

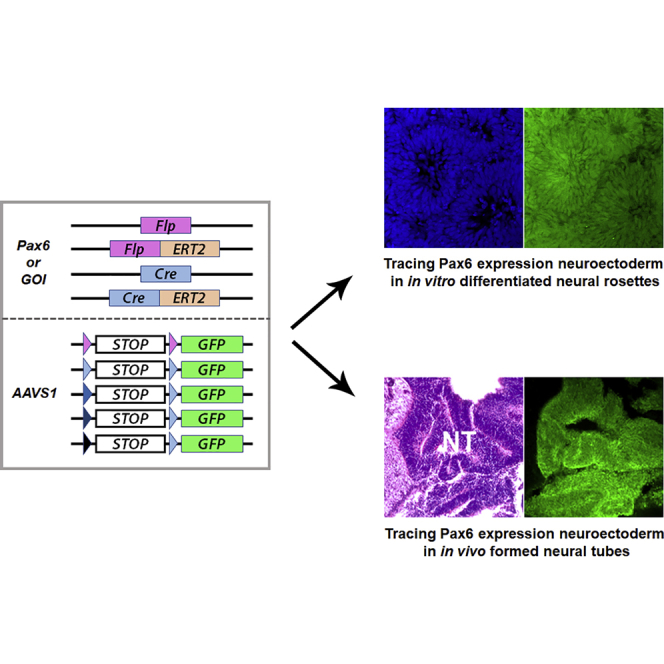
Summary
It is highly desirable to specify human developmental principles in an appropriate human model with advanced genetic tools. However, genetically engineering human cells with lineage-tracing systems has not been achieved. Here we introduce strategies to construct lineage-tracing systems in human embryonic stem cells (hESCs). The AAVS1 locus was suitable for the integration of the conditional reporter. The Cre-LoxP and Flp-FRT systems were highly sensitive, which may cause inaccurate lineage labeling in human cells. The recombination sensitivity and tracing fidelity could be finely tuned by modification of the LoxP recombination site. Moreover, tamoxifen-controllable CreERT2-LoxP and FlpERT2-FRT systems showed compelling advantages in tightly tracing human lineages temporally. In proof-of-principle experiments, we traced human PAX6+ neuroectoderm cells and revealed their full neural lineage differentiation potency both in vitro and in vivo. Devising and optimizing of lineage-tracing systems in hESCs will thus set up a solid foundation for human developmental studies.
Keywords: human embryonic stem cells, lineage-tracing, genetic engineering, development, PAX6, FOXA2
Graphical Abstract
Highlights
-
•
Two-step strategy for constructing lineage-tracing systems in human PSCs
-
•
Tracing fidelity could be shaped via modifying the LoxP sequences
-
•
Temporal tracing could be achieved by introducing inducible recombinases
-
•
Tracing PAX6-expressing neuroectoderm identifies its full neural lineage potency
In this article, Zhang and colleagues successfully constructed lineage-tracing systems in human pluripotent stem cells. Multiple strategies were designed in order to shaping the fidelity of the lineage-tracing system for studying human development both in vitro and in vivo. Tracing PAX6-expressing neuroectoderm identifies its full neural lineage potency in generating all spectrum of regional neural progenitors.
Introduction
Genetic recombination-based lineage-tracing is a method that labels a specific type of progenitor cells and all their future progeny. Lineage-tracing is therefore a powerful tool for investigating the hierarchy, organization, and location of a clonal of cell lineages in embryonic or postnatal animals (Blanpain and Simons, 2013, Hsu, 2015, Kretzschmar and Watt, 2012). Typically, a conditional reporter driven by a ubiquitously expressed promoter is integrated into an open chromosome region, such as the Rosa26 locus in mouse (Mao et al., 2001, Soriano, 1999). Permanent expression of the reporter in the progenitor cells and all their progeny is achieved after genetic recombination mediated by a recombinase enzyme, which is tightly controlled by a lineage-specific gene. The most commonly used recombination strategies are based on Cre-LoxP and Flp-FRT systems (Harrison and Perrimon, 1993, Hoess et al., 1982). Through the use of sophisticated transgenic technologies and selective breeding between transgenic mouse lines, lineage-tracing studies have now been widely used in studying lineage development in an embryo or stem cell properties in adult mouse tissues (Awatramani et al., 2003, Feil et al., 1997, Livet et al., 2007, Tabansky et al., 2013).
Despite recently accumulated knowledge relating to tissue development, maintenance, and repair under both normal and diseased conditions in model organisms, our direct evidence relating to human lineage development is exceptionally limited (Zhu and Huangfu, 2013). Although we can directly extrapolate conserved developmental principles learned from classical model organisms, it remains a big challenge in human biology to validate and distinguish those conserved and non-conserved developmental mechanisms (Niakan and Eggan, 2013, Pera and Trounson, 2004). To this end, human developmental biology would greatly benefit from a system that can faithfully recapitulate human tissue development and incorporate available genetic tools to verify and discover conserved or unique developmental principles in humans (Keller, 2005).
Human pluripotent stem cells (hPSCs), including human embryonic stem cells (hESCs) (Thomson et al., 1998) and human induced pluripotent stem cells (hiPSCs) (Takahashi et al., 2007, Yu et al., 2007), can be expanded indefinitely and differentiate into all human cell types (Williams et al., 2012). Because of their self-renewing capacity, hPSCs are endowed with an unlimited time window for in vitro genetic engineering. Gene targeting efficiency in hPSCs has now been substantially improved by the introduction of the site-specific double-strand breaks (Smih et al., 1995) within the genome through custom-engineered nucleases (CENs), such as the type II CRISPR/Cas9 system (Cong et al., 2013, Jinek et al., 2012, Mali et al., 2013), the transcription activator-like effector nucleases (TALENs) (Boch et al., 2009, Hockemeyer et al., 2011, Moscou and Bogdanove, 2009, Sanjana et al., 2012), and the zinc-finger nucleases (Hockemeyer et al., 2009, Kim et al., 1996, Urnov et al., 2005). To date, single- and multi-step gene targeting designs for hPSCs with the purpose of controlled gene ablation or expression, gene editing, or genetic reporter labeling have been achieved (Chen et al., 2015, González et al., 2014, Hockemeyer et al., 2009, Hockemeyer et al., 2011, Liu et al., 2016). Meanwhile, hPSCs retain their inherent developmental traits during in vitro differentiation and in vivo teratoma formation assays (Bulic-Jakus et al., 2016, Thomson et al., 1998). Indeed, through mirroring developmental cues, hPSCs have been successfully differentiated into most desired cell types of all three germ layers. Compelling evidence has also started to shed light on molecular mechanisms underpinning human embryonic development through studying the in vitro differentiation of hPSCs (Chi et al., 2016a, Zhang et al., 2010, Zhu and Huangfu, 2013). Here, we describe a two-step strategy in engineering a series of lineage-tracing systems in hPSCs for human developmental studies.
Results
Conditional Tracer Integrated in Human AAVS1 Locus Is Recombinase Controllable
A functional lineage-tracing system composes two necessary elements, a conditional tracer and a recombinase whose expression is temporally and/or spatially controlled in order to mark a specific lineage of interest. To devise a lineage-tracing system in human cells, we constructed a donor vector comprising a puromycin selection module, a CAG promoter-driven GFP cassette segmented by recombinase-controllable LoxP-STOP-LoxP (LSL) regulatory sequences, and 5′ and 3′ homology arms (Figure 1A). The AAVS1 locus in human cells is a relatively open locus epigenetically, supporting robust and sustainable transgene expression (Smith et al., 2008). We selected this region to incorporate the conditional GFP reporter to ensure reliable expression at both pluripotent and differentiated stages (Hockemeyer et al., 2011). The donor vector, and the left and right TALENs, were then co-electroporated into H9 hESCs. After puromycin selection, monoallelically (heterozygotic) and biallelically (homozygotic) recombined colonies were prescreened by genomic DNA PCR and further validated through Southern blot analysis (Figures 1B–1E, and hereafter referred as AAVS1-LSL-GFP lines). To test whether the controllable LSL regulatory sequences could be recombined by Cre recombinase at the AAVS1 locus, we infected these heterozygotic and homozygotic clones with lentiviruses expressing the Ef1α-driven Cre recombinase. As expected, either heterozygotic or homozygotic conditional GFP reporter lines started to express GFP 1 day after viral transfection (Figure 1F), suggesting efficient recombination and functional removal of the STOP cassette.
Figure 1.

Conditional Tracer Integrated in the Human AAVS1 Locus Is Recombinase Controllable
(A) Schematic diagram for constructing of AAVS1-LSL-GFP hPSCs lines through TALEN-mediated gene targeting. Genotyping PCR primer sets are labeled with arrows. Location of restriction enzyme cutting sites for Southern blot analysis are marked with black bars. The 5′ arm and GFP probes for Southern blot analysis are marked with blue bars at indicated genetic regions after homologous recombination (HR).
(B and C) Genomic DNA PCR results identify monoallelic (B) and biallelic (C) HR colonies after genetic engineering.
(D and E) Southern blot analyses of wild-type (WT), monoallelic, and biallelic HR colonies with 5′ arm probe (D) and GFP probe (E) show precise gene targeting.
(F) Bright-field (BF) and fluorescent GFP images of AAVS1-LSL-GFP hPSCs line infected with or without lentivirus expressing Cre. Scale bars, 100 μm.
Tight Control and Efficient Cleavage of PAX6-P2A-Cre or FOXA2-P2A-Cre Fusion Protein
The neuroectoderm (NE) is the earliest neural primordium specified from the epiblast during the gastrulation stage of a developing embryo. Patterning morphogens regionalize the NE into a spectrum of regional neural progenitors (NPs), which subsequently differentiate into all types of neurons and glia. We have previously demonstrated that differential transcriptional programs are used for NE development in humans, as compared with mice (Li et al., 2005, Tao and Zhang, 2016, Zhang et al., 2010). Human NE uniformly expresses PAX6, a transcription factor shown to have expression restricted to the dorsal part of the mouse telencephalon in later stages, and thought to be necessary for the dorsalization of the mouse forebrain. The advanced expression pattern of PAX6 in human NE may account for our large and sophisticated cerebral cortex, based on human dorsoventral patterning following a repression release model (Chi et al., 2016a). Ectopic expression of PAX6 represses ventral NP specification while ensuring cortical commitment in human even in the presence of the ventralization morphogen, sonic hedgehog (SHH) (Chi et al., 2016a). To obtain direct evidence that PAX6+ NE is the only source generating the various regional NPs, and not just dorsal forebrain progenitors as indicated in mouse studies, we selected PAX6 as a lineage-specific marker for tracing. FOXA2 is another early transcription factor expressed in floor plate (FP) and notochord, which communicates closely with various neural tissues and has an essential impact on neural fate induction as well as regional patterning (Placzek, 1995, Sasaki and Hogan, 1993). FOXA2 was thus also selected for our tracing study.
Short guide RNAs (gRNAs) recognizing genome regions neighboring the stop codons (TAA) of FOXA2 and PAX6 were designed (Figure S1). Targeting efficiencies of the gRNAs were evaluated and confirmed in HEK293 cells after transient transfection of the gRNAs and the Cas9 expression vector (Figure S1). Recombination donor plasmids comprising the P2A-Cre cassette, and the 5′ and 3′ recombination arms, were designed (Figure 2A). After homologous recombination (HR) of the P2A-Cre cassette in close juxtaposition in front of the stop codon, either the FOXA2-P2A-Cre or PAX6-P2A-Cre transcript was anticipated to be transcribed under the tight control of individual promoters. The FOXA2-P2A-Cre or PAX6-P2A-Cre fusion proteins were also expected to be generated and cleaved by endogenous proteases at the P2A recognition site (Figure 2A).
Figure 2.

Tight Control and Efficient Cleavage of PAX6-P2A-Cre or FOXA2-P2A-Cre Fusion Proteins
(A) Schematic diagram for constructing PAX6-P2A-Cre and FOXA2-P2A-Cre hPSCs lines through a gRNA-guided CRISPR/Cas9 system. Genotyping PCR primer sets are labeled with arrows.
(B) Genomic DNA PCR results identify five monoallelic HR colonies after genetic engineering.
(C) Immunolabeling of FOXA2 and Cre in FOXA2-P2A-Cre lines differentiated toward a cortical NP or FP fate for 6 or 12 days. Nuclei are counter stained with Hoechst. Scale bar, 100 μm.
(D) Western blot analysis of FOXA2-P2A-Cre lines differentiated toward a cortical NP or FP fate for 6, 12, or 20 days.
(E) Genomic DNA PCR results identify two monoallelically recombined PAX6-P2A-Cre colonies after genetic engineering.
(F) Immunolabeling of PAX6 and Cre in PAX6-P2A-Cre lines at hESC stage, day 10 NE, day 17 cortical NPs, or day 17 ventral NPs. Nuclei are counter stained with Hoechst. Scale bar, 100 μm.
(G) Western blot analysis of PAX6-P2A-Cre lines along with neural differentiation.
See also Figure S1.
Cas9 and gRNA expression vectors, donor and transient puromycin expression plasmids, were together electroporated into hESCs. After puromycin selection for 3–4 days, individual cell colonies were formed within the following 6–7 days. Genomic DNA PCR analyses were performed to confirm both the wild-type (WT) allele and HR allele's integration state (Figures 2B and 2E). The correctly recombined FOXA2-P2A-Cre lines were differentiated into adherent monolayer cultures of dorsal cortical NPs and FP cells in the absence or presence of SHH, respectively (Chi et al., 2016b, Fasano et al., 2010). FOXA2 and Cre were completely absent in early and late cortical (forebrain dorsal) NPs at both 6 and 12 days post differentiation (Figure 2C). With SHH patterning, both FOXA2 and Cre were now uniformly expressed in day 6 and day 12 FP progenitors (Figure 2C). Similar results were observed through western blot analysis, and substantial amounts of FOXA2-P2A-Cre fusion proteins were correctly cleaved (Figure 2D). There results confirm the efficiency of FP differentiation and the reliability of Cre in mirroring endogenous FOXA2 expression.
PAX6-P2A-Cre lines were also differentiated into human NE over 10 days through an embryoid body (EB)-based suspension culture protocol (Chen et al., 2016, Chi et al., 2016b, Tao and Zhang, 2016, Zhang et al., 2001). The day 10 NE cells were then guided to cortical NPs or ventral medial ganglionic eminence NPs over an extra 7 days in the absence or presence of SHH (Chi et al., 2016b, Liu et al., 2013). Immunostaining data verified that PAX6 and Cre were either not expressed or completely repressed in day 0 hESCs or day 17 ventral NPs (Figure 2F). Whereas PAX6 and Cre were uniformly expressed in day 10 human NE, their expression was only maintained in day 17 cortical NPs when SHH was not present (Figure 2F). Again, the PAX6-P2A-Cre fusion proteins were efficiently cleaved as revealed by western blot analysis (Figure 2G). These data suggest that Cre expression also tightly mirrors the endogenous PAX6 expression pattern under EB differentiation conditions, which mimics NE induction and subsequent regional patterning of the human forebrain.
Tracing PAX6 Expressing NE In Vitro and In Vivo
A PAX6-P2A-Cre/AAVS1-LSL-GFP double-engineered lineage-tracing line was developed for studying the developmental potential of PAX6-expressing NE. For this we electroporated a number of constructs into the seed AAVS1-LSL-GFP hESCs line, including a gRNA targeting the PAX6 locus near its stop codon region, the Cas9 expression plasmid, and the P2A-Cre donor plasmid. Surprisingly, almost all surviving colonies uniformly expressed GFP. This indicates that the Cre-LoxP recombination system is extremely sensitive in human cells and a minimal amount of Cre expression is sufficient to carry out the recombination and remove the STOP cassette, albeit lacking a typically driven promoter in the donor vector (data not shown). We then electroporated AAVS1 TALENs and the AAVS1-LSL-GFP donor plasmid into the characterized PAX6-P2A-Cre line instead. Interestingly, we found no colonies exhibiting GFP expression at the pluripotent stage, suggesting tight control of the Cre expression under the endogenous PAX6 promoter after local HR. Genomic PCR analysis identified multiple monoallelically or biallelically targeted colonies (Figure 3A). Clone no. 4 was a biallelically targeted line and its precise and single-copy targeting was further verified by Southern blot analysis (Figures 3A and 3B). We therefore chose line no. 4 for future tracing assays.
Figure 3.

Tracing PAX6 Expressing NE In Vitro and In Vivo
(A) AAVS1 left and right TALENs and AAVS1-LSL-GFP donor plasmids are electroporated into PAX6-P2A-Cre H9 hESCs. Genomic DNA PCR analysis identifies monoallelically and biallelically targeted colonies.
(B) Southern blot analysis of no. 4 biallelic HR colony shows precise engineering without off-target recombination.
(C) Immunostaining of GFP, PAX6, Cre, and NKX2.1 in PAX6-P2A-Cre/AAVS1-LSL-GFP H9 hESC line at day 17 dorsal or ventral NPs reveals that both regional NPs are developed from PAX6 expression NE. Scale bar, 100 μm.
(D) H&E and immunolabeling results of adjacent sections from PAX6-P2A-Cre/AAVS1-LSL-GFP H9 hESC line-derived teratomas. GFP labels all existing neural tubes (NT), but it is not present in cartilages (CT) or intestine tissues (IN). Scale bar, 200 μm.
(E and F) H&E and immunolabeling results of adjacent sections from PAX6-P2A-Cre/AAVS1-LSL-GFP H9 hESC line-derived teratomas. Both PAX6+ dorsal NPs and NKX2.1+ ventral NPs are labeled with GFP. Scale bar, 100 μm.
See also Figure S2.
In line with the Cre expression pattern, there was no GFP expression in the obtained PAX6-P2A-Cre/AAVS1-LSL-GFP line in day 0 hESCs even after 20 continuous passages. Under EB differentiation conditions driving toward an NE fate, GFP started to be expressed at day 6, and by the time the cells were committed to the NE at day 10 almost all cells were GFP positive. Without SHH treatment, the NE cells were specified to a cortical NP identity with PAX6 expression at day 17, while the ventral NP lineage marker, NKX2.1 (Xu et al., 2004), was not expressed (Figure 3C). The application of SHH to day 10 NE for 7 days completely ventralized the cells by repressing PAX6 while inducing NKX2.1 expression (Figure 3C). Notably, both dorsal and ventral NPs at day 17 universally bore GFP expression. Similar results were obtained in another hESC line, H7 (Figures S2A–S2C). These data indicate that both PAX6+/NKX2.1− dorsal and PAX6–/NKX2.1+ ventral NPs are directly developed from PAX6+ NE cells at an earlier stage. Moreover, the hindbrain and spinal cord NPs derived from PAX6-P2A-Cre/AAVS1-LSL-GFP H9 hESCs via RA caudalization at day 17 (Li et al., 2005) were also uniformly marked with GFP, again indicating their developmental origin from PAX6+ NE cells (Figure S2D). As a human NE determinant, PAX6 induces an NE fate in epiblast cells, and these cells have the differentiation potential to generate various regional NPs, opposing its expression pattern and developmental roles in mouse. Teratoma formation analysis is the gold standard for accessing the developmental potentials of hPSCs and an invaluable tool for studying human lineage development. We subcutaneously injected the PAX6-P2A-Cre/AAVS1-LSL-GFP hESCs into immunocompromised non-obese diabetic (NOD)/severe combined immunodeficiency (SCID) mice to form teratomas. H&E staining showed normal ectoderm, mesoderm, and endoderm tissues present in the teratomas, indicating that the two-step gene targeting does not interfere with the normal differentiation potencies of hPSCs (Figure 3D). Adjacent slices were then used for GFP analysis under a fluorescent microscope. GFP-positive tissues were exclusively presented in the neurotube (NT)-like structures, whereas the cartilage structures of a mesodermal origin and the intestine tissues of an endodermal origin were both lacking GFP expression (Figure 3D). These data reveal that the lineage-tracing system built into the PAX6 and AAVS1 loci is tight and efficient. PAX6-expressing cells represent a bona fide human NE which belongs to human pan neural stem cells and holds the capacity to generate the entire neuroepithelial cell population within the NT (Figure S2E).
Immunostaining PAX6 and NKX2.1 in teratoma slices also showed that both PAX6+ dorsal NPs and NKX2.1+ ventral NPs expressed GFP, again suggesting their common origin from the PAX6-expressing NE cells (Figures 3E and 3F). All these in vitro and in vivo lineage-tracing data, together with our previous functional analyses of human neural induction and regional patterning, have started to shed rudimental yet crucial light on mechanisms underlying human neural development, which could not have been directly extrapolated from mouse studies (Chi et al., 2016a, Tao and Zhang, 2016, Zhang et al., 2010).
Inaccurate Lineage Labeling Given Hypersensitivity of the Recombination System
We successfully obtained the FOXA2-P2A-Cre/AAVS1-LSL-GFP lines through electroporation of AAVS1 TALENs and AAVS1-LSL-GFP donor plasmids into the characterized FOXA2-P2A-Cre line. Genomic PCR analysis was performed, and heterozygotic and homozygotic HR colonies were subsequently identified (Figure 4A). Colony no. 3 was a homozygotically integrated line and Southern blot analysis validated its precise targeting at the AAVS1 locus without off-target recombination (Figures 4A and 4B). We then used the adherent culture protocol to generate FP cells from the no. 3 FOXA2-P2A-Cre/AAVS1-LSL-GFP line. The engineered hESCs at day 0 were Oct4 positive and lacked GFP expression at an early passage (Figure 4C). Nevertheless, 1 day after SHH administration, GFP expression was initiated in most of the cells which were still OCT4+, while Cre and FOXA2 were undetectable (Figure 4C). We observed the same results in H7 hESCs (Figures S3A–S3C). The OCT4+/FOXA2− nature of the cells 2 days post differentiation conferred a pluripotent identity on them; however, the FOXA2-P2A-Cre/AAVS1-LSL-GFP line failed to faithfully label the FP lineage in this case. We reasoned that this nonspecific labeling of the GFP reporter in FOXA2-P2A-Cre/AAVS1-LSL-GFP line may root from the supersensitivity of the recombination system, and that an undetectable, low-level FOXA2-P2A-Cre expression is sufficient to trigger recombination and remove the STOP cassette ahead of GFP. Indeed, when we continuously passaged the FOXA2-P2A-Cre/AAVS1-LSL-GFP line in vitro, more and more GFP cells emerged in the culture, and they could be maintained in an undifferentiated state (Figures 4D and S4D). Fluorescence-activated cell sorting (FACS) sorting experiments showed that there were more than 90% of cells which were inaccurately labeled with GFP in FOXA2-P2A-Cre/AAVS1-LSL-GFP line remained as hESCs or differentiated to an FP fate (Figure S3E). Similar inaccurate labeling was also observed in the FOXA2-P2A-Flp/AAVS1-FSF-GFP line, suggesting that both Cre-LoxP and Flp-FRT systems are hypersensitive in human cells (Figure S3F). To test whether the hypersensitivity of the recombination systems is unique in human cells, we generated fibroblast-like cells from AAVS1-LSL-GFP hESCs (Yen et al., 2011) and cultured skin fibroblasts from Rosa26-LSL-GFP mouse. Low amounts of pLenti-Cre plasmids were then electroporated to both type of cells, while FACS results showed no significant difference in them (Figure S3G).
Figure 4.

The Cre-LoxP Recombination System Is Hypersensitive in Human Cells
(A) Genomic DNA PCR results of retrieved colonies after recombination of AAVS1-LSL-GFP into FOXA2-P2A-Cre seed H9 hESC line.
(B) Southern blot analysis of no. 3 FOXA2-P2A-Cre/AAVS1-LSL-GFP colony with GFP probe.
(C) Immunolabeling with GFP, OCT4, Cre, and FOXA2 of no. 3 FOXA2-P2A-Cre/AAVS1-LSL-GFP H9 hESC line maintained in a pluripotent stage or 2 days post differentiation toward an FP fate. Scale bar, 100 μm.
(D) Bright-field (BF) and fluorescent images of FOXA2-P2A-Cre/AAVS1-LSL-GFP hESC line at different passages indicated. Scale bar, 100 μm.
See also Figure S3.
Shaping the Labeling Sensitivity and Specificity by Introducing Mutations in the LoxP Sequences
The completely different behavior of the lineage-tracing lines targeting the FOXA2 and PAX6 loci may stem from their differential expression patterns. Low levels of FOXA2 oscillation resulted in inaccurate reporter gene labeling that potentially could have misled our interpretation of underlying developmental mechanisms. It is therefore crucial to design a series of tracing systems with multiple levels of recombination sensitivities for lineage-specific genes with various expression profiles. Based on the native sequences which resemble the LoxP sites and hold partial sensitivity for Cre-triggered recombination (Thyagarajan et al., 2000), we designed four mutant LoxP sites corresponding to LSLm1-4, which then were built into the donor plasmids (Figure 5A). We first examined the recombination efficiencies of these LSL mutants by transient expression of each donor plasmid with the Cre expression vector in HEK293 cells. Analyzing the percentage of GFP-positive cells in each group revealed that the recombination efficiencies of each LoxP sequence followed a trend of LSL > LSLm4 > LSLm2 > LSLm1 > LSLm3 (Figure 5B).
Figure 5.

A Series of LSL Variants with Distinct Recombination Sensitivities
(A) Sequences of LSL and LSL variants. Mutated nucleotides are labeled in red.
(B) HEK29 cells are transfected with pLenti-Cre and AAVS1-LSL-GFP or AAVS1-LSLm1-4-GFP plasmids. GFP+ cells are calculated to evaluate the sensitivity of Cre-mediated recombination of each LSL variant. Data are presented as mean ± SEM of three independent experiments.
(C) Genomic DNA PCR results of retrieved colonies after recombination of AAVS1-LSLm1-GFP or AAVS1-LSLm2-GFP into FOXA2-P2A-Cre seed H9 hESC line.
(D) Bright-field (BF) and fluorescent images of FOXA2-P2A-Cre/AAVS1-LSLm2-GFP hESC line at passages 35 and 49. Scale bar, 100 μm.
(E and F) Immunolabeling of GFP, FOXA2, and Cre in FP differentiation derivatives of FOXA2-P2A-Cre/AAVS1-LSLm2-GFP (E) or FOXA2-P2A-Cre/AAVS1-LSLm1-GFP (F) lines at different days indicated. Scale bars, 100 μm.
See also Figure S4.
We next electroporated AAVS1 TALENs and AAVS1-LSLm1-GFP or AAVS1-LSLm2-GFP tracing donor plasmids into FOXA2-P2A-Cre hESCs. Genomic PCR successfully retrieved monoallelically or biallelically engineered colonies of both LSL variants (Figure 5C). FOXA2-P2A-Cre/AAVS1-LSLm2-GFP lines were passaged continuously in vitro to passage 49, where no GFP-positive cells were observed (Figure 5D), indicating an improvement of the specificity of the tracing reporter. Immunostaining results collected from the FP differentiation derivatives of the FOXA2-P2A-Cre/AAVS1-LSLm1-GFP and FOXA2-P2A-Cre/AAVS1-LSLm2-GFP lines showed that GFP-positive cells were absent at day 5, but gradually increased as differentiation proceeded, tightly following the induction of the FOXA2 and Cre proteins (Figures 5E and 5F). It was also notable that the recombination efficiency of LSLm1 in FP cells was lower than that of LSLm2 resembling what had been observed in HEK293 cells (Figures 5B, 5E, and 5F). Similar results were obtained in H7 cells (Figures S4A and S4B). These data suggest that the recombination efficiency as well as the tracing fidelity of the Cre-LoxP system could be adjusted by using different LSL sequences. This allowed the newly designed LSL mutants in the current study to be purposely selected for accurate tracing studies.
Improvement of the Tracing Fidelity by Introducing Controllable Recombinases
Controllable recombinases are constructed by the fusion of Cre or Flp with the ligand-binding domain of the human estrogen receptor (CreER) (Metzger et al., 1995). The fusion proteins are located in the cytoplasm, but will translocate into the nucleus where they fulfill their recombination activity when binding with tamoxifen. CreERT2 or FlpERT2 with specific mutations in the estrogen receptor improves the sensitivity of the recombination system in response to tamoxifen (Feil et al., 1997). In this way, temporal control of the recombinase through tamoxifen administration might offer additional choices for tracing those lineages with pleiotropic maker gene expression at various developmental stages.
We designed an additional gRNA targeting the ATG start codon region located in exon 1 of FOXA2 (Figures 6A and S1). A donor plasmid with a CreERT2 expression cassette and both left and right homology arms was also constructed (Figure 6A). The gRNA, Cas9 expression, and donor plasmids were concomitantly electroporated into hESCs with a transient puromycin expression vector for a short period of drug selection. Retrieved colonies underwent genomic PCR analysis (Figure 6B). Monoallelically engineered colonies were further electroporated with AAVS1 TALENs and the AAVS1-LSL-GFP donor plasmid. Correct integration of the conditional tracer at the AAVS1 locus was also validated by genomic PCR and Southern blot analysis (Figures 6C and 6D). The established FOXA2-CreERT2/AAVS1-LSL-GFP lines were then differentiated into dorsal NPs or FP cells accordingly. No GFP expression was observed in dorsal NPs, even with tamoxifen treatment at either day 6 or day 12 post differentiation (Figures 6E and 6F). In the absence of tamoxifen treatment, day 6 and day 12 FP cells were also negative for GFP. In contrast, most of the FP cells expressed GFP after 1 day of tamoxifen treatment in a dose-dependent manner (Figures 6E, 6F, and S5A). These data suggest that the tracing fidelity, the tightness, and the effectiveness of the tamoxifen-controllable elements meet the requirements for accurate tracing of human lineages. We also validated the FlpERT2-FRT recombination system in hESCs and this system could also be applied for human lineage developmental investigations (Figures S5B–S5F).
Figure 6.

Improvement of the Tracing Fidelity by Introducing Controllable Recombinases
(A) Schematic diagram for generating FOXA2-CreERT2 lines. Primer sets for genomic DNA PCR are labeled with arrows.
(B and C) Genomic DNA PCR results showing recombination of CreERT2 at the ATG region of FOXA2 (B) and CAG-LSL-GFP (C) cassettes within the AAVS1 intron.
(D) Southern blot analysis of no. 4 FOXA2-CreERT2/AAVS1-LSL-GFP colony with GFP probe.
(E and F) Immunolabeling of GFP, FOXA2, Cre, and PAX6 in dorsal NPs and FP cells with (E) or without (F) 4-OHT treatment at days 6 or 12 post differentiation. 4-OHT was administered 2 days before staining. Scale bars, 100 μm.
See also Figure S5.
Temporal Tracing of Pax6-Expressing Cells Identifies their Sequential NE and Dorsal NP Identities
Pax6 is expressed at both NE and cortical NP stages in humans. To examine whether the CreERT2-LoxP system is appropriate for tracing cell lineages temporally, we also constructed a PAX6-CreERT2/AAVS1-LSL-GFP line in hESCs (Figures 7A–7C). By adding tamoxifen through days 8–10, we reconfirmed that both PAX6+/NKX2.1− cortical NPs and PAX6−/NKX2.1+ ventral NPs were specified from PAX6+ NE (Figures 7D and S6A). Day 17 cortical NPs but not ventral cells were efficiently labeled with GFP after tamoxifen treatment through days 15–17, suggesting distinct cell fates were committed after day 15 when the NE cells were patterned with or without SHH (Figures 7E and S6B). These data suggest that the tamoxifen-controllable lineage-tracing system represents a precise and useful tool for modeling human lineage development in a temporal dimension.
Figure 7.

Temporal Tracing of PAX6 Expression Cells Identifies their Sequential NE and Dorsal NP Identities
(A) Schematic diagram for generating PAX6-CreERT2 lines. Primer sets for genomic DNA PCR are labeled with arrows.
(B and C) Genomic DNA PCR results showing recombination of CreERT2 at the ATG region of PAX6 (B) and CAG-LSL-GFP (C) cassettes within the AAVS1 intron.
(D) Immunolabeling of GFP, PAX6, Cre, and NKX2.1 in dorsal and ventral NPs at day 17 in PAX6-CreERT2/AAVS1-LSL-GFP H9 hESC line treated with 4-OHT through days 8–10. Scale bar, 100 μm.
(E) Immunolabeling of GFP, PAX6, Cre, and NKX2.1 in dorsal and ventral NPs at day 17 in PAX6-CreERT2/AAVS1-LSL-GFP H9 hESC line treated with or without 4-OHT through days 15–17. Scale bar, 100 μm.
See also Figure S6.
Discussion
In this study, we have successfully designed a series of lineage-tracing tools in a human system by incorporating of both Cre-LoxP and Flp-FRT recombination systems in hPSCs. The AAVS1 locus has been proved to be highly efficient for reporter integration and recombinase-triggered recombination. The recombinase expression cassette was designed to generate an in-frame fusion protein with either the lineage marker gene, or a substitute of the open reading frame, and both have proved to be functional. Moreover, both the constitutive and controllable (Cre-LoxP and Flp-FRT VS CreERT2-LoxP and FlpERT2-FRT) recombination systems have been validated to be suitable for human lineage studies.
A major concern of the lineage-tracing technology in human cells arises from the hypersensitivity of the recombination systems, which causes inaccurate reporter expression during lineage-tracing. We have proposed multiple ways to deal with this caveat. Practically, one can construct the recombinase expression line first, followed by building the conditional reporter in the AAVS1 locus to circumvent unnecessary leakage caused by transient recombinase expression noise during electroporation. A series of LSL mutants have also been designed in the current study to comply with various lineage-specific genes of interest, which may enhance the fidelity of lineage-tracing without scarifying too much of the recombination efficiency. Furthermore, involving of tamoxifen-controllable recombinases will not only solve the problem of inaccurately labeling, but also offer more sophisticated tracing systems in an additional temporal dimension.
As one of the most abundantly cultured human cell types, hPSCs represent themselves as an ideal platform to study human tissue homeostasis when combined with advanced genetic engineering tools. The fascinating self-renewal capacity of hPSCs enables long-term complex genetic engineering in vitro. Meanwhile, the full lineage differentiation potential of hPSCs in vitro and in vivo makes them novel cell models to mirror human embryonic development. We have reported that human, but not mouse NE within the neural plate of early embryos expresses PAX6, and that PAX6 is not only necessary, but also sufficient to specify human epiblast toward an NE fate (Zhang et al., 2010). Meanwhile, the advanced expression of PAX6 in human NE may facilitate preferential human neocortex formation, since PAX6 represses ventralization of human NE induced by SHH (Chi et al., 2016a). In the current study, we have also proved that PAX6-expressing cells are bona fide human NE cells, which generate all spectra of regional NPs within the NT. All these together point to a hypothesis that human neural development follows an evolutionarily novel pattern, which establishes our more sophisticated human brain and enabling our unique social behavior. All these observations emphasize the necessity to devise practical investigation systems specifically for studying human embryonic development, where hPSCs could indeed be used as a powerful tool for this purpose (Keller, 2005, Williams et al., 2012).
With the help of newly developed CENs, genetic engineering in hPSCs has become increasingly practical. To date, gene ablation, constitutive or inducible gene expression, genetic labeling, and gene editing have been successfully established. Together with these tools, the human lineage-tracing system described here is expected to have a profound impact on pioneering studies related to human embryonic development. This includes integration with in vitro lineage differentiation techniques, in vivo lineage development in teratomas, and, recently, in the development of human-animal chimera studies with primed or naive hPSCs (Wu et al., 2017).
Experimental Procedures
Plasmids Construction
AAVS1-LSL-GFP donor vector was obtained by insertion of an LSL cassette between the CAG promoter and the eGFP cassette within the AAVS1-CAGGS-eGFP plasmid (Addgene, no. 22212) (Hockemeyer et al., 2009). AAVS1-LSLm1-GFP, AAVS1-LSLm2-GFP, AAVS1-LSLm3-GFP, and AAVS1-LSLm4-GFP were obtained by replacing the 5′ LoxP site within the AAVS1-LSL-GFP donor vector with the mutant LoxP sequence shown in Figure 5A. gRNAs were constructed as described previously (Fang et al., 2017, Mali et al., 2013), and gRNA targeting sequences for PAX6 and FOXA2 are shown in Figure S1. PAX6-P2A-Cre, FOXA2-P2A-Cre, PAX6-CreERT2, and FOXA2-CreERT2 donor plasmids were constructed by ligating the left and right homologous arms with Cre or CreERT2 cassettes PCR amplified from pCAG-Cre-ERT2 (Addgene, no. 14797) (Matsuda and Cepko, 2007) into Oct4-2a-eGFP-PGK-puro vector (Addgene, no. 31938) (Hockemeyer et al., 2011) with BamHI and EcoRI. FOXA2-P2A-Flp or FOXA2-P2A-FlpERT2 donor plasmid was constructed by replacing Cre in FOXA2-P2A-Cre with Flp or FlpERT2 cassette PCR amplified from pCAG-Flp or pCAG-Flp-ERT2 (Addgene, no. 13787 and no. 14756) (Matsuda and Cepko, 2007). Primers for these donor vectors were as follows:
| PAX6-P2A-5′ arm-F | GAAT CCTGCAGG GAACAGTCAGCCAATGGGCA |
| PAX6-P2A-5′ arm-R | GTAC GCTAGC CTGTAATCTTGGCCAGTATTG |
| PAX6-P2A-3′ arm-F | GATT GGCGCGCC GGAAATATTGTGTTAATTCAGTCAG |
| PAX6-P2A-3′ arm-R | GTAT GGCCGGCC GGACCGTGAACAGTAATACAAC |
| FOXA2-P2A-5′ arm-F | GAAT CCTGCAGG ATCCAGCAGAGCCCCAACAAG |
| FOXA2-P2A-5′ arm-R | GTAC GCTAGC AGAGGAGTTCATAATGGGCCGG |
| FOXA2-P2A-3′ arm-F | GATT GGCGCGCC GAAGACGACGGCTTCAGGCCCGGC |
| FOXA2-P2A-3′ arm-R | GTAT GGCCGGCC GGGATAAAACCGGGTATAACAC |
| FOXA2-CreERT2-5′ arm-F | GGTCTC AGATC TGACAGAGAATGAGCACTGAGAGCG |
| FOXA2-CreERT2-5′ arm-R | GGTCTC AACAT GGCAGTTTAAAATTTAACAGCCAC |
| FOXA2-CreERT2-3′ arm-F | CGTCTC ACGCGGCGGCCGCATGCACTCGGCTTCCAGTATGCTG |
| FOXA2-CreERT2-3′ arm-R | CGTCTC AAATT CATGGAGTACTCCCAAGCCAGCA |
| PAX6-CreERT2-5′ arm-F | GGTCTC ACTAGCTCAGCTCTTGGCCTCTACTCCTTA |
| PAX6-CreERT2-5′ arm-R | GGTCTC AACATGCTGGCTCTGGCTGGGGGCC |
| PAX6-CreERT2-3′ arm-F | CGTCTC ACGCGCAGAACAGTAAGTGCCTCTGGTCT |
| PAX6-CreERT2-3′ arm-R | CGTCTC AAATTAGGCTCCCAGGTCGGAGCTCTAGA |
hESC Culture
hESCs line H9 and H7 (WA09, WA07, WiCell agreement no. 14-W0377, 17-W0044) were cultured as described previously (Zhang et al., 2010). In brief, hESCs were maintained on irradiated embryonic mouse fibroblasts (MEF) in hESC medium containing DMEM/F12, 20% knockout serum replacer, 1× non-essential amino acids, 1× GlutaMAX, and 0.1 mM β-mercaptoethanol. Fibroblast growth factor 2 (4 ng/mL) was added when refreshing medium. Cells were passaged every 5 days with dispase digestion.
Neural Differentiation
Detailed procedures for EB-based differentiation were described previously (Chi et al., 2016b, Zhang and Zhang, 2010). In brief, hESCs were lifted from the culture surface and suspended as floating EBs for 4 days in the hESC culture medium. EBs were then switched to a neural induction medium (NIM, DMEM/F12 with 1× N2 supplement) for 2 extra days and then plated onto a laminin-coated culture surface. NE formed at day 10 post differentiation. For ventralization or caudalization, SHH (100 ng/mL) (Miltenyi Biotechnology, no. 130095730) and purmorphamine (0.3 μM) (StemGent, no. 04–0009) or retinoic acid (0.1 μM) (Sigma, no. 302794) were added through days 10–17. For FP differentiation, hESCs were triturated into small clumps and plated onto MEF feeders. Cells were changed to NIM supplied with dual-Smad inhibitors, SB431542 (2 μM) (StemGent, no. 04–0010) and LDN193189 (0.2 μM) (StemGent, no. 04–0074), for 7 days. At day 1 post differentiation, SHH and purmorphamine were added to induce an FP fate.
Construction of Transgenic Cell Lines
hESCs were cultured in hESC medium with Rho kinase inhibitor, Y27632 (1 mM; Calbiochem) for 3 hr prior to electroporation. Cells were digested by trypsin into single cells and washed by PBS twice, and then 1 × 107 cells were electroporated with appropriate combination of Cas9 plasmids (5 μg), gRNAs (5 μg), TALENs (5 μg), donor plasmids (30 μg), or puromycin transient expression plasmids (5 μg) in 200 μL of electroporation buffer (KCl 5 mM, MgCl2 25 mM, HEPES 15 mM, Na2HPO4 102.94 mM, NaH2PO4 47.06 mM [pH 7.2]) using the Gene Pulser Xcell system (Bio-Rad) at 250 V, 500 μF in a 0.4-cm cuvette (Phenix Research Products). Cells were treated with puromycin (0.5 μg/mL) for 3–4 days. After puromycin selection, MEF-conditioned medium was supplied. Concrete colonies would be visible after 1 to 2 weeks.
Genomic DNA PCR
Genomic DNA was extracted with QuickExtract DNA Extraction Solution (Epicentre, QE09050). WT and HR alleles were identified by using the following primer sets:
| AAVS1-F1 | CTTCCGCATTGGAGTCGCTTTA |
| AAVS1-F2 | CAGCCGGTCCTGGACTTTGTC |
| AAVS1-R1 | ACAGGAGGTGGGGGTTAGAC |
| AAVS1-R2 | AGCCGGGAACCGCTCAACTC |
| PAX6-F1 | GTAACCACAGGTTTGCCTCT |
| PAX6-R1 | CCAGTGGTACAATACAGG |
| FOXA2-F1 | GCAGGGCTACTCCTCCGTGAGCAA |
| FOXA2-R1 | TTCCCGTTTTCCTCCTTATATAGAA |
| Cre-R1 | CTAATCGCCATCTTCCAGCAGG |
| FOXA2-F2 | GGTCGTTTGTTGTGGCTGTTAAA |
| FOXA2-F3 | CGTTCCGGGTCTGAACTGTAACAG |
| FOXA2-R2 | CATGGAGTACTCCCAAGCCAGCA |
| PAX6-F2 | CTTCCCCTGGTCTCCAAACTTCAG |
| PAX6-R2 | TCTCCAGTATCGAGAAGAGCCA |
| Cre-R2 | AGATATAGAAGATAATCGCGAACAT |
| Flp-R | TTGCCGGTCCTATTTACTCG |
Southern Blot Analysis
The 5′ arm probe and the GFP probe were PCR amplified from the AAVS1-LSL-GFP donor vector with the PCR DIG Probe Synthesis Kit (Roche). Primer sets for 5′ arm probe were: ACAGGTACCATGTGGGGTTC and CTTGCCTCACCTGGCGATAT; and primer sets for GFP probe were: AGGTTCCGTCTTCCTCCACT and GTCCAGGCAAAGAAAGCAAG. Genomic DNA (5–10 μg) was digested overnight with EcoRI for the 5′ arm probe and BsaI for the GFP probe, and subjected to electrophoresis. Gels were denatured, neutralized, and transferred overnight by capillarity on Hybond-N membranes (GE Healthcare) using 10× SSC transfer buffer. For membrane hybridization, 5 μL of denatured DIG-labeled probe was mixed with 20 mL hybridization buffer. Hybridization was carried out overnight at 65°C. Probes were detected by an AP-conjugated DIG-Antibody (Roche) using CDP-Star (Roche) as a substrate for chemiluminescence.
Lentivirus Production
pLenti-Cre (10 μg), 7.5 μg △8.9, and 5 μg VSVG were co-transfected into HEK293FT cells per 10-cm dish using calcium-phosphate precipitation protocol. Viral particles were concentrated through ultracentrifugation.
Immunocytochemistry
Cells cultured on coverslips were fixed in 4% paraformaldehyde for 10 min. Cells were then incubated in a blocking and permeabilization buffer (10% donkey serum and 0.2% Triton X-100 in PBS) for 1 hr followed by primary antibody incubation overnight at 4°C. On the next day, coverslips were incubated with corresponding fluorescent-conjugated secondary antibodies (1:1,000, Jackson ImmunoResearch) for 1 hr at room temperature. Nuclei were counter stained with Hoechst 33258. Primary antibodies used in this study were, Cre (1:500, mouse immunoglobulin G [IgG], Millipore, MAB3120), PAX6 (1:1,000, mouse IgG, DSHB, PAX6), PAX6 (1:1,000, rabbit IgG, Covance, PRB-278P), NKX2.1 (1:500, mouse IgG, Millipore, MAB5460), HOXB4 (1:50, mouse IgG, DSHB, I12), FOXA2 (1:2,000, rabbit IgG, Abcam, ab40874), and GFP (1:2000, chicken IgY, Aves, GFP-1020).
Western Blot Analysis
Cells were lysed with RIPA buffer supplied with protease inhibitors. Protein concentrations were calculated with the BCA kit (Thermo Scientific). Samples were subjected to SDS-PAGE. Primary antibodies included PAX6 (1:2,000, mouse IgG, DSHB, PAX6), FOXA2 (1:5,000, rabbit IgG, Abcam, ab40874), Cre (1:1,000, mouse IgG, Millipore, MAB3120), and β-actin (1:5,000, mouse IgG, Sigma, A5316). Secondary antibodies were horseradish peroxidase-conjugated IgG (1:10,000, Jackson ImmunoResearch).
Teratoma Formation Assay
hESCs were injected subcutaneously on the back of NOD/SCID immunodeficient mice. After 2 months, mice bearing teratomas were sacrificed and perfused with 4% paraformaldehyde. Cryostat sections of teratomas were stained with H&E or antibodies recognizing PAX6 or NKX2.1.
FACS Analysis
Cells were digested with trypsin-EDTA into single cell and resuspended with PBS. FACS analysis was performed with BD FACSVerse and fluorescence was excited with a 488-nm laser. Up to 10,000 events were captured and data were analyzed with FlowJo 7.
Author Contributions
Z.C., X.R., X.X., L.L., and X.Z. conceived the study. Z.C., X.R., and X.X. performed most of the experiments. X.Z., Y.H., Z.L., L.S., L.M., Y.F., and Y.L. helped with setting up the HR system in hPSCs. Z.C. collected and analyzed the data. Z.C., L.L., and X.Z. wrote the manuscript.
Acknowledgments
The present study was supported by the National Key Research and Development Program of China (grant no. 2018YFA0108000), the National Natural Science Foundation of China (grant nos. 31471040, 31872760, 31771132, 31400934, and 31801204), Science and Technology Commission of Shanghai Municipality (15JC1400202), Shanghai Municipal Education Commission (C120114), and Fundamental Research Funds for the Central Universities.
Published: October 25, 2018
Footnotes
Supplemental Information includes six figures and can be found with this article online at https://doi.org/10.1016/j.stemcr.2018.09.014.
Contributor Information
Ling Liu, Email: lliu@tongji.edu.cn.
Xiaoqing Zhang, Email: xqzhang@tongji.edu.cn.
Supplemental Information
References
- Awatramani R., Soriano P., Rodriguez C., Mai J.J., Dymecki S.M. Cryptic boundaries in roof plate and choroid plexus identified by intersectional gene activation. Nat. Genet. 2003;35:70–75. doi: 10.1038/ng1228. [DOI] [PubMed] [Google Scholar]
- Blanpain C., Simons B.D. Unravelling stem cell dynamics by lineage tracing. Nat. Rev. Mol. Cell Biol. 2013;14:489–502. doi: 10.1038/nrm3625. [DOI] [PubMed] [Google Scholar]
- Boch J., Scholze H., Schornack S., Landgraf A., Hahn S., Kay S., Lahaye T., Nickstadt A., Bonas U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- Bulic-Jakus F., Katusic Bojanac A., Juric-Lekic G., Vlahovic M., Sincic N. Teratoma: from spontaneous tumors to the pluripotency/malignancy assay. Wiley Interdiscip. Rev. Dev. Biol. 2016;5:186–209. doi: 10.1002/wdev.219. [DOI] [PubMed] [Google Scholar]
- Chen Y., Cao J., Xiong M., Petersen A.J., Dong Y., Tao Y., Huang C.T.-L., Du Z., Zhang S.-C. Engineering human stem cell lines with inducible gene knockout using CRISPR/Cas9. Cell Stem Cell. 2015;17:233–244. doi: 10.1016/j.stem.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Zhang K., Zhou L., Gao X., Wang J., Yao Y., He F., Luo Y., Yu Y., Li S. Coupled electrophysiological recording and single cell transcriptome analyses revealed molecular mechanisms underlying neuronal maturation. Protein Cell. 2016;7:175–186. doi: 10.1007/s13238-016-0247-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi L., Fan B., Feng D., Chen Z., Liu Z., Hui Y., Xu X., Ma L., Fang Y., Zhang Q. The dorsoventral patterning of human forebrain follows an activation/transformation model. Cereb. Cortex. 2016;27:2941–2954. doi: 10.1093/cercor/bhw152. [DOI] [PubMed] [Google Scholar]
- Chi L., Fan B., Zhang K., Du Y., Liu Z., Fang Y., Chen Z., Ren X., Xu X., Jiang C. Targeted differentiation of regional ventral neuroprogenitors and related neuronal subtypes from human pluripotent stem cells. Stem Cell Reports. 2016;7:941–954. doi: 10.1016/j.stemcr.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y., Liu Z., Chen Z., Xu X., Xiao M., Yu Y., Zhang Y., Zhang X., Du Y., Jiang C. Smad5 acts as an intracellular pH messenger and maintains bioenergetic homeostasis. Cell Res. 2017;27:1083–1099. doi: 10.1038/cr.2017.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasano C.A., Chambers S.M., Lee G., Tomishima M.J., Studer L. Efficient derivation of functional floor plate tissue from human embryonic stem cells. Cell Stem Cell. 2010;6:336–347. doi: 10.1016/j.stem.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Feil R., Wagner J., Metzger D., Chambon P. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem. Biophys. Res. Commun. 1997;237:752–757. doi: 10.1006/bbrc.1997.7124. [DOI] [PubMed] [Google Scholar]
- González F., Zhu Z., Shi Z.-D., Lelli K., Verma N., Li Q.V., Huangfu D. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell Stem Cell. 2014;15:215–226. doi: 10.1016/j.stem.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison D.A., Perrimon N. Simple and efficient generation of marked clones in Drosophila. Curr. Biol. 1993;3:424–433. doi: 10.1016/0960-9822(93)90349-s. [DOI] [PubMed] [Google Scholar]
- Hockemeyer D., Soldner F., Beard C., Gao Q., Mitalipova M., DeKelver R.C., Katibah G.E., Amora R., Boydston E.A., Zeitler B. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D., Wang H., Kiani S., Lai C.S., Gao Q., Cassady J.P., Cost G.J., Zhang L., Santiago Y., Miller J.C. Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 2011;29:731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoess R.H., Ziese M., Sternberg N. P1 site-specific recombination: nucleotide sequence of the recombining sites. Proc. Natl. Acad. Sci. U S A. 1982;79:3398–3402. doi: 10.1073/pnas.79.11.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu Y.C. Theory and practice of lineage tracing. Stem Cells. 2015;33:3197–3204. doi: 10.1002/stem.2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller G. Embryonic stem cell differentiation: emergence of a new era in biology and medicine. Genes Dev. 2005;19:1129–1155. doi: 10.1101/gad.1303605. [DOI] [PubMed] [Google Scholar]
- Kim Y.G., Cha J., Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. U S A. 1996;93:1156–1160. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar K., Watt F.M. Lineage tracing. Cell. 2012;148:33–45. doi: 10.1016/j.cell.2012.01.002. [DOI] [PubMed] [Google Scholar]
- Li X.-J., Du Z.-W., Zarnowska E.D., Pankratz M., Hansen L.O., Pearce R.A., Zhang S.-C. Specification of motoneurons from human embryonic stem cells. Nat. Biotechnol. 2005;23:215–221. doi: 10.1038/nbt1063. [DOI] [PubMed] [Google Scholar]
- Liu Y., Weick J.P., Liu H., Krencik R., Zhang X., Ma L., Zhou G.-M., Ayala M., Zhang S.-C. Medial ganglionic eminence-like cells derived from human embryonic stem cells correct learning and memory deficits. Nat. Biotechnol. 2013;31:440–447. doi: 10.1038/nbt.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Hui Y., Shi L., Chen Z., Xu X., Chi L., Fan B., Fang Y., Liu Y., Ma L. Efficient CRISPR/Cas9-mediated versatile, predictable, and donor-free gene knockout in human pluripotent stem cells. Stem Cell Reports. 2016;7:496–507. doi: 10.1016/j.stemcr.2016.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livet J., Weissman T.A., Kang H., Draft R.W., Lu J., Bennis R.A., Sanes J.R., Lichtman J.W. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 2007;450:56–62. doi: 10.1038/nature06293. [DOI] [PubMed] [Google Scholar]
- Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X., Fujiwara Y., Chapdelaine A., Yang H., Orkin S.H. Activation of EGFP expression by Cre-mediated excision in a new ROSA26 reporter mouse strain. Blood. 2001;97:324–326. doi: 10.1182/blood.v97.1.324. [DOI] [PubMed] [Google Scholar]
- Matsuda T., Cepko C.L. Controlled expression of transgenes introduced by in vivo electroporation. Proc. Natl. Acad. Sci. U S A. 2007;104:1027–1032. doi: 10.1073/pnas.0610155104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger D., Clifford J., Chiba H., Chambon P. Conditional site-specific recombination in mammalian cells using a ligand-dependent chimeric Cre recombinase. Proc. Natl. Acad. Sci. U S A. 1995;92:6991–6995. doi: 10.1073/pnas.92.15.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscou M.J., Bogdanove A.J. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- Niakan K.K., Eggan K. Analysis of human embryos from zygote to blastocyst reveals distinct gene expression patterns relative to the mouse. Dev. Biol. 2013;375:54–64. doi: 10.1016/j.ydbio.2012.12.008. [DOI] [PubMed] [Google Scholar]
- Pera M.F., Trounson A.O. Human embryonic stem cells: prospects for development. Development. 2004;131:5515–5525. doi: 10.1242/dev.01451. [DOI] [PubMed] [Google Scholar]
- Placzek M. The role of the notochord and floor plate in inductive interactions. Curr. Opin. Genet. Dev. 1995;5:499–506. doi: 10.1016/0959-437x(95)90055-l. [DOI] [PubMed] [Google Scholar]
- Sanjana N.E., Cong L., Zhou Y., Cunniff M.M., Feng G., Zhang F. A transcription activator-like effector toolbox for genome engineering. Nat. Protoc. 2012;7:171–192. doi: 10.1038/nprot.2011.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H., Hogan B.L. Differential expression of multiple fork head related genes during gastrulation and axial pattern formation in the mouse embryo. Development. 1993;118:47–59. doi: 10.1242/dev.118.1.47. [DOI] [PubMed] [Google Scholar]
- Smih F., Rouet P., Romanienko P.J., Jasin M. Double-strand breaks at the target locus stimulate gene targeting in embryonic stem cells. Nucleic Acids Res. 1995;23:5012–5019. doi: 10.1093/nar/23.24.5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.R., Maguire S., Davis L.A., Alexander M., Yang F., Chandran S., ffrench-Constant C., Pedersen R.A. Robust, persistent transgene expression in human embryonic stem cells is achieved with AAVS1-targeted Integration. Stem Cells. 2008;26:496–504. doi: 10.1634/stemcells.2007-0039. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Tabansky I., Lenarcic A., Draft R.W., Loulier K., Keskin D.B., Rosains J., Rivera-Feliciano J., Lichtman J.W., Livet J., Stern J.N.H. Developmental bias in cleavage-stage mouse blastomeres. Curr. Biol. 2013;23:21–31. doi: 10.1016/j.cub.2012.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tao Y., Zhang S.-C. Neural subtype specification from human pluripotent stem cells. Cell Stem Cell. 2016;19:573–586. doi: 10.1016/j.stem.2016.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson J.A., Itskovitz-Eldor J., Shapiro S.S., Waknitz M.A., Swiergiel J.J., Marshall V.S., Jones J.M. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Thyagarajan B., Guimarães M.J., Groth A.C., Calos M.P. Mammalian genomes contain active recombinase recognition sites. Gene. 2000;244:47–54. doi: 10.1016/s0378-1119(00)00008-1. [DOI] [PubMed] [Google Scholar]
- Urnov F.D., Miller J.C., Lee Y.-L., Beausejour C.M., Rock J.M., Augustus S., Jamieson A.C., Porteus M.H., Gregory P.D., Holmes M.C. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- Williams L.A., Davis-Dusenbery B.N., Eggan K.C. SnapShot: directed differentiation of pluripotent stem cells. Cell. 2012;149:1174–1174.e1. doi: 10.1016/j.cell.2012.05.015. [DOI] [PubMed] [Google Scholar]
- Wu J., Platero-Luengo A., Sakurai M., Sugawara A., Gil M.A., Yamauchi T., Suzuki K., Bogliotti Y.S., Cuello C., Morales Valencia M. Interspecies chimerism with mammalian pluripotent stem cells. Cell. 2017;168:473–486.e15. doi: 10.1016/j.cell.2016.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q., Cobos I., De La Cruz E., Rubenstein J.L., Anderson S.A. Origins of cortical interneuron subtypes. J. Neurosci. 2004;24:2612–2622. doi: 10.1523/JNEUROSCI.5667-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen M.L., Hou C.H., Peng K.Y., Tseng P.C., Jiang S.S., Shun C.T., Chen Y.C., Kuo M.L. Efficient derivation and concise gene expression profiling of human embryonic stem cell-derived mesenchymal progenitors (EMPs) Cell Transplant. 2011;20:1529–1545. doi: 10.3727/096368910X564067. [DOI] [PubMed] [Google Scholar]
- Yu J., Vodyanik M.A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J.L., Tian S., Nie J., Jonsdottir G.A., Ruotti V., Stewart R. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Zhang X.Q., Zhang S.C. Differentiation of neural precursors and dopaminergic neurons from human embryonic stem cells. Methods Mol. Biol. 2010;584:355–366. doi: 10.1007/978-1-60761-369-5_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.C., Wernig M., Duncan I.D., Brustle O., Thomson J.A. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 2001;19:1129–1133. doi: 10.1038/nbt1201-1129. [DOI] [PubMed] [Google Scholar]
- Zhang X., Huang C.T., Chen J., Pankratz M.T., Xi J., Li J., Yang Y., LaVaute T.M., Li X.-J., Ayala M. Pax6 is a human neuroectoderm cell fate determinant. Cell Stem Cell. 2010;7:90–100. doi: 10.1016/j.stem.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z., Huangfu D. Human pluripotent stem cells: an emerging model in developmental biology. Development. 2013;140:705–717. doi: 10.1242/dev.086165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.