

Abstract
The Hippo signaling pathway is a conserved regulator of organ size and has important roles in the development and cancer biology. Due to technical challenges, it remains difficult to assess the activity of this signaling pathway and interpret it within a biological context. The existing literature on large tumor suppressor (LATS) relies on methods that are qualitative and cannot easily be scaled-up for screening. Recently, we have developed a bioluminescence-based biosensor to monitor the kinase activity of LATS-a core component of the Hippo kinase cascade. Here, we describe procedures for how this LATS biosensor (LATS-BS) can be used to characterize Hippo pathway regulators. First, we provide a detailed protocol for investigating the effect of an overexpressed protein candidate (e.g., VEGFR2) on LATS activity using the LATS-BS. Then, we show how the LATS-BS can be used for a small-scale kinase inhibitor screen. This protocol can feasibly be scaled-up to perform larger screens, which undoubtedly will identify novel regulators of the Hippo pathway.
Keywords: Cancer Research, Issue 139, LATS, LATS biosensor, Hippo pathway, split luciferase assay, cell culture, drug screening, kinase
Introduction
The Hippo signaling pathway was first identified in Drosophila as a novel regulator of cell growth and animal size1,2. Since its initial discovery, mounting evidence has convincingly shown that the Hippo pathway plays critical roles in the development (e.g., early embryonic development, organ size control, and three-dimensional [3-D] morphology), tumorigenesis (e.g., tumor development, metastasis, angiogenesis, immune evasion, genomic instability, stress response, and drug resistance), and tissue homeostasis (e.g., stem cell renewal and differentiation and tissue regeneration after injury)3,4,5,6,7,8,9,10. Hippo signaling is frequently dysregulated in various cancers7,8,9,10,11,12. Therefore, elucidating functions of the Hippo pathway in cancer biology and therapeutics and regenerative medicine has become one of the hottest areas in biomedical research.
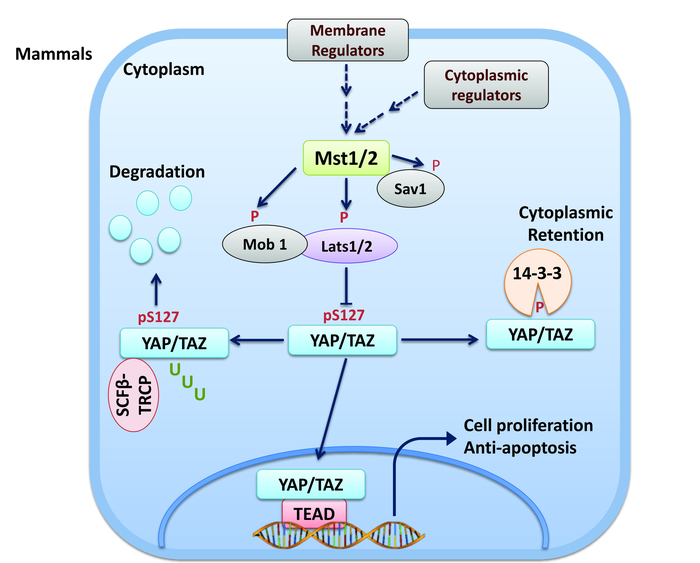
In brief, in the Hippo pathway, upon activation by upstream regulators (e.g., cell-cell contact, nutrient stress, and extracellular matrix [ECM]), MST1/2 (MST; mammalian homologs of Drosophila Hippo) serine/threonine (S/T) kinases phosphorylate/activate adaptor proteins hMOB1 and WW45, as well as LATS1/2 (LATS) kinases which, subsequently, phosphorylate transcriptional co-activator YAP and its paralog TAZ at conserved HX(H/R/K)XX(S/T) (H, histidine; R, arginine; K, lysine; S, serine; T, threonine; X, any amino acids) motifs, including YAP-S127 and TAZ-S8911,12. S127-phosphorylated YAP (YAP-pS127) and S89-phosphorylated TAZ (TAZ-pS89) are degraded by ubiquitination or bind to cytoplasmic protein 14-3-3 and are prevented from interacting with TEAD1-4 transcription factors in the nucleus to transactivate downstream genes involved in the cell proliferation and apoptosis (Figure 1). Despite the tremendous interest in the Hippo pathway, few tools for measuring Hippo signaling exist and those that do have been historically limited to reporters of YAP/TAZ/TEAD transcriptional output. Indeed, until very recently, there were no tools for measuring the dynamics and activity of the Hippo signaling components in a quantitative, real-time, high-throughput, and non-invasive manner both in vitro and in vivo.
Given our emerging understanding of the role of protein-protein interactions in physiology and pathology, there is great interest in the development of tools that can be used to study these interactions in a quantitative and real-time manner13,14,15,16. Indeed, there has been significant progress in the development of bio-analytical strategies, including the yeast two-hybrid (Y2H)17, the surface plasmon resonance (SPR)18, and Förster resonance energy transfer (FRET)19 assays, to evaluate protein-protein interactions. However, these approaches carry the limitation of requiring significant optimization of the reporter orientation, such that many constructs must be tested to find an efficient one. Further, these approaches also have a relatively low signal-to-noise ratio, such that discerning a true positive signaling can be challenging.
Protein complementation assays were developed to overcome these limitations. The first generation of protein complementation assays was based on split multicolor fluorescence proteins and could not solve the aforementioned limitations20. Multicolor fluorescence proteins consist of only one domain, making it difficult to split them into two separate stable fragments with low affinity and background noise21. Subsequently, firefly luciferase was identified as a new candidate for use in developing split protein complementation assays. In this approach, firefly luciferase is split into two fragments (N-terminal and C-terminal luciferase [NLuc and CLuc]) with each fragment fused to a target protein of interest. If the NLuc and CLuc are brought into proximity upon the interaction of the two target proteins, luciferase activity is reconstituted and bioluminescent light is generated in the presence of luciferin substrate and ATP22. In 2001, by performing a combinatorial screening using a library containing NLuc and CLuc fragments cut at various sites and attached to proteins with different linkers, Paulmurugan and Gambhir at Stanford University developed an optimized split-firefly luciferase fragment-assisted complementation system for protein-protein interactions23. In this system, firefly luciferase is cut at amino acid (aa) 398 to form NLuc and CLuc, which are attached to two proteins of interest using a flexible linker of eight glycine residues and two serine residues.
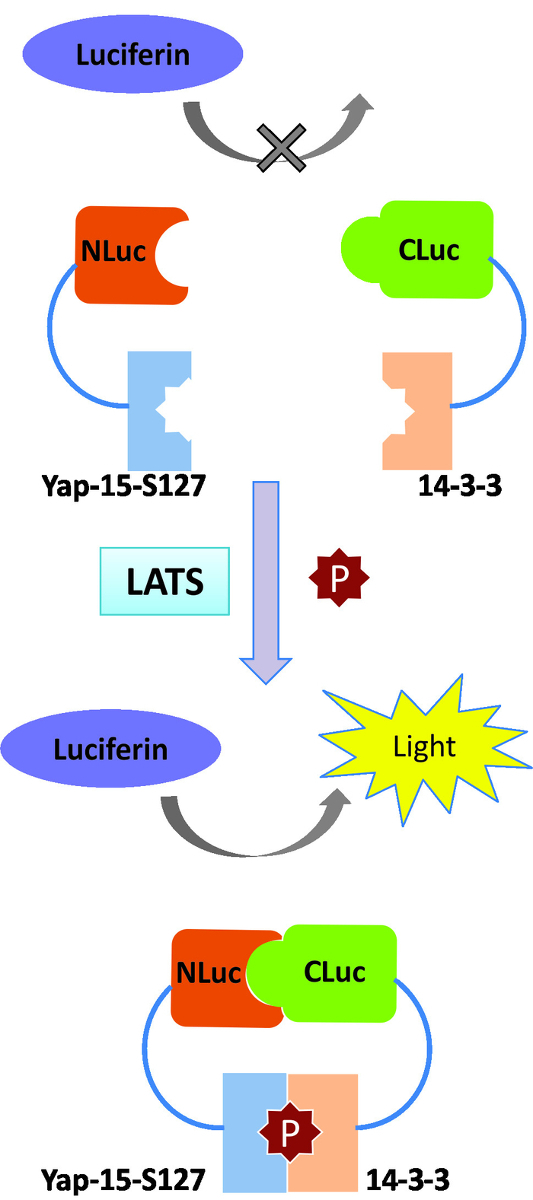
Using a similar approach, we recently developed a new LATS-BS by fusing NLuc to 15 aa of YAP surrounding the LATS phosphorylation site at S127 (YAP15) and CLuc with 14-3-3. The full-length YAP protein was not used, to avoid confounding signals by post-translational modifications of YAP (e.g., phosphorylation at other sites and ubiquitination) by other upstream regulators. The LATS-BS presented here can non-invasively monitor Hippo signaling activity both in vitro in living cells and in vivo in mice20,24 (Figure 2). Here, we describe a detailed protocol for measuring LATS kinase activity in vitro using the LATS-BS. First, we show how the LATS-BS can be used to investigate the effect of an overexpressed protein on LATS activity. Then, we show how the biosensor can be used to monitor the activity of the Hippo pathway after treatment with agents regulating the Hippo pathway. This protocol could be used to identify and characterize signaling pathways or stimuli regulating LATS kinase activity.
Protocol
1. Investigation of a Putative Regulator of Hippo Signaling Using the LATS-BS
- Plating and transfection of cells
- Preparation of cell culture
- Warm 1x PBS, DMEM containing 10% FBS and 1% penicillin/streptomycin, and 0.25% trypsin-EDTA to 37 °C in a water bath for approximately 30 min.
- Thoroughly clean a biosafety cabinet with 70% ethanol.
- Place tissue culture dishes, Pasteur pipettes, serological pipettes, and pipette tips into the tissue culture hood as required.
- Take out 1x PBS, the media, and the trypsin-EDTA from the water bath, clean them with 70% ethanol, and place them in the hood.
- Maintenance and plating of the cells for transfection
- Choose a cell line with a high transfection efficiency (e.g., HEK293) for the experiment. Check for the expression of Hippo pathway components in this cell line by western blot to make sure Hippo signaling is active.
- Grow HEK293 cells in DMEM in 10 cm Petri dishes in an incubator at 37 °C and 5% CO2. Monitor the cells under a microscope every day. Passage the cells as described below when 80–90% confluency is observed.
- Wash the cells by adding 5 mL of 1x PBS into a plate, gently swirling the plate, and aspirating the media completely.
- Add 1 mL of trypsin-EDTA to the plate. Swirl the plate to ensure that the trypsin evenly covers the cells.
- Incubate the cells at 37 °C in a CO2 incubator for 5 min or until all the cells are detached.
- Add 4 mL of media (with 10% FBS and 1% penicillin/streptomycin) to neutralize the trypsin, resuspend the cells by pipetting up and down several times, and dispense one-tenth (0.5 mL) of the cells into new 100 mm plates (for a passing ratio of 1:10) containing 9.5 mL of complete media.
- For LATS-BS transfection, count the cells using a hemocytometer and plate 2 x 105 cells into each well of a 12-well plate 24 h before transfection. NOTE: Using higher cell numbers may increase the background bioluminescent signal as Hippo signaling activity is regulated by cell confluency. Using lower cell numbers may increase cell sensitivity to transfection reagents and increase cell death24.
- Transfection of the LATS-BS alone or together with the candidate gene
- Miniprep LATS-BS plasmids (NLuc-YAP15-pcDNA3.1-Hygro and 14-3-3-CLuc-pcDNA3.1-Hygro) fresh from DH5a bacteria liquid culture (necessary to achieve an optimum LATS-BS expression).
- 1 h before transfection, aspirate the medium from the plate, add 500 µL of complete growth medium to each well of the 12-well plates and return the cells to the incubator. NOTE: The following protocol describes transfection methods using a commercially available polymer-based transfection reagent. Other transfection reagents may also be suitable for transfection; however, we have not yet tested them.
- Make solutions A and B as described in Table 1. For N transfections, make (N + 0.5) x solution B premix.
- Immediately add the diluted polymer-based transfection reagent (solution B) to DNA (solution A). Pipette up and down gently to mix.
- Incubate the sample at room temperature for 15 min to allow transfection complexes to form.
- Add the total volume (76 µL) of transfection complexes dropwise to each well of the 12-well plate with gentle swirling.
- Incubate the cells overnight (18–24 h) at 37 °C. Note: For cells that are sensitive to polymer-based transfection reagents, the incubation time can be reduced to 5 h.
- The day after transfection, remove the transfection-complex-containing media and replace it with fresh complete media. Culture the cells at 37 °C for another day to perform luciferase assay.
- Luciferase assay NOTE: The Luciferase Reporter Assay System detecting firefly and Renilla together in one assay is used for the luciferase assays. If Renilla was not used as an internal control, perform one-step luciferase assay by adding LARII without adding the Renilla detection reagent.
- Preparation of the cell lysates
- Dilute 5x passive lysis buffer (PLB) with distilled water as follows:
- Total volume of 1x PLB required = N x (50 μL of 5x PLB + 200 μL of dH2O) = N x 250 μL of 1x PLB per well of the 12-well plates (N = number of samples).
- Aspirate the media completely. Add 1 mL of 1x PBS into each well. Swirl the plate gently to remove detached cells and residual growth media. Aspirate the 1x PBS completely. Make sure no residual 1x PBS remains prior to the addition of 1x PLB.
- Add 250 μL of 1x PLB/well.
- Place the culture plates on a rocking platform with gentle rocking to ensure complete and even coverage of the cell monolayer with 1x PLB. Rock the culture plates at room temperature for 15 min to allow the cells to lyse.
- Transfer the lysate to a 1.5 mL microcentrifuge tube.
- Measuring the bioluminescent signal
- Take out the LARII (20 μL/sample) from -80 °C and equilibrate it to room temperature.
- Prepare Renilla detection reagent for N assays: add (N + 1) x 0.4 μL of 50x Substrate to (N + 1) x 19.6 μL of its Buffer.
- Predispense 10 μL of each cell lysate to 1.5-mL tubes.
- Program a luminometer to perform a 2-s premeasurement delay, followed by a 10-s measurement period for each dual luciferase assay. NOTE: The first and second measurements will be corresponding to the LATS-BS signal and the Renilla internal control signal, respectively. Any luminometer compatible with a single or dual luciferase can be used.
- Carefully transfer 20 μL of LARII reagent into one tube and quickly pipette up and down 3x to mix it.
- Immediately place the tube in the luminometer and initiate the reading.
- Remove the sample tube from the luminometer, immediately add 20 μL of Renilla Reagent, and mix by pipetting up and down 3x. Bring the sample in the luminometer and initiate the next reading. Record the measurements.
- Discard the reaction tube and proceed to the next sample.
2. A screen to Identify Hippo Pathway Regulators Using the LATS-BS
- Cell culture and transfections
- Culture HEK293AD cells as described in section 1.1. NOTE: HEK293AD is more adherent than other HEK293 cell lines, which makes it a good cell line for use in screens.
- Detach cells as described in section 1.1.2. Count and plate 2 x 106 cells in 100 mm plates 24 h before transfection.
- 1 h before transfection, aspirate the media from the plate and replace them with 5 mL of fresh complete growth media.
- Make solutions A and B as described in Table 2.
- Immediately add the diluted polymer-based transfection reagent (solution B) to DNA (solution A). Pipette up and down to mix.
- Incubate the sample at room temperature for 15 min to allow transfection complexes to form.
- Add the total volume (500 µL) of transfection complexes dropwise to cells with gentle swirling.
- Incubate the cells overnight at 37 °C.
- The next day, trypsinize and count the cells. Plate 1 x 104 to 2 x 104 cells into each well of a 96-well plate in a total volume of 100 µL of media per well. Incubate for another day.
- The next day, add agents/small molecules regulating the Hippo pathway at a final concentration of 10 µM to each well, 1–4 h before performing the luciferase assay. Note: For most small molecules/agents, a 4 h treatment at 10 µM does not affect cell viability. If a lower concentration is used, treatment may be extended for a longer period of time.
- Luciferase assay
- In vitro luciferase assay
- Aspirate the media from the cells completely. Wash the cells with 100 µL of 1x PBS in each well. Aspirate completely.
- Add 20 µL of 1x PLB (prepared as described in step 1.2.1.1) into each well of the 96-well plates.
- Place the culture plates on a rocking platform with gentle rocking at room temperature for 15 min.
- Carefully transfer 20 µL of LARII reagent into each well and pipette up and down 3x to mix.
- Immediately place the plate in a plate-reading luminometer and initiate the reading.
- Living cell luciferase assay
- Make 11x D-luciferin stock as follows: dissolve 1.5 mg of D-luciferin powder in 1 mL of normal culture media.
- Transfer 10 µL of 11x D-luciferin into each well and mix it gently with the media which is already in the well.
- Replace the cells into the incubator for 10 min.
- Place the plate in the luminometer and initiate the reading. Note: If the signal intensity is not strong enough at this concentration of D-luciferin, another 10 µL of the D-luciferin stock may be added to the cells.
Representative Results
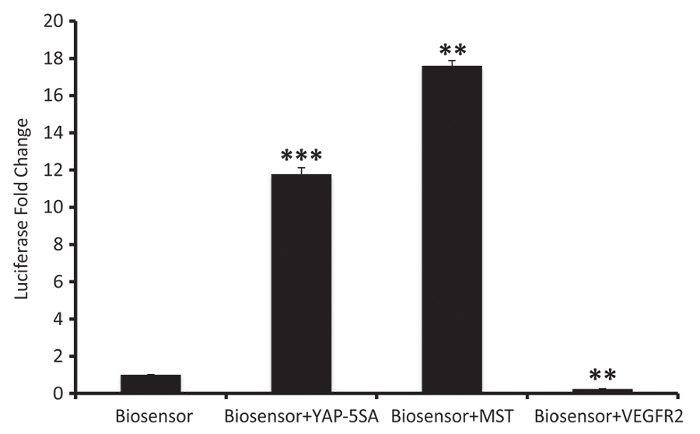
The LATS-BS was cotransfected with different genes to evaluate their effect on LATS activity (Figure 3). In this experiment, Renilla was used as an internal control. The transient expression of YAP-5SA, a constitutive active form of YAP, causes increasing levels of YAP transcriptional targets and a subsequent increase in LATS kinase activity through an established feedback pathway25. MST, the upstream activator of LATS in the Hippo pathway (Figure 1), also increases the biosensor signal intensity. However, VEGFR overexpression, which triggers PI3K/MAPK signaling, inhibits LATS and decreases the signal intensity of the LATS-BS.
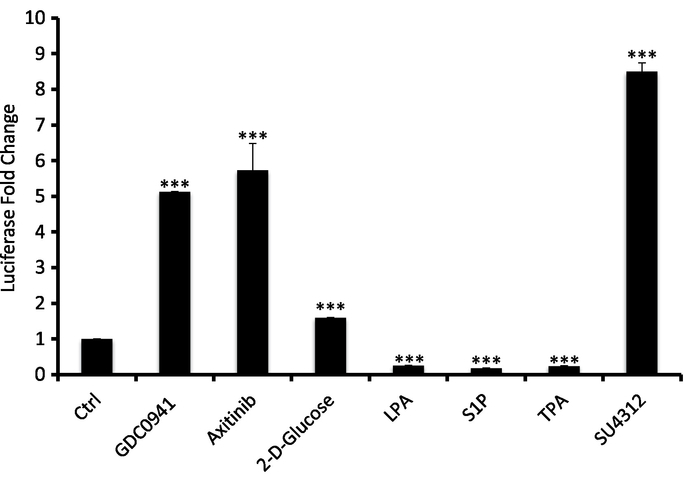
In the second protocol, LATS-BS was transfected into HEK293AD and, 24 hours after the transfection, cells were passed into 96-well plates. 40 h after the transfection, the cells were treated with different known small molecule regulators of Hippo signaling followed by a luciferase assay (Figure 4). VEGFR, PI 3-kinase, and cellular energy level are three known negative regulators of the LATS kinase24. In this experiment, GDS0941, a PI 3-kinase inhibitor, axitinib and SU4312, inhibitors of VEGFR, and 2-D glucose, which causes energy stress by an inhibition of glucose metabolism and ATP production, were used to activate the LATS-BS. Inversely, LPA, S1P, and TPA were used to inhibit the LATS-BS. This experiment shows that the LATS-BS can be used to measure both the activation and the inhibition effect of a small molecule on the LATS kinase activity.
| Solution A | |
| pCDNA3.1-Hygro-NLuc-YAP15 | 1 μL (50 ng) |
| pCDNA3.1-Hygro-14-3-3-CLuc | 1 μL (50 ng) |
| Plasmid expressing a candidate gene (e.g. MST) | 1 μL (400 ng) |
| or empty vector (e.g. pcDNA3.1-Hygro) | |
| Renilla vector (pRL-TK) (optional) | 1 μL (20 ng) |
| DMEM (no serum/antibiotics) | 34 μL |
| Total volume | 38 μL |
| Solution B | |
| Polymer-based transfection reagent | 1.5 μL |
| (3:1 ratio with total μg DNA) | |
| DMEM (no serum/antibiotics) | 36.5 μL |
| Total volume | 38 μL |
Table 1: Protocol for making solutions A and B using a polymer-based transfection reagent for investigating a putative regulator of the Hippo pathway with the LATS-BS.
| Solution A | |
| pCDNA3.1-Hygro-NLuc-YAP15 | 10 μL (3 μg) |
| pCDNA3.1-Hygro-14-3-3-CLuc | 10 μL (3 μg) |
| DMEM (no serum/antibiotics) | 230 μL |
| Total volume | 250 μL |
| Solution B | |
| Polymer-based transfection reagent | 18 μL |
| (3:1 ratio with total μg DNA) | |
| DMEM (no serum/antibiotics) | 232 μL |
| Total volume | 250 μL |
Table 2: Protocol for making solutions A and B using a polymer-based transfection reagent for screening to identify regulators of the Hippo pathway using the LATS-BS.
Figure 1: The Hippo pathway in mammals. The Hippo pathway restricts organ size by phosphorylating and inhibiting YAP and TAZ. When the Hippo pathway is activated, SAV, LATS1/2, and MOB are phosphorylated and activated by MST1/2. Activated LATS1/2 phosphorylates YAP/TAZ at conserved serine-containing sites. 14-3-3 interacts with phosphorylated YAP/TAZ, thereby inhibiting their nuclear localization and co-transactivation of downstream gene targets through TEADs. Please click here to view a larger version of this figure.
Figure 2: LATS-BS split-protein complementation assay based on luciferase. Luciferase N- and C-terminus domains (NLuc and CLuc) are attached to 15 amino acid surrounding YAP-S127 and 14-3-3, respectively. YAP15/14-3-3 binding after LATS phosphorylation promotes luciferase complementation and bioluminescence signal emission. Please click here to view a larger version of this figure.
Figure 3: Results from a representative experiment evaluating the effect of a gene on LATS activity. The biosensor can be expressed alone or together with other genes in HEK293AD cells to investigate their effect on LATS kinase activity (mean ± SD; n = 3, **p <0.01, ***p <0.001). Please click here to view a larger version of this figure.
Figure 4: Results from a representative experiment evaluating the effect of small molecules on LATS activity in HEK293AD cells. HEK293AD cells were transfected with the LATS-BS and treated with the following drugs: PI3K inhibitor (GDC0941), 10 μM for 4 h; VEGFR inhibitor (axitinib), 10 μM for 4 h; 2-deoxyglucose, 25 mM for 1 h; LPA, 10 μM for 1 h; sphingosine-1-phosphophate (S1P), 1 μM for 1 h; 12-O-tetradecanoylphorbol-13-acetate (TPA), 5 nM for 1 h; VEGFR inhibitor (SU4312), 10 μM for 4 h (mean ± SD; n = 3, **p <0.01, ***p <0.001). Please click here to view a larger version of this figure.
Discussion
While the Hippo pathway plays critical roles in various biological processes, and dysregulation of the Hippo pathway leads to cancer6, how the Hippo pathway is regulated in response to various stimuli is not completely understood. In addition, there has been no quantitative and real-time system to assess the activity of core Hippo components. Recently, we developed and validated a novel biosensor for measuring LATS kinase activity in the Hippo pathway24. This biosensor can be used not only for quantitatively and accurately monitoring Hippo signaling activity in living cells but also for screens of drugs targeting the Hippo pathway for cancer therapy. In addition, the biosensor-based screening can lead to the discovery of novel genes responsible for the regulation of the Hippo pathway. Given the importance of Hippo signaling in cancer, finding crucial crosstalk between other genes and the Hippo pathway would be a valuable study to undertake for all cancer biologists. Therefore, this biosensor has the potential to make innovations in the current treatment of cancer and might provide a useful new therapeutic strategy for successful treatments of cancer.
There are several important points that should be considered when using the LATS-BS. First, in our experience, the LATS-BS has the greatest sensitivity when the plasmids used for transfection are extracted fresh from bacteria. Second, the amount of LATS-BS plasmids transfected into cells will affect the LATS-BS sensitivity and signal intensity. The amount of plasmid DNA can be increased to 400 ng/fragment for the LATS-BS and to 600 ng for the gene of interest per well of a 12-well plate. In general, using more plasmid will give a higher signal intensity but less sensitivity. Third, cell confluency can affect the LATS-BS signal background. Consistent with the well-established role of Hippo signaling in cellular responses to confluency, we have observed that cells with higher confluency have a higher background LATS-BS signal due to the activation of endogenous LATS. Accordingly, sparsely-plated cells show a lower LATS-BS background signal. Optimal cell confluency can be selected depending on the purpose of the experiment (high confluency for observing LATS inhibition; low confluency for observing LATS activation). Fourth, Renilla may be used as an internal control for transfection efficiency, and for hard-to-transfect cells (e.g., MCF10A), the transfection reagent:DNA ratio can be modified to 4:1. Finally, we strongly recommend cotransfecting MST2 as a positive control and YAP-S127A as a negative control each time the biosensor is used.
Every tool has its limitations. The LATS-BS is the artificial substrate of an endogenous protein, which can always outcompete the true substrate of the protein of interest. This effect is known as a buffering effect of the biosensor. Improving the biosensor with higher signal intensity and lower expression levels may help to overcome this problem. Another limitation is that feedback pathways may complicate the interpretation of data from the LATS-BS. There are many linear and non-linear interacting signaling pathways that likely influence the LATS-BS signal. The LATS-BS only demonstrates the cumulative effect of a stimulus on LATS kinase activity. It should be noted, however, that that limitation is not unique to the LATS-BS but rather is commonly encountered in studies of molecular biology. Thus, LATS-BS data should be interpreted in the context of multiple probative experiments.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by grants from the Canadian Institute of Health Research (CIHR#119325, 148629) and the Canadian Breast Cancer Foundation (CBCF) to XY. TA is supported by the Vanier Canada Graduate Scholarship and the Ontario International Graduate Scholarship. HJJVR is supported by a Queen Elizabeth II Graduate Scholarship in Science and Technology.
References
- Justice RW, Zilian O, Woods DF, Noll M, Bryant PJ. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes & Development. 1995;9(5):534–546. doi: 10.1101/gad.9.5.534. [DOI] [PubMed] [Google Scholar]
- Xu T, Wang W, Zhang S, Stewart RA, Yu W. Identifying tumor suppressors in genetic mosaics: the Drosophila lats gene encodes a putative protein kinase. Development. 1995;121(4):1053–1063. doi: 10.1242/dev.121.4.1053. [DOI] [PubMed] [Google Scholar]
- Taha Z, Janse van Rensburg HJ, Yang X. The Hippo Pathway: Immunity and Cancer. Cancers. 2018;10(4):94. doi: 10.3390/cancers10040094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maugeri-Saccà M, De Maria R. The Hippo pathway in normal development and cancer. Pharmacology & Therapeutics. 2018;186:60–72. doi: 10.1016/j.pharmthera.2017.12.011. [DOI] [PubMed] [Google Scholar]
- van Rensburg HJJ, Yang X. The roles of the Hippo pathway in cancer metastasis. Cellular Signalling. 2016;28(11):1761–1772. doi: 10.1016/j.cellsig.2016.08.004. [DOI] [PubMed] [Google Scholar]
- Yeung B, Yu J, Yang X. Roles of the Hippo pathway in lung development and tumorigenesis. International Journal of Cancer. 2016;138(3):533–539. doi: 10.1002/ijc.29457. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Yang X. The Hippo pathway in chemotherapeutic drug resistance. International Journal of Cancer. 2015;137(12):2767–2773. doi: 10.1002/ijc.29293. [DOI] [PubMed] [Google Scholar]
- Yu F-X, Guan K-L. The Hippo pathway: regulators and regulations. Genes & Development. 2013;27(4):355–371. doi: 10.1101/gad.210773.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Xu T. Molecular mechanism of size control in development and human diseases. Cell Research. 2011;21(5):715. doi: 10.1038/cr.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser S, Yang X. LATS tumor suppressor: a new governor of cellular homeostasis. Cell Cycle. 2010;9(19):3892–3903. doi: 10.4161/cc.9.19.13386. [DOI] [PubMed] [Google Scholar]
- Zhao B, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes & Development. 2007;21(21):2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Chun A, Cheung K, Rashidi B, Yang X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. Journal of Biological Chemistry. 2008;283(9):5496–5509. doi: 10.1074/jbc.M709037200. [DOI] [PubMed] [Google Scholar]
- Ozawa T, Kaihara A, Sato M, Tachihara K, Umezawa Y. Split Luciferase as an Optical Probe for Detecting Protein. Protein Interactions in Mammalian Cells Based on Protein Splicing. Analytical Chemistry. 2001;73(11):2516–2521. doi: 10.1021/ac0013296. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Adams KW, Fan Z, McLean PJ, Hyman BT. Characterization of oligomer formation of amyloid-β peptide using a split-luciferase complementation assay. Journal of Biological Chemistry. 2011;286(31):27081–27091. doi: 10.1074/jbc.M111.257378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decock M, et al. Analysis by a highly sensitive split luciferase assay of the regions involved in APP dimerization and its impact on processing. FEBS Open Bio. 2015;5(1):763–773. doi: 10.1016/j.fob.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad T, Tashakor A, Rahmati F, Hemmati R, Hosseinkhani S. Oscillation of apoptosome formation through assembly of truncated Apaf-1. European Journal of Pharmacology. 2015;760:64–71. doi: 10.1016/j.ejphar.2015.04.008. [DOI] [PubMed] [Google Scholar]
- Brückner A, Polge C, Lentze N, Auerbach D, Schlattner U. Yeast two-hybrid, a powerful tool for systems biology. International Journal of Molecular Sciences. 2009;10(6):2763–2788. doi: 10.3390/ijms10062763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattnaik P. Surface plasmon resonance. Applied Biochemistry and Biotechnology. 2005;126(2):79–92. doi: 10.1385/abab:126:2:079. [DOI] [PubMed] [Google Scholar]
- Clegg RM. Fluorescence resonance energy transfer. Current Opinion in Biotechnology. 1995;6(1):103–110. doi: 10.1016/0958-1669(95)80016-6. [DOI] [PubMed] [Google Scholar]
- Azad T, Tashakor A, Hosseinkhani S. Split-luciferase complementary assay: applications, recent developments, and future perspectives. Analytical and Bioanalytical Chemistry. 2014;406(23):5541–5560. doi: 10.1007/s00216-014-7980-8. [DOI] [PubMed] [Google Scholar]
- Hu C-D, Kerppola TK. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nature Biotechnology. 2003;21(5):539–545. doi: 10.1038/nbt816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseinkhani S. Molecular enigma of multicolor bioluminescence of firefly luciferase. Cellular and Molecular Life Sciences. 2011;68(7):1167–1182. doi: 10.1007/s00018-010-0607-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulmurugan R, Gambhir SS. Combinatorial library screening for developing an improved split-firefly luciferase fragment-assisted complementation system for studying protein-protein interactions. Analytical Chemistry. 2007;79(6):2346–2353. doi: 10.1021/ac062053q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad T, et al. A LATS biosensor screen identifies VEGFR as a regulator of the Hippo pathway in angiogenesis. Nature Communications. 2018;9(1):1061. doi: 10.1038/s41467-018-03278-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroishi T, et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes & Development. 2015;29(12):1271–1284. doi: 10.1101/gad.262816.115. [DOI] [PMC free article] [PubMed] [Google Scholar]