169, doi: 10.1093/bioinformatics/btu638 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colaprico A et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res 44, e71, doi: 10.1093/nar/gkv1507 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.