

Abstract
What differentiates Mycobacterium abscessus from other saprophytic mycobacteria is the ability to resist phagocytosis by human macrophages and the ability to multiply inside such cells. These virulence traits render M. abscessus pathogenic, especially in vulnerable hosts with underlying structural lung disease, such as cystic fibrosis, bronchiectasis or tuberculosis. How patients become infected with M. abscessus remains unclear. Unlike many mycobacteria, M. abscessus is not found in the environment but might reside inside amoebae, environmental phagocytes that represent a potential reservoir for M. abscessus. Indeed, M. abscessus is resistant to amoebal phagocytosis and the intra-amoeba life seems to increase M. abscessus virulence in an experimental model of infection. However, little is known about M. abscessus virulence in itself. To decipher the genes conferring an advantage to M. abscessus intracellular life, a screening of a M. abscessus transposon mutant library was developed. In parallel, a method of RNA extraction from intracellular Mycobacteria after co-culture with amoebae was developed. This method was validated and allowed the sequencing of whole M. abscessus transcriptomes inside the cells; providing, for the first time, a global view on M. abscessus adaptation to intracellular life. Both approaches give us an insight into M. abscessus virulence factors that enable M. abscessus to colonize the airways in humans.
Keywords: Immunology and Infection, Issue 139, Microbiology, amoeba, Acanthamoeba castellanii, Mycobacterium abscessus, transposon library, co-culture amoeba-mycobacteria, RNA extraction of intracellular mycobacteria, RNAseq.
Introduction
The genus Mycobacterium includes species ranging from harmless saprophytic organisms to major human pathogens. Well-known pathogenic species such as Mycobacterium tuberculosis, Mycobacterium marinum and Mycobacterium ulcerans belong to the subgroup of slow-growing mycobacteria (SGM). In contrast, the subgroup of rapid-growing mycobacteria (RGM) is characterized by their ability to form visible colonies in less than 7 days on agar medium. The RGM group comprises more than 180 species, mainly non-pathogenic saprophytic mycobacteria. Studies on RGM interactions with their hosts have mainly focused on Mycobacterium smegmatis and demonstrate that these mycobacteria are rapidly eliminated by the bactericidal action of macrophages.
Mycobacterium abscessus is one of the rare RGM that are pathogenic to humans and is responsible for a wide range of infections ranging from the skin and soft tissue infections to the pulmonary and disseminated infections. M. abscessus is considered, along with Mycobacterium avium, to be the main mycobacterial pathogen in cystic fibrosis patients1.
Various studies performed on M. abscessus indicate that this mycobacterium behaves like an intracellular pathogen, capable of surviving the bactericidal response of macrophages and fibroblasts in the lungs and skin, which is not usually observed in RGM2,3,4. M. abscessus genome analysis has identified metabolic pathways typically found in environmental microorganisms in contact with the soil, plants and aquatic environments, where free amoebae are often present5. They have also demonstrated that M. abscessus is endowed with several virulence genes not found in the saprophytic and non-pathogenic RGM, probably acquired by the horizontal gene transfer in a niche favorable to genetic exchange that might gather various amoeba resistant bacteria.
Experimentally, one of the first striking results was the observation of intracellular growth of M. abscessus in macrophages as well as for M. tuberculosis6. M. abscessus also resists the acidification of the phagosome, apoptosis and autophagy, three essential mechanisms of the cellular resistance to the infection2. It has even been shown that M. abscessus is able to establish an immediate communication between the phagosome and the cytosol, a more nutrient-rich environment that might favor bacterial multiplication2. Very little is known about the genomic advantages that M. abscessus possesses or has acquired to allow survival in an intracellular environment. Amoeba coculture is an efficient method that allowed the isolation of many new amoeba resistant bacteria as Mycobacterium massiliense7,8. An ability to multiply within amoebae was observed, in a model of aerosolization of M. abscessus in mice, which can confer an increased virulence to M. abscessus4. One hypothesis is that M. abscessus had developed genetic traits encountered within this environment to survive in phagocytic cells, which are different from other non-pathogenic RGM. These acquisitions might favor the ability to spread and its virulence in the human host.
This report describes tools and methods to highlight the genomic advantages conferred to M. abscessus to survive in the amoebae environment. For this purpose, the screening of M. abscessus transposon mutants is first described, on the Acanthamoeba castellanii type strain, which allows the identification of mutant's defective for intracellular growth. A second screening in macrophages is also reported, to confirm if this defect persists in the human host. Secondly, to understand which mechanisms are harnessed in M. abscessus to adapt to life in phagocytic cells and increase its virulence in the animal host, a method specifically adapted for M. abscessus was developed, after co-culture in the presence of amoebae that allowed the extraction of total RNA from intra-amoebal bacteria. As a consequence, a comprehensive view of M. abscessus genes that are required for an intracellular life was developed.
Protocol
1. Library Screening
- Construction of the Tn mutant library
- Obtain a transposon library. NOTE: For this experiment, a transposon mutant library was obtained from E.J. Rubin, Harvard School of Public Health, Boston, USA. The library was constructed from a smooth clinical strain (43S) of the M. abscessus complex (M. abscessus subsp. massiliense) with a phagemid introduced into M. abscessus allowing the random insertion of a single Tn into a TA dinucleotide (91,240 TA sequences in the M. abscessus genome). For more details see reference of Rubin et al.9 and Laencina et al.10.
- Titration of the library and conservation of the M. abscessusTn mutants in plates NOTE: This takes one week.
- Thaw the library on the ice. Determine the bacterial concentration by performing serial dilutions in 1 mL of water (10-1 to 10-15). Plate a drop of each dilution on a Petri dish containing 7H11 agar medium with 250 mg/mL kanamycin. Incubate the plates at 37 °C for 3–4 days. NOTE: Here, a titration of 1011 bacteria/mL were recovered.
- After determining the concentration of the library, dilute the library to a final concentration of 3,000 bacteria/mL in 10 mL of 25% glycerol-water. Aliquot the library in 10 cryotubes with 1 mL of the library in each tube. Store the aliquots at -80 °C.
- When ready to use, thaw one aliquot of the diluted library on ice and add 100 µL of the library to 10 square Mueller-Hinton (MH) agar plates (size 12 cm) to recover 200 to 300 individual colonies after 3–4 days of incubation at 37 °C.
- With sterile toothpicks, transfer the individual colonies (about 6,000 colonies in 60 x 96-well plates) into 96-well plates filled with 200 µL of 7H9 medium supplemented with 0.2% glycerol, 1% glucose, 250 mg/mL of kanamycin. Incubate at 37 °C for 5 days without shaking.
- Transfer 100 µL of the culture (step 1.2.4) to a 96-well plate containing 100 µL of 7H9 medium with 40% glycerol. Store the ordered library at -80 °C.
- Repeat steps 1.2.3. to 1.2.5. with the rest of the library until 10,000 individual clones are recovered (around 100 x 96-well plate and 30 MH plates).
- Preparation of amoebae
- Culture amoeba Acanthamoeba castellanii (AC) in 75 cm2 tissue culture flasks at room temperature (RT) in 30 mL of PYG medium (Table 1) for amplification of the cell line11. NOTE: Mature trophozoites from large adhesive cells with numerous digestive vacuoles are clearly visible on a microscope. The cell doubling time is estimated to be 25 h. Cells were amplified every 3 days by spreading one third of the initial culture in 75 cm2 tissue culture flasks.
- Remove the PYG medium with a 25 mL pipette. Wash the cells with 10 mL of AC buffer (Table 2), which does not contain any carbon or nitrogen sources12. Repeat the wash twice more. NOTE: AC buffer that contains no carbon or nitrogen sources avoids the extracellular growth of mycobacteria. This minimum medium leads to the encystment of amoebae. To limit this, heat-inactivated E. coli were added as a substrate (step 1.4) to amoebae and very short culture times were promoted (48 h for mutant screening and 4 h or 16 h culture for RNAseq experiments). Alternatively, use this medium but enriched with PYG medium (usually 10%).
- Detach the amoeba from the bottom of the flask with a single vigorous shake of the tissue culture flask.
- Count cells with a hemocytometer to adjust the cell concentration to 5 x 105 amoebae/mL diluted in AC buffer.
- Preparation of heat-inactivated Escherichia coli bacteria to feed amoeba after infection
- Grow the E. coli clinical isolate strain from a glycerol stock in 100 mL of Luria Broth (LB) overnight at 37 °C.
- Harvest the bacteria by centrifugation at 1,835 x g for 10 min in 50 mL tubes. Discard the supernatant. Resuspend the pellet with 10 mL of 10% glycerol-water. Repeat the washing twice more.
- Resuspend the pellet with 5 mL of 10% glycerol-water. Aliquot 0.5 mL into 1.5 mL tubes. Determine the amount of bacteria/mL by performing serial dilutions and adding dilutions on LB agar plates. Store the aliquots at -80 °C.
- The day before the amoeba infection, thaw one aliquot on ice and proceed to heat-killing by incubating the tube at 70 °C for 60 min. NOTE: Check the inactivation by plating 100 µL of inactivated bacteria on LB agar plates overnight at 37 °C. No colonies should be visible after one night of culture.
- Dilute the heat-killed bacteria to a concentration of 107 bacteria in 50 µL of AC buffer and store the diluted bacteria at 4 °C for no more than one week.
- Co-culture of amoebae-M. abscessus Tn mutant: first screen NOTE: This takes 3–4 months for the screening of 6,000 mutants.
- Spread 5 x 104 amoebae/well to a 96-well plate in 100 µL of AC buffer. Incubate for 1 h at 32 °C without shaking. Allow the floating amoeba trophozoïtes time to adhere to the wells.
- While incubating, thaw bacteria on ice for 1 h.
- Add 5 µL of the thawed bacteria cultures with an electronic multichannel pipette. Infect for 1.5 h. Screen around 6,000 individualized clones from the 96-well plate Tn mutants stock. NOTE: The MOI is unknown at this step of the protocol; however, all the 96-well plates containing Tn mutants were cultivated in exactly the same way. This first screen aims to quickly identify intracellular deficient mutants, without considering the variations in inoculum density.
- After the 1.5 h of infection, invert the 96-well plate on sterile gauzes placed in a sterilized metal tray to remove the AC medium under a fume hood. Refill with 200 µL of AC medium. Repeat this wash twice more.
- Add 200 µL of fresh medium supplemented with 100 µg/mL amikacin to eliminate remaining extracellular mycobacteria.
- Incubate for 2 h before proceeding with 3 additional washings (as described in step 1.5.3). After washing, maintain the cultures with 50 µg/mL amikacin in 200 µL of AC buffer.
- Add 5 x 107 heat-inactivated Escherichia coli bacteria (from step 1.4) at the end of the infection and every 24 h to prevent encystment of amoebae.
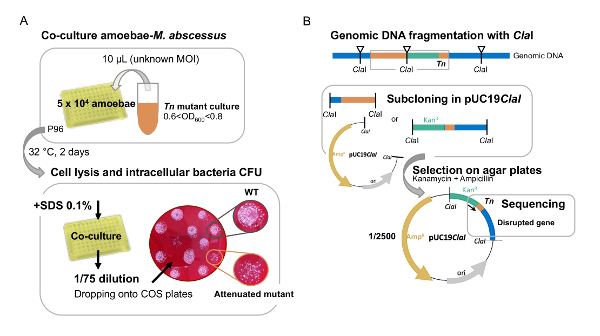
- After 48 h of co-culture, lyse the cells by adding 10 µL of 10% SDS (sodium dodecyl sulfate) to each well and incubate for 30 min at 32 °C. Proceed with the serial dilution and add on Columbia agar plates containing 5% sheep blood (COS plates) to evaluate the number of intracellular mycobacteria. Seal the plate with the parafilm and incubate at 37 °C.
- When colonies appear on the agar plates after 3-4 days, photograph the plate and identify the mutants impaired for intracellular growth by observing the deposits with the lowest number of bacterial colonies. NOTE: Do not mind about the shape, height or density of the colony (Figure 1A). 136/6000 mutants were identified.
- Co-culture of amoebae-M. abscessusTn mutant: second screen of the mutants identified during the first screen NOTE: This takes 2 weeks. A second screen must be performed with the 136 attenuated mutants obtained after the first screen to quantify precisely the number of bacteria inoculated and the number of intracellular survival bacteria 2 days post-infection.
- Grown 1 colony of each impacted mutant in 50 mL tubes filled with 20 mL of 7H9 medium supplemented with 0.2% glycerol, 1% glucose and 250 mg/mL of kanamycin. Allow the bacteria to reach the exponential phase (0.6<OD<0.8).
- Wash the culture by centrifugation (1,835 x g for 10 min) in AC medium. Discard the supernatant. Repeat this wash twice more with 20 mL of 7H9 medium supplemented with 0.2% glycerol, 1% glucose and 250 mg/mL of kanamycin.
- Resuspend the bacteria in 5 mL of AC buffer.
- Determine the colony forming unit (CFU) concentration of the mutants. In 24-well plates, add 1 mL of water to the two first rows. Add 100 µL of the bacterial suspension to the first well. With a micropipette, mix three times. Then, aspirate 100 µL and deposit to the second well.
- With a new tip, repeat step 1.6.4 from the second well to the third one, etc. until the twelfth well.
- Aspirate 30 µL of the 10-12 dilution and add it to a COS plate. Repeat for the other dilutions.
- Wrap the COS plate with parafilm and incubate for 3–4 days at 37 °C. Determine the concentration by counting the colonies.
- Perform co-culture assays as described above in triplicate in a 24-well plate with 5 x 104 amoeba per well in 1 mL of AC co-culture medium. Inoculate with a multiplicity of infection (MOI) of 10.
- After 48 h of co-culture, proceed with the cell lysis as described in step 1.5.7. Perform serial dilutions in water (10-1 to 10-6) and add 30 µL to the COS plates. Incubate the plates for 4 days at 37 °C before proceeding with CFU counting. NOTE: After this second screen, 60 of 136 mutants were conserved.
- Store the mutants impacted for their survival inside the cells. Add a colony to a cryotube containing LB medium with glycerol (20% final) or in a cryotube containing commercial solution and microbeads to favor a long-term conservation and facilitate future inoculation and then store at -80 °C.
- Assess the in vitro mutant growth by measuring the optical density (OD) at 600 nm every 2 days.
- Grow 1 colony of each impacted mutant in 50 mL tubes filled with 20 mL of 7H9 medium supplemented with 0.2% glycerol, 1% glucose and 250 mg/mL of kanamycin. Allow the bacteria to reach the exponential phase (0.6<OD<0.8) before adjusting the OD to 0.01 to begin the kinetics.
- Exclude mutants with an in vitro growth defect when compared with the WT strain from the set of intracellular deficient mutants.
- Co-culture of macrophages-M. abscessus Tn mutant defective for an intra-amoeba life NOTE: This takes 1 month.
- Spread 5 x 104 J774.2 murine macrophages per well of a 24-well plate in 1 mL of rich medium, DMEM supplemented with 10% of Fetal Calf Serum (FCS). Incubate the plates for 24 h at 37 °C and 5% CO2 to allow macrophage adhesion. Provide 3 replicates.
- Perform the infection. Inoculate Tn mutants, with a MOI of 10, diluted in DMEM with 10% FCS in a volume of 100 µL.
- After 3 h of infection, perform three washings with 1 mL of DMEM (a vacuum pump can be used). Then treat with 250 µg/mL amikacin (in DMEM with 10% FCS) for 1 h to eliminate remaining extracellular mycobacteria.
- After 3 additional washes with 1 mL of DMEM, maintain the cultures in 250 µg/mL amikacin (in DMEM with 10% FCS) to a final volume of 1 mL to prevent extracellular mycobacterial multiplication.
- 72 h after co-culture, remove the medium and lyse the macrophages by adding 1 mL of cold water. Incubate for 30 min at 4 °C.
- Perform CFU assays on COS plates to evaluate the number of intracellular bacteria.
- In a 24-well plate, add 1 mL of water to the two first rows. Add 100 µL of the co-cell lysate to the first well. With a micropipette, mix three times. Aspirate 100 µL and deposit them in the second well.
- With a new tip, repeat the dilution from the second well to the third one, etc. until the twelfth well. Then aspirate 30 µL of the 10-12 dilution and add it to a COS plate.
- Repeat for the other dilutions. Wrap the COS plate with parafilm and wait 3-4 days to observe and count the colonies.
- Identification and sequencing of the site of insertion of the Tn in M. abscessus mutants NOTE: This takes 1.5 months for 60 mutants.
- Genomic extraction of the identified M. abscessus mutants
- Grow the mutants in a 50 mL tube with 20 mL of 7H9 medium supplemented with 0.2% glycerol, 1% glucose and kanamycin 250 mg/mL to exponential phase (OD600=0.8).
- Harvest 10 mL of the cultures (2,396 x g, 5 min). Discard the supernatant. Suspend the bacteria in 500 µL of solution I (25% sucrose, 50 mM Tris pH 8, 10 mg/mL lysozyme, 40 mM thiourea). Transfer the solution into 2 mL tubes with screw cap and incubate for 2 h at 37 °C.
- Add 500 µL of solution II (25% sucrose, 50 mM Tris pH 8, 50 mM EDTA). After pipetting up and down 5-10 times, add zirconium beads until a volume of 500 µL in the tube.
- Proceed with cell lysis with a homogenizer (3 x 6,000 rpm, 45 s) with a 1 min incubation on ice between each round.
- Add 5 µL of 20 mg/mL Proteinase K (100 µg/mL final) and incubate the samples overnight at 55 °C.
- Add 1 mL of cold of phenol/chloroform/isoamyl alcohol (25:24:1) under a fume hood. Mix the samples by vortexing before proceeding with centrifugation at RT and 16,500 x g for 30 min.
- After centrifugation, recover the aqueous phase and add an equal volume of cold phenol/chloroform/isoamyl alcohol solution under a hood. Centrifuge the samples at 16,500 x g for 30 min to recover the aqueous phase again.
- Add 0.7 volume of RT isopropanol to the recovered aqueous phase under a fume hood. Invert slowly the tubes to allow DNA precipitation and incubate 5 min at RT. Then centrifuge for 10 min at 16,500 x g and RT.
- Remove the supernatant and wash the pellet with 500 µL of 70% ethanol under a hood. Centrifuge the tubes for 10 min at 16,500 x g at RT before removing the ethanol. Incubate for 10 min to allow remaining alcohol evaporation.
- Finally, suspend the DNA pellet in 100 µL of DNase/RNase free water. Determine DNA yield and purity DNA with a spectrophotometer.
- Remove 1 µg of genomic DNA and digest with 10 U of ClaI for one night for sub-cloning the Tn insertion site. Use manufacturer's protocol. NOTE: The ClaI enzyme is one of the most highly represented restriction enzymes in the M. abscessus genome (2,173 restriction sites). A ClaI restriction site is also found in the Tn sequence upstream of the kanamycin resistance cassette of the Tn mutant. Thus, the ClaI-mediated subcloning enables to identify the Tn insertion site.
- Sub-cloning in a plasmid of genomic DNA containing the Tn NOTE: Before proceeding with the Tn-subcloning in the 2,686 bp pUC19 ampicillin-resistant plasmid, add a ClaI restriction site in the multiple cloning site of the plasmid. The sequence of the pUC19 plasmid can be found at this link https://www.addgene.org/50005/sequences/.
- Follow manufacturer's protocol to digest the pUC19 plasmid with EcoRI and HindIII, which releases a fragment of 51 bp and a fragment of 2635 bp. Purify the double digested vector with a commercial PCR purification kit to eliminate the 51 bp released fragment.
- Mix 1 µL (concentration 25 pmol/µL) of two complementary oligonucleotides (5'AGCT TATA AGATCT TATA ATCGAT TATA3' and 5'AAT TAA TAA TCG ATT ATA AGA TCT TAT A3') of 28 bp each containing the ClaI restriction site and at the ends the HindIII and EcoRI restriction sites. Heat at 100 °C for 10 min and then cool in order to bind them.
- Digest the paired primers by EcoRI and HindIII according to manufacturer's protocol.
- Ligate 150 ng of EcoRI/HindIII-restricted pUC19 plasmid with different concentration of paired primers (50 pmol/µL, 25 pmol/µL, 2.5 pmol/µL) for 1 h at RT with 1 U of T4 DNA ligase in a final volume of 20 µL. Check the cloning by a different enzymatic digestion.
- Digest the modified plasmid with ClaI for 2 h at 37 °C.
- Ligate 50 ng of ClaI-restricted pUC19plasmid and 50 ng of genomic DNA digested by ClaI for 1 h at RT with 1 U of T4 DNA ligase in 10 µL.
- Proceed to E. coli electrocompetent bacteria transformation with 5-10 µL of the ligation (2500 V, 200 Ohms, 25 µF). Select the clones on LB agar plates supplemented with 50 µg/mL kanamycin and 50 µg/mL ampicillin to select the bacteria bearing plasmids with portions of Tn sequences.
- Grow resistant clones overnight in LB liquid medium with the appropriate antibiotic selection (50 µg/mL kanamycin and 50 µg/mL ampicillin).
- Extract plasmid DNA. Check the presence of the insert by enzymatic restriction and sequence the 3' end of the Tn to identify the disrupted gene with an oligonucleotide (5' TTGACGAGTTCTTCTGA 3') corresponding to the last 17 base pairs of the Tn kanamycin resistance cassette.
- Align the sequence against the M. abscessus GO 06 strain by performing a nucleotide blast (BLAST-N).
2. RNA Extraction of Intracellular Mycobacteria for Rnaseq Analysis
NOTE: This takes 4 months for experiments and 4 months for RNAseq analysis.
- Co-culture of amoebae-M. abscessus in batches NOTE: In the following experiment, co-culture is performed in 50 mL tubes with agitation to favor bacteria to cell contacts.
- Grow M. abscessus in 7H9 medium supplemented with 0.2% glycerol, 1% glucose to the mid-exponential phase at 120 rpm.
- Wash the bacteria twice with 10 mL of AC buffer.
- Estimate the bacteria concentration by measuring the OD600nm (1 OD600=4 x 108 mycobacteria). Add 8.5 x 108 mycobacteria to 8.5 x 106 amoebae in 10 mL of AC medium.
- Incubate for 1 h at 30 °C with gentle agitation (about 80 rpm).
- Harvest the cells by centrifuging at 253 x g for 12 min. Remove the supernatant and suspend the cells in 10 mL of AC buffer. Repeat this wash twice more.
- Resuspend the cells in 10 mL of AC buffer containing 50 µg/mL amikacin to eliminate extracellular bacteria and incubate for 4 h at 30 °C with gentle agitation (80 rpm). Wash the cells twice again.
- Extraction of total RNA from intracellular M. abscessus NOTE: Perform steps 2.2.1–2.2.3 under the fume hood due to the use of toxic reagents.
- After the final washings, resuspend the infected amoebae in 4 mL of cold solution III (Table 3) with extemporaneously 280 µL of β-mercaptoethanol to disrupt amoebae. This is the guanidinium thiocyanate (GTC) step. Maintain the suspension on ice for 10 min. Centrifuge the cell lysate for 30 min at 2,396 x g and 4 °C to pellet the released intracellular bacteria.
- Remove the supernatant and suspend the bacterial pellet in 1 mL of cold solution III. Harvest the bacteria at 2396 x g for 30 min at 4 °C. Resuspend the pellet in 1 mL of the extraction buffer and transfer into a 2 mL screw tube containing zirconium beads (until the 500 µL gradation of the tube). Store the tubes for at least 24 h at -80 °C. NOTE: Storage in the lysis reagent permits the inactivation of RNases and cell dissolution.
- When ready for use, thaw samples on ice and lyse them with a homogenizer by performing two rounds at 6,000 rpm for 25 s, followed by one round at 6,000 rpm for 20 s with a 1 min incubation on ice between each round.
- To cool the sample down, incubate them on ice for 10 min. Then add 200 µL of chloroform diluted in isoamyl alcohol. After vortexing the samples for 1 min, centrifuge for 15 min at 16,500 x g and 4 °C.
- Add 0.8 mL of cold isopropanol to the aqueous phase and incubate the samples for 2–3 h at 20 °C. Centrifuge the samples for 35 min at 16,500 x g and 4 °C. Discard the supernatant.
- Wash the RNA pellet by adding 500 µL of cold 70% ethanol (do not mix).
- Centrifuge the samples at 16,500 x g for 10 min to remove the ethanol. Aspirate the solution and dry in a centrifugal concentrator.
- Once dried, resuspend the pellets in 100 µL of DNase/RNase free water. NOTE: Samples must be treated twice with DNases to remove DNA contaminants according to the manufacturer's recommendations.
- Assess the RNA integrity on an agarose gel and confirm by measuring the RNA integrity number (RIN) and the ribosomal ratio with a chip-based capillary electrophoresis method. NOTE: The RIN takes the entire electrophoretic trace into account. The RIN software algorithm allows for the classification of total RNA, based on a numbering system from 1 to 10, with 1 being the most degraded profile and 10 being the most intact. To proceed with cDNA library preparation, the RINs must be over 8 and the ribosomal ratio [28S/18S] as high as possible, the ideal value being 2, depending of the organism and its specificities.
- Store RNA samples at -80 °C until preparation of the cDNA library for sequencing.
- Preparation of the cDNA library
- To proceed with library preparation, use a commercially available cDNA synthesis kit (Table of Materials) using the manufacturer's protocol.
- Check the library for concentration and quality on a DNA Chip. NOTE: More precise and accurate quantification may be performed with sensitive fluorescent-based quantitation assays.
- Library sequencing
- Normalize the samples to 2 nM and proceed to multiplexing according to the flow cell organization.
- Denature the samples at a concentration of 1 nM using 0.1 N NaOH for 5 min at RT.
- Dilute the samples at 10 pM. Load each sample on the flow cell at 10 pM.
- Proceed with sequencing with a high-throughput sequencing system that enables large-scale genomics. Here the run was an SRM run (SR: Single Read, PE: Paired-end Reads, M: multiplexed samples) of 51 cycles with 7 index bases read.
- Assess the quality of the sequencing with the external FastQC program13.
- After the trimming of adapter sequences, proceed with read alignments against a reference genome. Align against the eukaryotic cell genome to assess sample contamination and, if necessary, proceed with the trimming of the non-bacterial reads.
- Statistical analysis
- Use several software programs available online to define sets of differentially expressed genes: R software, Bioconductor packages including DESeq2 and the PF2tools package (version 1.2.12) developed at the platform "Transcriptome et Epigénome" (Pasteur Institute, Paris).
Representative Results
M. abscessus has the ability to resist and escape the bactericidal responses of macrophages and environmental protozoa such as amoebae. M. abscessus expresses virulence factors when grown in contact with amoebae, which makes it more virulent in mice4. The first objective of these methods was to identify the genes present in M. abscessus allowing its survival and multiplication within amoebae.
For this, a mutant library of M. abscessus subsp. massiliense, obtained by Tn delivery9, was screened following co-culture in the presence of amoebae, to identify mutants with attenuated growth in this intracellular environment (Figure 1). The behavior of the same mutants was also evaluated following co-culture with macrophages to analyze whether this growth attenuation was preserved in mice macrophages.
This blind transposon library screening approach, confirmed a defective replication phenotype for 47 of 6,000 mutants that had a survival of 50% or less in amoebae and/or macrophages, compared to controls10. To rule out mutants with attenuated intracellular survival that might be due to an intrinsic growth defect of the mycobacterium, the growth curves of all selected Tn mutants were evaluated in an in vitro liquid culture enriched medium (1% glucose-7H9). In vitro growth of all mutants was monitored by following OD600 of cultures every 2 days. The different mutants that displayed an in vitro growth defect compared to the corresponding wild-type strain were excluded from the study.
It was vitally important for this blind test to be reproduced each day using exactly the same protocol. The limitation of this technique was at the first visual screening, photographs were taken of each CFU to provide an accurate image for reference. In this group of mutants, 12 M. abscessus mutants (included duplicates) were identified in which the transposon was inserted into genes belonging to the ESX-4 locus of the type VII secretion system which underlines its importance in the intracellular growth of M. abscessus10.
Up until now, the analysis of M. abscessus virulence has essentially been based on the comparative analysis of the genome of M. abscessus with that of M. chelonae, a mycobacterium belonging to the same group but causing only infections of the skin in humans. The objective was to obtain a catalogue of the genes expressed during different co-culture conditions in order to better understand the adaptation and potentially virulence mechanisms involved during co-culture in the presence of environmental professional phagocytes. Analysis of this increased virulence through a comprehensive approach of total sequencing messenger RNAs of M. abscessus, was performed in order to detect the RNAs induced or repressed during amoeba co-cultures compared to the RNAs transcribed under in vitro conditions. The differential expression of these RNAs can confer to M. abscessus an enhanced "virulence" explaining the colonization of the upper airways in humans.
These co-cultures were again carried out in a minimum medium (no source of carbon or nitrogen) as described above, in order to prevent the extracellular growth of M. abscessus and to be representative of the environmental conditions faced by these mycobacteria and under the pressure of selection of an antibiotic throughout the duration of co-culture to select intracellular mycobacteria.
Therefore, a technique was developed to isolate the RNA from intracellular mycobacteria. The main difficulty with this extraction of mycobacterial RNA was avoiding contamination with amoebal RNA (Eukaryote type). To avoid this, lysis of amoeba was performed at different times post co-culture, using guanidinium thiocyanate (GTC); since intracellular mycobacteria are resistant. After this step, a mechanical extraction of the mycobacterial RNAs was carried out using a cell-homogenizer in the presence of zirconium beads. This technique allowed us to obtain mycobacterial RNA of good quality, with a RIN number of high quality, essential to improve the ability to undertake a complete analysis of the M. abscessus transcriptome, using the messenger RNA sequencing (mRNA) technique (Figure 2). This has never been done before and gave us a fundamental vision of the gene families induced or repressed, by grouping them according to their role: the response of M. abscessus to an environment limited in its source of nutrients and minerals, to a hypoxic or acidic environment and resistance of M. abscessus to oxidative and nitrosative stress and finally expression of the virulence of M. abscessus in environmental amoeba.
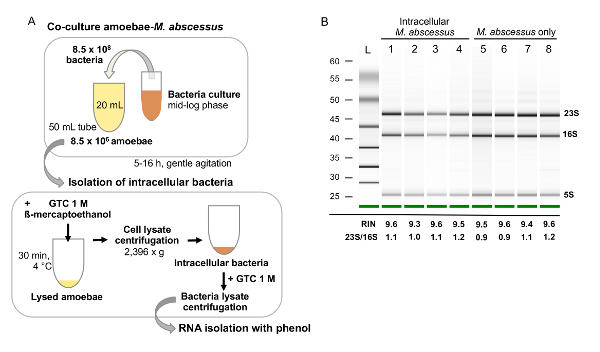
Figure 1: A. First visual screen of M. abscessus Tn mutants in amoeba. (A) large-scale screen of Tn mutants on amoebae is performed in 96-well plates with a random multiplicity of infection to quickly identify attenuated mutants. (B) Identification of the attenuated Tn mutants disrupted genes. Tn mutants' genomic DNA is fragmented with ClaI to clone the Tn insertion site. M. abscessus genomes harbors 2500 ClaI restriction sites which favors the obtaining of short DNA fragments but lowered the probability to clone the Tn insertion site (1/2500). The clones are selected with kanamycin, resistance bear by the transposon. The disrupted gene is identified by sequencing with a primer inside the Tn kanamycin resistance cassette (black arrow). Please click here to view a larger version of this figure.
Figure 2: Analysis of mycobacterial RNA extraction. (A) RNA extraction of intracellular M. abscessus during amoebae-M. abscessus co-culture. Amoebae co-cultures with M. abscessus are performed at a high multiplicity of infection (i.e., 100), in large volumes, with gentle agitation, to favor cell to bacteria contacts. After co-cultures, cells are harvested and lysed with a combination of GTC and ß-mercaptoethanol. The cell lysate-containing bacteria is treated with GTC again to weaken the bacteria cell wall to facilitate bacteria mechanic lysis prior to RNA extraction. (B) RNA quality and integrity assessment with a bioanalyzer. An electrophoresis gel is given on which rRNA is observed (23S, 16S and 5S). RIN must be above 8 to proceed with library preparation for sequencing and rRNA ratios (23S/16S) as high as possible depending on the organism, the ideal value being 2. Please click here to view a larger version of this figure.
Discussion
The behavior of M. abscessus is much more similar to the behavior of pathogenic SGM such as M. tuberculosis than any other mycobacteria belonging to RGM2. The key element in the pathogenicity of SGM is their ability to survive or even multiply within antigen-presenting cells, such as macrophages and dendritic cells.
M. abscessus has acquired certain genomic advantages as shown by the total sequence of its genome14 in order to survive within a eukaryotic phagocytic cell. The genome of M. abscessus has been shown to be rapidly evolving and very plastic, with many recently introduced insertion sequences such as prophages and new genes15. Although there is less data on virulence factors in M. abscessus compared to M. tuberculosis, M. abscessus seems to be able to evolve rapidly and actively towards increased virulence. Until now, the analysis of M. abscessus virulence was essentially based on the comparative analysis of the genome of M. abscessus with that of M. chelonae, a mycobacterium belonging to the same group but causing infections limited to the skin in humans. Some genes not present in M. chelonae but present in M. abscessus, such as phospholipase C, have partially explained the resistance of M. abscessus after phagocytosis by environmental amoebae4.
The development of an amoeba co-culture technique supplemented with environmental samples has demonstrated unambiguously amoeba-mycobacterial interactions16,17. Thus, mycobacteria could be isolated from lysates co-cultured in the presence of amoebae in a water treatment plant18, and M. massiliense was isolated from the sputum of a patient after co-culture with amoebae, whereas the culture alone did not allow the isolation of this mycobacteria8. Amoebae are increasingly considered as an incubator for mycobacteria4 and Acanthamoebasp. as a natural environmental host for many non-tuberculous mycobacteria. Amoeba plus mycobacteria co-culture systems are increasingly being proposed as simple and rapid models to characterize factors involved in the intracellular growth of mycobacteria19,20,21,22,23,24.
In the environment, the interaction between mycobacteria and amoebae is poorly documented. The first article describing evidence of an association between mycobacteria and amoebae in the field came out in 2014, and this study focused on an analysis of a drinking water network25.
To our knowledge, this is the first screening described during a co-culture with amoebae. This study allowed us to demonstrate the role played by environmental phagocytes in the acquisition of virulence of an opportunistic pathogen affecting humans. In order to highlight those genes required for the intracellular survival of M. abscessus, a screen of a mutant library by transposition was carried out in a subspecies of the M. abscessus complex and in a clinical strain. This mutant library was screened with amoebae, this last having been demonstrated as a "training ground" for increasing the virulence of M. abscessus in mice4, similar to that observed with M. avium26. The major result was the discovery for the first time in mycobacteria of the essential role of the ESX-4 locus encoding a type VII secretion system, and ancestor of the ESX-1 locus considered essential for virulence and intracellular survival of M. tuberculosis Mycobacterium microti and M. marinum27,28,29,30,31.
The second objective was to obtain a catalogue of the genes expressed during different co-culture conditions in order to better understand the adaptation and potential virulence mechanisms involved during co-culture in the presence of environmental professional phagocytes. Analysis of this increased virulence through a comprehensive approach of total sequencing of messenger RNAs of M. abscessus, was performed in order to detect the RNAs induced or repressed during amoeba co-cultures compared to the RNAs transcribed under in vitro conditions. The differential expression of these RNAs can confer to M. abscessus an enhanced "virulence" explaining the colonization of the upper airways in humans. Among RGM species, M. abscessus is an exception to the non-pathogenic phenotype along with M. chelonae. The ability of M. abscessus to cause similar pathologies to tuberculous mycobacteria makes it even more unique. Many studies have reported the intracellular adaptations of M. tuberculosis to life inside macrophages32,33 but none have focused on M. abscessus adaptations in vivo. Transcriptional response of M. abscessus to hypoxia has been described34 though, highlighting the role of mycobactins and the dosR regulon as described in M. tuberculosis. Analysis of the M. abscessus transcriptome inside amoebae and macrophages gives an insight into the bacterial adaptions to intracellular life per se. A comparison of those transcriptomes permits the assessment of the role of amoebae in triggering M. abscessus virulence in humans and to the deciphering of specific adaptations to mammalian phagocytic cells. A supposition was made that the amoebal environment might constitute a 'training ground' for M. abscessus before transfer to human cells, in order to describe M. abscessus virulence in terms of evolutionary adaptations, renders this approach original.
Acknowledgments
We greatly acknowledge Pr. E.J. Rubin (Harvard Medical School, Boston, USA) for the precious gift of the mutant library, and Dr. Ben Marshall (Faculty of Medicine, University of Southampton, UK) for the corrections of the manuscript. We greatly acknowledge the French Patient Association for Cystic Fibrosis "Vaincre la Mucoviscidose" and "L'Association Gregory Lemarchal" for their financial support (RF20150501377). We also thank the National Agency for Research (ANR program DIMIVYR (ANR-13-BSV3-0007-01)), and the Région Ile-de-France (Domaine d'Intérêt Majeur Maladies Infectieuses et Emergentes) for funding the postdoctoral fellowship to V.L-M. L. L. is a doctoral fellow from the "Ministère de L'Enseignement Supérieur et de la Recherche".
References
- Qvist T, et al. Comparing the harmful effects of nontuberculous mycobacteria and Gram negative bacteria on lung function in patients with cystic fibrosis. Journal of Cystic Fibrosis. 2016;15(3):380–385. doi: 10.1016/j.jcf.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux A-L, et al. The distinct fate of smooth and rough Mycobacterium abscessus variants inside macrophages. Open Biology. 2016;6(11):160185. doi: 10.1098/rsob.160185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castañeda-Sánchez J, et al. Defensin Production by Human Limbo-Corneal Fibroblasts Infected with Mycobacteria. Pathogens. 2013;2(4):13–32. doi: 10.3390/pathogens2010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakala N'Goma JC, et al. Mycobacterium abscessus phospholipase C expression is induced during coculture within amoebae and enhances M. abscessus virulence in mice. Infection and Immunity. 2015;83(2):780–791. doi: 10.1128/IAI.02032-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripoll F, et al. Non mycobacterial virulence genes in the genome of the emerging pathogen Mycobacterium abscessus. Public Library of Science One. 2009;4(6):5660. doi: 10.1371/journal.pone.0005660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tailleux L, et al. Constrained intracellular survival of Mycobacterium tuberculosis in human dendritic cells. Journal of Immunology. 2003;170(4):1939–1948. doi: 10.4049/jimmunol.170.4.1939. [DOI] [PubMed] [Google Scholar]
- Jacquier N, Aeby S, Lienard J, Greub G. Discovery of new intracellular pathogens by amoebal coculture and amoebal enrichment approaches. Journal of Visualized Experiments. 2013. p. e51055. [DOI] [PMC free article] [PubMed]
- Adékambi T, et al. Amoebal coculture of "Mycobacterium massiliense" sp. nov. from the sputum of a patient with hemoptoic pneumonia. Journal of Clinical Microbiology. 2004;42(12) doi: 10.1128/JCM.42.12.5493-5501.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin EJ, et al. In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(4):1645–1650. doi: 10.1073/pnas.96.4.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laencina L, et al. Identification of genes required for Mycobacterium abscessus growth in vivo with a prominent role of the ESX-4 locus. Proceedings of the National Academy of Sciences of the United States of America. 2018;115(5):1002–1011. doi: 10.1073/pnas.1713195115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowbotham TJ. Isolation of Legionella pneumophila from clinical specimens via amoebae, and the interaction of those and other isolates with amoebae. Journal of Clinical Pathology. 1983;36(9):978–986. doi: 10.1136/jcp.36.9.978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat JF, Tompkins LS. A quantitative model of intracellular growth of Legionella pneumophila in Acanthamoeba castellanii. Infection and Immunity. 1992;60(1):296–301. doi: 10.1128/iai.60.1.296-301.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FastQC program. 2018. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- Ripoll F, et al. Non mycobacterial virulence genes in the genome of the emerging pathogen Mycobacterium abscessus. Public Library of Science One. 2009;4(6):5660. doi: 10.1371/journal.pone.0005660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo SW, et al. Genomic reconnaissance of clinical isolates of emerging human pathogen Mycobacterium abscessus reveals high evolutionary potential. Science Reports. 2015;4 doi: 10.1038/srep04061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greub G, Raoult D. Microorganisms Resistant to Free-Living Amoebae. Clinical Microbiology Reviews. 2004;17(2):413–433. doi: 10.1128/CMR.17.2.413-433.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kicka S, et al. Establishment and Validation of Whole-Cell Based Fluorescence Assays to Identify Anti-Mycobacterial Compounds Using the Acanthamoeba castellanii - Mycobacterium marinum Host-Pathogen System. Public Library of Science One. 2014;9(1):87834. doi: 10.1371/journal.pone.0087834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas V, Loret J-F, Jousset M, Greub G. Biodiversity of amoebae and amoebae-resisting bacteria in a drinking water treatment plant. Environmental Microbiology. 2008;10(10):2728–2745. doi: 10.1111/j.1462-2920.2008.01693.x. [DOI] [PubMed] [Google Scholar]
- Lamrabet O, Medie FM, Drancourt M. Acanthamoeba polyphaga-enhanced growth of mycobacterium smegmatis. Public Library of Science One. 2012;7(1) doi: 10.1371/journal.pone.0029833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosson P, Soldati T. Eat, kill or die: when amoeba meets bacteria. Current Opinion in Microbiology. 2008;11(3):271–276. doi: 10.1016/j.mib.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Lelong E, et al. Role of magnesium and a phagosomal P-type ATPase in intracellular bacterial killing. Cellular microbiology. 2011;13:246–258. doi: 10.1111/j.1462-5822.2010.01532.x. November 2010. [DOI] [PubMed] [Google Scholar]
- Ouertatani-Sakouhi H, et al. Inhibitors of Mycobacterium marinum virulence identified in a Dictyostelium discoideum host model. Public Library of Science One. 2017;12(7):0181121. doi: 10.1371/journal.pone.0181121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trofimov V, et al. Antimycobacterial drug discovery using Mycobacteria-infected amoebae identifies anti-infectives and new molecular targets. Science Reports. 2018;8(1):3939. doi: 10.1038/s41598-018-22228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenal-Muñoz E, Barisch C, Lefrançois LH, López-Jiménez AT, Soldati T. When Dicty Met Myco, a (Not So) Romantic Story about One Amoeba and Its Intracellular Pathogen. Frontiers in Cellular and Infection Microbiology. 2017;7:529. doi: 10.3389/fcimb.2017.00529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delafont V, et al. First evidence of amoebae-mycobacteria association in drinking water network. Environmental Science & Technology. 2014;48(20) doi: 10.1021/es5036255. [DOI] [PubMed] [Google Scholar]
- Cirillo JD, Falkow S, Tompkins LS, Bermudez LE. Interaction of Mycobacterium avium with environmental amoebae enhances virulence. Infection and Immunity. 1997;65(9) doi: 10.1128/iai.65.9.3759-3767.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm LM, et al. Mycobacterium marinum escapes from phagosomes and is propelled by actin-based motility. Journal of Experimental Medicine. 2003;198(9):1361–1368. doi: 10.1084/jem.20031072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groschel MI, Sayes F, Simeone R, Majlessi L, Brosch R. ESX secretion systems: mycobacterial evolution to counter host immunity. Nature Reviews Microbiology. 2016;14(11):677–691. doi: 10.1038/nrmicro.2016.131. [DOI] [PubMed] [Google Scholar]
- Pym AS, et al. Recombinant BCG exporting ESAT-6 confers enhanced protection against tuberculosis. Nature Medicine. 2003;9(5):533–539. doi: 10.1038/nm859. [DOI] [PubMed] [Google Scholar]
- Hsu T, et al. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(21):12420–12425. doi: 10.1073/pnas.1635213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J, et al. Evidence for pore formation in host cell membranes by ESX-1-secreted ESAT-6 and its role in Mycobacterium marinum escape from the vacuole. Infection and Immunity. 2008;76(12):5478–5487. doi: 10.1128/IAI.00614-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnappinger D, et al. Transcriptional Adaptation of Mycobacterium tuberculosis. within Macrophages. Journal of Experimental Medicine. 2003;198(5):693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontan P, Aris V, Ghanny S, Soteropoulos P, Smith I. Global Transcriptional Profile of Mycobacterium tuberculosis during THP-1 Human Macrophage Infection. Infection and Immunity. 2008;76(2):717–725. doi: 10.1128/IAI.00974-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda-CasoLuengo AA, Staunton PM, Dinan AM, Lohan AJ, Loftus BJ. Functional characterization of the Mycobacterium abscessus genome coupled with condition specific transcriptomics reveals conserved molecular strategies for host adaptation and persistence. BMC Genomics. 2016;17(1):553. doi: 10.1186/s12864-016-2868-y. [DOI] [PMC free article] [PubMed] [Google Scholar]