

Abstract
A chelation-controlled and highly diastereoselective synthesis of syn-aldols is described. Aldol reaction of (S)-valinol-derived ester with a variety of aldehydes proceeded with high syn-diastereoselectivities (up to 99:1) and isolated yields (94%).
Optically active syn- and anti-2-alkyl-3-hydroxycarbonyl units are inherent to numerous biologically active natural products.1 As a consequence, a number of stereoselective methodologies have been developed for their syntheses.2,3 In our recent work on ester-derived titanium enolate aldol reactions4 we have demonstrated that the aldol reactions of phenylalaninol-derived sulfonamido esters with a number of bidentate oxyaldehydes provided syn-aldols diastereoselectively. The corresponding reactions with monodentate aldehydes, however, have shown little syn-selectivities. The chirality transfer presumably proceeds through chelation by the β-sulfonamide functionality. In our continuing effort to further develop these ester-derived titanium enolate aldol reactions, we have subsequently investigated the effect of a β-chiral substituent on aldol stereochemistry by replacing the phenylmethyl substituent of phenylalaninol with other alkyl groups. Herein, we report that the aldol reaction of an (S)-valinol-derived sulfonamido ester with a variety of mono- and bidentate aldehydes proceeded with good to excellent syn-diastereoselectivities and isolated yields. Removal of the chiral auxiliary by mild saponification provided optically active syn-α-alkyl-β-hydroxy acids and full recovery of the chiral auxiliary. The ready availability of valinol and use of inexpensive TiCl4 make this methodology practical and provide rapid access to syn-aldols in optically active form.
Optically active N-p-tosyl-(2S)-valinol 1 was prepared in multigram quantities by reduction of l-valine with LiAlH4 and followed by sulfonylation of the amine functionality with p-toluenesulfonyl chloride and tri ethylamine in the presence of DMAP at 0°C for 2 h (89% yield). As shown in Scheme 1, reaction of sulfonamide 1 with propionylchloride and triethylamine afforded propionyl ester 2 in 85% yield after silica gel chromatography (mp 78°C, (c 0.92, CHCl3)). The corresponding titanium enolate of 2 was prepared by treatment with TiCl4 (1.1 equiv., 1 M solution in CH2Cl2) in CH2Cl2 at 0°C followed by addition of N,N’-diisopropylethylamine (3 equiv.) after 10 min and stirring of the resulting mixture at 0°C for 1 h. The enolate so formed was reacted with a variety of aldehydes precomplexed with TiCl4 (3 equiv.) at −78°C for 1.5–2 h. Interestingly, aldol reaction of 2 with various aldehydes proceeded with excellent diastereoselectivity and isolated yields; the results are summarized in Table 1. Out of four possible diastereomers, formation of syn-diastereomer 3 (major) and anti-diastereomer 4 were observed by 1H and 13C NMR as well as HPLC analysis before and after chromatography. Reaction of 2 with monodentate aldehydes exhibited high syn-diastereoselectivity (entries 1–5)8 except with phenylpropargyl aldehyde which afforded the anti-isomer as the major product (entry 6). Furthermore, reaction with bidentate aldehydes such as benzyloxyacetaldehyde and benzyloxypropionaldehyde provided a single syn-aldolate in high yield (entries 7 and 8). We have also carried out double stereodifferentiating experiments in which an oxyaldehyde bearing an α-chiral center was reacted with the chiral enolate derived from propionate ester 2. Aldol reaction of 2 and 2(S)-benzyloxypropionaldehyde (stereochemically matched case) under identical conditions afforded virtually a single (by HPLC and 400 MHz 1H NMR analysis) aldol product 3h (entry 9) in 77% yield after silica gel chromatography. However, the reaction of 2 and 2(R)-benzyloxypropionaldehyde, a mismatched case, afforded a 34:64 mixture of syn and anti isomers in 82% isolated yield (entry 10). Because of the marked stereo-chemical preference (matched isomer), we then attempted reaction of 2 (1 equiv.) with racemic benzyloxypropionaldehyde (2 equiv.) at −78°C for 0.5 h (Scheme 2).
Scheme 1.

Reagents and conditions: (a) CH3CH2COCl, Et3N, CH2Cl2, 0°C, 2 h; (b) TiCl4, iPr2NEt, 0°C, 1 h, then RCHO and TiCl4, CH2Cl2, −78°C, 2 h; (c) LiOH, THF–H2O, 23°C, 2–3 h; (d) CH2N2, Et2O, 23°C, 30 min; (e) LiBH4, THF–MeOH, 23°C, 2 h.
Table 1.
Aldol reaction of ester 2 with representative aldehydes
| Entry | Aldehyde | Compda | Yield (%)b | syn:anti (3/4)c |
|---|---|---|---|---|
| 1 | Me2CHCHO | 3a | 89 | 90:10 |
| 2 | Me2CHCH2CHO | 3b | 93 | 95:5 |
| 3 | trans-PhCH=CHCHO | 3c | 89 | 96:4 |
| 4 | PhCHO | 3d | 94 | 88:12 |
| 5 | Me(CH2)6CHO | 3e | 96 | 82:18 |
| 6 | Ph-C≡C-CHO | 3f | 86 | 36:64d |
| 7 | BnOCH2CHO | 3g | 74 | 99:1 |
| 8 | BnOCH2CH2CHO | 3h | 81 | 99:1 |
| 9 | 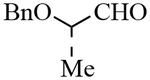 |
3i | 77 | 99:1 |
| 10 | 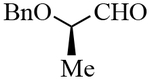 |
3j | 82 | 34:66d |
Major isolated product.
Isolated yield after chromatography.
Ratios determined by 1H MR and HPLC analysis before and after chromatography. Reaction time=1.5–2 h.
Ratio after removal of chiral auxiliary.
Scheme 2.

Interestingly, 2-derived Ti-enolate reacted exclusively with the 2(S)-benzyloxypropionaldehyde (1 equiv.) under these reaction conditions providing only diastereomer 3i (matched case) in 82% isolated yield after silica gel chromatography. Separation and purification of the corresponding unreacted enantioenriched 2(R)-benzyloxypropionaldehyde was difficult due to overlapping by-product. Subsequently, the crude aldehyde was reduced with NaBH4 in ethanol at 23°C to afford 2(R)-(benzyloxy)propanol with an enantiomeric excess of 81% ee (41% recovered, (c 1.9, CHCl3); lit.:9 (c 1.0, CHCl3).
The relative and absolute stereochemistry of various syn-aldolates (3) were established based upon comparison of optical rotation as well as 1H and 13C NMR spectra of the resulting acids, esters or diols with literature values.5 Thus, saponification of the above aldolates with aqueous lithium hydroxide in THF at 23°C for 2 h furnished the corresponding β-hydroxy acids (5). Treatment of these acids with CH2N2 afforded the corresponding methyl esters (6). Various aldolates were converted to diols 7 by treatment with LiBH4 at 23°C for 2–4 h. In either case, the chiral auxiliary was fully recovered without loss of optical activity.
We subsequently investigated substituent effects on the chiral auxiliary as well as the influence of various achiral and chiral bases on diastereoselectivity. As shown in Scheme 3, we have examined ester enolate aldol reactions of N-p-tosyl-(S)-tert-leucinol and N-p tosyl-(S)-leucinol-derived esters and a number of aldehydes. The results of these various aldol reactions are illustrated in Table 2. As can be seen, the sterically demanding tert-leucinol-derived chiral auxiliary exhibited lower yield over leucinol-derived auxiliary; however, stereoselectivities were comparable (entries 1 and 2). The phenylalaninol-derived chiral auxiliary has also shown comparable syn-diastereoselectivity under the reaction conditions described above (entry 3). Interestingly however, the same aldol reaction with fewer equivalents of TiCl4 precomplexed to isovaleraldehyde displayed anti-diastereoselectivity.10 Such reversal in diastereoselectivity is not totally unexpected as the Lewis acid to aldehyde ratio is known to effect aldol stereoselectivity.11 The choice of base is quite important for reaction yield but does not seem to effect observed stereoselectivity (entries 3–5). The chirality on the base has little influence on diastereoselectivities. Bidentate oxyaldehydes are in general very good substrates for ester enolate aldol reactions, providing syn-aldolates in excellent yields and diastereoselectivities (entry 8). The valinol-derived chiral auxiliary has also shown very good syn-diastereoselectivity and reaction yield with isocaproate ester (entry 9).
Scheme 3.

Table 2.
Aldol reaction of various esters with representative aldehydes
| Entry | Ester | Aldehyde | Base (equiv.) | Yield (%)a | syn:anti (9/10)b |
|---|---|---|---|---|---|
| 1 | R1=tBu, R2=Me | Me2CHCHO | DIPEA (3) | 35 | 90:10 |
| 2 | R1=iBu, R2=Me | Me2CHCHO | DIPEA (2.5) | 73 | 85:15 |
| 3 | R1=Bn, R2=Me | Me2CHCHO | DIPEA (2.5) | 86 | 86:14 |
| 4 | R1=iBu, R2=Me | Me2CHCHO | (−)-Spartein (2.2) | 63 | 88:12 |
| 5 | R1=iBu, R2=Me | Me2CHCHO | Et3N (2.5) | 83 | 88:12 |
| 6 | R1=iBu, R2=Me | PhCHO | Et3N (2.5) | 77 | 85:15 |
| 7 | R1=iBu, R2=Me | Ph-C≡C-CHO | Et3N (2.5) | 83 | 14:86 |
| 8 | R1=iBu, R2=Me | BnOCH2CHO | DIPEA (3) | 80 | 99:1 |
| 9 | R1=iPr, R2=iBu | Me2(CH2)2CHO | DIPEA (3) | 97 | 87:13 |
Isolated yield after chromatography.
Ratios determined by 1H NMR before and after chromatography.
In summary, we devised a chelation-controlled ester-derived titanium enolate-based highly diastereoselective syn-aldol reaction with various aldehydes. The current methodology is quite practical due to the ready availability of optically pure chiral auxiliary and use of inexpensive TiCl4 as the key reagent. Further mechanistic investigations, effects of various sulfonamido functionalities and synthetic applications are underway in our laboratories.
Acknowledgements
Financial support for this work was provided by the National Institutes of Health (GM 53386).
References
- 1. (a).Crimmins ST; Choy AL J. Am. Chem. Soc 1999, 121, 5653; [Google Scholar]; (b) Sulikowski GA; Lee W-M; Jin B; Wu B Org. Lett 2000, 2, 1439; [DOI] [PubMed] [Google Scholar]; (c) Roush WR; Pfeifer LA Org. Lett 2000, 2, 859; [DOI] [PubMed] [Google Scholar]; (d) Evans DA; Trotter BW; Coleman PJ; Côté B; Dias LC; Rajapakse HA; Tyler AN Tetrahedron 1999, 55, 8671; [Google Scholar]; (e) Ghosh AK; Fidanze S Org. Lett 2000, 2, 2405; [DOI] [PubMed] [Google Scholar]; (f) Ghosh AK; Bischoff A Org. Lett 2000, 2, 1537; [DOI] [PubMed] [Google Scholar]; (g) Kobayashi S; Hamada T; Nagayama S; Manabe K Org. Lett 2001, 3, 165; [DOI] [PubMed] [Google Scholar]; (h) Andrus MB; Meredith EL; Soma Sekhar BB V. Org. Lett 2001, 3, 259. [DOI] [PubMed] [Google Scholar]
- 2.For syn-aldol reactions, see:Masamune S; Bates GS; Corcoran JW Angew. Chem., Int. Ed. Engl 1977, 16, 585;Evans DA; Bartroli J; Shih TL J. Am. Chem. Soc 1981, 103, 2127;Roder H; Helmchen G; Peters E-M; von Schmering H-G Angew. Chem., Int. Ed. Engl 1984, 23, 898;Masamune S; Choy W; Peterson JS; Sita LR Angew. Chem., Int. Ed. Engl 1985, 24, 1;Jackson RFW; Sutter MA; See-bach D Liebigs Ann. Chem 1985, 2313;Paterson I; Lister MA; McClure CK Tetrahedron Lett 1986, 27, 4787;Corey EJ; Imwinkelreid R; Pikul S; Xiang YB J. Am. Chem. Soc 1989, 111, 5493;Oppolzer W; Blagg J; Rodriguez I; Walther EJ J. Am. Chem. Soc 1990, 112, 2767;Evans DA; Rieger DL; Bilodeau MT; Urpi FJ Am. Chem. Soc 1991, 113, 1047;Ghosh AK; Duong TT; McKee SPJ Chem. Soc., Chem. Commun 1992, 1673;Abiko A; Liu J-F; Masamune SJ Org. Chem 1996, 61, 2590;Crimmins MT; Chaudhary K Org. Lett 2000, 2, 775 and references cited therein.
- 3.For anti-aldol reactions, see:Meyers AI; Yamamoto Y Tetrahedron 1984, 40, 2309;Helm-chen G; Leikauf U; Taufer-Knopfel I Angew. Chem., Int. Ed. Engl 1985, 24, 874;Gennari C; Bernardi A; Colombo L; Scolastico CJ Am. Chem. Soc 1985, 107, 5812;Oppolzer W; Marco-Contelles J Helv. Chim. Acta 1986, 69, 1699;Masamune S; Sato T; Kim BM; Wollman TA J. Am. Chem. Soc 1986, 108, 8279;Danda H; Hansen MM; Heathcock CH J. Org. Chem 1990, 55, 173;Corey EJ; Kim SS J. Am. Chem. Soc 1990, 112, 4976;Corey EJ; Kim SS Tetrahedron Lett 1990, 31, 3715;Meyers AG; Widdowson KL J. Am. Chem. Soc 1990, 112, 9672;Duthaler RO; Herold P; Helfer S-W; Riediker M Helv. Chim. Acta 1990, 73, 659;Walker MA; Heathcock CH J. Org. Chem 1991, 56, 5747;Braun M; Sacha H Angew. Chem., Int. Ed. Engl 1991, 30, 1318;Gennari C; Moresca D; Vieth S; Vulpetti A Angew. Chem., Int. Ed. Engl 1993, 32, 1618;Paterson I; Wren SPJ Chem. Soc., Chem. Com-mun 1993, 1790;Abiko A; Liu J-F; Masamune SJ Am. Chem. Soc 1997, 119, 2586;Kurosu M; Lorca MJ Org. Chem 2001, 66, 1250 and references cited therein.
- 4. (a).Ghosh AK; Kim J-H Tetrahedron Lett 2001, 42, 1227; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghosh AK; Onishi MJ Am. Chem. Soc 1996, 118, 2527; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ghosh AK; Fidanze S; Hussain KA Tetrahedron Lett 1997, 38, 7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Optical rotation of corresponding carboxylic acids, diols and methylesters (CHCl3 solvent for all, unless otherwise noted), 5g: (c 1.1), lit.:4c (c 3.7); 7a: (c 0.68), lit.:6 (c 0.2); 7c: (c 0.83, MeOH), lit.:7 (c 0.55, MeOH); 6b: (c 0.48), sample prepared from Evans aldol: (c 0.13).
- 6.Wood RD; Ganem B Tetrahedron Lett 1982, 23, 707. [Google Scholar]
- 7.Meyer HH Liebigs Ann. Chem 1984, 791. [Google Scholar]
- 8. All new compounds gave satisfactory spectroscopic and analytical results. Preparation of syn-aldol 3b: To a stirred solution of propionate ester 2 (313 mg, 1 mmol) in CH2Cl2 (10 mL) at 0°C was added a 1 M solution of TiCl4 (1.1 mL, 1.1 mmol) dropwise under a N2 atmosphere. The resulting solution was stirred for 10 min. To this solution was added N,N-diisopropylethylamine (0.52 mL, 3 mmol) in a dropwise manner. The mixture was stirred for 1 h at 0°C and then warmed to 23°C. In a separate flask, to a stirred solution of isovaleraldehyde (172 mg, 2 mmol) in CH2Cl2 (20 mL) at −78°C under N2 atmosphere, was added a 1 M solution of TiCl4 (3 mL, 3 mmol). After stirring for 10 min, the above enolate solution was added to this aldehyde solution dropwise via cannula over 30 min. The mixture was stirred at −78°C for 1.5 h before quenching by aqueous NH4Cl. The resulting mixture was warmed to 23°C and the layers were separated. The aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure to afford the aldol products. Silica gel chromatography of the crude product yielded the aldol product (363 mg, 93%) as a viscous oil. Compound 3b: (c 1.58, CHCl3); 1H NMR (400 MHz, CDCl3): δ 0.81 (d, 6H, J=6.9 Hz), 0.84 (d, 6H, J=6.9 Hz), 1.10 (d, 3H, 7.1 Hz), 1.43 (m, 1H), 1.77 (m, 2H), 2.38 (m, 2H), 2.41 (s, 3H), 2.48 (br, 1H), 3.34 (m, 1H), 3.95 (dd, 1H, J=11.6, 4.5 Hz), 4.01 (m, 1H), 4.06 (dd, 1H, J=11.6, 5.9 Hz), 5.17 (d, 1H, J=9.0 Hz), 7.28 (d, 2H, J=8.3 Hz), 7.75 (d, 2H, J=8.3 Hz); 13C NMR (100 MHz, CDCl3): δ 10.1, 18.3, 18.9, 21.5, 21.9, 23.4, 24.6, 30.0, 42.9, 44.7, 57.9, 64.0, 69.6, 127.0, 129.6, 138.2, 143.3, 175.8; IR (neat): 3521, 3286, 1730, 1327, 1161 cm−1.
- 9. (a).Takai K; Heathcock CH J. Org. Chem 1985, 50, 3247; [Google Scholar]; (b) Fuganti C; Grasselli P; Servi S; Spreafico F; Zirotti CJ Chem. Res 1984, 976. [Google Scholar]
- 10. Reaction of titanium enolate of 8 (R1=Bn, R2=Me) with isovaleraldehyde (2 equiv.) precomplexed with 1.05 equiv. of TiCl4 at −78°C afforded a mixture of anti and syn diastereomers (70% yield). Removal of chiral auxiliary provided anti/syn ratio of 68:32. We have previously reported a 70:30 anti/syn ratio and 70% yield for this reaction. See: Ref.4a.
- 11. (a).Walker MA; Heathcock CH J. Org. Chem 1991, 56, 5747; [Google Scholar]; (b) Oppolzer W; Lienard P Tetrahedron Lett 1993, 34, 4321. [Google Scholar]