

Abstract
Binary transcription systems are powerful genetic tools widely used for visualizing and manipulating cell fate and gene expression in specific groups of cells or tissues in model organisms. These systems contain two components as separate transgenic lines. A driver line expresses a transcriptional activator under the control of tissue-specific promoters/enhancers, and a reporter/effector line harbors a target gene placed downstream to the binding site of the transcription activator. Animals harboring both components induce tissue-specific transactivation of a target gene expression. Precise spatiotemporal expression of the gene in targeted tissues is critical for unbiased interpretation of cell/gene activity. Therefore, developing a method for generating exclusive cell/tissue-specific driver lines is essential. Here we present a method to generate highly tissue-specific targeted expression system by employing a "Clustered Regularly Interspaced Short Palindromic Repeat/CRISPR-associated" (CRISPR/Cas)-based genome editing technique. In this method, the endonuclease Cas9 is targeted by two chimeric guide RNAs (gRNA) to specific sites in the first coding exon of a gene in the Drosophila genome to create double-strand breaks (DSB). Subsequently, using an exogenous donor plasmid containing the transactivator sequence, the cell-autonomous repair machinery enables homology-directed repair (HDR) of the DSB, resulting in precise deletion and replacement of the exon with the transactivator sequence. The knocked-in transactivator is expressed exclusively in cells where the cis-regulatory elements of the replaced gene are functional. The detailed step-by-step protocol presented here for generating a binary transcriptional driver expressed in Drosophila fgf/branchless-producing epithelial/neuronal cells can be adopted for any gene- or tissue-specific expression.
Keywords: This Month in JoVE, Issue 139, Binary expression system, targeted gene expression, CRISPR/Cas9, homology-directed repair, exon replacement, Drosophila, transactivator, LexA, fgf
Introduction
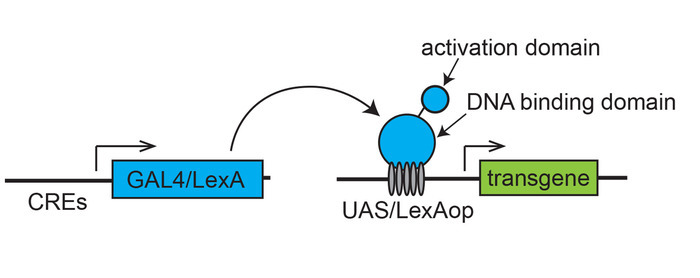
The genetic toolbox for targeted gene expression has been well developed in Drosophila, making it one of the best model systems to investigate the function of genes involved in a wide variety of cellular processes. Binary expression systems, such as yeast Gal4/UAS (upstream activation sequence), was first adopted for tissue-specific enhancer trapping and gene misexpression in the Drosophila genetic model1 (Figure 1). This system facilitated the development of a large number of techniques such as spatiotemporal regulation of gene overexpression, misexpression, knockout in selected groups of cells as well as in cell ablation, cell marking, live tracing of cellular and molecular processes in embryo and tissues, lineage tracing and mosaic analyses during development. A number of binary transcription system, such as the bacterial LexA/LexAop system (Figure 1) and Neurospora Q-system, are powerful genetic tools that are now widely used in Drosophila, in addition to the original Gal4/UAS system for targeted gene expression1,2,3.
Here, we present a method to generate highly reliable tissue-specific binary expression system by employing a genome-editing technique. The recent advancements in CRISPR/Cas9 genome editing technology have allowed unprecedented opportunities to make directed genome changes in a broad range of organisms. Compared to the other available genome editing techniques, the CRISPR/Cas9 system is inexpensive, efficient, and reliable. This technology utilizes components of the bacterial adaptive immune system: a Cas9 endonuclease of Streptococcus pyogenes that creates a double-strand break (DSB), and a chimeric guide RNA (gRNA), which guides the Cas9 to a particular genome site for targeted DSB4. The cells contain the machinery to repair the DSB using different pathways. Non-homologous end joining (NHEJ) leads to small insertions or deletions to disrupt gene function, while homology-directed repair (HDR) introduces a defined directed/desirable genomic knock-in/knock-out by using an exogenous HDR donor as a template. The HDR-based replacement strategy can efficiently be utilized to generate a highly reliable tissue-specific binary expression system, which can overcome all the limitations of the traditional enhancer trap methods. We describe a step-by-step procedure for utilization of CRISPR/Cas9-based HDR repair in generating a binary transcription driver line that is expressed under the control of endogenous transcriptional and post-transcriptional regulation of a Drosophila gene. In this protocol, we demonstrate the generation of a driver line specific for branchless (bnl) gene encoding an FGF family protein that regulates branching morphogenesis of tracheal airway epithelium5. In this example, the first coding exon of the bnl gene was replaced by the sequence of a bacterial LexA transactivator sequence without altering any endogenous cis-regulatory sequences of the bnl gene. We show that the strategy generated a bnl-LexA driver line that spatiotemporally controls the expression of a reporter gene placed downstream of LexAoperator (LexAop or LexO) exclusively in bnl-expressing epithelial/mesenchymal/neuronal cells.
Protocol
1. Designing and Constructing the gRNA Expression Vector
To precisely replace a long defined region of an exon, use a dual gRNA approach6, in which each gRNA can specifically target two ends of the selected region of interest. To obtain an accurate gene-specific spatiotemporal expression of the driver, select two gRNA target sites within the first coding exon of the gene.
- For Drosophila melanogaster, select the gRNA target sites using the flyCRISPR Optimal Target Finder tool (http://tools.flycrispr.molbio.wisc.edu/targetFinder/). Other available CRISPR design tools can be used as well, including the Optimized CRISPR Design tool (http://crispr.mit.edu), FlyCas9 (https://shigen.nig.ac.jp/fly/nigfly/cas9), and E-CRISP (www.e-crisp.org/E-CRISP). NOTE: The following protocols of gRNA design and cloning was adopted from methods previously described6,7.
- Copy the actual sequence into the box provided in the online tool. Select the most recently released Drosophila melanogaster genome annotation version, “r_6,” in the drop-down menu for “Select genome.” In the field “Select guide length (nt),” input“20.” Select to find “All CRISPR targets” and click “Find CRISPR Targets”.
- Evaluate all the candidate gRNA targets by setting “Maximum” for “Stringency” and “NGG Only” for “PAM”. Try to select the gRNA sites without any potential off-targets or a minimum number of off-targets possible. Use the web-tool described in Doench et al. 2014 8 to select gRNAs with a potential “good” activity score.
- Simultaneously, ensure that there is no single nucleotide polymorphism in the gRNA targets in the genome of the parent fly line selected for injection (e.g., nos-Cas9 on X chromosome, BL# 54591). Follow the protocol described in Gratz et al. 20157 to extract the genomic DNA from the parent fly line. PCR amplify, approximately, the 500–1,000 nt regions using primers that flank the sequence of interest and a high-fidelity DNA polymerase; sequence-verify the PCR amplified products.
- For generating a tandem gRNA expression vector, follow a ligation-independent cloning protocol6 to introduce two protospacer sequences into a pCFD4 RNA expression vector. Note that an improved multi-gRNA expression vector (pCFD5) is now available where both the sgRNAs are expressed from the strong U6:3 promoters9 (https://www.crisprflydesign.org). Use a DNA Assembly method to clone the gRNAs into the pCFD4 vector.
- Design and order the forward and reverse primers for introducing two protospacer sequences into the RNA expression vector pCFD4 (Table 1A). As previously described6, the forward primer contains the first protospacer sequence flanked by regions corresponding to the U6-1 promoter and gRNA core in pCFD4; the reverse primer contains the reverse complement sequence of the second protospacer flanked by regions corresponding to the gRNA core and U6-3 promoter in pCDF4.
- Resuspend primers to 100 μM with DNase and RNase-free double distilled water (ddH2O). Make a 10 μM working concentration primer. Use pCFD4 plasmid as a template and set up PCR reaction using a high-fidelity polymerase and recommended settings for PCR reaction6.
- To clone the amplified product into pCFD4, digest pCFD4 plasmid with BbsI enzyme by setting up the following reaction: 2–5 μg of pCFD4 plasmid, 5 μL of 10x digestion buffer, 1 μL of BbsI enzyme, and ddH2O to bring the final volume to 50 μL. Mix the reaction contents by gently tapping the tube and collecting all the scattered droplets from the wall of the tube to the bottom by a brief spin. Incubate the reaction mix at 37 °C for 2 h.
- Run the PCR product from step 2 and the digested product from step 3 on a 1% agarose gel. Perform the electrophoresis for enough time to completely separate the DNA bands. Cut the correct sized DNA bands under a UV trans-illuminator before purifying the expected products using gel elution column following the manufacturer’s instruction (see Table of Materials). The PCR product should be 600 bp, and the linearized pCFD4 plasmid should be ~6.4 kb 6.
- Set up the DNA assembly reaction in a PCR tube, per the manufacturer’s instruction (see Table of Materials).
- Transform 2 μL of the assembly product into competent bacteria (DH5α or similar strains with ΔrecA1, ΔendA1), plate on Ampicillin (100 μg/mL) containing LB (LB/Amp) agar plates, and incubate at 37 °C overnight. Note that the DNA assembly mix might be toxic to certain bacteria strains, but diluting the assembly mix can reduce the toxicity.
- Pick 3–4 ampicillin-resistant colonies from the overnight-incubated plate, inoculate the colony in 3 mL LB containing 100 μg/mL ampicillin, and grow inoculated bacteria in a 37 °C shaker overnight. Extract plasmid from the overnight-cultured bacteria using the plasmid mini-prep kit following the manufacturer’s instruction (see Table of Materials).
- Sequence the plasmids with T3 universal primer to screen for the correct clones containing the tandem gRNA insertion. Establish a glycerol stock of bacteria transformed with a sequence-verified pCFD4-gRNA in 20% glycerol and store in a -80 °C freezer for future use.
2. Designing and Constructing the HDR Donor
- For genomic knock-in of a Gal4 or LexA sequence, design a double-stranded HDR donor containing the transactivator sequence flanked by two homology arms.
- Use >1.5 kb left and right homology arms flanking the gRNA targeting site(s). Extended homology arms increase the efficiency of HDR during the repair process 10.
- PCR amplify the homology arms from the genomic DNA (gDNA) extracted from the parent fly line selected for injection (e.g., nos-Cas9 (on X chromosome), BL# 54591). Use a proof-reading hot-start Taq-DNA polymerase enzyme suitable for long PCR extension (see Table of Materials). Sequence-verify.
To avoid retargeting of the gRNA to the engineered locus, design the replacement donor in such a way that the gRNA recognition sites are disrupted by the exogenous sequence introduced. NOTE: to avoid altering the putative cis-regulatory elements, always select the gRNA recognition sites within the coding exon region.
- For generating a replacement cassette, plan the DNA Assembly strategy to join four segments together: the 5’ and 3’ homology arms, the middle exogenous transactivator DNA sequence, and the linearized cloning vector backbone (e.g., pUC19 or other commonly used cloning vectors). Following the manufacturer’s guideline for DNA assembly (see Table of Materials), design the suitable primers for PCR amplification and assembly of each segment. Resuspend primers to 100 μM with ddH2O. Make a 10 μM working concentration primer. Use high-fidelity polymerase for all the PCR reactions. Follow the following protocol:
- PCR amplify a Gal4 or LexA expression cassette from an available vector (e.g., nls-LexA:p65; the ideal Gal4/LexA/QF2 expression plasmids can be found and obtained from common resources such as Addgene) using the primers listed in Table 1B.
- To retain all the original transcriptional and post-transcriptional regulations of the replaced exon on the expression of the transactivator DNA sequence, design the cassette in such a way that the edited genomic allele would express a chimeric mRNA of the transactivator and the gene. Preserve the 5’ and 3’ end of the targeted exon to retain any splicing signal.
- Incorporate a T2A self-cleaving peptide sequence between the residual 5’ coding exon and the transactivator sequence to prevent translation into a chimeric protein. Add a translation stop codon after the exogenous transactivator sequence (Figure 5).
- PCR amplify the homology arms from the genomic DNA (gDNA) extracted from the parent fly line selected for injection (e.g., nos-Cas9 (on X chromosome), BL# 54591) using the primers listed in Table 1B. Use high-fidelity polymerase for all the PCR reactions using the systems as follows: 5 μL of 5x Reaction Buffer, 0.5 μL of 10 mM dNTPs, 1.25 μL of 10 μM Forward Primer, 1.25 μL of 10 μM Reverse Primer, 0.5 μL of Template DNA, 0.25 μL of High-Fidelity DNA Polymerase, 16.25 μL of ddH2O.
- Use linearized cloning vector such as pUC19. A number of donor vectors are now available. See a comprehensive list at the following website- http://flycrispr.molbio.wisc.edu.
- Run the PCR products on a 1% agarose gel. Perform the electrophoresis for enough time to ensure a clear separation of the desired band from the undesired non-specific bands (if any). Gel-purify the expected DNA fragments. Measure the concentration of each purified DNA fragment using a spectrophotometer.
- Set up the DNA assembly reaction following the manufacturer’s instruction. Mix the linearized cloning vector and target fragments from step 3 and step 4 in the following reaction: 1 μL of linear cloning vector (50 ng/μL), 2–3 fold excess of each target fragment, 10 μL of 2x DNA assembly master mix, bring the final reaction volume to 20 μL with ddH2O. Incubate the reaction mix for 1 hour at 50 °C.
- Transform 2 μL of the assembly product into competent bacteria, plate on LB/Amp agar plates containing 100 μg/mL ampicillin. Also, add X-Gal + IPTG on the plate for blue/white screening.
- Pick 3–4 white colonies from the overnight-incubated plate, inoculate the colony in 3 mL LB containing 100 μg/mL ampicillin, and grow at 37 °C overnight in a shaker. Extract plasmid from the overnight-cultured bacteria using the plasmid mini-prep kit following the manufacturer’s manual (see Table of Materials).
- Screen the positive colony by PCR or restriction digestion.
- Sequence verify the HDR donor region of the final plasmid. Save a bacterial stock transformed with the correct clones in 20% glycerol at -80 °C for future inoculation.

3. Embryo Injection, Fly Genetics and Screening for Genome Editing
Prepare the high purity endotoxin-free gRNA expression vector as well as the HDR donor plasmid using a plasmid maxiprep kit (see Table of Materials).
Co-inject the gRNA expression plasmid (100 ng/μL) and the replacement donor (500 ng/μL) into the germline of nos-Cas9 embryos6. Note: we use a commercial service for injection, but this procedure can be performed in the laboratory as well.
- Fly genetics and screening (Figure 2 and Figure 3):
- When the injected embryos develop into adults, cross each single G0 flies to balancer flies. Select suitable balancer for the chromosome containing the targeted allele.
- Anesthetize the F1 offspring from each G0 cross on a CO2 pad and randomly pick 10-20 males under a stereomicroscope. Cross them individually to the balancer females as shown in Figure 3.
- When the F2 larvae hatch, pick the single F1 father from each cross and extract gDNA using the single fly genomic DNA preparation protocol:
- Prepare gDNA extraction buffer: 10 mM Tris-Cl pH 8.2, 1 mM EDTA, 25 mM NaCl, store at room temperature. Prepare 20 mg/mL Proteinase K stock solution and store in the freezer.
- Put each fly in a 1.5 mL micro-centrifuge tube and label the tube. Keep in the -80 °C freezer overnight.
- Prepare a fresh working volume of gDNA extraction buffer containing 200 µg/mL final concentration of Proteinase K. Note: Do not use an old buffer for this step.
- Squish each fly for 10–15 s with a pipette tip containing 50 μL of squishing buffer without dispensing the liquid. Dispense the remaining buffer into the tube and mix well. Incubate at 37 °C for 20–30 min.
- Put tubes in 95 °C heat block for 1–2 min to inactivate the Proteinase K.
- Spin down for 5 min at 10,000 x g. Store the preparation at 4 °C for further PCR analysis.
Use the same method to prepare gDNA from a nos-Cas9 fly, which serves as a negative control. Perform three-step PCR based screens to identify the correct “ends out” HDR (Figure 2, Figure 5) using gDNA of each F1 male as a template5. Use 1 μL of the DNA prep in the following PCR reaction system: 10 μL of 2x PCR Master Mix with Dye, 1 μL of each primer (10 μM), 1 μL of DNA template, and 7 μL of ddH2O.
As shown in Figure 5A, perform PCR using primers fwd1 and rev1 to screen for the existence of the insertion or replacement; perform PCR using fwd2 and rev2 primers to verify the insertion or replacement from 3’ region; perform PCR using primers M13F and rev3 to check “ends-in” HDR (Table 1C).
Keep the fly lines with the confirmed ends-out HDR and establish balanced stocks from the F2 generation. Outcross to the balancer flies again to remove any unintended mutations on other chromosomes.
- Prepare high-quality gDNA from the established stocks for amplifying long PCR (>800–1,000 nt) amplicon with high-fidelity Taq polymerase and fully sequence verify the amplicons obtained from the engineered genomic regions. Alternatively, use crude gDNA extract as described earlier to amplify shorter (<800 nt), overlapping PCR products to sequence-validate the edited genome. Use the following protocol to prepare the good quality Drosophila genomic DNA:
- Put about 25 adult flies in a 1.5 mL microcentrifuge tube, and freeze in -20 °C or -80 °C freezer for at least 1 hour.
- Add 250 μL of Solution A (pH 9.0 Tris HCl 0.1 M, EDTA 0.1 M, SDS 1%).
- Homogenize the flies using the homogenizing pestles in microcentrifuge tubes, and put the tube on ice.
- Incubate at 70 °C for 30 min.
- Add 35 μL of KOAc (5 M), shake, and mix well, but do not vortex.
- Incubate on ice for 30 min.
- Spin at 12,000 x g for 15 min.
- With a 1 mL micropipette, carefully transfer only the clear supernatant to a new tube, leaving back any precipitate or interphase.
- Add 150 μL of isopropanol to the supernatant. Gently mix by inversion.
- Spin at 10,000 x g for 5 min.
- Carefully remove the supernatant and leave the pellet in the tube.
- Wash the pellet with 1 mL 70% EtOH.
- Spin at 12,000 x g for 5 min.
- Remove the supernatant. Air dry the pellet for 15 to 20 min. Do not over dry the pellet.
- Dissolve the pellet in 100 μL of ddH2O.
- Use high fidelity DNA polymerase suitable for long amplicon (>800 bp) to amplify the complete edited region. Ideally, take two primers outside the inserted cassette to amplify the genomic region. Gel purify the PCR product, and as described earlier, sequence verify the product with multiple overlapping primers.
- Validate the correct spatiotemporal expression patterns from dissected tissues/embryos of the engineered fly lines:
- Perform RT-PCR from the total RNA to verify the expression of the hybrid mRNA product (Table 1D).
- Perform an in situ hybridization of the mRNA product of the interest in the larval tissues/embryo to validate the expression.
- To screen for the accurate tissue-specific spatiotemporal expression patterns among the sequence-verified ends-out lines, cross each of the edited lines obtained for the binary expression driver with the LexO-GFP or LexO-RFP (for LexA driver) line (available from the Bloomington stock centers) and observe the LexA or Gal4 driven reporter-gene expression in embryo, larval, and adult tissues under a fluorescence microscope.

Representative Results
This protocol was successfully used to generate a targeted binary expression reporter system specific for bnl expressing cells5. The cis-regulatory elements (CREs) that control complex spatiotemporal bnl expression are not characterized. Therefore, to achieve spatiotemporal expression under the control of the endogenous bnl regulatory sequence, only the first coding exon of bnl was designed to be replaced with the sequence of the bacterial LexA transactivator. An improved nls-LexA::p65 cassette known to provide optimal expression in Drosophila was selected.
The replacement strategy aimed to generate a chimeric nls-LexA:p65-bnl mRNA under endogenous transcriptional and post-transcriptional control. This chimeric mRNA contained an intact bnl 5' UTR, part of the 5' end and the 3' end of the edited bnl coding exon (first exon), and the complete downstream bnl introns and exons. To preserve the bnl RNA-specific splicing of the replaced exon, small 5' and 3' ends of the replaced coding exon were retained. However, to prevent synthesis of a chimeric Bnl protein fused to nls-LexA:p65, a T2A self-cleaving peptide sequence was incorporated between the residual 5' bnl coding region and the ATG of nls-LexA:p65. Also, a translation stop codon was added after the nls-LexA:p65 sequence to avoid co-translation of a truncated Bnl protein5 (Figure 4 and Figure 5A).

A replacement donor containing all these features was used, which had the T2A-nls-LexA:p65 sequence and 2 kb 5'- and 1.8 kb 3'- homology arms. Long homology arms were used for efficient HDR and insertion of a large DNA fragment at the site of repair (T2A-nls-LexA:p65 is 1.8 kb) (Figure 5A).
Two gRNAs were used to create DSBs at the defined positions to delete most of the first coding exon of bnl. Two gRNAs that matched all criteria described in section 1.3 (PAM sequence underlined) were: gRNA1: TGTATCTGCGATGCCCCTCATGG gRNA2: ATCCTTCAGATATTGCGGGATGG
Both gRNA recognition sites were disrupted in the replacement donor by the exogenous T2A-nls-LexA:p65 sequence, which is the easiest way to avoid retargeting of the gRNAs to the engineered locus. The disrupted gRNA recognition sequences in the replacement donor (the exogenous sequences that disrupted gRNA recognition sites in italics) were: gRNA1: TGTATCTGCG-GGCTCCGGCGAAGGAC… gRNA2: …AAAAACTCGTTTAGA-CGGGATGG
The pCFD4 gRNA expression vector containing these gRNAs, along with the replacement donor, was co-injected into the germline of nos-Cas9 embryos. Subsequently, the protocol described here was followed to screen for the intended replacement (Figure 5B, C). About 67% of injected G0 animals were fertile. And 56% of the fertile G0s were the founders, which gave rise to HDR-positive progeny. Among the F1 progeny checked, about 23% were positive for successful HDR, and 73% of the positive HDR were "ends-out" (Table 2). Since the first coding exon of bnl was "knocked out", all the HDR lines were found to be homozygous lethal, and the stocks were maintained over balancers.
The generation of a chimeric LexA-bnl mRNA in the cells was validated by RT-PCR analyses (Figure 5D). To verify if the obtained bnl-LexA lines were expressed in the endogenous bnl expression pattern, each "ends-out" HDR line was crossed to LexO-mCherryCAAX transgenic flies5, and the reporter expression pattern was examined in the progeny embryos and larvae. 42 out of 46 lines showed accurate spatiotemporal expression consistent with the previously reported bnl patterns5 (Figure 6). Four lines showed either weak or non-specific expression patterns. We predict that these lines might have accumulated mutations, which we need to verify with thorough sequencing analyses. Together these results confirmed that the HDR-mediated exon replacement strategy could successfully be employed to generate a targeted binary expression system for a gene. The tool can successfully be used to express reporter or ectopic genes in the spatiotemporal patterns of the bnl gene.

Figure 1: Scheme of a binary transcription system for targeted gene expression. A transactivator (GAL4 or LexA), expressed under the control of cis-regulatory elements (CREs) of a gene, drives an effector transgene placed downstream of the specific transcription binding sites (UAS for Gal4 or LexAop for LexA). Please click here to view a larger version of this figure.
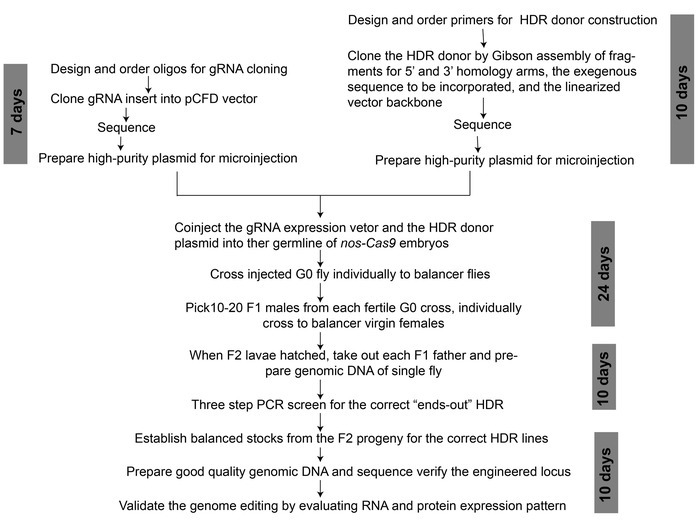
Figure 2: An overview of workflow for CRISPR/Cas9-mediated genome editing to generate a binary transcription system. The approximate time duration required for each step is indicated. Please click here to view a larger version of this figure.
Figure 3: An illustration of the CRISPR screening and genetic cross scheme for establishing genome-edited fly lines. In genetic crosses, R stands for the edited allele that is on the 3rd chromosome. MKRS (Tp(3;3)MRS, M(3)76A[1] kar[1] ry[2] Sb[1]), is a 3rd chromosome marker; TM6B (In(3LR)TM6B, Antp[Hu] e[1] Tb[1]) is a 3rd chromosome balancer. Genome edited fly stocks are verified by PCR amplifying the target regions of interest from the genomic DNA extracted from either the F2 or F3 generation flies and sequence determining the PCR products. Please click here to view a larger version of this figure.
Figure 4: Generation of the replacement cassette by DNA assembly. A schematic drawing depicting PCR amplification and assembly of different PCR products (a, b, and c) into the donor vector (d) using a DNA assembly method. Please click here to view a larger version of this figure.
Figure 5: Generation of bnl-LexA by CRISPR/Cas9-mediated exon replacement. (A) Schematic drawing depicting the strategy of the CRISPR/Cas9 mediated HDR for exon replacement in the bnl locus. Box - exon; line- intron; replacement donor (pDonor-bnl:LexA) and two possible outcomes of the HDR were shown. The pDonor-bnl:LexA had the following features: (1)T2A-nls-LexA:p65 (~1.8kb) sequence flanked by 2 kb and 1.8 kb long homology arms (dashed lines), (2) a T2A self-cleaving peptide between the residual N terminal bnl exon and the nls-LexA:p65, and (3) a translation stop codon (red *) after the nls-LexA:p65 sequence. The HDR product retained all the transcriptional and post-transcriptional control of bnl,and the LexA:p65 protein is expected to be produced in the same pattern as endogenous Bnl. Small black arrows show the relative binding sites (not in scale) of the PCR primers (Table 1) used for 3-step screening or RT-PCR validation. (B) Agarose gel pictures showing results of the 3-step PCR screening. PCR products amplified from the genomic DNA of four successful ends-out HDR lines are shown; negative control, the genomic DNA of nos-Cas9 parental line; positive control, pDonor-bnl:LexA plasmid; M, Marker (SL2K DNA ladder). (C) An example of the screening gel showing the expected PCR product using primers M13F and rev3 for ends-in lines; M8-7 and M9-6 are two ends-in lines; negative and positive controls, the same as in B; M, Marker (NEB 1 kb DNA ladder). (D) RT-PCR analysis on total RNA from bnl-LexA and the nos-Cas9 control flies. Forward primer binds to a LexA specific region, reverse primer binds to a downstream bnl exon region; ~440 bp (base-pair) amplification band (*) was detected from RT-PCR on bnl-LexA mRNA, but not from the control RNA. M, 100 bp Marker (NEB). Adapted from Figures 2 and S1 in Du et al. 20175. Please click here to view a larger version of this figure.
Figure 6: Validation of tissue-specific and conditional bnl-LexA expression in different tissues. (A-D) bnl expression (red) in different larval tissues, with the btl expressing cells shown in green. (A,A') Wing disc bnl source ahead of the growing air sac primordium (ASP). (B, C) bnl expression in TR5 transverse connective (TC) (B) and TR2 dorsal branch (DB) (C). (D) bnl expression in genital discs. (E-J) Dynamic bnl expression (red) during embryonic tracheal branch (green) patterning; Small arrows, five bnl sources surrounding the tracheal placode at stage 10. Genotypes: A-J; btl-Gal4, UAS-CD8GFP/+; bnl-LexA,LexO-CherryCAAX/+. (K-L") Hypoxia-induced bnl-LexA expression profile in wing discs and associated TR2 tracheal metamere (TC, DB, dorsal trunk-DT). Genotype: bnl-LexA,LexO-CherryCAAX/+. (K) control discs from ex vivo cultured organs without CoCl2. (L-L") wing discs (L, L') and trachea (DT, (L'')) from cultured ex vivo organs with CoCl2 induced hypoxia. Star, ectopic expression induced by hypoxia. Scale bars = 30 µm; 50 µm (K-L"). Adapted from Figure 3 in Du et al. 20175. Please click here to view a larger version of this figure.
| A. gRNA cloning and sequencing | |
| bnl-lexA gRNA fwd | TATATAGGAAAGATATCCGGGTGAACTTCgTGTATCTGCGAT GCCCCTCAGTTTTAGAGCTAGAAATAGCAAG |
| bnl-lexA gRNA rev | ATTTTAACTTGCTATTTCTAGCTCTAAAACTCCCGCAATATCTGAAGG ATcGACGTTAAATTGAAAATAGGTC |
| T3 primer used for sequencing | CAATTA ACCCTCACTAAAGG-3' |
| Note: nucleotides underlined anneal to U6 promoter or gRNA core on pCFD4 vector, the lowercase g/c was added to aid U6 promoter-dependent transcription | |
| B. HDR donor construction | |
| bnl N-F_pUC19 | AATTCGAGCTCGGTACtgtggtctttgaggctggaac |
| bnl-lexA-N-R | tCCGcaagtCagtAGgctgccgcgtccttcgccggaGCCCGCAGATACAAGGCCC C |
| lexA-F | CTactGacttgCGGaGAtGTcGAaGAGAACCCtGGCCCtATGCCACCCAA GAAGAAGC |
| lexA-R | CTAAACGAGTTTTTAAGCAAACTCACTC |
| bnl lexA-C Fwd | TAAAAACTCGTTTAGACGGGATGGCGTTGTCAAC |
| bnl C-R_pUC19 | GCCAAGCTTGCATGCCtcgcataattgccgcctgg |
| Note: nucleotides in capital overlap with pUC19 vector for Gibson Assembly, nucleotides underlined were sequence overhang for T2A peptide addition. | |
| C. HDR screening and sequencing | |
| bnl-lexA scr fwd1 | GTGGCGCACGCCCAATAAAC |
| bnl-lexA scr rev1 | GATCCCAGCCAATCTCCGTTG |
| bnl-lexA scr fwd2 | CAACGGAGATTGGCTGGGATC |
| bnl-lexA scr rev2 | CTGGCCAACTGTAGGGAAGTC |
| ends-in check rev3 | GCAATGTTATGCAATGCGTTGAC |
| bnl-lexA seq fwd3 | CACTTGTCGCCCATATTGATACAATTG |
| NOTE: These primers were used for PCR screening and sequencing, the approximate locations of primer binding sites are shown in Figure 5A as fwd1-2 and rev1-3. | |
| D. RT-PCR analysis | |
| RT-f | GATATGGATTTCTCCGCTTTGCTG |
| RT-r | CCATGCAGAGATACAGGCAAGTG |
Table 1: Primers used in this study. (A) Primers for cloning gRNA expression vector and sequencing. (B) Primers for cloning HDR donor template. (C) Primers for screening and sequencing of the HDR products. (D) Primers used for RT-PCR verification of the chimeric LexA-bnl mRNA product.
| genotype | HDR transmission rate % (# HDR-yielding G0/# fertile G0) | # HDR-positive F1/# total F1 checked (%) | # "ends-out" HDR/# total HDR (%) |
| bnl-LexA | 56 (15/27) | 23 (60/259) | 73 (46/60) |
Table 2: The efficiency of CRISPR/Cas9-mediated HDR. The HDR transmission rate is calculated as the # of HDR-yielding G0/# of total fertile G0. Successful HDR% is calculated as the # of HDR-positive F1/# of total F1 screened. "Ends-out"% is calculated as the # of "ends-out" HDR/# of total positive HDR.
Discussion
Traditionally, Drosophila enhancer traps were generated by two different methods. One of the ways includes random insertion of a driver (eg., Gal4) sequence in the genome by transposition (e.g., P-element transposition)1 . Alternatively, the driver sequences can be placed under the transcriptional control of a putative enhancer/promoter region in a plasmid construct, which would then be integrated into an ectopic site of the genome3,11. Although these methods have generated a large pool of Gal4 or LexA enhancer traps for Drosophila, they have several limitations. The P-element-mediated random insertion in the genome may disrupt the regulatory activity of critical cis-regulatory sequences. Due to the random transposition in the genome, transactivator expression may report patterns of multiple neighboring genes. On the other hand, a prior knowledge of the cis-regulatory sequences of a gene is necessary for an enhancer-trap plasmid construct. There is also a limit to the length of a sequence that can be cloned and tested in a plasmid construct. Isolating and cloning only a partial fragment of DNA out of the endogenous genomic context might not reproduce a complete gene-specific expression patterns. For instance, the bnl-Gal4 (NP2211) line, which was generated by P-element insertion of an enhancer trap Gal4 element just ahead of the bnl gene, does not completely reproduce the gene-specific expression patterns in the embryo5. Therefore, under such contexts, the repurposing techniques, such as HACK (homology assisted CRISPR knock-in), that can convert the existing Gal4 lines into another LexA or Q-system based driver line12 might not be very useful. To overcome these limitations, here, we described an efficient and alternative method for generating binary expression systems by genome editing. In this method, the first coding exon of a gene is swapped with the transactivator (eg., LexA or Gal4) sequence so that the expression of the transcription driver exclusively mimics the spatiotemporal patterns of the gene expression. Although the strategy and protocols described here were optimized for bnl-expression, the method can easily be adopted for other genes.
Determining an insertion site within the targeted gene region is the most critical step in this experimental strategy. The method we presented was designed to retain all the original transcriptional as well as the post-transcriptional regulations of the bnl gene5. Therefore, we planned to delete most of the first coding exon of the bnl gene and replace it with the LexA cassette. The replacement was planned in such a way that the edited allele expresses a hybrid LexA-bnl mRNA from the endogenous transcription and translation start site of bnl while retaining all the intronic regions. However, the hybrid mRNA was engineered to produce only LexA protein, but not Bnl. We showed that the strategy had successfully generated a bnl-LexA /LexO-based transcription system and that the system could accurately report the dynamic, spatiotemporal bnl expression patterns (Figure 5)5. A limitation of this process is the homozygous lethality in the Drosophila lines if the targeted gene is essential for survival. Further, use of a dual gRNA strategy may increase the chance of off-target editing. An alternative strategy can be employed to avoid these problems. In this strategy, a LexA or Gal4 cassette can be placed right at the ATG start site of the replaced gene and a T2A self-cleaving peptide sequence can be placed between the LexA/Gal4 cassette and the start of the intact coding exon of the target gene. This strategy is expected to produce a chimeric mRNA that can be translated into both the transctivator and the functional gene product. Such a design could possibly reduce the chance of homozygous lethality. In this strategy, a single gRNA is sufficient for genome editing. However, in the presence of two copies of functional genes, expression of the transgenic protein variants driven by the Gal4/LexA driver (eg., bnl-LexA driven expression of LexO-Bnl:GFP fusions, or Bnl proteins with deletions) will not provide information of the functionality of the variant proteins.
A limitation of the genome-editing strategy is the laborious process of targeting and editing of a single gene at a time. However, the simplicity and efficiency of CRISPR/Cas9-mediated genome editing method have revolutionized the targeted genomic knock-in/knock-out and replacement technology that can easily be adopted in any standard laboratory set up. In the last several years, Drosophila researchers have developed convenient toolkits for genome editing that can be employed to expand the genetic toolsets essential for understanding fundamental biological processes7,13. The genome editing and screening processes described here is relatively faster and takes about two months in comparison to any other homologous recombination-based replacement. For successful and efficient genome-editing results, it is important to follow several technically critical steps: 1) each gRNA is selected by the maximum stringency and activity without potential off-target; 2) the HDR donor contains ~1.5 kb homology arms flanking the gRNA targeting sites. Longer homology arms (1.8–2 kb) are used to increase the efficiency of HDR when a large exogenous DNA fragment needs to be inserted; 3) the HDR donor is designed to be free of the gRNA recognition sites to avoid retargeting of the gRNA to the engineered locus; and 4) a foolproof elaborate plan and coordination of the timing of genetic crosses with 3-step PCR-based screening to select an "ends-out" HDR line.
Unlike random transposition or cloned enhancer constructs, the strategy and protocols we described here ensures generation of an exclusively target gene-specific binary transcription system. Since the expression of the driver replaces a native gene exon, gene-specific expression of the driver is also versatile and is expected to mimic gene expression patterns under all developmental, metabolic, and physiological conditions5. Gene/cell-specific expression is the most desirable criteria of all the binary driver lines when they are used in various cell and developmental biology methods. For instance, gene-specific expression of reporter genes by a transcription driver facilitates reliable lineage analyses of the cells expressing the target gene. It also enables live imaging and tracking of the spatiotemporal expression, migration, and reorganization of cells during development. To investigate the cellular and molecular events during tissue morphogenesis, Gal4 or LexA driven overexpression/misexpression of various transgenes and RNAi-mediated gene knock-down in specific sets of cells are essential. Highly specific targeted expression system provides an unbiased analyses and interpretation of the results. Therefore, CRISPR/Cas9-based targeting of a gene exon and its replacement with a transactivator sequence provides a better alternative for generating highly specific targeted gene expression systems.
Disclosures
The authors have no conflicts of interest to disclose.
Acknowledgments
We thank Dr. F. Port, Dr. K. O'Connor-Giles, and Dr. S. Feng for discussions on CRISPR strategy; Dr. T.B. Kornberg, and the Bloomington Stock Center for reagents; UMD imaging core facility; and funding from NIH: R00HL114867 and R35GM124878 to SR.
References
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118(2):401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Lai S-L, Lee T. Genetic mosaic with dual binary transcriptional systems in Drosophila. Nature Neuroscience. 2006;9(5):703–709. doi: 10.1038/nn1681. [DOI] [PubMed] [Google Scholar]
- Potter CJ, Tasic B, Russler EV, Liang L, Luo L. The Q system: a repressible binary system for transgene expression, lineage tracing, and mosaic analysis. Cell. 2010;141(3):536–548. doi: 10.1016/j.cell.2010.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L, Zhou A, Patel A, Rao M, Anderson K, Roy S. Generation of a targeted expression system for branchless and characterization of novel cellular expression patterns of the gene in Drosophila. Developmental Biology. 2017;427(1):35–48. doi: 10.1016/j.ydbio.2017.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Port F, Chen H-M, Lee T, Bullock SL. Optimized CRISPR/Cas tools for efficient germline and somatic genome engineering in Drosophila. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(29):E2967–E2976. doi: 10.1073/pnas.1405500111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratz SJ, Rubinstein CD, Harrison MM, Wildonger J, O'Connor-Giles KM. CRISPR-Cas9 Genome Editing in Drosophila. Current Protocols in Molecular Biology. 2015;111(1) doi: 10.1002/0471142727.mb3102s111. 31.2.1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Hartenian E, et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature Biotechnology. 2014;32(12):1262–1267. doi: 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Port F, Bullock SL. Augmenting CRISPR applications in Drosophila with tRNA-flanked sgRNAs. Nature Methods. 2016;13(10):852–854. doi: 10.1038/nmeth.3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beumer KJ, Trautman JK, Mukherjee K, Carroll D. G3. 4. Vol. 3. Bethesda, Md: 2013. Donor DNA Utilization During Gene Targeting with Zinc-Finger Nucleases; pp. 657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BD, Ngo T-TB, et al. Refinement of tools for targeted gene expression in Drosophila. Genetics. 2010;186(2):735–755. doi: 10.1534/genetics.110.119917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C-C, Potter CJ. Editing Transgenic DNA Components by Inducible Gene Replacement in Drosophila melanogaster. Genetics. 2016;203(4):1613–1628. doi: 10.1534/genetics.116.191783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Köster J, et al. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biology. 2015;16(1):281. doi: 10.1186/s13059-015-0843-6. [DOI] [PMC free article] [PubMed] [Google Scholar]