

Abstract
Droplet digital polymerase chain reaction (ddPCR) is a highly sensitive quantitative polymerase chain reaction (PCR) method based on sample fractionation into thousands of nano-sized water-in-oil individual reactions. Recently, ddPCR has become one of the most accurate and sensitive tools for circulating tumor DNA (ctDNA) detection. One of the major limitations of the standard ddPCR technique is the restricted number of mutations that can be screened per reaction, as specific hydrolysis probes recognizing each possible allelic version are required. An alternative methodology, the drop-off ddPCR, increases throughput, since it requires only a single pair of probes to detect and quantify potentially all genetic alterations in the targeted region. Drop-off ddPCR displays comparable sensitivity to conventional ddPCR assays with the advantage of detecting a greater number of mutations in a single reaction. It is cost-effective, conserves precious sample material, and can also be used as a discovery tool when mutations are not known a priori.
Keywords: Genetics, Issue 139, Drop-off ddPCR, Hotspot mutations, Cancer, Circulating tumor DNA, Diagnostics
Introduction
Thousands of somatic mutations related to cancer development have been reported1. Among these, a few are predictive markers of the efficacy of targeted therapy 2,3 and genetic screening of these mutations is now routine clinical practice. Droplet digital PCR (ddPCR) technology can be used to monitor for the presence or the absence of mutations with high detection accuracy and is highly compatible with non-invasive liquid biopsies4,5. However, current ddPCR assays are primarily designed to detect mutations known a priori. This severely limits the use of ddPCR as a discovery tool when the mutation is unknown. Indeed, in the conventional ddPCR design, a hydrolysis probe recognizing the wild-type allele (WT Probe) competes with a specific probe recognizing the mutant allele (MUT Probe) (Figure 1A). The probe with higher affinity hybridizes to the template and releases its fluorophore, indicating the nature of the corresponding allele. The fluorescence data obtained for each droplet can be visualized in a scatter plot representing the fluorescence emitted by the WT and MUT probes in different dimensions. A schematic representation of a typical result for a conventional ddPCR assay is presented in Figure 1A. In this example, the blue cloud corresponds to the droplets containing WT alleles identified by the WT probe, whereas the red cloud corresponds to droplets in which the MUT allele has been amplified and identified by the MUT probe. Depending on the quantity of DNA loaded in the reaction, double positive droplets containing both WT and MUT amplicons can appear (green cloud). The light grey cloud corresponds to empty droplets.
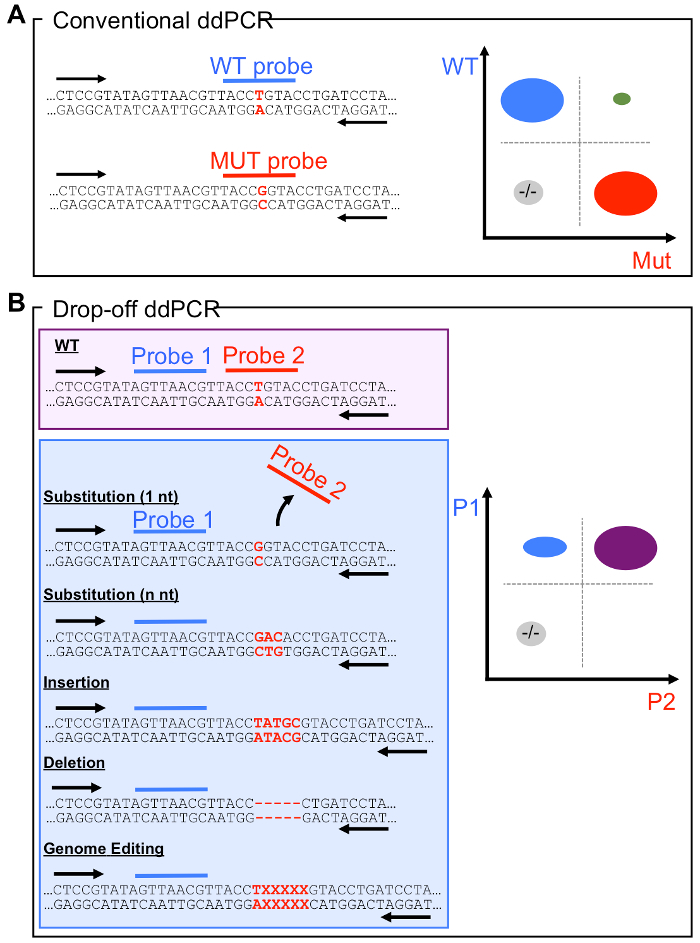
Since most platforms allow for the detection of a limited number of fluorophores (usually 2, but up to 5), throughput of conventional ddPCR is limited. Therefore, targeting regions with multiple adjacent mutations requires design of technically challenging multiplex ddPCR assays or the use of multiple reactions targeting each mutation in parallel. The drop-off ddPCR strategy, overcomes this limitation as it can potentially detect any genetic alteration within a target region using a unique pair of WT hydrolysis probes. The first probe (Probe 1) covers a non-variable sequence, adjacent to the target region and the second probe (Probe 2) is complementary to the WT sequence of the target region where mutations are expected (Figure 1B). While the first probe quantifies the total amount of amplifiable molecules in the reaction, the second probe discriminates WT and MUT alleles by sub-optimal hybridization to mutant sequences. Probe 2 can identify multiple types of mutations in the target region (single or multiple nucleotide substitutions, deletion, etc.)6. As in conventional ddPCR, the light grey cloud corresponds to droplets containing no DNA molecules. It is important to recall that in this type of assay, optimal separation of WT and MUT clouds is dependent on the quantity of DNA loaded in the reaction, since droplets containing both WT and MUT target alleles cannot be distinguished from droplets containing only the WT form. Therefore, this assay requires that most droplets contain no more than one copy of the targeted gene.
Protocol
The protocol presented here follows the ethics guidelines of the Institut Curie. All human samples were obtained from patients enrolled, after informed consent, in studies approved by the Institutional Review Board at Institut Curie.
1. Blood Collection, Plasma Storage and Cell-free DNA Extraction
NOTE: DNA extracted from any type of “tissue” can be used (e.g., fresh or formaldehyde-fixed paraffin embedded (FFPE) tissues, cells in culture or blood samples). Here, we provide detailed instructions for blood collection, plasma isolation and storage, and cell-free DNA (cfDNA) extraction.
- Blood collection and plasma storage ( Figure 2B )
- Collect blood samples in 7 mL EDTA tubes. Two tubes are recommended to collect around 4 mL of plasma.
- Centrifuge the tubes for 10 min at 820 x g within 3 h of the blood draw to separate red blood cells (RBC), white blood cells (WBC) and plasma. The time between the sampling and the centrifugation is critical to obtain high quality cfDNA (i.e., cfDNA not contaminated with DNA released from WBC).
- Carefully pipet the supernatant (plasma) using a 1 mL pipet without touching the WBC layer; around 2 mL of plasma are usually recovered per EDTA tube.
- Transfer the plasma to 2 mL tubes and centrifuge the aliquots at 16,000 x g for 10 min at 15 °C to remove cell debris.
- Carefully pipet the supernatant (plasma) without disturbing the pellet. Distribute the plasma in 2 mL cryogenic tubes and store at -80 °C until needed.
- cfDNA Extraction (Figure 2B) NOTE: Here, we present the procedure using manual extraction kit. An automated version following the manufacturer's instructions can also be performed for cfDNA isolation.
- Put 400 µL of Proteinase K into a 50 mL tube. Add 4 mL of plasma (volume routinely used) and 3.2 mL of lysis buffer ACL (containing 1.0 µg carrier RNA) from the kit. Close the tube and mix by vortexing for 30 s to obtain a homogeneous solution. Incubate at 60 °C for 30 min.
- Add 7.2 mL of buffer ACB (from the kit) to the lysate to optimize the binding of the circulating nucleic acids to the silica membrane and mix by vortexing for 15–30 s. Incubate the tube for 5 min on ice.
- Place the column and the 20 mL extender into the vacuum pump. Close the unused positions to allow vacuum aspiration.
- Carefully apply the lysate-buffer ACB mixture into the tube extender (briefly centrifuge the tube to collect the maximum of lysate-buffer ACB mixture), switch on the vacuum pump and allow the lysate to be drawn through the columns completely. Remove and discard the tube extender.
- Apply 600 µL of buffer ACW1 to the column. When buffer ACW1 has drawn through the column, add 750 µL Buffer ACW2 and finally add 750 µL of ethanol (96–100%). Then, place the column in a clean 2 mL collection tube and centrifuge at full speed (20,000 x g) for 3 min to eliminate ethanol.
- Place the column into a new 2 mL collection tube. Open the lid and incubate at 56 °C for 10 min to dry the membrane completely.
- Place the column into a clean 1.5 mL DNA low binding tube. Carefully apply 36 µL of RNase-free water with 0.04% Sodium azide (buffer AVE). Close the lid and incubate at room temperature for 3 min.
- Centrifuge at full speed (20,000 x g) for 1 min to elute the nucleic acids and store at -20 °C until needed.
- cfDNA quantification NOTE: cfDNA quantification is performed with a fluorometer using the manufacturer’s protocol immediately after extraction or thawing the samples at room temperature (RT), before use. Avoid multiple freeze/thaw cycles.
- Use the high-sensitivity assay according to the expected cfDNA quantity, which is usually less than 100 ng/mL plasma. cfDNA concentrations range from 2–10 ng/mL for healthy donors but can be as high as 100 ng/mL in patients with metastatic disease.
- Ensure all reagents are at RT and mix 199 µL of fluorometer dilution buffer with 1 µL of dye (200x) per reaction (N number of samples + 2 standards to calibrate the fluorometer) to obtain a working solution.
- Vortex the cfDNA solution thoroughly to ensure homogeneity and briefly centrifuge to collect contents at the bottom of the tube.
- Mix 5 µL of each cfDNA sample with 195 µL of the working solution.
- Mix 10 µL of each standard with 190 µL of the working solution.
- Vortex thoroughly all tubes for 2–3 s to ensure homogeneity and briefly centrifuge to collect contents at the bottom of the tubes.
- Incubate at RT for 2 min in the dark.
- Read the tubes in the fluorometer, using the ‘ng/µL’ output mode
- Calculate the final concentration in ng/mL of plasma as follows: ng/µL x 36 (volume of cfDNA eluate) / 4 (volume of plasma extracted).
2. ddPCR Probe and Primer Design (Figure 2A)
NOTE: The amplification of the targeted molecules in the drop-off ddPCR assay follows similar principles of qPCR. Each primer and probe is designed with the conventional dedicated software Primer37 (http://primer3.ut.ee/).
- Primer design
- In the general settings window, change 'concentration of divalent cations' to 3.8, 'concentration of dNTPs' to 0.8, and 'mispriming/repeat Library' to the correct organism.
- In the advance settings window, change both the 'Table of thermodynamic parameters' and 'salt concentration formula' to Santa Lucia 1998.
- Choose a Tm between 55 and 70 °C (with 50 mM for salt concentration and 300 nM for oligonucleotide concentration).
- Check the specificity of primers using tools such as PrimerBlast.
- Avoid regions that have secondary structures.
- Choose a sequence that has a GC content of 50–60%.
- Avoid repeats of Gs and Cs longer than 3 bases.
- Choose primers with Gs and Cs at their 3’ end when possible.
- Ensure that 3’ ends of paired primers do not have significant complementarity to avoid primer-dimers. Amplicons should be smaller than 150 bp to efficiently amplify cfDNA (mean size is 170 bp8) and DNA extracted from FFPE, which is often fragmented.
- Probe design NOTE: Hydrolysis probes are labeled with a fluorescent reporter at the 5’ end and a quencher at the 3’ end. They provide high specificity, high signal-to-noise ratio and allow multiplexing if needed. Two-channel droplet readers are compatible with FAM and HEX or VIC dyes as well as analysis of FAM/HEX or FAM/VIC coupled probes.
- Design hydrolysis probes complementary to the WT sequence of the target region and the adjacent non-variable region.
- Choose probes both located within the amplicon defined by the primer pair previously designed. Primer sequences cannot overlap with the probe sequences, although they may sit next to one another.
- Choose the strand that has more Cs than Gs. Probes must have a GC content of 30-80%.
- Select probes that are between 13 and 30 nucleotides in length. Longer probes tend to display higher background fluorescence because the distance between the fluorophore and the quencher affects quenching of the fluorophore reporter.
- Select Tm of the probes to be 3 to 10 °C higher than the Tm of the primers to ensure probe hydrolysis during primer annealing and extension. Tm enhancers are recommended to reach the required Tm while keeping the probe short, as shorter probes discriminate single nucleotide variants more efficiently. Probes should not have a G at the 5’ end because this quenches the fluorescence signal even after hydrolysis.
- Design drop-off probes as follows: 5’-(VIC)-complementary WT sequence of the mutated region-(MGB NFQ)-3’ and reference probe as: 5’-(6-FAM)-complementary sequence of a non-variable region-(MGB NFQ)-3’. Other combinations of reporter/quencher are also possible.
3. Optimization of the ddPCR Reaction (Figure 2A)
NOTE: Wild-type and mutant DNA controls used in this step can be obtained from cell lines, tumor samples or commercially available DNA reference standards, for example.
- Perform a conventional PCR on a thermal cycler using the thermal gradient feature to validate the specificity of the PCR product amplified by the primers designed.
- Prepare a unique PCR reaction mix as described below in step 4.1 (without probes) on a wild-type DNA control template for a minimum of 8 reactions to have at least one reaction per temperature.
- Run the amplification using the program described at step 4.3.2 and a range of annealing temperatures around the theoretical annealing temperature of the primers.
- Load PCR products on a 3% agarose gel to select for annealing temperatures providing specific products.
- Perform a series of drop-off ddPCRs using the parameters described above but this time including both probes.
- Use 100% WT DNA, 100% MUT DNA and a mixture of WT and MUT DNA (e.g., 50% MUT/50% WT) to determine the best conditions for clear separation of WT and MUT droplet clouds.
- Choose a range of annealing temperatures above the temperature chosen in step 4.1.
- Select the annealing temperature which allows for specific detection of WT or MUT signals when using 100% WT DNA or 100% MUT DNA and best separation of WT and MUT droplet clouds.
- Annealing time and primer/probe concentrations can also be adjusted to improve separation of clouds and to decrease the number of droplets with intermediate fluorescence (rain effect).
- Evaluate the background or limit of blank (LOB) of the assay.
- Perform a minimum of 48 individual drop-off ddPCR reactions with a WT control DNA using the annealing parameters and primers/probes concentrations chosen in 3.2.
- For each reaction, calculate the mutant allele frequency (MAF) as follows: (number of mutant droplets/number of filled droplets) x 100. NOTE: The LOB is defined as the false-positive MAF mean and associated SD from which the 95% confidence interval (95% CI) is calculated. LOB = (mean of false-positive MAF) + 95% CI.
- Evaluate the limit of detection (LOD) of the assay.
- Perform drop-off ddPCR reactions using serial dilutions of MUT DNA in WT DNA, with MAFs ranging from 5% to 0.01% (≥ 8 replicates per condition). Use annealing parameters and primers/probes concentrations chosen in 3.2. NOTE: The LOD is defined as the smallest MAF values for which replicates present average values and SD above the LOB (see Decraene et al.6 for more details).
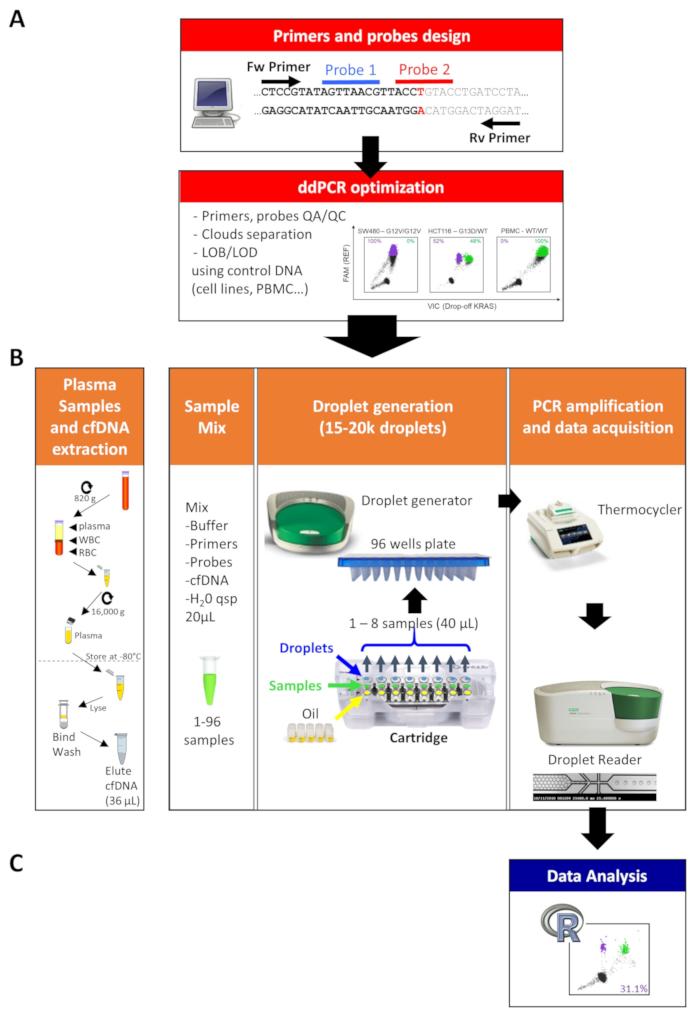
4. ddPCR Protocol (Figure 2B)
NOTE: Similar to conventional ddPCR, the drop-off ddPCR protocol consists of 4 steps: (i) PCR mix preparation, (ii) droplet generation, (iii) PCR amplification and (iv) data acquisition.
- PCR mix preparation
- Follow standard precautions, such as wearing gloves, using a clean PCR hood, clean pipettes, and RNase, DNase-free distilled water to avoid contamination.
- Thaw and equilibrate the reaction components at RT.
- Prepare 20x primers/probes mix in distilled water, with primers at 18 µM each and probes at 5 µM each.
- For all individual PCR reactions, prepare a sample mix by combining 2x ddPCR supermix (10 μL), 1 µL of 20x primers/probes mix and up to 10 ng DNA sample qsp 20 µL of distilled water.
- Vortex the reaction mix thoroughly to ensure homogeneity and briefly centrifuge to collect contents at the bottom of the tube before dispensing. Do not hesitate to repeat this step for each reaction mix before use.
- Droplet generation
- Insert the ddPCR cartridge into the holder and load 20 μL of reaction mix containing samples into the middle wells of the cartridge for droplet generation (green positions in Figure 2B) using an 8-channel pipette.
- Add 70 μL of droplet generator oil into the bottom wells (yellow positions).
- Insert the cartridge, with a gasket, in the droplet generator; droplets will be produced in the top wells of the cartridge (blue positions).
- Aspirate and dispense slowly and gently (to avoid shearing) 40 μL of the droplets from the cartridges into a 96-well PCR plate. Use a multi-channel pipette to optimize the transfer of the droplets.
- PCR amplification
- Seal the final PCR plate with an aluminum foil using a plate sealer immediately after transferring droplets to avoid evaporation. Briefly centrifuge the plate to collect contents at the bottom of the wells.
- Run a conventional PCR amplification on a thermal cycler using the following program: 95 °C 10 min, 40 cycles of [94 °C 30 s, validated annealing T° for 60 s], 98 °C 10 min (ramp 2.5°C/s). Include controls with no input DNA and wild type DNA (≥3 replicates) in each run.
- When the run is completed, briefly centrifuge the plate to collect contents at the bottom of the wells.
- Data acquisition
- After PCR amplification, use the droplet reader to count PCR-positive and PCR-negative droplets, following the manufacturer’s instructions. NOTE: The droplet reader usually offers a two-color optical detection system compatible with FAM, VIC or HEX fluorophores.
5. Data analysis (Figure 2C and Figure 3)
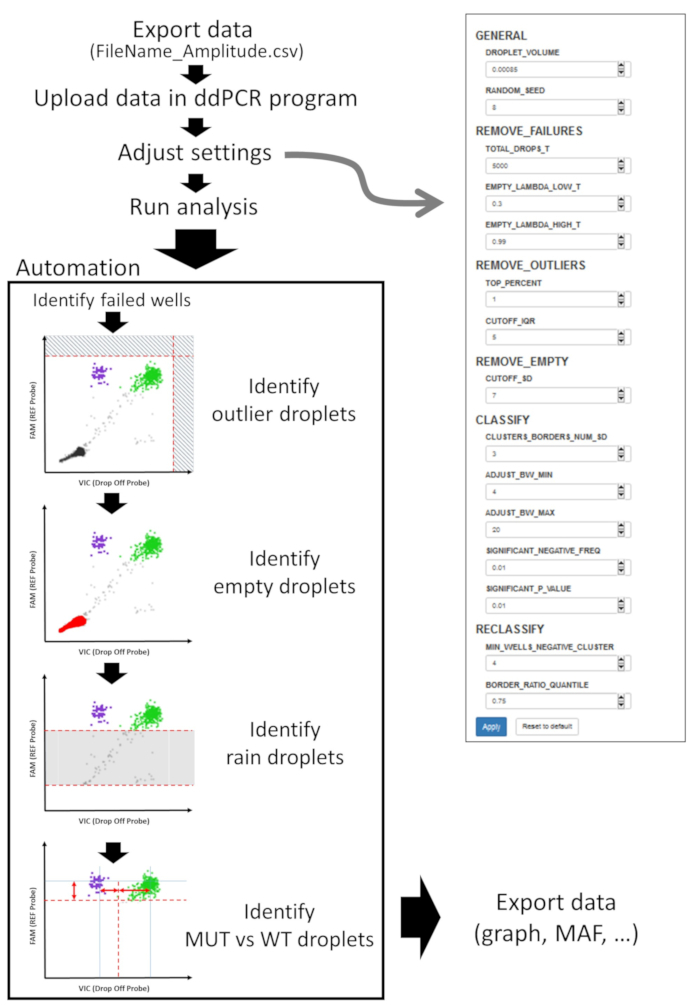
NOTE: PCR-positive and PCR-negative droplets are counted to provide an absolute quantification of the MUT and the WT alleles at the targeted region. The assigned droplets are used to compute the MAF in each well. Droplet quantification and analysis of drop-off assays can be performed using the ddPCR R package (https://github.com/daattali/ddpcr) developed by Attali and colleagues9,10. This R package automatically classifies droplets as empty, part of the “rain” (due to inefficient amplification), or as filled with a real positive signal (Figure 3). The following section presenting the different steps of the analysis is a brief version of the descriptive vignette of the algorithm written by its authors (see https://github.com/daattali/ddpcr/blob/master/vignettes/ algorithm.Rmd for more details). Data are exported to comma-separated values files (FileName_Amplitude.csv) and uploaded in the ddPCR package to be analyzed directly in R or through the dedicated web interface.
- Parameter adjustments NOTE: Each drop-off assay design results in specific final droplet distributions and variations in the cloud form, separations or positions is to be expected. To ensure efficient automated analysis, a set of parameters needs to be adjusted. If a well has an unexpected profile, it might cause the analysis of the whole plate to fail, resulting in an error message. The problematic well will have to be identified and removed from the automated analysis.
- Identify failed wells using (REMOVE_FAILURES). Use TOTAL_DROPS_T (default value is 5,000) to adjust the minimal number of droplets in each well. Use EMPTY_LAMBDA_LOW_T (default value is 0.3) and EMPTY_LAMBDA_HIGH_T (default value is 0.99) to adjust the metrics that evaluate the expected droplet clouds (empty, MUT, WT) and their quality. Note: Having too many empty droplets is a sign that there was not enough amplifiable template in the reaction.
- Identify outlier droplets using (REMOVE_OUTLIERS). Use TOP_PERCENT (default is p = 1%) to adjust the top p percent of droplets having the highest signal value in each signal dimension (FAM, VIC/HEX) out of droplets in the entire plate. Use CUTOFF_IQR (default is 5) to adjust the outlier threshold of the top p percent of highest signal.
- Identify empty droplets using (REMOVE_EMPTY). Use CUTOFF_SD (default is 7) to adjust the standard deviation of the lower (empty droplets) Gaussian distribution of the Probe 1 values.
- Gate droplets using (CLASSIFY). Use CLUSTERS_BORDERS_NUM_SD (default is 3) to adjust the standard deviation of the higher (filled droplets) Gaussian distribution of the Probe 1 values. Use ADJUST_BW_MIN (default is 4) and ADJUST_BW_MAX (default is 20) to adjust the k_min and k_max parameters to estimate the kernel density of the Probe 2 values and discriminate the MUT and the WT cloud of droplets.
- Use SIGNIFICANT_NEGATIVE_FREQ (default is p=1%) and SIGNIFICANT_P_VALUE (default is significance level s=0.01%) to classify wells as either mutant or wildtype. Note: In this algorithm, a mutant well is defined as having a MAF that is statistically significantly higher than p%.
- Automated analysis NOTE: For each parameter presented in this section, the authors of the ddPCR R package have proposed default values based on their expertise and their data. However, all parameters can be modified to increase the quality of the analysis (Figure 3).
- Identify failed wells.
- Use four quality control metrics (settings: REMOVE_FAILURES) to identify failed wells; the first one is the minimum number of droplets that a well should contain. The other three focus on identifying valid candidate populations for empty, possibly FAM+ and HEX+/VIC+ droplets. Remove wells that fail to meet these criteria from downstream analysis.
- Identify the outlier droplets.
- In order to avoid skewing droplet gating, remove outliers (i.e., droplets with an abnormally high HEX/VIC or FAM value) from further analysis by setting an upper boundary threshold (settings: REMOVE_OUTLIERS).
- Identify empty droplets.
- Identify empty droplets to reduce the dimension of the data (in a typical well, they will account for most of the droplets) and eliminate statistical bias due to the presence of a large portion of useless information as only template-containing droplets remain (settings: REMOVE_EMPTY).
- Perform droplet gating.
- Classify the remaining droplets as either mutant, wildtype or rain (settings: CLASSIFY).
- Identify rain droplets.
- As rain droplets result from inefficient amplification, exclude from FAM+ or HEX+/VIC+ clouds. Determine the borders of their clusters using their Probe 1 values (setting: CLUSTERS_BORDERS_NUM_SD).
- Identify mutant versus wild-type droplets.
- As the highlight of the analysis is discrimination between mutant and wildtype templates, identify all remaining droplets at this stage as either one or the other. Since they should have similar Probe 1 values, use Probe 2 values to best predict their respective nature (settings: ADJUST_BW_MIN and ADJUST_BW_MAX).
- Classify wells as mutant versus wild-type.
- After determining which droplets are MUT and WT, calculate the MAF. Using the mutant frequency and associated p-value of each well, it is possible to classify them (settings: SIGNIFICANT_NEGATIVE_FREQ, SIGNIFICANT_P_VALUE) as WT (containing mostly WT droplets) or MUT wells (containing a statistically significant number of MUT droplets).
- Explore and export the data
- Explore and export the data as number of droplets, outliers, empty droplets, concentrations or graphs that can be further customized for optimal representation.
Representative Results
In a proof-of-concept study, KRAS exon 2 mutations (codons 12 and 13) and EGFR exon 19 deletions were investigated in FFPE tissues and plasma samples from cancer patients using the drop-off ddPCR strategy6.
The KRAS drop-off probe interrogated a 16 bp region encompassing multiple mutations in exon 2 of the KRAS gene, which harbor more than 95% of the known KRAS mutations in human cancers11. The reference probe was 20 bp in length and located 3 bp downstream. To optimize our assay, we used DNA extracted from cell lines containing 2 of the most frequently mutated KRAS amino acids: G12V (c.35GT, SW480) and G13D (c.38GA, HCT 116).
The EGFR drop-off assay was designed to scan for all activating deletions of EGFR exon 19. The assay used a 25 bp-long drop-off probe placed over the recurrent deleted region together with a reference probe of 21 bp, located 31 bp downstream in the same amplicon. To optimize our assay, we used a cfDNA reference standard set including the EGFR exon 19 deletion c.2235_2249del, p.Glu746_Ala750del.
Sensitivity, specificity and location of droplet clouds have been defined with control MUT samples and WT PBMC genomic DNA as shown in Figure 4A for KRAS. We further demonstrated that this type of assay works on multiple types of genetic alterations such as single substitution, multiple substitutions or deletions (Figure 4B) as well as on various sample types6.
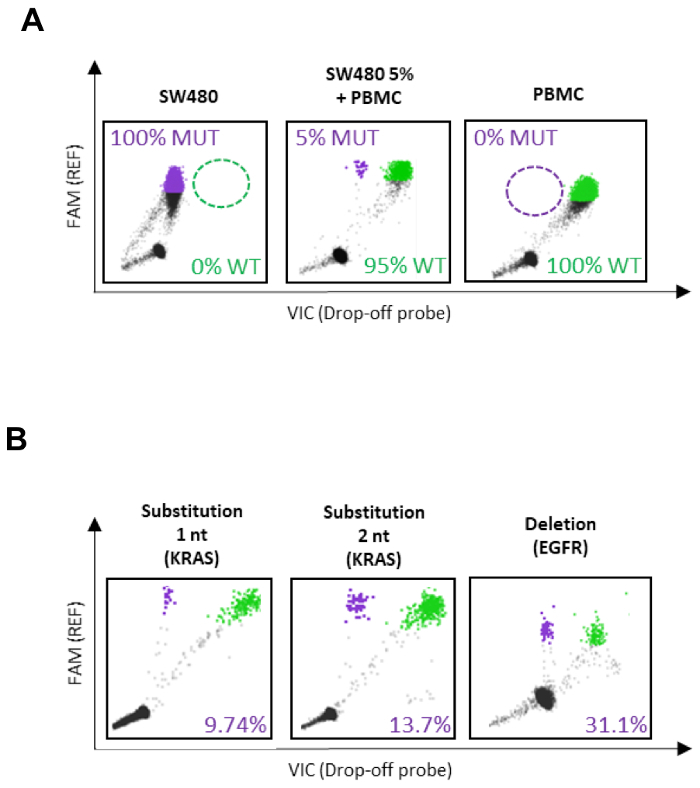
Figure 1: The drop-off ddPCR requires a unique pair of hydrolysis oligo-probes to scan all mutations of a hotspot region. Assay designs with a schematic result displayed as two-dimensional scatter plots showing droplet fluorescence intensity. (A) Conventional ddPCR with mutant (MUT probe) and wild-type (WT probe) probes targeting the exact same region. (B) The drop-off ddPCR contains a reference probe (Probe 1) that anneals to a non-variable region and a drop-off probe (Probe 2) targeting the mutated region but complementary to the WT sequence, within the same amplicon. Please click here to view a larger version of this figure.
Figure 2: Drop-off ddPCR workflow for plasma DNA mutation profiling. The full workflow is composed of 5 major steps: (A) (1) design of primers and probes, (2) optimization of the assay, (B) (3) plasma isolation and cfDNA extraction, (4) ddPCR run and (C) (5) data analysis. Please click here to view a larger version of this figure.
Figure 3: Drop-off ddPCR analysis. Drop-off ddPCR analysis workflow highlighting important parameters that need to be fine-tuned. This ensures efficient automated identification of outliers, empty, rain or positive droplets; assigning MUT or WT droplets and computing the MAF for each well. Please click here to view a larger version of this figure.
Figure 4: Representative results - KRAS and EGFR drop-off ddPCR assays. (A) Example of results obtained during optimization steps of the drop-off ddPCR assay. Detection of mutant and/or wild-type DNA in a reaction containing 100% MUT DNA, 5% MUT DNA and 100% WT DNA. (B) Examples of plasma samples analyzed with the KRAS and the EGFR drop-off ddPCR assays showing the detection of single nucleotide and multiple substitutions in exon 2 of KRAS and a deletion in EGFR exon 19. This figure has been adapted from Decraene et al.6. Please click here to view a larger version of this figure.
Discussion
To design an efficient drop-off ddPCR assay, optimization is crucial, and the protocol must be followed carefully. Each combination of primers and probes has a unique PCR reaction efficiency. Thus, an individual assay has to be carefully validated on control samples before being used on valuable test samples. Optimization and validation are important to certify peak signal detection and assess specificity and sensitivity. As described in the protocol, all mutations located in a target region covered by the "probe 2" can potentially be interrogated. Nonetheless, validation is recommended for mutations located at either 5' or 3' ends of "probe 2", since these may affect the efficiency of the drop-off assay.
The method presented here can be performed with DNA extracted from any type of "tissue", including FFPE tissues, cells in culture or blood samples. However, the quality of the nucleic acid extraction can dramatically impact ddPCR results. PCR inhibitors such as heparin, proteins, heme, phenol, proteases, collagen, melanin, detergents or salts have to be eliminated as much as possible during the sampling and the purification steps. Although DNA samples can be diluted to reduce the impact of PCR inhibitors on the ddPCR, dilution decreases sensitivity because fewer copies of the target will be tested per reaction. This should be taken into consideration when a certain level of sensitivity is to be reached, as a minimum of copies must be tested.
Drop-off ddPCR can be applied to many types of molecular alterations (single and multiple nucleotide substitutions, deletions, etc.). Besides using only two probes to detect all alterations (thereby increasing practicality and cost-effectiveness of the assay), drop-off ddPCR generates less background noise6 and shows increased sensitivity compared to multiplex assays. Additionally, since it is performed in a single reaction, minimal amounts of patient samples are consumed. This assay is particularly appealing in the context of liquid biopsy because the amount of ctDNA is often low in these samples.
Indeed, the method presented here is applicable for mutation screening in liquid-biopsy material and has already proven its utility for clinical practice (ESR1 mutation screening in the PADA-1 clinical trial - NCT03079011). However, it is crucial to use amplicons as short as possible in this setting because of the fragmentation of cfDNA8.
Downstream characterization still needs to be performed separately to identify the exact mutation carried by mutant alleles. However, because therapeutic decisions are primarily based on the mutational status of hotspot regions of targetable oncogenes (WT vs mutant), the ability of drop-off ddPCR to immediately identify mutations makes it a powerful diagnostic tool. Moreover, the exact mutation does not need to be known a priori to design an assay in contradistinction to current commercial solutions. In addition, drop-off ddPCRs can be combined with specific conventional ddPCR assays to further increase the number of mutations screened for (data not shown). Finally, drop-off ddPCR can be applied to other fields of research. In particular, it may prove to be a useful tool for screening positive colonies in genome editing experiments.
Disclosures
Several authors are named inventors of two patent applications (EP17305920.5, EP18305277.8) related to ctDNA detection.
Acknowledgments
This work was supported by Institut Curie SiRIC (grant INCa- DGOS-4654). The authors wish also to thank Caroline Hego for her contribution to the video.
References
- Alexandrov LB, Nik-Zainal S, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amado RG, Wolf M, et al. Wild-Type KRAS Is Required for Panitumumab Efficacy in Patients With Metastatic Colorectal Cancer. Journal of Clinical Oncology. 2008;26(10):1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- Karapetis CS, Khambata-Ford S, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. New England Journal of Medicine. 2008;359(17):1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- Postel M, Roosen A, Laurent-Puig P, Taly V, Wang-Renault S-F. Droplet-based digital PCR and next generation sequencing for monitoring circulating tumor DNA: a cancer diagnostic perspective. Expert Review of Molecular Diagnostics. 2018;18(1):7–17. doi: 10.1080/14737159.2018.1400384. [DOI] [PubMed] [Google Scholar]
- Saliou A, Bidard F-C, et al. Circulating tumor DNA for triple-negative breast cancer diagnosis and treatment decisions. Expert Review of Molecular Diagnostics. 2015;16(1):39–50. doi: 10.1586/14737159.2016.1121100. [DOI] [PubMed] [Google Scholar]
- Decraene C, Silveira AB, et al. Multiple Hotspot Mutations Scanning by Single Droplet Digital PCR. Clinical chemistry. 2018;64(2):317–328. doi: 10.1373/clinchem.2017.272518. [DOI] [PubMed] [Google Scholar]
- Untergasser A, Cutcutache I, et al. Primer3--new capabilities and interfaces. Nucleic Acids Research. 2012;40(15):e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell. 2016;164(1-2):57–68. doi: 10.1016/j.cell.2015.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidshahri R, Attali D, et al. Quantitative Detection and Resolution of BRAF V600 Status in Colorectal Cancer Using Droplet Digital PCR and a Novel Wild-Type Negative Assay. The Journal of Molecular Diagnostics. 2016;18(2):190–204. doi: 10.1016/j.jmoldx.2015.09.003. [DOI] [PubMed] [Google Scholar]
- Attali D, Bidshahri R, Haynes C, Bryan J. ddpcr: an R package and web application for analysis of droplet digital PCR data. F1000Research. 2016;5 doi: 10.12688/f1000research.9022.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Beare D, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Research. 2017;45(D1):D777–D783. doi: 10.1093/nar/gkw1121. [DOI] [PMC free article] [PubMed] [Google Scholar]