

Abstract
Enterochromaffin (EC) cells in the gastrointestinal (GI) epithelium constitute the largest subpopulation of enteroendocrine cells. As specialized sensory cells, EC cells sense luminal stimuli and convert them into serotonin (5-hyroxytryptamine, 5-HT) release events. However, the electrophysiology of these cells is poorly understood because they are difficult to culture and to identify. The method presented in this paper outlines primary EC cell cultures optimized for single cell electrophysiology. This protocol utilizes a transgenic cyan fluorescent protein (CFP) reporter to identify mouse EC cells in mixed primary cultures, advancing the approach to obtaining high-quality recordings of whole cell electrophysiology in voltage- and current-clamp modes.
Keywords: Biology, Issue 139, Gastrointestinal, epithelium, enterochromaffin cell, primary culture, whole cell electrophysiology, voltage clamp, current clamp
Introduction
The gastrointestinal (GI) epithelium is a diverse community consisting of several cell types. Enteroendocrine cells comprise roughly 1% of all epithelial cells, and enterochromaffin (EC) cells are the largest enteroendocrine cell population1. Recent studies show that enteroendocrine2 and EC3,4 cells are electrically excitable. We are interested in understanding primary EC cell electrophysiology. Thus, the purpose of this study was to establish primary EC cell cultures optimized for whole cell electrophysiology.
Existing EC cell lines that produce and secrete 5-HT (e.g., QGP-15, BON6, KRJ-17) and have been used to examine electrophysiology5,8 are typically generated from immortalized neoplastic tissues. While the information acquired from these cell lines is valuable5,8, studies of primary cell electrophysiology are necessary to properly understand EC cell physiology. The electrophysiology of primary EC cells requires the isolation and culture of single epithelial cells, which has been limited by low viability of epithelial cultures.
The culture method presented in this study relies on transgenic mice with fluorescently labeled EC cells, like Tph1-CFP9, used in this study. The method optimizes mixed primary epithelial cultures developed previously2,10 for single cell electrophysiology3. Previous methods used combinations of Trypsin/EDTA plus Collagenase A or EDTA and DTT for enzymatic digestion and used density gradients to specifically isolate and culture guinea-pig11 and rat EC cells12. More recently, intestinal organoids were generated and mechanically disrupted for electrophysiological recordings4. Cultures using these methods are well suited for serotonin release experiments, RT-qPCR analysis and, while they could be used for electrophysiology, are reliant on the success of time consuming organoid generation, density gradients to identify EC cells, and the cellular disruptive effects of Trypsin and EDTA. By contrast, the protocol described here improves enzymatic and mechanical treatments, optimizes culturing conditions, and streamlines procedures to produce single and healthy EC cells suitable for the high cellular standards needed for electrophysiology.
This method will be useful for scientists who want to work on primary EC cells in mixed cultures rather than immortalized cultures and want to investigate the electrophysiology of single cells. However, it may prove less appropriate for studying cell populations that need sorting or long-term cultures that require survival past 72 h.
Protocol
All methods described here have been approved by the Institutional Animal Care and Use Committee (IACUC) of Mayo Clinic. The primary cell culture portion of this protocol is based on previously published methods that can be referenced for further details10. All experimental procedures must be approved by the (IACUC), and all experiments must be performed in accordance with relevant guidelines and regulations.
1. Culture Preparation
- Media.
- Prepare EC cell complete culture media with high glucose [4500mg/L] Dulbecco's Modified Eagle Medium (DMEM) supplemented with 5% Fetal Bovine Serum (FBS), 1% L-Glutamine and 1% Penicillin-Streptomycin (Table 1). NOTE: Media can be stored at 4 °C for up to one month.
- Filter the EC cell complete culture media and place 25 mL of media into a 50 mL tube at 37 °C.
- Prepare digestion media with high glucose [4,500 mg/L] DMEM supplemented with 0.1% Bovine Serum Albumin (BSA) (Table 1).
- Filter the digestion media and place four 10 mL aliquots of media into separate 50 mL tubes and incubate in a 37 °C water bath. NOTE: Media can be stored at 4 °C for up to one month.
- Enzyme.
- Weigh out 4 mg (for jejunum) or 24 mg (for colon) of Collagenase XI [≥800 units/mg] in a 1.5 mL microcentrifuge tube. NOTE: Collagenase is stable at -20 °C for up to one year.
- Resuspend the collagenase aliquot in 1 mL of ice cold PBS with Ca2+ and Mg2+. Vortex the solution to thoroughly combine, and place on ice.
- Coating Culture Plates.
- Thaw 150 µL of extracellular matrix overnight at 4 ˚C on ice.
- Make a 5% w/v extracellular matrix solution by mixing 100 µL of thawed extracellular matrix with 2 mL of ice-cold serum-free high-glucose DMEM. NOTE: Extracellular matrix solidifies at room temperature and should be handled with pre-chilled pipettes, tubes, and media until ready to coat dishes.
- Immediately coat the glass inner circle of glass-bottom 35 mm dishes with 250 µL of the 5% extracellular matrix solution. This recipe will cover up to 8 dishes.
- Place the glass-bottom dishes in a 37 °C incubator during the remaining cell culture isolation steps. The extracellular matrix takes a minimum of 1 h to set but can be incubated for up to 2.5 h while performing other isolation steps. NOTE: Incubations longer than 3 h can create a build-up of extracellular matrix which could impede seal formation during whole cell electrophysiology.
2. Tissue Isolation
According to ethical guidelines, sacrifice a 3to 6 week-old male or female Tph1-CFP transgenic mouse (The Jackson Laboratory Stock: 028366)9 or another reporter strain. NOTE: We use a fatal dose of rising levels of carbon dioxide and a cervical dislocation as a secondary means to ensure death. The AVAM Committee on Euthanasia also considers cervical dislocation an allowable primary euthanasia method by individuals trained in its use if the animals are first sedated.
Pin the euthanized mouse out on a polystyrene foam dissection stage using 21 G needles. Decontaminate the mouse abdomen with 70% ethanol.
Pull mouse skin taut using micro-dissection forceps (#5), and then use sharp surgical scissors to make a transverse incision at the bottom of the mouse abdomen to expose the intraperitoneal cavity. Make another incision vertically up the abdomen until the rib cage is exposed.
Use the micro-dissection forceps to move the cecum to the left of the mouse to attain a clear view of the colon.
Use the micro-dissection scissors to cut the most distal portion of the colon (but still proximal to the rectum) and make a second cut distal to the stomach. Place the extracted intestines into a 100 x 15 mm2 Petri dish filled with 20 mL of ice-cold phosphate buffered saline (PBS).
Use a small ruler to measure and cut out 10 cm of either mid colon or jejunum. Extract colon by measuring out the 10 cm segment of intestine located proximally to the first cut taken above the rectum. Identify the jejunum by folding the entire small intestine in half and making a first incision at the halfway point and a second incision 10 cm proximal to the first cut. NOTE: Continue to perform all subsequent isolation steps on ice.
Fill a 6 mL syringe with ice cold PBS and place the syringe needle into the lumen of the extracted intestinal segment. Use the micro-dissection forceps to clamp and seal the intestines around the syringe needle, and gently flush 10 mL of PBS through the lumen to rinse away fecal matter.
Invert the intestinal tissue by gently weaving the micro-dissection forceps through the lumen, pinching an intestinal wall, and retracting the tissue proximal to the tip of the forceps. Repeat this process until the segment is inside-out with the lumen facing outward.
Use micro-dissection scissors to cut the tissue segment into 1 cm pieces.
Use micro-dissection forceps to transfer the tissue segments in a 50 mL beaker filled with 5 mL sterile PBS. Use the micro-dissection scissors to mince the tissue pieces to a liquified consistency.
Place the minced tissue and 15 mL of ice cold PBS in a clean 50 mL tube by transfer pipette. Triturate 2 times with the transfer pipette, wait for the tissue to settle, remove 15 mL of the PBS supernatant, and replace with fresh PBS. Repeat this process three times until the PBS is clear.
Place the 50 mL tube with washed tissue pieces on ice and bring the ice bucket into a tissue culture hood.
3. Enzymatic and Mechanical Digestion
- First digestion
- From the water bath, retrieve one of the 50 mL tubes containing 10 mL of pre-warmed digestion media. To the 50 mL tube, add 250 µL of the PBS collagenase solution and minced intestinal tissue pieces. NOTE: Take care to avoid including any PBS from previous washing steps.
- Place the 50 mL tube in a water bath for 5 minutes (colon or jejunum) at 37 °C. Shake the tube 2x per min. NOTE: The optimal timing and intensity of mechanical digestion may vary and will need to be determined by the user. To determine the optimal digestion conditions, a 10 µL aliquot of cells can be viewed after each digestion. During Digestion 1, the supernatant should consist of clumps of cells.
- Slowly triturate the tissue one time with a 10 mL serological pipette. Briefly let the tissue pieces settle, then use a transfer pipette to remove the entire 10 mL of supernatant.
Second digestion: Repeat steps 3.1.1 to 3.1.3. Increase the incubation time to 10 min if digesting colon and keep the incubation time at 5 min for jejunum. NOTE: Upon observation, the supernatant should still consist of large clumps of cells.
- Third digestion
- Repeat step 3.1.1
- Place the 50-mL tube in a 37 ˚C water bath for 10 min (colon or small jejunum). Shake the tube 2-3x per min at a higher intensity than Digestions 1 and 2.
- Slowly pipette the tissue up and down with a 10-mL serological pipette. Allow the tissue pieces to settle for a short time. NOTE: The supernatant should consist of a combination of single cells and small clumps.
- Use a transfer pipette to place 10 mL of the supernatant into a new 15 mL tube, add 2 mL of warmed EC cell complete culture media, and invert once to mix.
- Spin at 100 x g for 5 min at room temperature, then use a transfer pipette to remove the supernatant. Use a transfer pipette to resuspend the remaining pellet in 2.5 mL of warmed EC cell complete culture media.
Fourth digestion: Repeat steps 3.3.1 to 3.3.5. Increase the incubation time to 15 min for colon and remain at 10 min for jejunum. NOTE: The supernatant should now consist of single cells.
4. Cell Culture
Use a transfer pipette to combine the cell suspensions collected from Digestions 3 and 4 into a new 15 mL tube (5 mL total volume).
Remove a 10 µL aliquot of cells to count with a hemocytometer and spin the remaining cell suspension at 100 x g for 5 min at room temperature. NOTE: The final cell suspension will consist of small clumps and single cells.
Remove the supernatant with a transfer pipette and resuspend the pellet in EC cell complete culture media at a density of 1,000,000 cells per mL. NOTE: The final volume of cell suspension will depend on the final cell count. Typical cell counts range from 2,000,000 to 4,000,000.
Remove coated glass-bottom culture dishes from the incubator. Use a P1000 pipette to replace extracellular matrix from each culture dish with 250 µL of the final cell suspension. NOTE: Extracellular matrix coating tends to stick to the edges of the glass-bottom dish. Special care must be taken to remove the coating from along the edge to prevent gel buildup.
Make a stock solution of ROCK inhibitor Y-27632 at 1 mM. Add 2.5 µL of stock solution to each glass bottom dish to achieve a working concentration of 10 µM ROCK inhibitor in each dish. NOTE: This step is critical for the survival of jejunum cultures but is optional for colon.
Place each culture dish in a 37 °C and 5% CO2 incubator for 24 to 72 h (Figure 1).
5. Preparation of EC Cells for Whole Cell Electrophysiology
Line the inner diameter of the microscope stage with two 5 x 0.5-cm strips of wax film to create an O-ring. Mount the 35-mm cell culture dish within the O-ring (Figure 2A-B).
- Rinse both floating debris, such as unattached extracellular matrix or dead cells, and culture media serum completely away from EC cells to prevent either from impeding seal formation between the EC cell and electrode. Use of the three following options to achieve thorough cell washing sufficient for electrophysiology:
- Option 1: Solution exchange is more efficient in a long chamber more so than a circular chamber. Since the EC cells were cultured in round 35 mm dishes, create a plastic elliptical insert for the dish.
- Create the insert by 3D printing or by traditional milling methods. The insert we used was milled from acrylic plastic with the following dimensions (in mm): outer diameter, 34.5; inner ellipse, 20x9; outer height, 10; inner height, 1.5; bridge span, 3x3; bridge clearance, 1.5; inlet and outlet, 4 x 4 each.
- With a plastic transfer pipette, slowly add extracellular solution to one side of the dish (inlet) while aspirating from the other side (outlet).
- Option 2: Without an engineered plastic insert, add extracellular solution by transfer pipette from one side of the dish (inlet) while aspirating from the opposite side (outlet). Slowly rotate the location of the transfer pipette (e.g., N to E to S) while mirroring the placement of the aspiration needle on the opposite side (e.g., S to W to N).
- Option 3: Coat extracellular matrix onto rectangular coverslips in step 1.3.3., and culture the cell suspension on these coverslips in step 4.4. Transfer this coverslip to a rectangular chamber filled with extracellular solution, omitting step 5.1 and skipping step 5.3. Rinse the chamber with extracellular solution from a plastic transfer pipette, as described in Option 1 (5.2.1.3).
Re-attach the stage with the cell culture above the inverted microscope (Figure 2D). Incubate the EC cell culture in serum-free extracellular solution at room temperature.
After 4 hours, rinse the culture again with extracellular solution as described above. Proceed with whole cell electrophysiology.

6. Whole Cell Electrophysiology of EC Cells from Primary Culture
- Achieving a Whole Cell Giga-seal
- Pull one dozen electrodes to a resistance of 1–5 MΩ. NOTES: Pipettes of 1–2 MΩ resistance that yield 10-100 GΩ seals perform best. Fire polishing the electrode tips is not necessary to achieve GΩ seals.
- Coat evenly the nose cone of all pipettes with polymer. Hold the pipette horizontal to the ground and perpendicular to the front of a heat gun. Slowly rotate the pipette until the polymer hardens.
- Pour an aliquot of intracellular solution into a 15-mL conical tube. Transfer 1.2 mL of this solution into a 1.5 mL microcentrifuge tube via 1-mL syringe. Withdraw another 0.7 mL of intracellular solution into the syringe. Dispel air from the end of the syringe. Attach a flexible 34 G, 67 mm needle to the syringe, and flush out any remaining air from this needle.
- Identify a Tph1-CFP+ cell that is not in contact with any other cell or debris.
- Fill the electrode tip with intracellular solution. Backfill the pipette by syringe. Carefully flick the pipette with a fingernail to dispel the air bubble interface.
- With the plunger drawn up with 0.3 mL of air, attach a 6 mL syringe to the outlet of the holder tubing. Mount the electrode into the holder. NOTE: Connect the syringe to the tubing before the electrode enters the bath solution to maintain neutral or positive pressure within the intracellular solution prior to sealing.
- Load a two-step voltage-clamp protocol that records steady-state activating currents by a voltage ladder on the first step and records the available currents by a single voltage on the second step. Open the seal test (5 mV at 50 kHz).
- Lower the electrode into the bath solution. With the micromanipulators, maneuver the electrode tip in-plane with and directly horizontal from the cell. Expel the 0.3 mL of air from the syringe.
- Gently move the electrode horizontally along the x-axis to touch the cell. Watch for a dimple to appear on the EC cell and/or an increase in membrane resistance on the seal test. If necessary, readjust the electrode down along the z-axis and/or horizontally along the x-axis, not moving past the midline of the cell.
- Apply 0.1–0.2 mL suction on the 6-mL syringe. Hold the plunger steady until 100 MΩ is achieved, then release grip from the plunger. Wait for the cell to giga-seal. Gently disconnect the syringe with an unscrewing motion. NOTE: If an EC cell does not seal initially, it might be possible to re-seal it with a subsequent attempt. To do this, gently pull the electrode away from an EC cell with <20 MΩ seal resistance. Steering the spent electrode with the micromanipulators, circumnavigate the targeted EC cell to manually clear away matrix or debris. Then, attempt to seal the EC cell with a fresh electrode.
- Record whole cell voltage-gated Na+ current.
- Attach a 3 mL syringe drawn up with 0.5–1.0 mL air. Apply a quick tap of suction (0.1–0.5 mL) on a 3 mL syringe. Repeat until whole cell access is achieved, then gently disconnect the syringe. NOTE: As the performance of a syringe can vary initially and change gradually with time and use, practice beforehand with the 3 mL syringe (sealing the end with an index finger) to ensure that the plunger will recoil within 0.1–0.2 mL of its starting position. If not, then exchange the syringe for a new one.
- Turn on whole cell capacitance compensation. Adjust capacitance and series resistance. Turn on series resistance compensation. With lag set at 60 ms, adjust predicted then corrected series resistance compensation. Close the seal test.
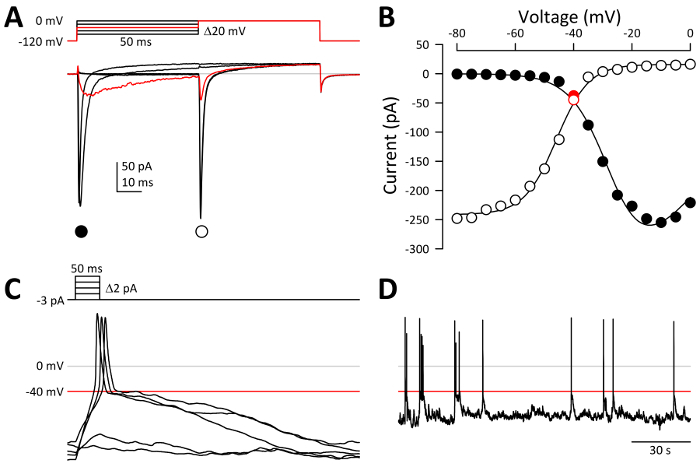
- Record an average of 5 runs of whole-cell voltage-gated Na+ current (Figure 3A).
- Quickly take note of two parameters of voltage-gated Na+ current: the voltage at window current (Figure 3B, red symbols, the intersection of steady-state activation and inactivation curves) and the peak Na+ current density. NOTE: EC cells with peak Na+ currents more than -50 pA in voltage clamp mode commonly will go on to fire elicited potentials in current clamp mode. Additionally, currents greater than -200 pA in voltage clamp (Figure 3A) then usually fire action potentials spontaneously in current clamp from membrane potentials (red line, Figure 3D) near the voltage of the window current (red symbols, Figure 3B).
- Record elicited action potentials.
- Turn off the series resistance compensation. Turn off the external command. Switch the mode from V-clamp to I-clamp. Adjust the gain. Load an episodic current clamp protocol. Adjust the amount of holding current to 0 pA. Turn on the external command. CAUTION: Do not switch from V-clamp to I-clamp (or vice versa) without first turning off the external command.
- Note the resting membrane potential. Adjust the holding current input so that the membrane potential reads below the window current (e.g., -80 to -100 mV). Adjust the episodic current clamp protocol so that the step interval of current input is less than the value of the holding current (Figure 3C inset, +2 pA steps for -3 pA holding current).
- Record elicited action potentials. Note the least amount of current injected to fire off an action potential (Figure 3C, sweep 3 of 5, a +4 pA step from -3 pA holding = +1 pA).
- Record spontaneous action potentials.
- Turn off the external command. Load a gap-free (non-episodic) current clamp protocol. NOTE: Do not turn the external command back on until the amount of holding current in the gap-free current clamp protocol has been verified to be suitable for the cell.
- Change the holding current to be the amount of current injected in the last sweep in the elicited protocol that did not fire an action potential (Figure 3D, the current input during the second sweep, a +2 pA step from -3 pA holding = -1 pA).
- Turn on the external command. Double check that the predicted holding current brings the membrane potential below the threshold to fire an action potential. Adjust the holding current if necessary. Record spontaneous activity.
Representative Results
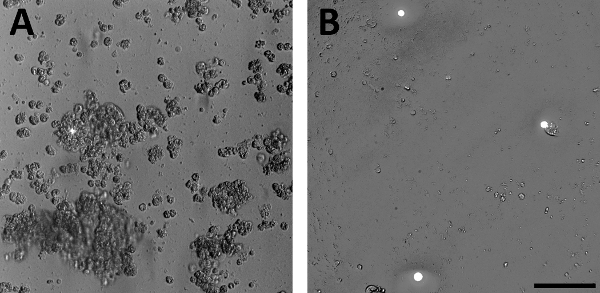
Cultures: Primary murine epithelial cells made from a transgenic Tph1-CFP model attach to dishes after 4 hours and are ready for physiological experiments between 24 to 72 h. When culture conditions had not been optimized, the epithelial cell culture consisted of large clumps, floating cellular debris, damaged membranes, and weak CFP signal in the EC cells (Figure 1A). Cultures that have been optimized for electrophysiology consisted of single cells and small clumps. Healthy EC cells were readily identified by bright CFP fluorescence and had still adhered to the dish after vigorous washing (Figure 1B).
Electrophysiology: EC whole cells were obtained on just 15 ±4% of attempts from colon cultures without ROCK inhibitor (29 whole cells out of 182 attempts from 22 cultures) but 30 ±7% of attempts from colon cultures with ROCK inhibitor (27 whole cells out of 102 attempts from 50 cultures) (Figure 4A; p <0.05 by a non-parametric two-tailed t-test, colon cultures without vs. with ROCK inhibitor). Similarly, jejunum EC cells without ROCK inhibitor did not survive long enough for electrophysiology, while EC whole cells were obtained on 41 ±3% of attempts from jejunum culture with ROCK inhibitor (186 whole cells out of 447 attempts from 50 cultures) (Figure 4A; p >0.05 by a non-parametric two-tailed t-test, jejunum vs. colon with ROCK inhibitor).
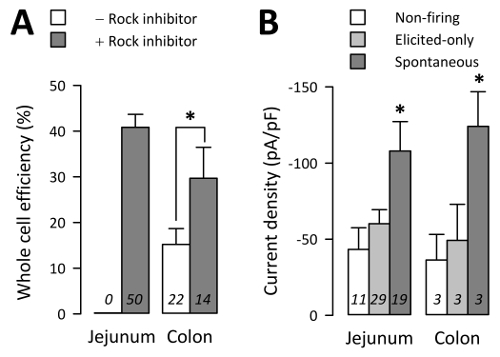
By whole cell electrophysiology, Na+ currents were recorded in voltage-clamp mode and action potentials (AP) in current-clamp mode. Na+ currents were recorded from 81.3 ±4.0% of TPH1-CFP+ cells from jejunum or 64.1 ±9.2% from colon3. Of 59 EC cells from jejunum cultures confirmed by voltage-clamp to have Na+ peak currents larger than -25 pA, 19 EC cells in current-clamp mode fired spontaneous AP ("spontaneous"), 29 EC cells fired AP only when elicited by a step protocol in current-clamp mode ("elicited-only"), and 11 EC cells did not fire AP spontaneously or by step protocol ("non-firing") (Figure 4B). Spontaneous cells had an average peak Na+ current density (IPEAK) of -108.0 ±19.3 pA/pF, elicited-only cells had an IPEAK of -60.2 ±9.2 pA/pF, while non-firing cells had an IPEAK of -43.3 ±14 pA/pF (Figure 4B; p < 0.05, spontaneous vs. elicited-only or non-firing groups; p >0.05 elicited-only vs. non-firing groups). The difference in current densities were not a reflection of whole cell capacitance, as the spontaneous (3.3 ±0.2 pF), elicited-only (2.9 ±0.2 pF), and non-firing (3.1 ±0.3 pF) cells had similar whole cell capacitances (P >0.05). EC cell Na+ currents and elicited AP were sensitive to [Na+]o concentration and NaV1.3 inhibition3.
Figure 1: Representative range of culture results. DIC with epifluorescence images of a primary epithelial jejunum culture from a Tph1-CFP mouse plated on a glass bottom dish and cultured for 24 h. These two images demonstrate a range of culture outcomes from this protocol. CFP fluorescent cells (white) are enterochromaffin (EC) cells. (A) representative image from a culture that is poor quality for electrophysiology. The culture consists of large clumps, damaged cell membranes, low fluorescence signal, and floating groups of dead cells. (B) representative image from a culture that has been optimized for electrophysiology. Culture consists mainly of single, healthy cells and small clumps that have adhered to the plate. EC cells in this culture maintain a strong fluorescence signal. Scale bar = 100 µm; panels are scaled 1:1. Please click here to view a larger version of this figure.
Figure 2: Preparation of cell cultures for electrophysiology. A strategy for efficient and thorough rinsing of EC cell cultures with serum-free extracellular solution is critical for reducing extracellular matrix and serum which would impede seal formation. (A) materials. Clockwise, from top left: Stage, two 5 mm wide strips of wax film, two 200 mg pieces of modeling clay, plastic transfer pipette, #5 forceps, elliptical recording chamber, 35 mm culture dish with glass bottom. (B) adhere wax film to the inner diameter of the stage, lower the elliptical chamber into the culture dish, and flatten the clay into rectangular strips. (C) gently press the culture dish into the wax film-lined stage and immobilize the recording chamber by stretching the modeling clay over the edge of the dish, from the chamber to the stage. Rinse EC cells with several volumes of extracellular solution. (D) return the stage to the mounting brackets above the inverted microscope. Please click here to view a larger version of this figure.
Figure 3: Representative electrophysiology results. (A) whole cell Na+ currents, recorded by a two-step voltage-clamp protocol (inset). 50 ms depolarizations from -120 mV activate channels during step 1 (●), while a further depolarization to 0 mV during step 2 (○) activates channels that had not yet inactivated (i.e., are still available to open). Red trace, current elicited by voltage steps from -120 to -40 to 0 mV. (B) current-voltage relationship of peak Na+ currents from panel A, step 1 (●, steady-state activation) or step 2 (○, steady-state inactivation). The intersection of the steady-state activation and inactivation curves reveals a small window current at -40 mV (red symbols). (C) action potentials elicited by depolarizing current stimuli. An action potential can fire past 0 mV (gray line) when the membrane potential reaches the voltage of Na+ window current (red line, -40 mV). Inset, current-clamp protocol, in which 0 to +8 pA was injected for 50-ms depolarizations from a baseline of -3 pA. (D) action potentials fire from atop spontaneous events that have depolarized to -40 mV. Please click here to view a larger version of this figure.
Figure 4: Throughput of whole cell electrophysiology. (A) mean percentage of whole cells per attempt for n days of work in the absence (-) or presence (+) of ROCK inhibitor (10 µM). EC cells from jejunum did not survive 24 h in culture without ROCK inhibitor. (B) Na+ current densities measured in voltage-clamp from n EC cells, in which action potentials did not fire (Non-firing), fired only when elicited by an episodic current-clamp protocol (Elicited-only), or fired spontaneously in gap-free current-clamp mode (Spontaneous) (error bars are SEM). Please click here to view a larger version of this figure.
Discussion
Fully understanding EC cell function requires a high-quality primary culture method to generate cells for whole cell electrophysiology. Primary cultures of GI epithelium had traditionally been difficult due to low survival. Alleviating this confounding factor, the present method yields cultures of singular EC cells from either small or large bowel that are capable of surviving for several days.
We found that the following were critical for improving the quality of cultures to be suitable for electrophysiology: (1) inverting the mucosa rather than peeling it, (2) adding a small amount of BSA to the digestion media and FBS to the culture media, (3) lowering the Collagenase XI concentration, (4) adjusting the duration and intensity of mechanical agitation to increase the number and health of single cells, (5) plating the extracellular matrix at a low concentration, and (6) culturing with ROCK inhibitor.
Similarly, the following adaptations were critical for obtaining whole cells while patch clamping: (1) thoroughly rinsing the cultures with serum-free extracellular solution, (2) letting the EC cells stand in the extracellular solution for at least 4 h, (3) re-purposing any spent electrodes-like those from failed seals-to clear away debris en route to the next EC cell, (4) adding 10 µM Gd3+ to the extracellular solution to block leak currents (if present) and to facilitate adherence of mammalian cell suspensions to glass. We caution, however, that Gd3+ could block voltage-gated Ca2+ and/or mechanosensitive channels expressed in EC cells.
Despite having carefully optimized the culturing conditions, we found a few limitations: EC cells need 24 h to fully attach yet expire after ~72 h in culture; therefore, they have a 48-hour window when they would be suitable for electrophysiological experiments. Because clumped cells often introduce capacitance transients that cannot be compensated from recordings, generation of single EC cells are critical. However, by separating EC cells from the epithelium, the cells then live in a non-native environment (i.e., high glucose media with fetal bovine serum and ROCK inhibitor atop the extracellular matrix as an artificial basement membrane) and could respond to stimuli not entirely representative of EC cell function in vivo. Despite these limitations, the primary culturing of EC cells still provides insight into their physiology.
EC cells play a critical role in maintaining a well-functioning GI tract and body, and as such, there have been many methods developed to gain an understanding about EC cell electrophysiology. Enteroendocrine and EC cell electrophysiology was investigated in immortalized cell models5,8,13, primary EC cell cultures from guinea pig11, and mouse3,4, Our method utilizes cell quality-preserving methods to create a culture that has been minimally processed and reflects the native EC cells. We use a transgenic mouse model, Tph1-CFP, that shows a cyan fluorescence in the cytoplasm when Tph1 is present9. This method can also be used for other reporter or lineage-traced enteroendocrine cell types. Our method also accomplishes the isolation of single EC cells in minimal time and enzymatic treatment, as the cells are only exposed to a gentle Collagenase XI at a minimal yet effective concentration of 0.1 mg/mL (jejunum) and 0.6 mg/mL (colon) for less than one hour. The result of these modifications is a culture that has been optimized for electrophysiological studies of the EC cells.
We have successfully used this method to study electrophysiology and serotonin release in murine EC cells3. The ability to successfully isolate, identify, and culture single EC cells facilitates important future directions in studies on EC cell physiology. This method can be used to further determine critical pathways and channels involved in the EC cell exocytosis of serotonin and other EC cell-specific transmitters. The method could also be modified to isolate and characterize the physiology of the human EC cell along with the use of acridine orange for EC cell identification13.
As with most scientific protocol development, communication and unbiased observations were critical in the troubleshooting of this protocol. When we tested new culture methods for the survival of our cultures, it was helpful to count the number of fluorescent cells as a measure of viability, estimate cell densities in each dish, and collect representative pictures and videos of the cultures across many time points. After the cells were able to survive past 24 h in culture, we worked on modifying our cultures for electrophysiology experiments. Along with collecting images and detailed observations, it was important to maintain clear communication between lab personnel regarding adjustments to mechanical digestion and supplement and/or substrate concentrations, so that healthy EC cells were ready for electrophysiological characterization.
Disclosures
None
Acknowledgments
The authors thank Mrs. Lyndsay Busby for administrative assistance and Mr. Robert Highet from Mayo Clinic Division of Engineering for design and 3D printing of the culture dish insert. This work was supported by NIH K08 to AB (DK106456), Pilot and Feasibility Grant to AB from Mayo Clinic Center for Cell Signaling in Gastroenterology (NIH P30DK084567) and 2015 American Gastroenterological Association Research Scholar Award (AGA RSA) to AB and NIH R01 to GF (DK52766).
References
- Rindi G, Leiter AB, Kopin AS, Bordi C, Solcia E. The "normal" endocrine cell of the gut: changing concepts and new evidences. Annals of the New York Academy of Sciences. 2004;1014:1–12. doi: 10.1196/annals.1294.001. [DOI] [PubMed] [Google Scholar]
- Rogers GJ, et al. Electrical activity-triggered glucagon-like peptide-1 secretion from primary murine L-cells. The Journal of Physiology. 2011;589:1081–1093. doi: 10.1113/jphysiol.2010.198069. Pt 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strege PR, et al. Sodium channel NaV1.3 is important for enterochromaffin cell excitability and serotonin release. Scientific Reports. 2017;7(15650) doi: 10.1038/s41598-017-15834-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellono NW, et al. Enterochromaffin cells are gut chemosensors that couple to sensory neural pathways. Cell. 2017;170(1):185–198. doi: 10.1016/j.cell.2017.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcaino C, Knutson K, Gottlieb PA, Farrugia G, Beyder A. Mechanosensitive ion channel Piezo2 is inhibited by D-GsMTx4. Channels. 2017;11(3):245–253. doi: 10.1080/19336950.2017.1279370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Javed NH, Yu JG, Christofi F, Cooke HJ. Mechanical stimulation activates Galphaq signaling pathways and 5-hydroxytryptamine release from human carcinoid BON cells. Journal of Clinical Investigation. 2001;108(7):1051–1059. doi: 10.1172/JCI12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modlin IM, Kidd M, Pfragner R, Eick GN, Champaneria MC. The functional characterization of normal and neoplastic human enterochromaffin cells. The Journal of Clinical Endocrinology and Metabolism. 2006;91(6):2340–2348. doi: 10.1210/jc.2006-0110. [DOI] [PubMed] [Google Scholar]
- Wang F, et al. Mechanosensitive ion channel Piezo2 is important for enterochromaffin cell response to mechanical forces. The Journal of Physiology. 2017;595(1):79–91. doi: 10.1113/JP272718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HJ, et al. Distinct cellular origins for serotonin-expressing and enterochromaffin-like cells in the gastric corpus. Gastroenterology. 2014;146(3):754–764. doi: 10.1053/j.gastro.2013.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psichas A, Tolhurst G, Brighton CA, Gribble FM, Reimann F. Mixed primary cultures of murine small intestine intended for the study of gut hormone secretion and live cell imaging of enteroendocrine cells. Journal of Visualized Experiments. 2017. p. 55687. [DOI] [PMC free article] [PubMed]
- Raghupathi R, et al. Identification of unique release kinetics of serotonin from guinea-pig and human enterochromaffin cells. The Journal of Physiology. 2013;591:5959–5975. doi: 10.1113/jphysiol.2013.259796. Pt 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa K, et al. TRPA1 regulates gastrointestinal motility through serotonin release from enterochromaffin cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(9):3408–3413. doi: 10.1073/pnas.0805323106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann F, et al. Characterization and functional role of voltage gated cation conductances in the glucagon-like peptide-1 secreting GLUTag cell line. The Journal of Physiology. 2005;563:161–175. doi: 10.1113/jphysiol.2004.076414. Pt 1. [DOI] [PMC free article] [PubMed] [Google Scholar]