

Abstract
Accurate transcription is required for the faithful expression of genetic information. Surprisingly though, little is known about the mechanisms that control the fidelity of transcription. To fill this gap in scientific knowledge, we recently optimized the circle-sequencing assay to detect transcription errors throughout the transcriptome of Saccharomyces cerevisiae, Drosophila melanogaster, and Caenorhabditis elegans. This protocol will provide researchers with a powerful new tool to map the landscape of transcription errors in eukaryotic cells so that the mechanisms that control the fidelity of transcription can be elucidated in unprecedented detail.
Keywords: Genetics, Issue 139, Genetic Processes, Gene Expression, Transcription, Genetic, Reverse Transcription, Transcriptome, Mutagenesis, Amino Acid Substitution, Phenomena and Processes, Genetic Phenomena, mutagenesis, transcription, RNA-Seq, S. cerevisiae, C. elegans, D. melanogaster, sequencing, RNA polymerase, errors
Introduction
The genome provides a precise biological blueprint of life. To implement this blueprint correctly, it is important for the genome to be transcribed with great precision. However, transcription is unlikely to be error free. For example, RNA polymerases have long been known to be error-prone in vitro1,2, and recently it was shown that they commit errors in vivo as well3,5,6, particularly when confronted with DNA damage7,8,9,10. Taken together, these observations indicate that transcription errors occur continuously in all living cells, suggesting that they could be a potent source of mutated proteins.
This process, termed transcriptional mutagenesis, differs from classical mutagenesis in two ways. First, in contrast to genetic mutations, transcription errors affect both mitotic and post-mitotic cells, as they do not depend on DNA replication. Studying the mechanisms that impact the fidelity of transcription will, therefore, provide valuable insight into the mutation load of both mitotic and post-mitotic cells. Interestingly, transcription errors have recently been implicated in the promotion of protein aggregation11,12,13 and have been hypothesized to contribute to both carcinogenesis10 and the development of antibiotic resistance in bacteria14.
Second, in contrast to genetic mutations, transcription errors are transient in nature. Their temporary existence is particularly challenging because it makes transcription errors exceedingly difficult to detect. For example, while several labs have devised valuable reporter assays for the study of transcriptional mutagenesis, these assays are only able to measure transcription errors in a limited number of contexts and model organisms4,15. To overcome these limitations, many researchers have turned to RNA sequencing technology (RNA-seq), which theoretically allows transcription errors to be recorded throughout the transcriptome of any species. However, these studies are easily confounded by library construction artifacts, such as reverse transcription errors, PCR amplification errors, and the error-prone nature of sequencing itself. For example, reverse transcriptases commit approximately one error every ~20,000 bases, while RNA polymerases (RNAPs) are expected to make only one error every 300,000 bases5,6. Because the error rate of reverse transcription alone dwarfs the error rate of RNA polymerases inside cells, it is virtually impossible to distinguish true transcription errors from artifacts caused by the library preparation in traditional RNA-Seq data (Figure 1a).
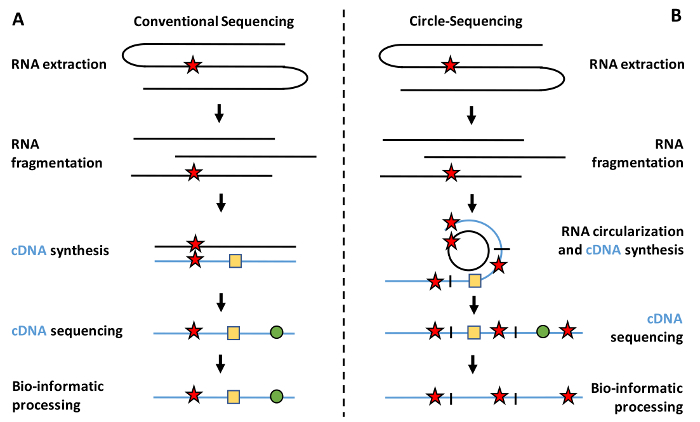
To solve this problem, we developed an optimized version of the Circle-Sequencing (Cirseq, or C-seq henceforth) assay5,16. This assay allows the user to detect transcription errors and other rare variants in RNA throughout the transcriptome5. The circular-sequencing assay carries this name because a key step in this assay revolves around RNA circularization. Once the RNA targets are circularized, they are reverse transcribed in a rolling circle fashion, to produce linear cDNA molecules that contain numerous copies of the same RNA template. If an error was present in one of these templates, this error would also be present in every single repeat contained within the cDNA molecule. In contrast, errors introduced by reverse transcription, PCR amplification, or sequencing tend to arise randomly, and will thus be present in only one or two repeats. Thus, by generating a consensus sequence for each cDNA molecule, and distinguishing random errors from errors that occur in all repeats, library construction artifacts can effectively be separated from true transcription errors (Figure 1b).
If used properly, the C-seq assay can be used to accurately detect the rate of base substitutions, insertions, and deletions in RNA throughout the transcriptome of any species (for example, see Traverse and Ochman17). For example, we have used the C-seq assay to provide genome-wide measurements of the error rate of transcription in Saccharomyces cerevisiae, Drosophila melanogaster, and Caenorhabditis elegans with a single base resolution5 (unpublished observations). Originally used to accurately sequence RNA virus populations, this optimized version of the C-seq assay has been streamlined to minimize harsh conditions during the library preparation that contribute to library construction artifacts. In addition, by using a number of commercially available kits, the throughput of the assay is greatly improved, as well as its user-friendliness. If used properly, this assay can accurately detect thousands of transcription errors per replicate, thereby greatly improving on previous studies6. Overall, this method provides a powerful tool to study transcriptional mutagenesis and will allow the user to gain novel insights into the mechanisms that control the fidelity of transcription in a wide range of organisms.
Protocol
1. Preparation
RNases are omnipresent; therefore, clean the workspace thoroughly by spraying it down with a decontamination reagent (e.g., RNase AWAY) and 70% ethanol. Spray down any pipettes, pens, or tube racks to remove potential sources of RNase contamination.
To further prevent RNases from contaminating the samples, wear a lab coat or long-sleeve T-shirt during the experimentation. Avoid contact between the gloves and the inside of any tubes that will be used, especially when retrieving them from a box or bag.
Prior to the reverse transcription, change gloves frequently. NOTE: For optimal results, do not pause the experiment prior to the reverse transcription.
2. Cell and Animal Culture and Collection
- Saccharomyces cerevisiae growth and collection
- If a C-seq library is desired from yeast, first inoculate a single colony of S. cerevisiae in 3 mL of medium containing yeast extract, adenine, peptone, and dextrose (YAPD), and incubate it overnight at 30 °C in a rolling drum.
- In the morning, re-inoculate the culture in 50 mL of YAPD medium to an optical density (OD) of 0.1 and grow the cells until they reach an OD of 0.5–0.7 (~7 x 106 cells/mL).
- Collect the entire 50 mL culture (approximately 350 x 106 cells) in a 50 mL conical tube and spin the cells down for 3 min at 2,100 x g. Carefully remove the YAPD medium and wash the pellet once with 1x PBS.
- Then, resuspend the pellet in 480 µL of lysis buffer, 48 µL of 10% SDS, and 480 µL of Phenol:Chloroform:Isoamyl alcohol (25:24:1), pH 6.8. Vortex the sample at maximum speed for 5 s, and transfer it to a 1.5 mL screw cap tube with 750 µL of ice-cold zirconia beads (supplied with the kit). Proceed to step 3 of this protocol for the cell lysis and total RNA extraction. NOTE: All of the reagents required for step 2.1.4 are provided in the recommended yeast RNA extraction kit. These reagents work equally well for yeast, worms, and flies so that after the collection of the yeast (step 2.1), worms (step 2.2) or flies (step 2.3), a unified approach can be used to generate C-seq libraries (steps 3–10).
- Caenorhabditis elegans culture and collection
- If a C-seq library is desired from worms, deposit 30 adult worms on a fresh plate of agar spotted with OP50 bacteria. NOTE: Five plates are sufficient for 1 replicate.
- Culture the worms for 4 days and collect the worms by gently washing the plates with 5 mL of M9 media, and pool the worms into a single 50 mL conical tube.
- Spin the worms down at 100 x g for 2 min to create a pellet. Decelerate the centrifuge at the lowest speed possible, so as not to disturb the pellet.
- Gently move the worms into a 1.5 mL tube, wash them twice in 1.5 mL of M9 media, and centrifuge the tube at 100 x g for 1 min to remove bacteria and generate a clean 100 µL pellet of worms.
- Resuspend the worms in 480 µL of lysis buffer, 48 µL of 10% SDS, and 480 µL of Phenol:Chloroform:Isoamyl alcohol. Vortex the sample at maximum speed for 5 s and transfer it to a 1.5 mL screw cap tube containing 750 µL of ice-cold zirconia beads. Proceed to step 3 of this protocol for the lysis of the worms and the total RNA extraction.
- Drosophila melanogaster culture and collection
- If a C-seq library is desired from flies, culture 20–25 flies in vials that contain either dextrose or a molasses-based medium. Aliquot the flies over 3 vials, so that each vial contains approximately 8 flies.
- To collect the flies, place the vial into an ice bucket until the flies are sedated. Then, collect them with a brush in a 1.5 mL tube. Do not collect the flies by CO2 treatment.
- Let the flies warm to room temperature (RT), and quickly add a premade mixture of 500 µL of lysis buffer, 50 µL of 10% SDS, and 500 µL of Phenol:Chloroform:Isoamyl alcohol to the tube. Use a micropestle to grind up the flies with approximately 15 strokes, accompanied by a firm twisting motion when the pestle reaches the bottom of the tube.
- Pour the mixture of flies and lysis media into a 1.5 mL screw cap tube containing 750 µL of ice-cold zirconia beads, and vortex it at maximum speed for 5 s. Proceed to step 3 of this protocol for the lysis of the flies and the total RNA extraction.
3. Total RNA Purification
NOTE: At this point, all three protocols converge, and a single, unified approach can be used to generate C-seq libraries.
Screw the cap on tightly and attach the tube to a vortexer with an adapter or sticky tape. Vortex the tube at maximum speed for 10 min to lyse the samples. Then, centrifuge the sample at 16,100 x g for 5 min to separate the organic solvents from the aqueous phase.
Carefully remove 400–500 µL of the aqueous phase without disturbing the white, cloudy interphase, and add it to a 15 mL conical tube containing 1.9 mL of binding buffer. Mix them thoroughly by vortexing.
Add 1.25 mL of 100% ethanol and mix the sample by vortexing. Transfer 700 µL of the sample to a filter cartridge assembled in a collection tube and spin the sample at 14,500 x g for 30 s. Repeat until all of the sample is passed through the cartridge. NOTE: At this point, the sample may turn slightly opaque. If too many cells, worms, or flies were lysed, precipitates could appear that may clog up the filter.
Wash the filter once with 700 µL of wash solution 1, and twice with 500 µL of wash solution 2 or 3. Finish the washes by spinning the samples at 14,500 x g for 2 min to remove any excess ethanol.
Transfer the filter cartridge to a 1.5 mL dolphin tube and add 108 µL of prewarmed elution solution (65 °C). NOTE: Never expose RNA samples to temperatures higher than 65 °C prior to a reverse transcription, as doing so will provoke cytosine to uracil deamination events that are impossible to distinguish from transcription errors.
Degrade the DNA contamination in the isolated RNA (see steps 2.1–3.4) by adding 11 µL of 10x DNase I buffer, 2 µL of RNase inhibitor, and 4 µL of DNase I (8 U), and incubate them at 37 °C for 30 min.
After digestion, add 11 µL of DNase inactivation reagent. Vortex the mixture and let it sit at RT for 5 min. Then, centrifuge the sample at 15,000 x g for 3 min to pellet the inactivation reagent and collect 110 µL of the supernatant without disturbing the pellet. Transfer the supernatant to a 1.5 mL tube. NOTE: The DNase inactivation reagent can be quite difficult to pipet properly, so visually inspect the pipette tip after each pipetting action. If the inactivation reagent becomes too viscous to pipet, add 10 mM Tris-HCl (pH 7.4) equal to 1/5 of the remaining volume.
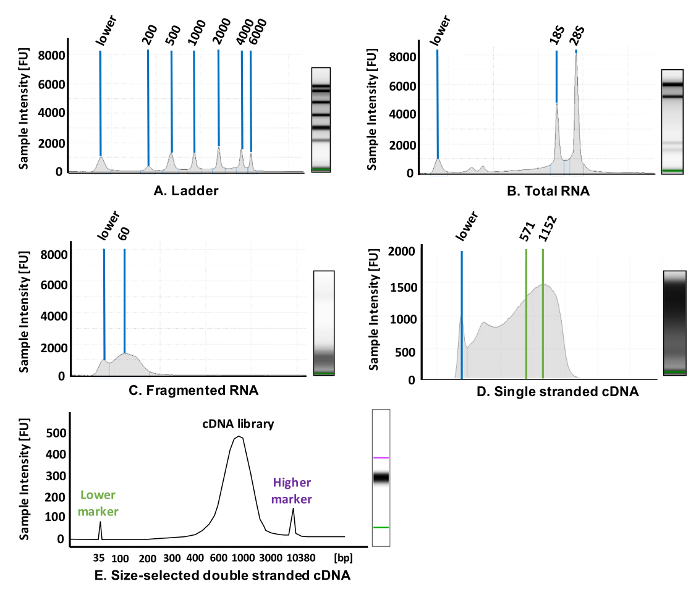
(Quality check) Measure the concentration of the total RNA using a spectrophotometer. Then set aside 1 µL of total RNA and dilute it to a concentration of 10 ng/µL in nuclease-free (NF) water. Run the RNA on an appropriate nucleotide analysis instrument with a High Sensitivity RNA Tape to ensure that the RNA is not degraded and has an eRIN value of at least 8.5 or higher (Figure 2B).
4. mRNA Enrichment
Perform the mRNA enrichment with an mRNA miniprep kit (see Table of Materials). Add 140 µL of NF water to the sample for a total volume of 250 µL. Then, add 250 µL of 2x binding buffer, mix the sample thoroughly by vortexing, and add 15 µL of oligo(dT)beads.
Mix the sample by pipetting it up and down and incubate it at 65 °C for 2 min. Then, place the sample at RT for 10 min. Finally, centrifuge the sample at 16,100 x g for 2 min to pellet the bead:mRNA complexes.
Discard the supernatant and resuspend the pellet in 500 µL of wash solution. Transfer the solution to a spin column and spin it for 1 min at 15,000 x g. Discard the flow-through and wash the sample with an additional 500 µL of wash solution. Then, spin the sample for 2 min at 15,000 x g to remove any excess ethanol from the spin filters.
Transfer the spin filter to a dolphin tube and resuspend the pellet in the column with 80 µL of prewarmed elution solution (65 °C). Incubate the sample for 1.5 min at 65 °C and elute it by spinning it for 1 min at 15,000 x g.
Measure the concentration of purified RNA using a spectrophotometer or similar tool that allows for the quantification of RNA concentration and quality.
5. RNase III Fragmentation and RNA Clean Up
NOTE: (Important) To prepare circular RNA molecules appropriate for generating C-seq libraries, the RNA must be fragmented to roughly 60–80 bases in length. While previous methods have used a chemical fragmentation to fragment RNA, chemical fragmentation with heavy metals introduces damages to the RNA samples that can be misinterpreted as transcription errors during the final analysis. To circumvent this problem, rely instead on a fragmentation using RNase III to generate small fragments. An additional advantage of this enzymatic approach is that it creates compatible ends required for ligation, obviating the need for an end-repair after the chemical fragmentation.
Set up the RNA fragmentation reaction with 1 µg of purified RNA, 10 µL of reaction buffer, 5 µL of RNase III, and enough NF water to bring the volume up to 100 µL (see Table of Materials). Add the enzyme last and pipette the mixture up and down at least 10 times to mix. Incubate the sample at 37 °C for 25 min, which tends to produce RNA fragments of approximately 60 - 80 bases in length.
Once the 25 min incubation is complete, clean up the reaction immediately with an oligo clean-up and concentrator kit (see Table of Materials) to prevent any further digestion. Add 200 µL of oligo-binding buffer to the sample, mix it thoroughly by vortexing, and add 800 µL of 100% ethanol. Mix the sample by vortexing and transfer it to a provided spin column in a collection tube.
Centrifuge the sample at 10,000 x g for 30 s and wash it with 750 µL of wash buffer to remove RNase III. Centrifuge the sample at 10,000 x g for 30 s, discard the flow-through, and wash the sample once more with 750 µL of wash buffer as described above. After the second flow-through has been discarded, centrifuge the column at 15,000 x g for 2 min to remove any excess ethanol.
Transfer the column to a fresh 1.5 mL tube and elute the sample in 24 µL of NF water by spinning it at 10,000 x g for 1 min.
QUALITY CHECK: Take 2 µL of the purified RNA fragments and dilute it 2-fold by adding 2 µL of NF water to it. Run the fragmented RNA on a screen tape to ensure that the fragmented RNA is in the range of 50–100 bases in length (Figure 2c).
6. RNA Circularization and Rolling Circle Reverse Transcription
To circularize the RNA fragments, heat-denature 20 µL of the sample at 65 °C for 1 min and place it on ice immediately for 2 min. Then, add 4 µL of 10x T4 RNA ligase buffer, 4 µL of 10 mM ATP, 1 µL of RNase inhibitor, 1 µL of NF water, and 8 µL of 50% polyethylene glycol (PEG).
Mix the sample thoroughly by vortexing, then add 2 µL of 10 U/µL of T4 RNA Ligase I. Mix the sample again by pipetting it and incubate it at 25 °C for 2 h in a thermocycler with the temperature of the lid set at 30 °C.
After 2 h, add 10 µL of NF water to the sample and clean up the sample with an oligo clean-up and concentration kit according to the manufacturer’s specifications. Elute the sample in 20 µL of NF water.
To reverse transcribe the circular RNA molecules, add 4 µL of 10 mM dNTPs, 4 µL of 50 ng/µL random hexamers, and 9 µL of NF water to 9 µL of RNA. Mix everything thoroughly by pipetting, denature the sample at 65 °C for 1 min, and place it on ice for 2 min. Store the remaining RNA as back-up.
Add 8 µL of 5x first-strand synthesis buffer, 2 µL of 0.1 mM DTT, and 4 µL of 200 U/µL reverse transcriptase and mix everything by pipetting. Incubate the sample at 25 °C for 10 min, followed by a 20 min incubation at 42 °C with the lid set at 5 °C higher than the incubation temperature. NOTE: A reverse transcriptase with strand displacement capabilities must be used at this step to allow for a rolling circle reverse transcription.
Add 10 µL of NF water to the sample and clean it up with the oligo clean-up and concentration kit according to the manufacturer’s recommendations. Elute the sample in 42 µL of elution solution.
QUALITY CHECK: Dilute 2 µL of the single-stranded cDNA in 2 µL of NF water and run this sample on a nucleotide analysis instrument with a High Sensitivity RNA Tape. If the reverse transcription worked properly, a library of cDNA molecules, ranging from roughly 60–1,500 bases in length, should be visible. Ideally, most cDNA is in the range of 500–1,000 bases (Figure 2d).
7. Second Strand cDNA Synthesis and End Repair
Use a second strand synthesis kit to generate a double-stranded cDNA library. Place the samples on ice and add 30 µL of NF water to 38 µL of your sample. Then, add 8 µL of 10x Second Strand Buffer and 4 µL of Second Strand Enzyme, and mix the sample by gentle pipetting before its incubation at 16 °C for 2.5 h.
Clean up the samples with the oligo clean-up and concentration kit according to the manufacturer’s recommendations for 100 µL reactions and elute the samples in 38 µL of NF water.
Set up the end-repair reaction by adding 20 µL of NF water to 35.5 µL of double-stranded cDNA, 6.5 µL of End Repair Reaction Buffer and 3 µL of End Prep Enzyme Mix. Carefully check the reaction buffer for white precipitates and dissolve them by hand-warming if present.
Mix the end-repair reaction by pipetting and incubate it at 20 °C for 30 min, followed by a second 30 min incubation at 65 °C. Set the temperature of the thermocycler lid at 5 °C higher than the incubation temperature.
8. Adapter Ligation and Size Selection of Prepared Libraries
Use a multi-step library preparation module for the final library preparations (see Table of Materials). First, dilute the adaptors for next-generation sequencing 10-fold in 10 mM Tris-HCl to a final concentration of 1.5 µM. Then, add 2.5 µL of diluted adapter and 1 µL of ligation enhancer to each sample and mix them by vortexing.
Add 15 µL of Blunt/TA Ligase Master Mix, mix it by pipetting and incubate it at 20 °C for 15 min. Then, add 3 µL of uracil-specific excision reagent enzyme and incubate it at 37 °C for 15 min. After the ligation is complete, add 13.5 µL of NF water to the sample for a total volume of 100 µL and proceed to the size selection. NOTE: It is best to size-select for cDNA molecules that are in the range of 600–800 bases in length. If the fragmented RNA molecules were approximately 60–80 bases in length, selecting for molecules that are 600–800 bases in length will ensure that most of the selected molecules contain at least three tandem repeats, which is ideal for the downstream processing and analysis.
To select for molecules that are 600–800 bp in length, add 30 µL of resuspended magnetic beads (see Table of Materials), acclimated to RT, to each sample and mix by pipetting. Transfer the sample to a 1.5 mL tube and incubate it at RT for 5 min.
Place the sample on a magnetic stand for 5 min to separate the beads from the supernatant. Transfer the supernatant (which contains the desired cDNA molecules) to a new 1.5 mL tube and dispose of the magnetic beads (which are bound to large cDNA molecules).
Add a fresh aliquot of 15 µL of magnetic beads to each samples, mix thoroughly by pipetting, and incubate for 5 min at RT. Place the samples on a magnetic rack and incubate them for 5 min at RT. Then, carefully remove and dispose of the supernatants, making sure not to disturb the beads. NOTE: Do not dispose of the magnetic beads, which now contain the desired target cDNA.
Add 200 µL of 80% freshly prepared ethanol to each of the samples without disturbing the pelleted magnetic beads and incubate for 30 s. Discard the ethanol and wash the samples once more with 80% ethanol. Then, fully remove any trace of ethanol from the samples and air-dry the beads for 5 min but be careful not to over-dry the beads. If the beads are overdried, cracks will appear in the pellets.
To elute the samples from the beads, remove the tubes from the magnetic stand and add 19 µL of 10 mM Tris-HCl. Pipet it up and down multiple times to resuspended the beads and incubate the samples for 5 min at RT. If the beads were overdried, incubate them for >10 min.
Place the samples back on the magnetic stand and incubate them for 5 min to separate the beads from the supernatant. Carefully remove 15 µL of the supernatant containing the purified, size-selected cDNA libraries from the tubes and transfer it to a fresh 1.5 mL tube.
9. PCR Amplification and Final Bead Purification
- Add 5 µL of universal primer and 5 µL of a unique index primer (see Table of Materials) to each purified cDNA library and mix them thoroughly by pipetting. Carefully check the polymerase master mix for crystals and other precipitates, and warm the master mix by hand until the precipitates dissolve.
- Add 25 µL of the polymerase master mix to the sample, mix them by pipetting, and follow the cycling conditions below for the PCR amplification. Run the initial denaturation at 98 °C for 30 s, and then perform a dual step PCR reaction by denaturating the samples at 98 °C for 10 s and an annealing/extension of the PCR products at 65 °C for 75 s (12x), followed by a final extension at 65 °C for 5 min and an indefinite hold at 4 °C.
Clean up the final libraries by adding 45 µL of resuspended magnetic beads directly to the PCR reaction, mix them by pipetting, and incubate them at RT for 5 min. Transfer the sample to a 1.5 mL tube and place it in a magnetic stand. Incubate it for 5 min, discard the supernatant and wash it twice with freshly prepared 80% ethanol for 30 s.
After the second wash, fully remove the ethanol from the sample, and air-dry the bead pellet for 5 min and elute it in 35 µL of 0.1x TE. Resuspend the beads by pipetting them and incubate them at RT for 5 min. Place the sample on the magnetic stand and after 5 min, transfer 30 µL of the supernatant to a fresh 1.5 mL tube. Store the libraries at -80 °C.
QUALITY CHECK: Dilute 1 µL of the final library in 9 µL of NF water and run the sample on a dedicated DNA analysis instrument with a high-sensitivity chip; the majority of cDNA should be in the range of 600–800 bp in length (Figure 2e).
After the elution, send the samples off for sequencing on a massively parallel sequencing platform using 250 bp paired-end reads, or 300 bp single-end reads.
10. Bio-informatic Analysis of Circle Sequencing Data
NOTE: Analyzing and interpreting raw data from the C-seq assay requires a dedicated bio-informatic pipeline. A schematic of the pipeline that was used for our analyses is depicted in Figure 3. Download this pipeline at https://github.com/LynchLab/MAPGD.
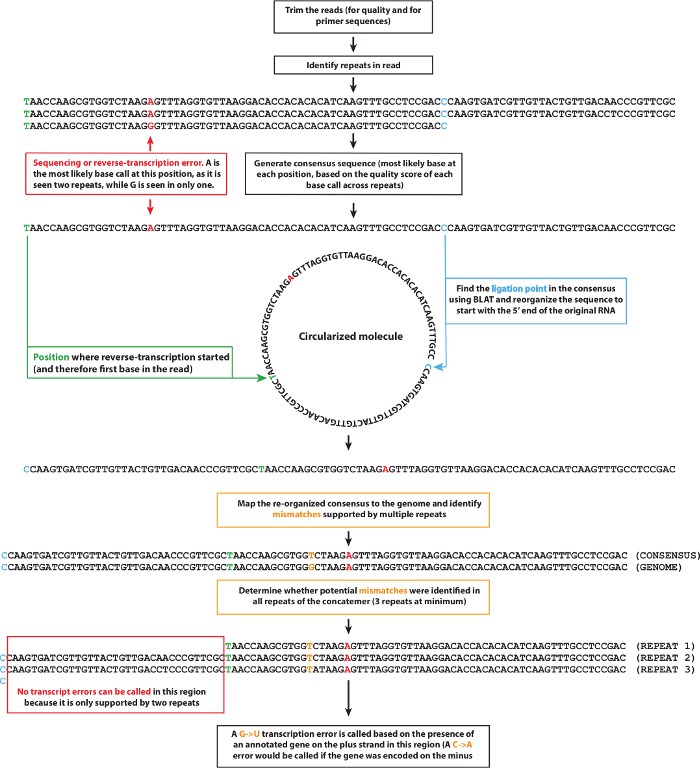
First, trim the reads to remove adapter sequences and low-quality base calls with cutadapt18, using a quality score threshold of 20.
Then, identify the repeats encoded by the concatemerized RNA molecule using an appropriate algorithm to detect any repeats with a minimum size of 30 nucleotides and a maximum of 2 mismatches between repeats18. Derive a consensus sequence from these repeats and keep track of all the base calls and associated quality scores at each position. NOTE: Because reverse transcription can start at any position on the circularized RNA molecule, the start of a repeat does not necessarily correspond to the 5′-end of the fragmented RNA molecule (hereafter referred to as the ligation point). Accordingly, the consensus sequence will not map continuously against the corresponding transcriptome, which necessitates the identification of the ligation point.
- To identify the ligation point, map a concatemer of the consensus sequence against the reference transcriptome using the BLAST-like alignment tool (BLAT)19.
- Concatenate two instances of the consensus sequence to ensure that the mapped sequence contains at least one full uninterrupted occurrence of the initial RNA fragment sequence. Then, rotate the consensus sequence to start at the predicted ligation point position.
Map the rotated consensus sequences against the genome using TopHat20 and use the alignments to detect any single nucleotide polymorphisms (SNPs) that could be mistaken for transcription errors.
- Call the transcription errors from the mapped rotated consensus sequences and test whether the candidate errors pass four additional tests.
- First, ensure that a transcription error does not overlap with an SNP (a typical requirement is that at least 20 reads need to cover a particular base and no more than 5% of the reads may support an alternative base call).
- Second, check that the consensus sequence is derived from at least 3 tandem repeats, each of which calls the same base pair; and thirdly, make sure that the sum of the quality scores of that position must be over 100.
For a fourth and final check, rotate the consensus sequence in all possible combinations (simulating all possible positions of the ligation point) and map each version against the reference genome using TopHat. NOTE: If one of the rotated sequences finds a perfect match in the genome, the corresponding ligation point is now considered the correct one and the transcription error call should be rejected.
Representative Results
Like all massively parallel sequencing approaches, each C-seq experiment produces an unwieldy, large dataset. For first-time users, it can be difficult to handle these datasets; thus, it is recommended that all users contact an experienced bio-informatician prior to the experimentation. On average, the expectation is that users will generate approximately 55–70 Giga bases (Gbases) per run on most massively parallel sequencing platforms. For this protocol, typically, 12–30 samples were multiplexed per run so that for each sample approximately 2–6 Gbases were acquired. After trimming the adaptor sequences and low-quality base calls (< 20) from this dataset, ~70% of the initial data size remained.
These bases are then analyzed further to investigate the efficiency of the RNA fragmentation and circularization by determining the size of the repeats that were generated. Most repeats tend to be 45–80 bases in length (Figure 4A) and approximately 50% of the bases that were sequenced are part of these repeats (Figure 4B). Since most of these bases are present in reads that contain 3 repeats or more, the number of unique bases that are sequenced is about one-third of the total number of bases sequenced. On average, >75% of these consensus sequences can be mapped back to the reference genome. Finally, approximately 25% of these bases are covered by 20 reads or more, which ultimately means that about 10% of the data can be used for error detection. An example of this analysis is given in Figure 4, which represents the sequencing information that was acquired during the set-up of this protocol for 1 replicate of a single C-seq library that was sequenced relatively shallowly and represents the lower limit of the data that users can expect to acquire.
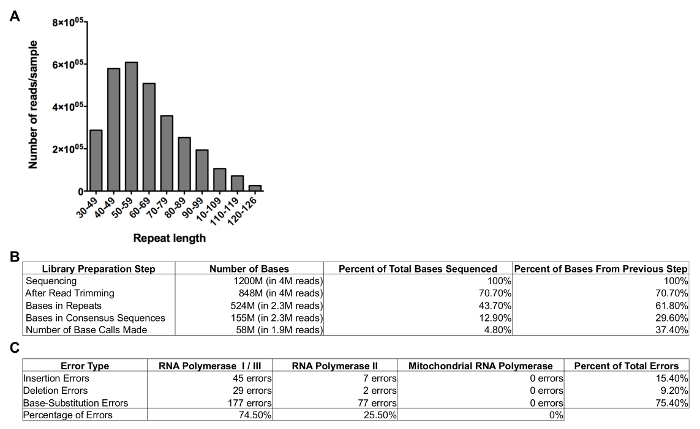
Approximately 5,000–25,000 errors per run tend to be identified, although these numbers can vary significantly depending on the error rate itself (the higher the error rate, the more errors will be detected), the size of the transcriptome (the larger the transcriptome, the fewer bases will be covered by 20 reads, limiting the sequencing data that can be used for error detection), and the depth at which it is sequenced (deeper sequencing will make it more likely that a given base will be covered by 20 reads or more). These errors tend to be distributed across the entire genome, so that on average ~47% of the errors are located in mRNA molecules generated by RNA polymerase II (RNAP II), ~49% are located in rRNA molecules generated by RNAP I, and the remaining ~3% are located in RNAs generated by RNAP III and the mitochondrial RNA polymerase. However, these ratios can vary significantly depending on the cell type or organism under investigation (Figure 4C). For example, cell types that rely heavily on mitochondrial function, such as cardiomyocytes, contain significantly more mitochondrial RNA than other cell types, greatly increasing the number of mitochondrial RNA molecules that are sequenced, and thus the number of errors detected.
Once a list of errors has been compiled, and their locations across the genome are known, these errors can be used to identify the parameters that control the error rate of transcription in each organism. For example, the location of these transcription errors can be correlated with numerous features of the genome, such as the presence of DNA repeats, specific genetic contexts, or the expression rate, to understand how these features alter the fidelity of transcription5. The expectation is that in the future, though, users will be able to determine how countless additional features affect transcriptional fidelity, including epigenetic markers, the 3-D organization of the genome, nutrient availability, age, or exposure to toxic compounds, to elucidate the contribution of genetic and environmental factors to the fidelity of transcription.
Figure 1: Schematic representation of RNA-seq versus C-seq. (A) Traditional RNA-seq experiments isolate RNA from a sample of interest, fragment the RNA, and reverse transcribe it prior to the final library preparation and sequencing. However, these preparation procedures introduce numerous technical artifacts into the library in the form of reverse transcription errors, PCR amplification errors, and sequencing errors. (B) This optimized C-seq assay allows for the correction of these technical artifacts by circularizing the fragmented RNA molecules prior to reverse transcription, which allows them to be reverse transcribed in a rolling circle fashion to produce linear cDNA molecules that contain several copies of the original RNA template in tandem repeat. These tandem repeats can then be used to distinguish true transcription errors from artifacts, as true transcription errors (star) will be present in all repeats at the same location, whereas artifacts such as reverse transcription errors (square) and PCR amplification errors (circle) are only present in one or two repeats of any given cDNA molecule. Please click here to view a larger version of this figure.
Figure 2: Representative results for library preparation. These panels show an electrophoretogram for (A) a high sensitivity RNA screen tape ladder (see Table of Materials), (B) the total RNA purified from Saccharomyces cerevisiae, (C) RNase III-fragmented RNA, and (D) rolling circle reverse-transcribed cDNA. (E) The final size-selected cDNA library runs on a high sensitivity double-stranded DNA analysis chip (see Table of Materials). Please click here to view a larger version of this figure.
Figure 3: Schematic of the bio-informatic pipeline used to analyze circle sequencing data. After trimming the sequencing reads, repeats are identified, and a consensus sequence is generated using the most likely base call at any given position. Then, the ligation point of the initial RNA template is identified and the consensus sequence is aligned to the reference genome so that potential transcription errors can be identified. Please click here to view a larger version of this figure.
Figure 4: Example of results for C-Seq pipeline. (A) This panel shows the size distribution of the consensus sequences obtained from a typical C-Seq experiment. (B) This panel shows the number of bases, reads, and percentage of reads sequenced in each step of the C-Seq bioinformatics pipeline. (C) This panel shows the number of transcription errors detected and the percentage of errors attributed to RNA polymerase I, RNA polymerase II, RNA polymerase III, and mitochondrial RNA polymerase. Please click here to view a larger version of this figure.
Discussion
Here, we describe an optimized protocol for the preparation of C-seq libraries for the detection of transcription errors in Saccharomyces cerevisiae, Drosophila melanogaster, and Caenorhabditis elegans. This protocol has numerous advantages over existing protocols, as well as alternative techniques.
Over the past 15 years, numerous reporter systems have been developed that rely on luciferase7,8 or Cre-Lox recombination3,4,15,21 to detect transcription errors. These reporter systems have been invaluable to researchers' understanding of transcriptional fidelity because they allowed genes, alleles, and molecular mechanisms to be identified that directly regulate the fidelity of transcription. However, they only report on errors in artificially damaged templates, or within highly specific genetic contexts, which limits the scientific questions they can answer. An important advantage of the C-seq protocol is that it monitors the fidelity of transcription throughout the entire transcriptome5, greatly expanding the scientific knowledge of the accuracy with which genetic information is expressed. Secondly, because the C-seq assay utilizes RNA as its source material, it is likely that this assay can be adapted to any organism of choice, obviating the need to generate complicated reporter constructs for each transgenic model. This protocol also has advantages over existing massively parallel sequencing approaches17,22. Most notably, most of these approaches make use of heavy metals to fragment RNA libraries. However, these fragmentation methods introduce artifacts into the RNA that are indistinguishable from true transcription errors5. To solve this problem, this protocol fragments RNA enzymatically with RNase III, which has the added advantage that it leaves compatible ends for self-ligation, greatly simplifying RNA circularization and increasing the sensitivity of the assay ~10-fold.
At the same time, the C-seq assay has its own share of limitations. First, even though the assay measures transcription errors throughout the entire genome, a large component of the data tends to be derived from genes that are highly expressed, as transcripts from these genes tend to dominate most RNA libraries. Another factor that skews the analysis towards highly expressed genes is the threshold built into the bio-informatic pipeline that prevents genetic mutations and RNA editing events from confounding the final measurements. This threshold dictates that only bases that were sequenced 20 times or more can be used for the final analysis. A second limitation concerns library diversity. Like most kits used for RNA extraction, the kit used here does not efficiently purify small RNA molecules, which limits the number of reads derived from molecules synthesized by RNAP III. The diversity of the final sequencing library is further limited by the mRNA purification step employed here. Even though molecules generated by RNAP I can be efficiently sequenced, most of the remaining mitochondrial RNAs, tRNAs, and non-coding RNAs are lost during this step. To capture these molecules more frequently, a different kit that specifically purifies small RNA molecules should be used, or cell types should be targeted that are enriched for these molecules. Please note, though, that most of these problems can be solved by sequencing deeper than usual, or simultaneously sequencing the DNA from the same cells, although both of these solutions will require a larger financial investment by the investigators.
In addition, future technological developments are poised to improve the C-seq assay at a fundamental level. For example, by extending the read-length of existing sequencing technology, it will be possible to sequence longer molecules, which means that more repeats can be used to ascertain the fidelity of transcription. Such improvements will automatically result in a greater sequencing depth and an increase in the percentage of reads that can be used for downstream analysis. A similar argument can be made for any sequencing technology that allows for more molecules to be sequenced in parallel. In addition, numerous components of the C-seq assay remain to be optimized, including the efficiency of RNA circularization, the stringency of size selection, and the time-consuming nature of the assay itself. Finally, it is strongly recommended that all users gain access to a dedicated, high-sensitivity tool for nucleic acid analysis and potential troubleshooting (see Table of Materials). Without this tool, it can be extremely difficult to determine at what point potential problems arise, thereby making them impossible to fix.
Although the entire protocol is fairly unforgiving (small mistakes tend to have significant consequences), there are several steps that are absolutely critical to the success of the C-seq assay. One of the most important requirements is that the isolated RNA is treated as gently as possible. Most RNA extraction methods and downstream processing kits care little about the chemical composition of the RNA beyond basic industry standards, which is sufficient for most molecular analyses. However, the C-seq assay is tasked with identifying a single transcription error among hundreds of thousands of WT bases, greatly increasing the need to reduce molecular stress. Even stress that impacts only 1 in 10,000 bases can introduce artifacts that completely invalidate the results. For example, users must never avoid/limit expose the RNA to any temperature over 65 °C. Secondly, each user must carefully optimize the time required for the RNA fragmentation with RNase III. It is absolutely crucial for the success of the assay that the final molecules are approximately 60–80 bases in length. Shorter molecules may be difficult to map to the genome, and longer molecules will not allow for 3 independent repeats to be sequenced within a 250 bp read. Finally, it is recommended not to freeze and thaw the RNA more than once during the entire protocol. Instead, isolate the RNA and synthesize the cDNA in a single day. Because of the time and effort involved with this commitment, the complexity of the protocol itself, and the number of samples that are frequently processed at once, it is recommended that two investigators work together to generate these libraries.
When successful, these experiments will make it possible to accurately detect transcription errors in any organism, under any experimental condition. For example, by comparing control and diseased individuals to each other, it may be possible to determine whether certain diseases are associated with transcription errors, which may reveal a new component of the etiology of numerous human pathologies. Other parameters that could be probed are organismal aging, nutrition, genotype, and environmental factors such as exposure to toxic chemicals. Moreover, because this assay detects transcription errors throughout the transcriptome, it will allow researchers to dissect the different mechanisms that contribute to the fidelity of RNA polymerase I, II, and III, as well as the mitochondrial RNA polymerase. Accordingly, we expect that this protocol will open up a new field of mutagenesis to widespread experimentation, characterized by the study of mutations in RNA rather than in DNA.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This publication was made possible by funding from grant T32ES019851 (to C. Fritsch), R01AG054641, and an AFAR young investigator grant (to M. Vermulst).
References
- Kireeva ML, et al. Transient reversal of RNA polymerase II active site closing controls fidelity of transcription elongation. Molecular Cell. 2008;30:557–566. doi: 10.1016/j.molcel.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walmacq C, et al. Rpb9 subunit controls transcription fidelity by delaying NTP sequestration in RNA polymerase II. The Journal of Biological Chemistry. 2009;284:19601–19612. doi: 10.1074/jbc.M109.006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strathern J, et al. The fidelity of transcription: RPB1 (RPO21) mutations that increase transcriptional slippage in S. cerevisiae. The Journal of Biological Chemistry. 2013;288:2689–2699. doi: 10.1074/jbc.M112.429506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvin JD, et al. A genetic assay for transcription errors reveals multilayer control of RNA polymerase II fidelity. PLoS Genetics. 2014;10:e1004532. doi: 10.1371/journal.pgen.1004532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout JF, et al. The landscape of transcription errors in eukaryotic cells. Science Advances. 2017;3:e1701484. doi: 10.1126/sciadv.1701484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout JF, Thomas WK, Smith Z, Okamoto K, Lynch M. Large-scale detection of in vivo transcription errors. Proceedings of the National Academy of Science of the United States of America. 2013;110:18584–18589. doi: 10.1073/pnas.1309843110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan A, You HJ, Doetsch PW. Phenotypic change caused by transcriptional bypass of uracil in nondividing cells. Science. 1999;284:159–162. doi: 10.1126/science.284.5411.159. [DOI] [PubMed] [Google Scholar]
- Bregeon D, Doddridge ZA, You HJ, Weiss B, Doetsch PW. Transcriptional mutagenesis induced by uracil and 8-oxoguanine in Escherichia coli. Molecular Cell. 2003;12:959–970. doi: 10.1016/s1097-2765(03)00360-5. [DOI] [PubMed] [Google Scholar]
- Saxowsky TT, Doetsch PW. RNA polymerase encounters with DNA damage: transcription-coupled repair or transcriptional mutagenesis. Chemical Reviews. 2006;106:474–488. doi: 10.1021/cr040466q. [DOI] [PubMed] [Google Scholar]
- Saxowsky TT, Meadows KL, Klungland A, Doetsch PW. 8-Oxoguanine-mediated transcriptional mutagenesis causes Ras activation in mammalian cells. Proceedings of the National Academy of Science of the United States of America. 2008;105:18877–18882. doi: 10.1073/pnas.0806464105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermulst M, et al. Transcription errors induce proteotoxic stress and shorten cellular lifespan. Nature Communications. 2015;6:8065. doi: 10.1038/ncomms9065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen FW, Burbach JP, Hol EM. Mutations in RNA: a first example of molecular misreading in Alzheimer's disease. Trends in Neurosciences. 1998;21:331–335. doi: 10.1016/s0166-2236(98)01280-6. [DOI] [PubMed] [Google Scholar]
- van Leeuwen FW, et al. Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer's and Down patients. Science. 1998;279:242–247. doi: 10.1126/science.279.5348.242. [DOI] [PubMed] [Google Scholar]
- Morreall JF, Petrova L, Doetsch PW. Transcriptional mutagenesis and its potential roles in the etiology of cancer and bacterial antibiotic resistance. Journal of Cellular Physiology. 2013;228:2257–2261. doi: 10.1002/jcp.24400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strathern JN, Jin DJ, Court DL, Kashlev M. Isolation and characterization of transcription fidelity mutants. Biochimica et Biophysica Acta. 2012;1819:694–699. doi: 10.1016/j.bbagrm.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acevedo A, Andino R. Library preparation for highly accurate population sequencing of RNA viruses. Nature Protocols. 2014;9:1760–1769. doi: 10.1038/nprot.2014.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traverse CC, Ochman H. Genome-Wide Spectra of Transcription Insertions and Deletions Reveal That Slippage Depends on RNA:DNA Hybrid Complementarity. mBio. 2017;8 doi: 10.1128/mBio.01230-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnet.journal. 2011;17(1):10–12. [Google Scholar]
- Kent WJ. BLAT--the BLAST-like alignment tool. Genome Research. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YN, et al. Isolation and characterization of RNA polymerase rpoB mutations that alter transcription slippage during elongation in Escherichia coli. The Journal of Biological Chemistry. 2013;288:2700–2710. doi: 10.1074/jbc.M112.429464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid-Bayliss KS, Loeb LA. Accurate RNA consensus sequencing for high-fidelity detection of transcriptional mutagenesis-induced epimutations. Proceedings of the National Academy of Science of the United States of America. 2017;114:9415–9420. doi: 10.1073/pnas.1709166114. [DOI] [PMC free article] [PubMed] [Google Scholar]