

Abstract
A first approach to study the function of an unknown gene in bacteria is to create a knock-out of this gene. Here, we describe a robust and fast protocol for transferring gene deletion mutations from one Escherichia coli strain to another by using generalized transduction with the bacteriophage P1. This method requires that the mutation be selectable (e.g., based on gene disruptions using antibiotic cassette insertions). Such antibiotic cassettes can be mobilized from a donor strain and introduced into a recipient strain of interest to quickly and easily generate a gene deletion mutant. The antibiotic cassette can be designed to include flippase recognition sites that allow the excision of the cassette by a site-specific recombinase to produce a clean knock-out with only a ~100-base-pair-long scar sequence in the genome. We demonstrate the protocol by knocking out the tamA gene encoding an assembly factor involved in autotransporter biogenesis and test the effect of this knock-out on the biogenesis and function of two trimeric autotransporter adhesins. Though gene deletion by P1 transduction has its limitations, the ease and speed of its implementation make it an attractive alternative to other methods of gene deletion.
Keywords: Genetics, Issue 139, Antibiotic resistance cassette, deletion mutagenesis, FLP recombinase, P1 transduction, translocation and assembly module, trimeric autotransporter adhesin
Introduction
A common first approach to study the function of a gene is to perform knock-out mutagenesis and observe the resulting phenotype. This is also termed reverse genetics. The bacterium E. coli has been the workhorse of molecular biology for the last 70 years or so, due to the ease of its culturing and its amenability to genetic manipulation1. Several methods have been developed to produce gene deletions in E. coli, including marker exchange mutagenesis2,3 and, more recently, recombineering using the λ Red or Rac ET systems4,5,6.
In a widely used system, coding sequences of individual genes are replaced by an antibiotic resistance cassette that can later be excised from the chromosome5,7. The coding sequences are replaced, for instance by a kanamycin (Kan) resistance cassette, which is flanked by flippase (FLP) recognition target (FRT) sites on either side. The FRT sites are recognized by the recombinase FLP, which mediates site-specific recombination between the FRT sites leading to the deletion of the Kan cassette. In this way, a full deletion of a given gene's coding sequence can be achieved, leaving behind only a minimal scar sequence of approximately 100 base pairs (bp) (Figure 1).
Just over a decade ago, the so-called Keio collection was developed. This is a bacterial library based on a standard laboratory E. coli K12 strain, where almost all non-essential genes were individually deleted by λ Red recombination7,8. The clones within this collection each have one coding sequence replaced with an excisable Kan resistance cassette. The Keio collection has proven to be a useful tool for many applications9. One such application is the production of deletion mutants in other E. coli strains. The Kan cassette from a given deletion clone can be mobilized by generally transducing bacteriophages, such as P110,11,12,13,14. A phage stock prepared from such a strain can then be used to infect a recipient E. coli strain of interest, where at a low but reliable frequency the Kan cassette-containing region can be incorporated into the recipient genome by homologous recombination (Figure 2). Transductants can be selected for the growth on the Kan-containing medium. Following this, if removal of the antibiotic resistance cassette is desired, the FLP recombinase can be supplied to the transductant strain in trans. After curing the FLP-containing plasmid, which carries an ampicillin (Amp) resistance marker, Kan and Amp-sensitive clones are screened for, and the correct excision of the wild-type coding sequence and the Kan cassette are verified by colony PCR.
Here, a detailed protocol is presented, describing each of the steps in producing a knock-out E. coli strain based on the strategy outlined above. As an example, a deletion of the tamA gene is demonstrated. tamA encodes an outer membrane β-barrel protein that is a part of the Transport and Assembly Module (TAM), which is involved in the biogenesis of certain autotransporter proteins and pili15,16,17. This knock-out strain was then used to examine the effect of the tamA deletion on the biogenesis of two trimeric autotransporter adhesins (TAAs), the Yersinia adhesin YadA and the E. coli immunoglobulin (Ig)-binding TAA EibD18,19.
Protocol
1. Strains and Plasmids
- Bacteriophages
- Use the phage P1vir for the general transduction. Store the phage as a liquid stock with a few drops of chloroform (see step 2.2). For more information, see Table of Materials.
- Growth conditions
- Propagate bacteria in a lysogeny broth (LB) medium25 with vigorous shaking (180–200 rpm) at 37 °C or 30 °C in the case of BL21ΔABCF and strains containing pCP20.
- Perform plasmid curing at 43 °C.
- For a solid medium, supplement LB with 1% agar (w/v).
- For top agar, supplement LB with 0.7% agar and 10 mM CaCl2 and autoclave the medium. Use SOC medium for the recovery after electroporation26.
- Use the following concentrations for antibiotics: 100 μg/mL for Amp and 25 μg/mL for Kan.
2. Preparing a Phage Lysate
- Infection of the donor strain
- Grow the donor strain JW4197 in 5 mL of LB medium supplemented with 10 mM CaCl2 and optionally with Kan (25 µg/mL) to an optical density at 600 nm (OD600) of ~1.0. Measure the OD600 value using a spectrophotometer.
- Make a dilution series of an existing P1 phage stock in the LB medium: recommended dilutions are between 10-3 to 10-7.
- Mix 200 μL of the bacterial suspension and 100 μL of a given phage dilution in a 15 mL centrifuge tube or equivalent. Prepare as many tubes as phage dilutions. Incubate the tubes for 20 min at 37 °C without shaking.
- Add ~3 mL of molten top agar (~50 °C) supplemented with 10 mM CaCl2 to the tubes, mix the contents thoroughly by vortexing the tubes shortly, and pour the mixtures onto prewarmed LB plates to make even layers.
- Incubate the plates overnight at 37 °C.
- Lysate preparation
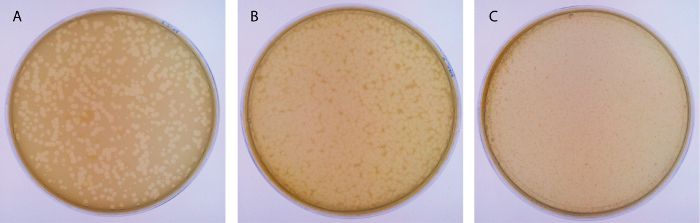
- The following day choose a plate with a semi-confluent growth of phage plaques. On a semi-confluent plate, approximately half the surface area of the plate is clear (Figure 3).
- Scrape the top agar layer from such a plate using an inoculation loop or a similar tool and place the top agar in a centrifuge tube. Add 1–2 mL of LB and a drop of chloroform and vortex the tube vigorously for ~1 min. Add the chloroform in a fume hood.
- Centrifuge the tube for 15 min at 4,000 x g or faster to pellet the agar and bacterial cells.
- Move the supernatant to a fresh microcentrifuge tube, avoiding carrying over any debris from the pellet. Add 2 drops of chloroform and store the lysate at 4–10 °C. Add chloroform in a fume hood. Do not freeze the phage lysate as this will result in a significant reduction of the number of infectious particles.
- Determining the lysate titer
- Grow BW25113 in LB supplemented with 10 mM CaCl2 at 37 °C till the culture reaches an OD600 of ~1.0.
- Prepare a dilution series of the phage lysate in LB (e.g., 10-6–10-9). NOTE: Be careful to pipet the sample from the top of the lysate to avoid transferring chloroform to the dilutions.
- Mix 200 μL of the bacterial suspension and 100 μL of a given phage dilution in a 15 mL centrifuge tube or equivalent. Prepare as many tubes as phage dilutions. Incubate the tubes for 20 min at 37 °C without shaking.
- Add ~3 mL of molten top agar (~50 °C) supplemented with 10 mM CaCl2 to the tubes, mix the contents thoroughly by vortexing the tubes shortly, and pour the mixture onto prewarmed LB plates to make even layers.
- Incubate the plates overnight at 37 °C.
- The following day count the number of plaques (clear regions in the bacterial mat) for each plate and calculate the titer of the lysate using the following formula:
 Example: On plates with the dilutions 10-7, 10-8, and 10-9, there are 231, 27, and 2 plaques, respectively. As 100 μL of each dilution was used for the infection, and because the titer is expressed as plaque-forming units (pfu)/mL, the plating factor is 10. These numbers are entered into the formula:
Example: On plates with the dilutions 10-7, 10-8, and 10-9, there are 231, 27, and 2 plaques, respectively. As 100 μL of each dilution was used for the infection, and because the titer is expressed as plaque-forming units (pfu)/mL, the plating factor is 10. These numbers are entered into the formula: 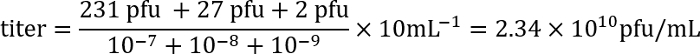 Thus, the titer is approximately 2 x 1010 infectious phage particles/mL.
Thus, the titer is approximately 2 x 1010 infectious phage particles/mL.
3. P1 Transduction
- Preparing the recipient cells
- Grow the recipient strain BL21ΔABCF in LB supplemented to an optical density at 600 nm (OD600) of ~1.0. Use a spectrophotometer to measure the OD600 value.
- Calculate the volume of the phage lysate needed to achieve a multiplicity of infection (MOI) value of 0.5. To calculate the MOI, estimate the number of bacteria based on the OD600 of the culture. Assume that an OD600 value of 1.0 corresponds to ~109 cfu/mL. Calculate the volume required based on the known titer of the lysate. Example (based on the titer calculated in the example after step 2.3.6):
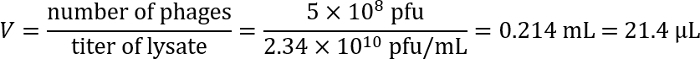
- Add CaCl2 to the recipient strain culture to 10 mM and mix. Take 1 mL of the culture for transduction.
- Performing transduction
- Add the appropriate volume of phage lysate to 1 mL of recipient culture (including CaCl2 at 10 mM) as calculated in step 3.1.2 and mix gently. NOTE: Be careful to pipette the sample from the top of the lysate to avoid transferring chloroform to the mix.
- Statically incubate the mix for 20 min at 37 °C.
- Stop the infection by adding sodium citrate, pH 5.5, to 100 mM.
- Centrifuge the bacteria (5,000 x g for 2 min) and remove the supernatant, then resuspend them in 1 mL of fresh LB supplemented with 100 mM sodium citrate, pH 5.5.
- Wash the cells twice more as in step 3.2.4 to ensure the removal of free phages and calcium.
- Resuspend the bacteria in 1 mL of fresh LB supplemented with 100 mM sodium citrate, pH 5.5. Incubate the bacteria at 30 °C for 1 h with shaking (> 100 rpm).
- Collect the bacteria by centrifugation (5,000 x g for 2 min) and resuspend them in ~100 μL of LB with 100 mM sodium citrate, pH 5.5.
- Spread the bacteria on an LB plate supplemented with Kan at 25 µg/mL and 10 mM sodium citrate, pH 5.5, and grow the bacteria at 30 °C until colonies appear (~24 h).
- Selecting the transductants
- Once colonies have grown on the selection plate, restreak them on LB + Kan for single colonies and grow at 30 °C till single colonies appear.
4. Excising the Kan cassette
- The transformation with recombination plasmid
- Make the Kan-resistant BL21ΔABCF strain electrocompetent.
- Grow the strain from a single colony in fresh LB (5 mL) supplemented with Kan (25 µg/mL) at 30 °C until the OD600 is ~0.5–0.7.
- From here on, carry out the following steps at 4 °C or on ice. Centrifuge the bacteria (5,000 x g for 10 min) and remove the supernatant.
- Wash the cell pellet with ice-cold distilled water twice by repeating the previous centrifugation step and, finally, resuspend cell pellet in 100 µL of ice-cold 10% glycerol.
- Cool down 1 mm electroporation cuvettes on ice.
- Add 1 pg of plasmid DNA (pCP20) to the cell suspension, mix the suspension gently, and transfer it to a cooled electroporation cuvette.
- Set the electroporator to 1.8 kV and electroporate the cells.
- Rescue transformed cells by adding 1 mL of SOC medium and growing them for 1 h at 30 °C.
- Plate 100 µL of the cells on LB + Amp plates and grow the cells at 30 °C overnight.
- Induction of the recombination
- Pick single colonies from the LB + Amp plate and inoculate fresh LB omitting all antibiotics.
- Grow the cells at the non-permissive temperature (43 °C) overnight to induce the expression of FLP recombinase.
- Selecting recombinants
- Make serial dilutions and plate 50 µL of a 105-106 dilution on non-selective plates and grow it overnight at 30 °C.
5. Verification of the Gene Deletion
- Verification of the plasmid loss and successful recombination
- Streak single colonies from the plates prepared in step 4.3.1 on LB + Kan, LB + Amp, and LB plates without antibiotics, in this order. To aid in the streaking, use a colony grid (see Supplementary File 1). Grow the plates at 30 °C until colonies appear (~24 h).
- Pick 10–20 clones that have grown on the non-selective plate but failed to grow on the selective media for further verification.
- Additional verification by colony PCR
- Perform a colony PCR with primers flanking the tamA coding sequence (see Table of Materials).
- Prepare a master PCR mix. The amount of mix needed depends on the number of colonies to be tested (see the example in Table 1). Mix the reagents on ice, add the polymerase last.
- Mix the PCR master mix thoroughly, then dispense 20 µL into tubes of a PCR strip. Pick a small amount of each colony to be screened using a sterile pipette tip and add it to a tube. Remember to include the original recipient strain for comparison. Optionally, also include the donor strain.
- Run a PCR reaction (see Table 2 for the program used).
- Prepare a 1% agarose gel.
- For a 50 mL gel, measure 0.5 g of agarose and add 50 mL of TAE buffer (40 mM Tris, 20 mM acetic acid, and 1 mM EDTA, pH 8.0). Heat the mixture in a microwave oven until all agarose has dissolved.
- Once the agarose has dissolved, cool the solution to approximately 50 °C, and then add 5 μL of DNA staining dye. Mix the solution well and pour it into a gel tray in a casting chamber. Insert one or more rows of well combs so that there are sufficient wells for all samples, as well as molecular size markers. Allow the gel to set for 30 min.
- Once the PCR run is complete, add 4 µL of 6x DNA loading dye to each sample. Place the gel in the electrophoresis chamber and add TAE buffer till the wells are covered.
- Apply 10–15 µL from each PCR reaction to a well in the gel. Also add 5 µL of a molecular size marker. Then, run the gel for 30 min at 75 V.
- Once the run is complete, image the gel using a blue-light imager.
6. Other Techniques
- Protein expression NOTE: The expression of test proteins (YadA and EibD) has been described in detail elsewhere23,24. This is a brief summary of the main steps.
- Transform BL21ΔABCF ΔtamA with the necessary plasmids (pIBA2-YadA and pEibD10 and the corresponding control plasmids) and select for transformants on LB + Amp.
- For the protein expression, inoculate 100 mL of LB medium + Amp with 1 mL of an overnight culture of transformed bacteria and grow these at 30 °C until mid-log phase, at which time, induce the protein production with either anhydrotetracycline (at 100 ng/mL) or isopropyl thiogalactoside (at 0.5 mM).
- After 2 h of induction at 30 °C, measure the turbidity of the cultures and collect a number of cells corresponding to 50 mL at OD600 = 1.0 by centrifuging the cultures for 15 min at 4,000 x g. Wash the pellet 1x with 10 mM HEPES, pH 7.4, and then either store it at -20 °C or process it further as in step 6.2.
- Outer membrane extraction NOTE: Outer membrane extraction is performed as described in detail by Leo et al.27. The main steps are summarized below.
- Resuspend the cell pellet in 1 mL of 10 mM HEPES, pH 7.4, supplemented with 10 mM MgCl2 and MnCl2, lysozyme (0.1 mg/mL), and a pinch of DNase I.
- Lyse the cells (e.g., using a bead beater).
- Centrifuge the cells shortly (2 min at 15,600 x g) to remove cell debris and move the supernatant to a fresh tube.
- Centrifuge the cells for 30 min at 16,000 x g, after which, resuspend the pellet in 400 μL of 1% N-lauroyl sarcosine in 10 mM HEPES, pH 7.4.
- Incubate the cells with agitation for 30 min at room temperature, after which, centrifuge them for 30 min as above.
- Wash the translucent pellet 1x with 200 μL of 10 mM HEPES, pH 7.4, and then resuspend it in 30 μL of 10 mM HEPES and add 10 μL of 4x SDS-PAGE sample buffer.
Activity assays NOTE: Perform SDS-PAGE and activity assays for YadA and EibD as described28. The main steps are summarized below.
- SDS-PAGE
- Heat the samples at 50 °C for 5 min before loading them onto a polyacrylamide gel, to avoid denaturing the proteins.
- After the separation in SDS-PAGE, transfer the proteins to a polyvinylidene difluoride (PVDF) membrane.
- After the transfer, block the membrane with 2% skimmed-milk powder in PBS.
- YadA-collagen far-western blot
- After the blocking, add bovine collagen type I diluted in blocking buffer to a concentration of 10 μg/mL and incubate the membrane for 1 h.
- Wash the membrane 2x with PBS + 0.05% Tween20 (PBS-T).
- Add the primary antibody (monoclonal anti-collagen COL-1) to the membrane, diluted 1:2,000 in blocking buffer.
- After incubating the membrane for 1 h, wash 2x as mentioned in step 6.3.2.2 and then add the secondary antibody [goat anti-mouse IgG-horseradish peroxidase (HRP) conjugate], diluted 1:10,000 in blocking buffer.
- Incubate the membrane for 1 h, then wash it 2x with PBS-T. Add an enhanced chemiluminescent substrate to the membrane according to the manufacturer’s instructions and detect the band using a CCD camera.
- EibD-IgG binding assay
- After the blocking, add a secondary antibody (goat anti-rabbit HRP), diluted 1:2,000 in blocking buffer.
- Incubate the membrane for 1 h, then wash it 2x with PBS. Perform chemiluminescent detection as mentioned in step 6.3.2.5.
Representative Results
Generation of a tamA Knock-out of BL21ΔABCF:
The strategy outlined above has previously been used to produce a derivative strain of BL21(DE3), a standard laboratory strain used for protein production, which is optimized for outer membrane protein production and called BL21ΔABCF21. This strain lacks four genes coding for abundant outer membrane proteins and, consequently, is able to produce more heterologously expressed outer membrane proteins than the wild-type strain. To test whether the TAM is involved in TAA biogenesis, the tamA gene was deleted in this background.
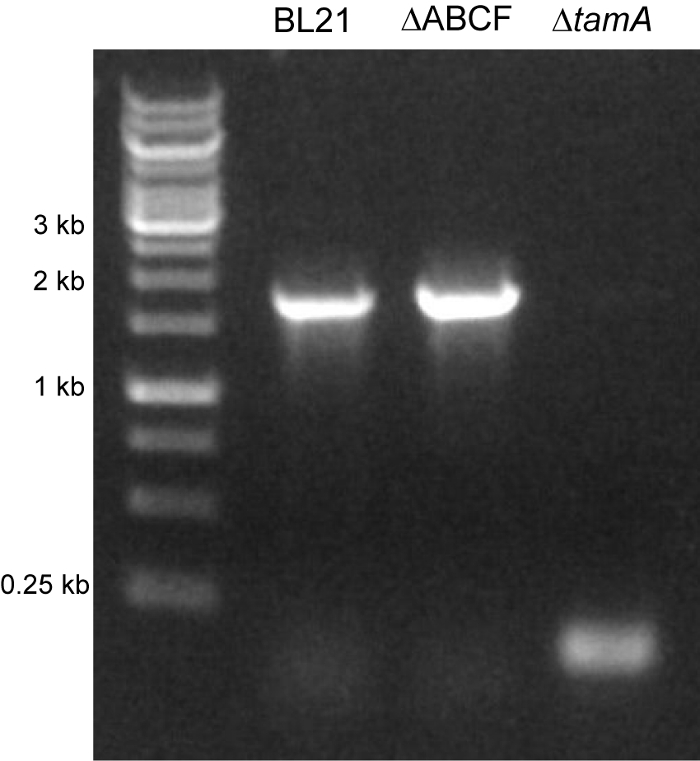
A P1 lysate was prepared from the Keio collection strain JW4179, where the tamA gene (previously called yftM) coding sequence is replaced by a Kan resistance cassette. Then, a transduction experiment was performed with BL21ΔABCF as the recipient strain. Several Kan-resistant colonies were obtained, after which two were chosen for the excision of the Kan cassette. The plasmid pCP20, encoding the FLP recombinase, was introduced into these clones and, subsequently, cured by growing at 43 °C in the absence of an antibiotic selection. A number of clones were screened for sensitivity to both Amp (which is a marker of pCP20) and Kan, and several clones sensitive to both were obtained. These clones were verified by colony PCR using primers flanking the tamA coding sequence and found that the tamA gene had successfully been deleted (Figure 4).
TamA's Role in TAA Biogenesis:
TamA has been shown to be involved in the biogenesis of some classical autotransporters16 and the inverse autotransporter intimin15. To test whether TamA is important for the biogenesis of TAAs, BL21ΔABCF ΔtamA was transformed with plasmids encoding two test proteins, the Yersinia adhesin YadA and the E. coli Ig-binding protein EibD. These proteins are known to express well in E. coli and have been used as models for TAA biogenesis in earlier studies23,28.
After inducing protein production, the outer membrane fractions of the expression cultures were isolated and analyzed by SDS-PAGE. The samples were not boiled to demonstrate trimerization of the proteins. The trimers run at sizes above 100 kDa, whereas the monomers have expected sizes of 45 kDa (YadA) and 51 kDa (EibD). No major differences were observed between the expression levels in BL21ΔABCF and BL21ΔABCF ΔtamA, although YadA seems to be produced at somewhat lower levels in the ΔtamA strain (Figure 5A). However, the opposite appears to be the case for EibD.
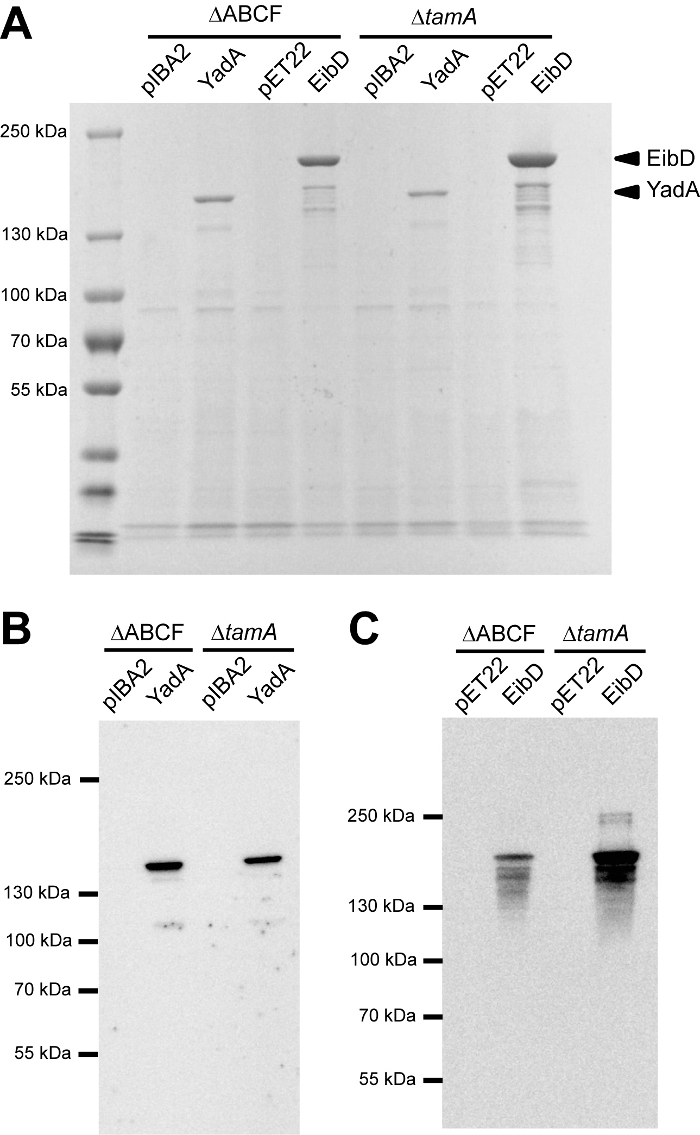
To examine whether the lack of TamA might influence the correct folding or transport of the proteins, their ability to bind to ligands was tested. For YadA, this was accomplished by a collagen far-western blot (Figure 5B). YadA, in both strains, bound collagen at a similar level, demonstrating that the protein is correctly folded and functional. Similarly, IgG-binding activities of EibD in the two strains correlated with the expression level (Figure 4C). These results demonstrate that the deletion of tamA does not have a significant effect on TAA biogenesis, at least not for these two model TAAs.
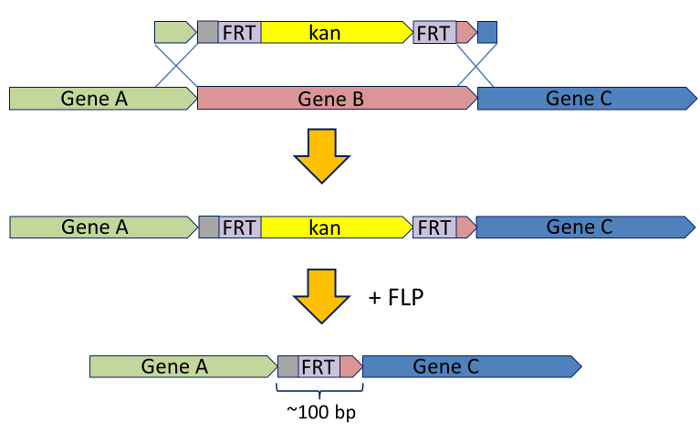
Figure 1: Generation of knock-outs with excisable antibiotic cassettes. For producing the gene knock-outs, a gene of interest (Gene B in this example) is replaced by a Kan resistance cassette flanked by FRT sites. The FRT-Kan cassette, in turn, is flanked by short (~50 bp) stretches of sequence homologous to the upstream and downstream regions of the Gene B. The coding sequence of Gene B is exchanged for the FRT-Kan cassette by λ Red recombination. Once this is accomplished, the Kan cassette itself can be removed by introducing the FLP recombinase, which will mediate a site-specific recombination between the FRT sites. This excises the Kan cassette, leaving a minimal (~100 bp) scar sequence in the B locus. For full details, see Baba et al.7. Please click here to view a larger version of this figure.
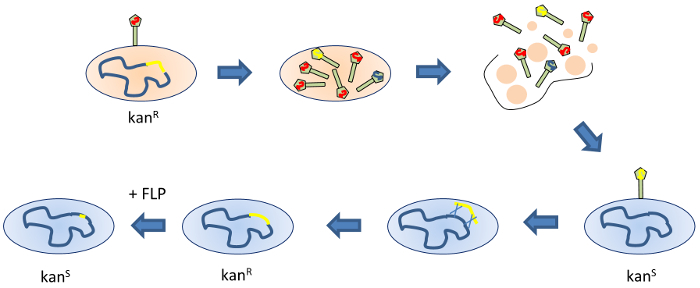
Figure 2: Gene deletion by P1 transduction. The donor strain (beige) carries a Kan cassette that has replaced the gene of interest (yellow) on the chromosome (blue). The donor is infected with P1 bacteriophage. The phage multiplies in the donor strain, producing a large number of progenies. Most are wild-type (red genome), but a fraction are transducing phages that have incorporated a portion of the donor strain chromosome rather than phage DNA (blue genome). A proportion of these will contain the Kan cassette (yellow genome). Eventually, the infected host cell lyses and releases the phages into the medium. These are used to prepare a lysate. In the transduction experiment, the recipient strain (light blue) is infected with the lysate prepared from the donor strain. In a minority of cases, the recipient is infected by a transducing phage carrying the Kan cassette (shown here). If the regions adjacent to the Kan cassette undergo homologous recombination, the Kan cassette is incorporated into the recipient chromosome replacing the endogenous allele, resulting in Kan-resistant clones that can be selected for. The addition of FLP recombinase on a curable plasmid excises the Kan cassette, leaving only a short scar sequence (shown here in yellow), which reverts the clone to the Kan-sensitive phenotype. Please click here to view a larger version of this figure.
Figure 3: Examples of plates after a P1 infection. (A) This panel shows a plate with individual plaques. (B) This panel shows a semi-confluent plate. (C) This panel shows an over-infected plate, where almost all the bacteria have been lysed by phage. Some resistant colonies have grown out of the top agar layer. Please click here to view a larger version of this figure.
Figure 4: Deletion of the tamA coding sequence. The tamA::kan deletion allele was introduced into the strain BL21ΔABCF by P1 transduction. After the excision of the Kan cassette, a scar sequence (~100 bp) is all that remains at the tamA locus. This was verified by PCR, using primers flanking the deletion site. In BL21ΔABCF and its parent strain, BL21(DE3), the PCR gives a product the length of the tamA coding sequence (the expected size is 1.7 kilobase pairs). In BL21ΔABCF, where the Kan cassette has been excised, the product corresponds to the expected scar sequence (145 bp). Please click here to view a larger version of this figure.
Figure 5: Expression of TAAs by a tamA deletion strain. (A) This panel shows the SDS-PAGE of outer membranes prepared from cells expressing TAAs (YadA or EibD) and vector controls (pIBA2 and pET22). The strains are BL21ΔABCF and its derivative strain lacking TamA (ΔtamA). (B) This panel shows a collagen far-western blot of YadA samples. The YadA samples and pIBA2 controls from panel A were transferred to a PVDF membrane and incubated with collagen type I. They were then probed with an anti-collagen antibody and detected by ECL. (C) This panel shows an antibody binding assay for EibD samples. The EibD samples and pET22 controls from panel A were transferred to a PVDF membrane and incubated with an HRP-conjugated secondary antibody and then detected by ECL. Please click here to view a larger version of this figure.
| Reagent | 1x mix | 7x mix |
| PCR-grade water | 17 µL | 119 µL |
| 10 x polymerase buffer | 2 µL | 14 µL |
| 10 mM deoxyribonucleotide mix | 0.4 µL | 2.8 µL |
| 100 µM forward primer | 0.2 µL | 1.4 µL |
| 100 µM reverse primer | 0.2 µL | 1.4 µL |
| Taq DNA polymerase | 0.2 µL | 1.4 µL |
| Total | 20 µL | 140 µL |
Table 1: Colony PCR master mix. The amount of mix depends on the number of colonies to be screened. In addition, prepare a control reaction (original recipient strain). For instance, if screening five clones, six reactions are needed, including the control. It is worth preparing an extra reaction to make sure there is enough PCR mix for all samples (repeated pipetting amplifies small pipetting errors). In this example, prepare a 7x mix (5 colonies, 1 control, and 1 extra reaction).
| Step | Temperature | Time | Notes |
| 1. | 94 °C | 3 min | |
| 2. | 94 °C | 30 s | |
| 3. | 50 °C | 30 s | |
| 4. | 70 °C | 2 min | 1 min/kb to be amplified, round up |
| Return to step 2 24 times | |||
| 5. | 70 °C | 5 min | |
| 6. | 12 °C | for ever | final hold |
Table 2: Colony PCR program.
Supplementary File 1: An example of colony grid. Please click here to download this file.
Discussion
P1 transduction is a fast, robust, and reliable method for generating gene deletions in E. coli. This is demonstrated here by transducing a tamA deletion mutant from a Keio donor strain to a BL21-derived recipient. The major stages in the transduction process are the production of the transducing lysate, the transduction itself, the excision of the Kan resistance cassette, and the verification of the knock-out by PCR. In total, the process takes approximately 1 week and requires no molecular biology methods to be used, apart from the final PCR for the verification. Thus, P1 transduction can compete in expended effort and time with λ Red recombination and is much faster than traditional marker exchange mutagenesis.
The presented protocol is very robust and allows for modifications of many of the steps. There are, however, a few critical parameters. For P1 infections, it is necessary to add calcium ions to the medium. Calcium is needed for the adsorption of the phage to the bacteria, and failure to add sufficient calcium to the medium will significantly reduce the efficiency of the infection. Some bacterial strains such as BL21ΔABCF tend to aggregate in the presence of CaCl221. In such cases, the bacteria can be grown without CaCl2, which can be added to the suspension shortly before the infection. However, for more conventional strains, 10 mM CaCl2 can be included in the growth medium from the beginning.
Conversely, it is important to remove the free calcium from the medium after the transduction. Citrate is a chelator of calcium ions, and because these are needed for the adsorption of P1 to host cells, removing the free calcium from the medium prevents further infections. If the calcium is not removed, the phages will infect further cells throughout the culture, at best reducing the efficiency of the transduction and at worst lysing the whole culture, including the transductants.
Another critical step is culturing bacteria carrying the plasmid pCP20. pCP20 is a conditionally replicating plasmid that does not replicate at temperatures of 37 °C or higher; thus, to establish the plasmid in the cells, incubations with this plasmid must be performed at 30 °C (or lower), a temperature permissive for the replication of pCP20. For curing pCP20, a high temperature (43 °C) is used. Some strains do not grow well at this temperature; in such cases, 37 °C should suffice, although the plasmid curing will be somewhat less efficient at this temperature.
When plating bacteria to test for antibiotic sensitivity, the order of plate streaking is important. The protocol calls for clones to be streaked first on antibiotic-containing plates and finally on non-selective medium. In this way, the investigator can be sure that any lack of growth on the selective media will be due to antibiotic sensitivity and rather than to a potential lack of material transferred on the plates. Following the protocol, no growth on the LB + Kan plate validates the excision of the Kan resistance cassette; no growth on the LB + Amp plate validates the loss of the recombination plasmid pCP20; strains growing on the LB plate (which did not grow in the same streaking experiment on the selection plates) will contain the positive recombinants.
Most of the other steps allow for considerable leeway. In the protocol as presented, BL21ΔABCF is cultured at 30 °C, as this strain does not grow well at 37 °C. However, E. coli strains without growth defects may be cultured at 37 °C (except when transformed with pCP20).
The number of bacteria used for infections can be varied to some extent. The relationship between OD600 and a viable count is roughly linear between an OD600 value of 0.1 and 1.0, where the former corresponds to approximately 108 cfu/mL and the latter to ~109 cfu/mL. However, this relationship may vary to some extent depending on the strain in question, the growth medium, and other factors. It is recommended that the relationship between OD600 and the viable count should be established for each laboratory and strain, particularly for calculating MOI values. The number of bacteria used in infection experiments is not particularly critical, and 109 cfu/mL represents the early stationary phase, which is a reasonable compromise between cell density and the proportion of viable cells in the culture. If a lower number of viable bacteria are used for transduction, the number of phages simply needs to be adjusted accordingly. The MOI itself can also be varied to some extent. The protocol calls for an MOI value of 0.5, although anything between 0.1 and 0.5 should result in a good efficiency. At an MOI of 0.5, the ratio of phages to bacteria is 1:2 (i.e., half the number of phages compared to the number of bacteria). In the example in the protocol, the OD600 of the culture is 1.0, and 1 mL of the culture is used for the transduction; thus, the number of phages required is 5 x 108. An MOI value of 0.5 gives a high level of infection but reduces the number of bacteria that are doubly infected, which would reduce the efficiency of the transduction as the wild-type (infectious) phages far outnumber the transducing phages. A double infection with a transducing phage and an infectious phage would lead to cell lysis, thus eliminating this transductant from the pool of survivors. Therefore, an MOI of 0.5 should not be exceeded.
The protocol also allows for some shortcuts. Rather than preparing lysates from plates, some authors advocate preparing phage lysates in liquid medium29. This can save time as an overnight incubation step is not needed. Similarly, titrating the phage lysate might not be necessary. As noted above, the MOI is not very critical, and reasonable efficiency can be achieved by simply assuming a titer of 1010 pfu/mL for a standard lysate.
Despite the ease of the technique, P1 transduction is not universally applicable, and several conditions must be met for it to be useful. Firstly, a donor strain with a selectable knock-out allele must be available. This is usually accomplished by using a deletion where an antibiotic resistance cassette has replaced the gene of interest. The Keio collection is particularly useful in this regard, as it offers a ready-to-use library of antibiotic cassette-based gene knock-outs covering almost all non-essential genes in E. coli K12. This collection is particularly useful for knocking out conserved genes found in most E. coli strains (i.e., constituents of the E. coli core genome). For less common genes, such as virulence factors of pathogenic E. coli strains or rare metabolic pathway genes, the mutation may need to be created de novo. In such cases, P1 transduction may well not be the method of choice. In addition to essential genes, genes that are required for P1 infection, such as galU, mutations of which lead to P1 resistance30, are poor targets for a deletion by P1 transduction. Another note when using the Keio collection, specifically, is that a few strains carry a duplication of the targeted gene, where only one copy was disrupted by the Kan resistance cassette8. In such cases, the gene of interest may be essential; investigators are recommended to check updated annotations for such genes8. However, given these restraints, P1 transduction allows the deletion of most genes in laboratory E. coli strains. For example, the differences between BW25113 and BL21(DE3) are small and affect only a handful of protein-coding genes31.
Secondly, it is important to emphasize that the recipient strain must be capable of a homologous recombination for the transduction to work. A strain lacking the recombinase RecA can, therefore, not be modified by this method. This excludes all standard cloning strains, such as DH5α, HB101, TOP10, and XL-1 Blue. RecA can mediate recombinations between identical stretches as short as 8 bp; however, a longer region of high similarity will increase the probability of recombination significantly32,33. Another problem, particularly with clinical E. coli strains, is that long O-antigen chains may mask the receptor for P1, which lies in the core oligosaccharide of lipopolysaccharide34. In addition to these requirements for the recipient strain, the P1 strain used for transduction should be a vir mutant; this mutation is required for a full lytic infection35.
A third caveat of P1 transduction is that the gene to be knocked out should reside in a region with high similarity in the flanking regions between the donor and recipient strains. If there is a lack of synteny between the strains, the replacement of the target gene coding sequence with the Kan cassette may well fail, due to differences in the content of the flanking regions. Therefore, before embarking on P1-mediated gene deletion, investigators should check the sequences of the donor and recipient strains to make sure the flanking sequences are homologous. Of course, this is only possible if the strains have been sequenced. In the case of strains with an unknown sequence, P1 transduction may either fail or cause other problems, such as a gene conversion in the flanking regions. P1 can transduce approximately 90 kilobase pairs of DNA; if there are differences in the gene content in the regions around the target gene (e.g., small deletions or insertions), it is likely that these will revert to the donor sequence. This might have unintended consequences on the phenotype of the recipient strain. Therefore, where possible, the sequences of the donor and recipient around the gene of interest should always be compared prior to P1 transduction.
In conclusion, P1 transduction is a rapid way of transferring a specific gene knock-out to a number of strains once the initial knock-out mutation has been generated. Though the technique has its limitations, the ease and speed of its implementation make it an attractive alternative to other methods of gene deletion. P1 only infects E. coli, which normally restricts its use to this species. However, some variants of P1 have been developed that have a broader host range and can infect other species within the family Enterobacteriaceae, and even some other γ-proteobacterial species, albeit at reduced efficiency36,37. Even the transduction of cloned DNA from E. coli to Myxococcus xanthus, a δ-proteobacterium, has been reported38. In future experiments, these variants could be augmented with the vir mutation to broaden the range of recipient strains used in antibiotic cassette-based gene deletion by general transduction.
Disclosures
The authors have nothing to disclose.
Acknowledgments
Keio collection strains were obtained from the National BioResource Project (NIG, Japan): E. coli. We thank Dirk Linke (Department of Biosciences, University of Oslo) for his continuing support. This work was funded by the Research Council of Norway Young Researcher grant 249793 (to Jack C. Leo).
References
- Blount ZD. The unexhausted potential of E. coli. eLife. 2015;4:e05826. doi: 10.7554/eLife.05826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton CM, Aldea M, Washburn BK, Babitzke P, Kushner SR. New method for generating deletions and gene replacements in Escherichia coli. Journal of Bacteriology. 1989;171(9):4617–4622. doi: 10.1128/jb.171.9.4617-4622.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link AJ, Phillips D, Church GM. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. Journal of Bacteriology. 1997;179(20):6228–6237. doi: 10.1128/jb.179.20.6228-6237.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawitzke JA, Thomason LC, Costantino N, Bubunenko M, Datta S, Court DL. Recombineering: in vivo genetic engineering in E. coli, S. enterica, and beyond. Methods in Enzymology. 2007;421:171–199. doi: 10.1016/S0076-6879(06)21015-2. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(12):6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyrers JP, Zhang Y, Stewart AF. Techniques: Recombinogenic engineering--new options for cloning and manipulating DNA. Trends in Biochemical Sciences. 2001;26(5):325–331. doi: 10.1016/s0968-0004(00)01757-6. [DOI] [PubMed] [Google Scholar]
- Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Molecular Systems Biology. 2006;2 doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto N, et al. Update on the Keio collection of Escherichia coli single-gene deletion mutants. Molecular Systems Biology. 2009;5:335. doi: 10.1038/msb.2009.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Huan H-C, Datsenko K, Wanner BL, Mori H. Methods in Molecular Biology. Vol. 416. Clifton, NJ: 2008. The applications of systematic in-frame, single-gene knockout mutant collection of Escherichia coli K-12; pp. 183–194. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay MK, Tabor CW, Tabor H. Polyamines are not required for aerobic growth of Escherichia coli: preparation of a strain with deletions in all of the genes for polyamine biosynthesis. Journal of Bacteriology. 2009;191(17):5549–5552. doi: 10.1128/JB.00381-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Wong WW, Tang Y. Improving simvastatin bioconversion in Escherichia coli by deletion of bioH. Metabolic Engineering. 2007;9(4):379–386. doi: 10.1016/j.ymben.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Maeda T, Sanchez-Torres V, Wood TK. Escherichia coli hydrogenase 3 is a reversible enzyme possessing hydrogen uptake and synthesis activities. Applied Microbiology and Biotechnology. 2007;76(5):1035–1042. doi: 10.1007/s00253-007-1086-6. [DOI] [PubMed] [Google Scholar]
- Meehan BM, Landeta C, Boyd D, Beckwith J. The disulfide bond formation pathway is essential for anaerobic growth of Escherichia coli. Journal of Bacteriology. 2017;199(16) doi: 10.1128/JB.00120-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niba ETE, Naka Y, Nagase M, Mori H, Kitakawa M. A genome-wide approach to identify the genes involved in biofilm formation in E. coli. DNA Research. 2008;14(6):237–246. doi: 10.1093/dnares/dsm024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz E, et al. Conserved features in the structure, mechanism, and biogenesis of the inverse autotransporter protein family. Genome Biology and Evolution. 2016;8(6):1690–1705. doi: 10.1093/gbe/evw112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkrig J, et al. Discovery of an archetypal protein transport system in bacterial outer membranes. Nature Structural & Molecular Biology. 2012;19(5):506–510. doi: 10.1038/nsmb.2261. [DOI] [PubMed] [Google Scholar]
- Stubenrauch C, et al. Effective assembly of fimbriae in Escherichia coli depends on the translocation assembly module nanomachine. Nature Microbiology. 2016;1(7):16064. doi: 10.1038/nmicrobiol.2016.64. [DOI] [PubMed] [Google Scholar]
- Leo JC, Goldman A. The immunoglobulin-binding Eib proteins from Escherichia coli are receptors for IgG Fc. Molecular Immunology. 2009;46(8-9):1860–1866. doi: 10.1016/j.molimm.2009.02.024. [DOI] [PubMed] [Google Scholar]
- Mühlenkamp M, Oberhettinger P, Leo JC, Linke D, Schütz MS. Yersinia adhesin A (YadA) - Beauty & beast. International Journal of Medical Microbiology. 2015;305(2):252–258. doi: 10.1016/j.ijmm.2014.12.008. [DOI] [PubMed] [Google Scholar]
- Studier FW, Moffatt BA. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. Journal of Molecular Biology. 1986;189(1):113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- Meuskens I, Michalik M, Chauhan N, Linke D, Leo JC. A new strain collection for improved expression of outer membrane proteins. Frontiers in Cellular and Infection Microbiology. 2017;7:464. doi: 10.3389/fcimb.2017.00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov PP, Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158(1):9–14. doi: 10.1016/0378-1119(95)00193-a. [DOI] [PubMed] [Google Scholar]
- Grosskinsky U, et al. A conserved glycine residue of trimeric autotransporter domains plays a key role in Yersinia adhesin A autotransport. Journal of Bacteriology. 2007;189(24):9011–9019. doi: 10.1128/JB.00985-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo JC, et al. Structure. 7. Vol. 19. London, UK: 1993. The structure of E. coli IgG-binding protein D suggests a general model for bending and binding in trimeric autotransporter adhesins; pp. 1021–1030. [DOI] [PubMed] [Google Scholar]
- Bertani G. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. Journal of Bacteriology. 1951;62(3):293–300. doi: 10.1128/jb.62.3.293-300.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D. Studies on transformation of Escherichia coli with plasmids. Journal of Molecular Biology. 1983;166(4):557–580. doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- Leo JC, Oberhettinger P, Linke D. Methods in Molecular Biology. Clifton, NJ: 2015. Assessing the outer membrane insertion and folding of multimeric transmembrane β-barrel proteins; pp. 157–167. [DOI] [PubMed] [Google Scholar]
- Mikula KM, Leo JC, Łyskowski A, Kedracka-Krok S, Pirog A, Goldman A. The translocation domain in trimeric autotransporter adhesins is necessary and sufficient for trimerization and autotransportation. Journal of Bacteriology. 2012;194(4):827–838. doi: 10.1128/JB.05322-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason LC, Costantino N, Court DL, et al. Ausubel FM, et al., editors. E. coli genome manipulation by P1 transduction. Current Protocols in Molecular Biology. 2007. Chapter 1, Unit 1.17. [DOI] [PubMed]
- Franklin NC. Mutation in gal U gene of E. coli blocks phage P1 infection. Virology. 1969;38(1):189–191. doi: 10.1016/0042-6822(69)90144-5. [DOI] [PubMed] [Google Scholar]
- Jeong H, et al. Genome sequences of Escherichia coli B strains REL606 and BL21(DE3) Journal of Molecular Biology. 2009;394(4):644–652. doi: 10.1016/j.jmb.2009.09.052. [DOI] [PubMed] [Google Scholar]
- Greene EC. DNA Sequence Alignment during Homologous Recombination. Journal of Biological Chemistry. 2016;291(22):11572–11580. doi: 10.1074/jbc.R116.724807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt VM, Ingles CJ, Urdea MS, Rutter WJ. Homology requirements for recombination in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 1985;82(14):4768–4772. doi: 10.1073/pnas.82.14.4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TD, Waldor MK. Enterohemorrhagic Escherichia coli O157:H7 gal mutants are sensitive to bacteriophage P1 and defective in intestinal colonization. Infection and Immunity. 2006;75(4):1661–1666. doi: 10.1128/IAI.01342-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda H, Tomizawa JI. Transducing fragments in generalized transduction by phage P1. I. Molecular origin of the fragments. Journal of Molecular Biology. 1965;14(1):85–109. doi: 10.1016/s0022-2836(65)80232-7. [DOI] [PubMed] [Google Scholar]
- Murooka Y, Harada T. Expansion of the host range of coliphage P1 and gene transfer from enteric bacteria to other Gam-negative bacteria. Applied and Environmental Microbiology. 1979;38(4):754–757. doi: 10.1128/aem.38.4.754-757.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg RB, Bender RA, Streicher SL. Direct selection for P1-sensitive mutants of enteric bacteria. Journal of Bacteriology. 1974;118(3):810–814. doi: 10.1128/jb.118.3.810-814.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor KA, Zusman DR. Coliphage P1-mediated transduction of cloned DNA from Escherichia coli to Myxococcus xanthus: use for complementation and recombinational analyses. Journal of Bacteriology. 1983;155(1):317–329. doi: 10.1128/jb.155.1.317-329.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]