

Abstract
Light-sheet fluorescence microscopy has been widely used for rapid image acquisition with a high axial resolution from micrometer to millimeter scale. Traditional light-sheet techniques involve the use of a single illumination beam directed orthogonally at sample tissue. Images of large samples that are produced using a single illumination beam contain stripes or artifacts and suffer from a reduced resolution due to the scattering and absorption of light by the tissue. This study uses a dual-sided illumination beam and a simplified CLARITY optical clearing technique for the murine heart. These techniques allow for deeper imaging by removing lipids from the heart and produce a large field of imaging, greater than 10 x 10 x 10 mm3. As a result, this strategy enables us to quantify the ventricular dimensions, track the cardiac lineage, and localize the spatial distribution of cardiac-specific proteins and ion-channels from the post-natal to adult mouse hearts with sufficient contrast and resolution.
Keywords: Bioengineering, Issue 139, Light sheet fluorescence microscopy, LSFM, murine, optical clearing, cardiomyocytes, ventricle, heart, dual illumination, CLARITY
Introduction
Light-sheet fluorescence microscopy was a technique first developed in 1903 and is used today as a method to study gene expression and also to produce 3-D or 4-D models of tissue samples1,2,3. This imaging method uses a thin sheet of light to illuminate a single plane of a sample so that only that plane is captured by the detector. The sample can then be moved in the axial-direction to capture each layer, one section at a time, and render a 3-D model after the post-processing of the acquired images4. However, due to the absorption and scattering of photons, LSFM has been limited to samples that are either a few microns thick or are optically transparent1.
The limitations of LSFM have led to extensive studies of organisms that have tissues that are optically transparent, such as the zebrafish. Studies involving cardiac development and differentiation are often conducted on zebrafish since there are conserved genes between humans and zebrafish5,6. Although these studies have led to advances in cardiac research related to cardiomyopathies6,7, there is still a need to conduct similar research on higher-level organisms such as mammals.
Mammalian cardiac tissue presents a challenge due to the thickness and opacity of the tissue, the absorption due to hemoglobin in red blood cells, and the striping that occurs due to single-sided illumination of the sample under traditional LSFM methods1,8. To compensate for these limitations, we proposed to use dual-sided illumination and a simplified version of the CLARITY technique9 combined with a refractive index matching solution (RIMS). Therefore, this system allows for the imaging of a sample that is greater than 10 x 10 x 10 mm3 while maintaining a good quality resolution in the axial and lateral planes8.
This system was first calibrated using fluorescent beads arranged in different configurations within the glass tubing. Then, the system was used to image post-natal and adult murine hearts. First, the post-natal mouse heart was imaged at 7 days (P7) to reveal the ventricular cavity, the thickness of the ventricular wall, the valve structures, and the presence of trabeculation. Secondly, a study was conducted to identify cells that would differentiate into cardiomyocytes by using a post-natal mouse heart at 1 day (P1) with Cre-labeled cardiomyocytes and yellow fluorescent protein (YFP). Finally, adult mice at 7.5 months were imaged to observe the presence of renal outer medullary potassium (ROMK) channels after gene therapy8.
Protocol
All the procedures involving the use of animals have been approved by the Institutional Review Committees (IACUC) at the University of California, Los Angeles, California.
1. Imaging System Setup
Note: See Figure 1 and Figure 2.
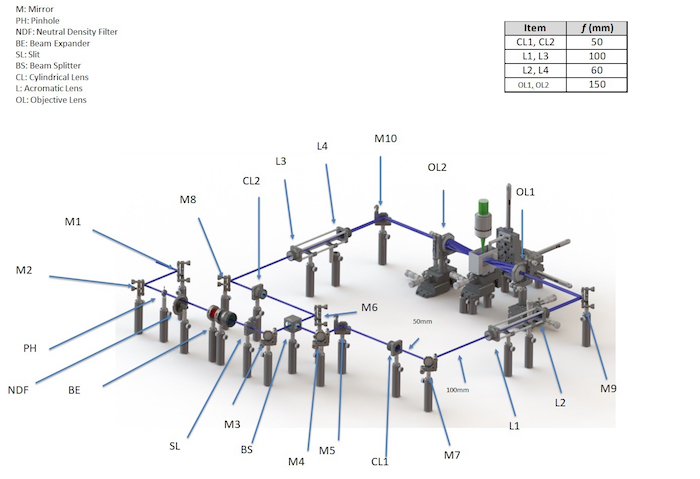
Retrieve a continuous wave (CW) laser with 3 wavelengths: 405 nm, 473 nm, and 532 nm. Place 2 mirrors (M1 and M2) 150 mm apart and align them with their mirror planes at 45° to the beam. Note: This step is performed to redirect the laser away from the dual-sided illumination setup. The resulting beam will be in the same direction as the initial beam.
Pass the beam through a 25-mm diameter iris diaphragm/pinhole (PH), a 50-mm diameter neutral density filter (NDF) with an optical density range of 0 - 4.0, a beam expander (BE), a 30 mm slit (S) with the width of ~0 - 6 mm, and a mirror (M3), all positioned 150 mm from each other. Place the mirror with its mirror plane at 45° to the beam.
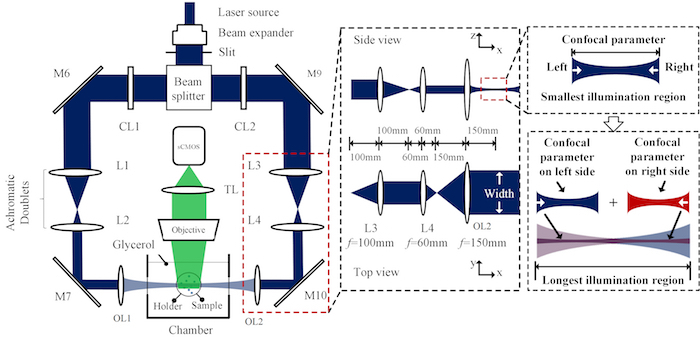
Pass the beam through a 50:50 beam splitter placed 150 mm from M3. Place a mirror (M6) 150 mm from the beam splitter and align it so that its mirror plane is at 45° to the beam that is emitted in the forward direction. Use the reflected beam to form one side of the dual-illumination light sheet.
Place a mirror (M4) 150 mm from the beam splitter and align it so that its mirror plane is at 45° to the beam that was emitted at a 90° angle from the beam splitter. Place a second mirror (M5) 150 mm from M4 and align it so that its mirror plane is at 45° to the beam reflected from M6. Use the beam emitted from M5 to form the second side of the dual-illumination light sheet.
Set up the dual-sided illumination system in a symmetric fashion (see Figure 1). Place one cylindrical lens (CL1) [diameter (d) = 1 in; focal length (f) = 50 mm] 150 mm in line with the beam emitted from M5 on one side and another identical cylindrical lens (CL2) 150 mm in line with the beam emitted from M6 on the other side of the dual-illumination setup. Place 2 mirrors (M7 and M8) each in line with the cylindrical lenses at distances of 50 mm to reflect the beam at 90°.
Form an achromatic doublet from a pair of lenses. Place the first lens, L1 (d = 1 in; f = 100 mm), 100 mm from M7, and the second lens, L2 (d = 1 in; f = 60 mm), 160 mm from L1. Repeat this on the other side of the dual-illumination with identical lenses (L3, L4) placed the same distance from M8.
Place mirrors, M9 and M10, 60 mm from lenses L2 and L4, respectively, and in line with the beam. Place the illumination objectives, OL1 and OL2 (d = 2 in; f = 150 mm), 150 mm from M9 and M10 and in line with the beam. The beam emitted from the objectives forms the light sheet for imaging the samples.
Use a 3-D printer to print the sample holder from acrylonitrile butadiene styrene (ABS). Embed a piece of cover glass [refractive index (RI) = 1.4745] on each side of the chamber, perpendicular to the illumination beam, to minimize refractive index mismatching. Evenly place the chamber in between the beams emitted from the objective lenses.
Install a stereo microscope with a 1X magnification objective and a scientific complementary metal oxide semiconductor (sCMOS) camera perpendicular to the illumination plane (see Figure 2).
2. Imaging System Calibration
Retrieve fluorescent polystyrene beads that are 0.53 μm in diameter.
Prepare the fluorescent bead sample by diluting the bead solution in a refractive index matching solution (RIMS) with 1% low-melt pointing agarose to 1:150,000. Cut a piece of borosilicate glass tubing with an inner diameter of 12 mm and an outer diameter of 18 mm to a length of 30 mm. Note: The borosilicate glass tubing is used to match the refractive index (1.47).
Mix the diluted bead sample with 1% agarose and pipette the bead/agarose solution into the borosilicate tubing. Allow the agarose to solidify at room temperature (23 °C).
Fill the ABS chamber (from step 1.7) with a 99.5% glycerol solution. Place the borosilicate glass tubing containing the beads inside the chamber. Place the ABS chamber in the imaging system so that it is in the center of the Gaussian beam created by the dual illumination system.
Attach a 3-D motorized translational stage to the borosilicate glass tubing to control the movement and orientation of the sample within the ABS chamber (see Supplemental Figure 1).
Acquire images using the sCMOS camera at a rate of 30 frames per second (fps). Using the motor controller, move the sample 1 mm in the lateral direction and acquire images at each 1-mm increment using the sCMOS camera. Continue until the entire sample has been imaged.
Stack the acquired images using a visualization software (see Table of Materials). Measure the point spread function (PSF) of the system using these bead images. Use this PSF for deconvolution during the image processing in later steps.
3. Sample Preparation
Dissect hearts from a wild type P1 mouse and from a double heterozygous sarcolipin-Cre knockin mouse with the Rosa26-YFP gene (SlnCre/+; R26YFP reporter/+) at P7. Note: Euthanasia was performed using pentobarbital and the technique described by Robbins et al.10. The dissection was performed according to the technique described by the National Heart, Lung, and Blood Institute (NHLBI)10,11.
- Perform a chemical clearing of the murine hearts as described by Kevin Sung et al.12. Note: The following steps should be performed in a fume hood since the chemicals being used are toxic.
- Rinse the hearts in 1x phosphate-buffered saline (PBS) 3x for 10 min. After rinsing, place the hearts in a 4% paraformaldehyde solution and incubate these samples at 4 °C overnight.
- Place the samples in a 4% acrylamide solution containing 0.5% w/v of 2,2’-Azobis dihydrochloride. Allow the samples to incubate at 4 °C overnight. After the overnight incubation, remove the samples and incubate them at 37 °C for 2 - 3 h.
- Rinse the samples with PBS and then place them in a solution of 8% w/v sodium dodecyl sulfate and 1.25% w/v boric acid. Incubate the samples at 37 °C until they are clear. This can take several hours depending on the thickness of the sample. After clearing, remove the samples and place them in 1x phosphate-buffered saline for 1 day.
- Prepare a refractive index matching solution with 40 g of nonionic density gradient medium (see Table of Materials) in 30 mL of 0.02 M phosphate buffer (PB), 0.1% Tween-20, and 0.01% sodium azide. Bring the solution to a pH of 7.5 with sodium hydroxide.
Place the cleared sample in RIMS with a refractive index of 1.46 - 1.48 and 1% agarose. Insert the sample into borosilicate glass tubing and allow the agarose to solidify at room temperature (23 °C).
Attach a 3-D motorized translational stage to the borosilicate glass tubing to control the movement and orientation of the sample within the 3-D printed ABS chamber (see Supplemental Figure 1).
4. Adult Heart Imaging
Inject 8.7 x 1012 viruses of type adeno-associated virus vector 9 (AAV9) containing a cardiac-specific troponin T promoter (cTnT) and green fluorescent protein (GFP) in a 100-μL volume into the tail vein of a 2-month wild type (C57BL/6) mouse. Note: The AAV9 was developed according to the technique described by Yuan et al.13. This causes GFP to bind to the renal outer medullary potassium channel (ROMK) producing AAV9-cTnT-ROMK-GFP (see Table of Materials).
Dissect the adult mouse hearts at 7.5 months of age according to the technique described by the NHLBI11. Prepare the samples using steps 3.1 - 3.3.
Connect the motor controller to the motor actuator. Attach the actuator to the borosilicate glass tubing to control the movement and orientation of the sample within the 3-D printed ABS chamber.
Position the sample so that it is in the center of the Gaussian beam created by the dual illumination system. Acquire images using the sCMOS camera at a rate of 100 fps.
Using the motor controller, move the sample 1 mm in the axial direction and acquire images at each 1-mm increment with the sCMOS camera. Continue until the entire sample has been imaged. Note: The system is controlled by a custom-developed instrument-control software.
Stack the acquired images using a visualization software (see Table of Materials). Develop 3-D images using these image stacks and the visualization software14. Deconvolve the PSF from step 2.6 with the acquired image stack. Set a pixel threshold intensity value to observe the contours of the heart and add pseudo-color to the images based on this gray-scale intensity.
Representative Results
The technique described here used a dual-sided illumination beam combined with an optical clearing of mouse heart tissue samples to achieve a deeper imaging depth and a larger imaging volume with sufficient imaging resolution (Figure 1). To calibrate the system, fluorescent beads were placed inside the glass tubing and within the imaging system. The beads were then imaged and visualized in the x-y plane, y-z plane, and in the x-z plane, to obtain the point spread function (PSF) for the system8. The system calibration produced a resolution in the lateral direction of dlateral ≈ 2.8 µm and a resolution in the axial directions of dXZ ≈ 17.4 µm and dYZ ≈ 17.9 µm when using glycerol as the embedding medium8.
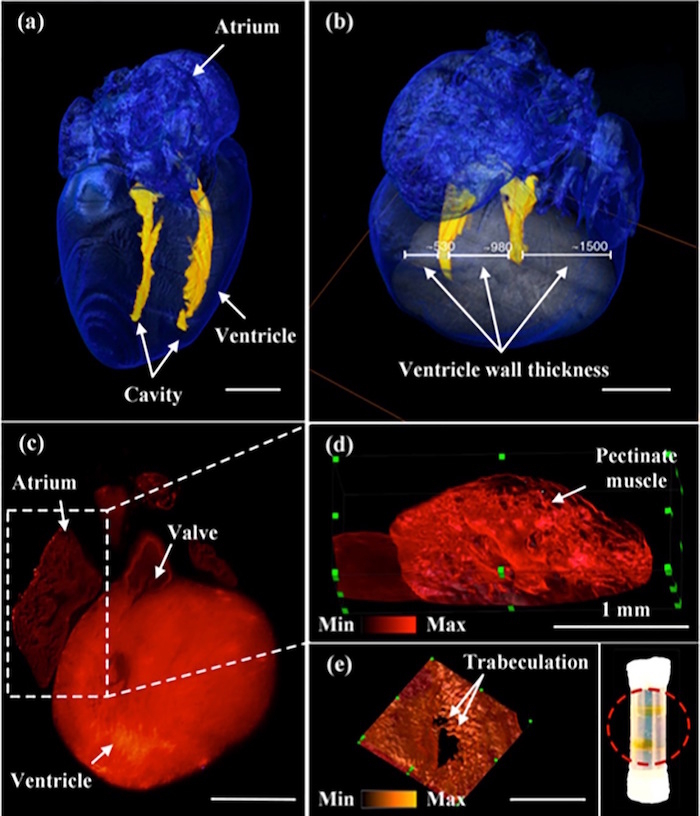
After the fluorescent beads were used for calibration, the imaging system was used to image mouse hearts at 1 day and at 7 days post-natal (Figure 2)8. The ventricular cavity dimensions, heart valves, and ventricular trabeculation are evident in these images of the mouse heart. To study a cardiomyocyte differentiation within the heart, heterozygous knockin mice with Cre-labeled cardiomyocytes were imaged post-natal. The images of the Cre-labeled cardiomyocytes are in each plane direction along with a 3-D rendering of the heart8 (Figure 3).
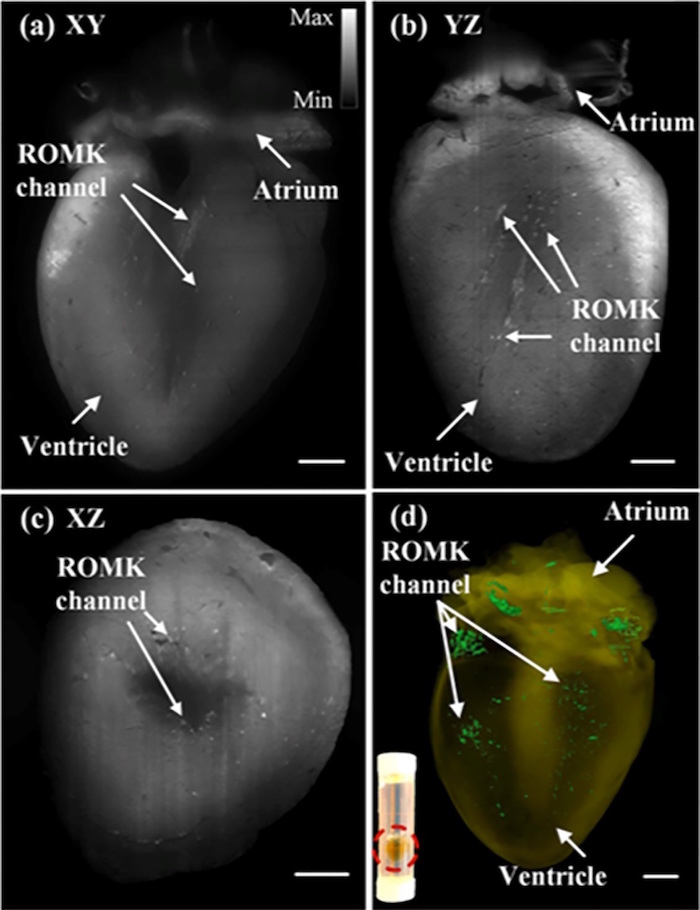
In addition to studying post-natal mice, the imaging system was also used to study the adult mouse heart. Initially, gene therapy was used to introduce GFP-tagged ROMK channels into the mouse heart. At 7.5 months, the mouse heart was imaged and these ROMK channels were visualized in the ventricular wall (Figure 4). This step within the procedure illustrates the ability of this system to image large samples and distinguish structures not seen with histological staining within the heart. Figure 4 shows the ROMK channels from each plane direction along with a 3-D rendering of the heart8.
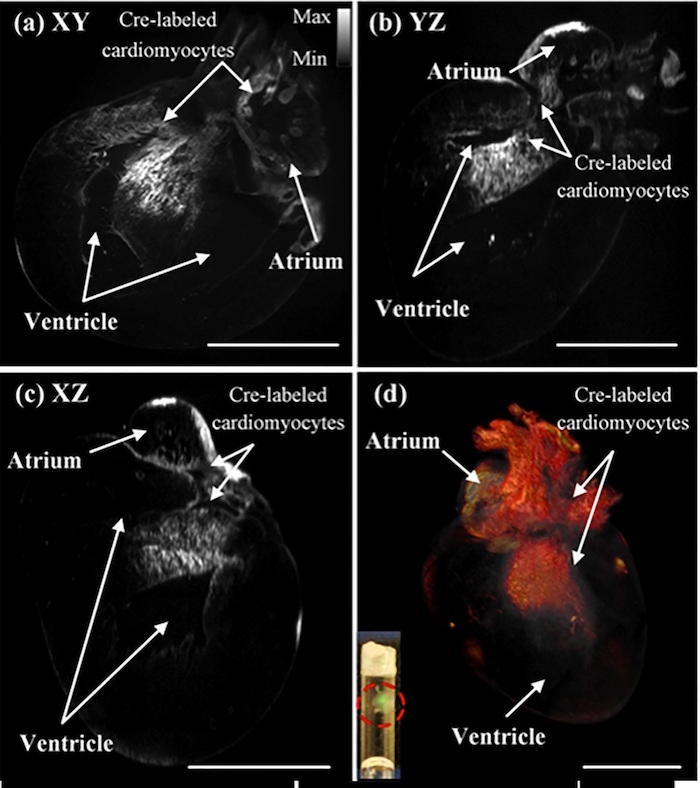
Figure 1: Schematic diagram of the light-sheet fluorescence microscopy system. This is a 3-dimensional rendering of the light sheet fluorescence microscopy system, including the distances between the components and the focal lengths of the components used in the system. This figure has been modified from Yichen Ding et al.8. Please click here to view a larger version of this figure.
Figure 2: Top view of light sheet microscopy system with detection. This is the top view of the dual-sided illumination microscopy system where the darkened blue region represents the beam at each location within the system. Included is a 2-dimensional enlarged section that shows one of the regions consisting of the achromatic doublets and illumination objective. This enlarged section also shows the principle of light sheet formation and how the beams are combined to form a dual-sided illumination. This figure has been modified from Yichen Ding et al.8. Please click here to view a larger version of this figure.
Figure 3: 3-D images of post-natal murine heart. (a) This panel shows an image of the post-natal mouse heart at day 7 showing the ventricle cavity. (b) This panel shows an image of the post-natal mouse heart at day 7 showing the thickness of the ventricle wall. (c) This panel shows the image of the post-natal mouse heart at day 1 showing the location of the valve structures. (d) This image shows an enlarged section from the post-natal mouse heart at day 1 showing the pectinate muscle located in the atrium of the heart. (e) This image shows trabeculation in the ventricle within the post-natal mouse heart at day 1. Scale bar: 1 mm. The red-circled region within the inset shows two translucent mouse hearts after undergoing the CLARITY technique and being inserted into the borosilicate glass tubing. This figure has been modified from Yichen Ding et al.8. Please click here to view a larger version of this figure.
Figure 4: Images of the Cre-labeled cardiomyocytes in a post-natal mouse heart from each plane. The P1 mouse heart was imaged with Cre-labeled cardiomyocytes in each cross-sectional plane: (a) XY, (b) YZ, and (c) XZ. These cross-sectional images show the presence of Cre-labeled cardiomyocytes in the atrium and the ventricle of the P1 mouse. (d) The images were also merged to create a 3-D rendering of the heart. The red-circled region within the inset shows two translucent mouse hearts after undergoing the CLARITY technique and being inserted into the borosilicate glass tubing. Scale bar: 1 mm. This figure has been modified from Yichen Ding et al.8. Please click here to view a larger version of this figure.
Figure 5: Images of the adult mouse heart showing the ROMK channels from each plane. The mouse heart was imaged at 7.5 months of age to show the presence and location of ROMK channels. The heart was imaged in each cross-sectional plane: (a) XY, (b) YZ, and (c) XZ. The gray scale value indicates the optical intensity within the image, with the lighter regions corresponding to more light intensity and the darker regions corresponding to less light intensity. (d) The images were also merged to create a 3-D rendering of the heart, and the original grayscaled images were pseudo-colored so that the ROMK channels appear green. The red-circled region within the inset shows a single translucent adult mouse heart after undergoing the CLARITY technique and being inserted into the borosilicate glass tubing. Scale bar: 1 mm. This figure has been modified from Yichen Ding et al.8. Please click here to view a larger version of this figure.
Supplemental Figure 1: Image of the ABS chamber with the borosilicate tubing within the light sheet of the imaging system. Please click here to download this file.
Discussion
The LSFM system and technique described here utilizes a dual-sided illumination beam combined with an optical clearing to image the mouse heart at post-natal and adult stages of its development. A traditional single illumination beam suffers from photon scattering and absorption through thicker and larger tissue samples1,3. The dual-sided beams provide a more even illumination of the sample, thereby minimizing the effect of striping and other artifacts that are often seen in images produced by a one-sided illumination. This technique also increases the size of the sample that can be imaged by adjusting the position of the dual Gaussian beams8. The maximum separation of these beams allows for imaging a tissue sample several millimeters in size, such as the adult mouse heart, whereas a single illumination limited the size of the sample to smaller hearts such as the hearts of zebrafish and Drosophila.
Fluorescence imaging in tissues that are not optically transparent is more challenging since light scattering may not be uniform throughout the tissue sample. This limits the depth to which a sample can be imaged and the effectiveness of fluorescence imaging. For this reason, it is common to use organisms such as the zebrafish, which naturally have optically transparent tissue5. However, to perform fluorescence imaging studies on organisms that do not naturally have optically transparent skin, there are different optical clearing techniques that have been developed to remove the coloration of the tissue. One such optical clearing technique is CLARITY, where the tissue is placed in a hydrogel, then incubated for a long period of time, and finally placed in a clearing solution that reduces the amount of reflection and refraction between the sample and other media1,9. In this study, this technique was modified to reduce the amount of clearing reagent used and did not require the addition of vacuum pumps or nitrogen gas8.
Although this technique provides an improved image resolution for thicker opaque samples, there are limitations to this strategy that could be improved in future studies. Additional studies have been conducted with Bessel beams and a two-photon nonlinear excitation to provide a better resolution at sufficient imaging depths15,16. These methods have currently been used to study smaller cardiac models but could be adapted to study the dynamic processes within a developing embryo. Also, changes to the CLARITY method for larger tissue samples could improve the effectiveness of fluorescent imaging by reducing the number of fluorophores that can potentially be lost during the clearing process8. Recently, we have integrated this LSFM system with virtual reality to elucidate developmental cardiac mechanics17.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors would like to express gratitude to Thao Nguyen and Atsushi Nakano from UCLA for providing the mice sample to image. This study was supported by grants NIH HL118650 (to Tzung K. Hsiai), HL083015 (to Tzung K. Hsiai), HD069305 (to N. C. Chi and Tzung K. Hsiai.), HL111437 (to Tzung K. Hsiai and N. C. Chi), HL129727 (to Tzung K. Hsiai), and University of Texas System STARS funding (to Juhyun Lee).
References
- Richardson DS, Lichtman JW. Clarifying tissue clearing. Cell. 2015;162(2):246–257. doi: 10.1016/j.cell.2015.06.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, et al. 4-Dimensional light-sheet microscopy to elucidate shear stress modulation of cardiac trabeculation. Journal of Clinical Investigation. 2016;126(5):1679–1690. doi: 10.1172/JCI83496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisken J, Swoger J, Del Bene F, Wittbrodt J, Stelzer E. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science. 2004;305(5686):1007–1009. doi: 10.1126/science.1100035. [DOI] [PubMed] [Google Scholar]
- Huisken J, Stainier DY. Selective plane illumination microscopy techniques in developmental biology. Development. 2009;136(12):1963–1975. doi: 10.1242/dev.022426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkers J. Zebrafish as a model to study cardiac development and human cardiac disease. Cardiovascular Research. 2011;91(2):279–288. doi: 10.1093/cvr/cvr098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- High FA, Epstein JA. The multifaceted role of Notch in cardiac development and disease. Nature Reviews Genetics. 2008;9(1):49–61. doi: 10.1038/nrg2279. [DOI] [PubMed] [Google Scholar]
- Sachinidis A. Cardiac specific differentiation of mouse embryonic stem cells. Cardiovascular Research. 2003;58(2):278–291. doi: 10.1016/s0008-6363(03)00248-7. [DOI] [PubMed] [Google Scholar]
- Ding Y, et al. Light-sheet fluorescence imaging to localize cardiac lineage and protein distribution. Scientific Reports. 2017;7:42209. doi: 10.1038/srep42209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomer R, Ye L, Hsueh B, Deisseroth K. Advanced CLARITY for rapid and high-resolution imaging of intact tissues. Nature Protocols. 2014;9(7):1682–1697. doi: 10.1038/nprot.2014.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins N, Thompson A, Mann A, Blomkalns AL. Isolation and excision of murine aorta; a versatile technique in the study of cardiovascular disease. Journal of Visualized Experiments. 2014. p. e52172. [DOI] [PMC free article] [PubMed]
- National Heart L, Blood Institute Standard Operating Procedures (SOP's) for Duchenne Animal Models. 2015.
- Sung K, et al. Simplified three-dimensional tissue clearing and incorporation of colorimetric phenotyping. Scientific Reports. 2016;6:30736. doi: 10.1038/srep30736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z, Qiao C, Hu P, Li J, Xiao X. A versatile adeno-associated virus vector producer cell line method for scalable vector production of different serotypes. Human Gene Therapy. 2011;22(5):613–624. doi: 10.1089/hum.2010.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FEI . Amira User's Guide. Berlin: Konrad-Zuse-Zentrum fur Informationstechnik; 2017. pp. 59–138. [Google Scholar]
- Gao L, et al. Noninvasive imaging of 3D dynamics in thickly fluorescent specimens beyond the diffraction limit. Cell. 2012;151(6):1370–1385. doi: 10.1016/j.cell.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong TV, Supatto W, Koos DS, Choi JM, Fraser SE. Deep and fast live imaging with two-photon scanned light-sheet microscopy. Nature Methods. 2011;8(9):757–760. doi: 10.1038/nmeth.1652. [DOI] [PubMed] [Google Scholar]
- Ding Y, et al. Integrating light-sheet imaging with virtual reality to recapitulate developmental cardiac mechanics. JCI Insight. 2017;2(22) doi: 10.1172/jci.insight.97180. [DOI] [PMC free article] [PubMed] [Google Scholar]