

Abstract
Neural crest cells (NCCs) are migrating multipotent stem cells that can differentiate into different cell types and give rise to multiple tissues and organs. The O9-1 cell line is derived from the endogenous mouse embryonic NCCs and maintains its multipotency. However, under specific culture conditions, O9-1 cells can differentiate into different cell types and be utilized in a wide range of research applications. Recently, with the combination of mouse studies and O9-1 cell studies, we have shown that the Hippo signaling pathway effectors Yap and Taz play important roles in neural crest-derived craniofacial development. Although the culturing process for O9-1 cells is more complicated than that used for other cell lines, the O9-1 cell line is a powerful model for investigating NCCs in vitro. Here, we present a protocol for culturing the O9-1 cell line to maintain its stemness, as well as protocols for differentiating O9-1 cells into different cell types, such as smooth muscle cells and osteoblasts. In addition, protocols are described for performing gene loss-of-function studies in O9-1 cells by using CRISPR-Cas9 deletion and small interfering RNA-mediated knockdown.
Keywords: Genetics, Issue 140, CRISPR, Cas9, O9-1 cells, gene knockout, gene knockdown, neural crest cells
Introduction
Neural crest cells (NCCs) are multipotent stem-like cells with a remarkable migratory ability and transient existence during embryonic development. NCCs originate between the surface ectoderm and the neural tube and migrate to other parts of the embryo during embryonic development1. Based on their functional domains, NCCs can be classified into several different types, including cranial, trunk, vagal, sacral, and cardiac NCCs. In addition, NCCs can differentiate into multiple cell lineages, such as smooth muscle cells, bone cells, and neurons, and give rise to various tissues2,3. The development of NCCs is characterized by a complex series of morphogenetic events that are fine-tuned by various molecular signals. Given the complex regulation of NCCs and their important contributions to numerous structures, the dysregulation of NCC development can commonly lead to congenital birth defects, which account for nearly 30% of all human congenital birth defects. Abnormalities during the neural crest development can lead to cleft lip/palate, flawed nose formation, syndromes, defects such as a defective cardiac outflow tract, or even infant mortality1,4,5,6,7. Understanding the molecular mechanisms of NCC development is important for developing treatments for diseases caused by defects in NCC development. With the use of various in vitro and in vivo approaches8,9,10,11,12,13,14,15, considerable progress has been made in NCC research. In vivo, animal models, including chickens, amphibians, zebrafish, and mice, have been used to investigate NCCs1. Furthermore, human embryos have been used to study the process of NCC migration in early human embryo development16. In vitro, cell models for NCCs, such as human NCCs that originated from patient subcutaneous fat, have been used to investigate Parkinson's disease17. The O9-1 NCC line, which was originally derived from mass cultures of endogenous NCCs isolated from E8.5 mouse embryos18, is a powerful cell model for studying NCCs. Importantly, under non-differentiating culture conditions, O9-1 cells are multipotent stem-like NCCs. However, under varying culture conditions, O9-1 cells can be differentiated into distinguished cell types, such as smooth muscle cells, osteoblasts, chondrocytes, and glial cells18. Given these properties, O9-1 cells have been broadly used for NCC-related studies, such as investigating the molecular mechanism of cranial-facial defects19,20.
Here, detailed protocols are provided for maintaining O9-1 cells, differentiating O9-1 cells into different cell types, and manipulating O9-1 cells by performing gene loss-of-function studies with CRISPR-Cas9 genome editing and small interfering RNA (siRNA)-mediated knockdown technologies. As a representative example, the use of O9-1 cells to study Yap and Taz loss-of-function is described. Yap and Taz are the downstream effectors of the Hippo signaling pathway, which plays a critical role in the cell proliferation, differentiation, and apoptosis. The Hippo pathway has also been shown to be important in the development and homeostasis of several different tissues and organs, as well as in the pathogenesis of different diseases20,21,22,23,24,25,26,27,28. The core components of Hippo signaling include the tumor suppressor sterile 20-like kinases Mst1/2, WW domain-containing Salvador scaffold protein, and the large tumor suppressor homolog (Lats1/2) kinases. Hippo signaling inhibits Yap and Taz activity and promotes their degradation in the cytoplasm. Without repression from Hippo, Yap and Taz can translocate into nuclei and function as transcriptional co-activators. We recently showed that specifically inactivating the Hippo signaling effectors Yap and Taz in mouse NCCs by using the Wnt1cre and Wnt1Cre2SOR drivers resulted in embryonic lethality at E10.5 with severe craniofacial defects20. We have also performed studies using O9-1 cells to investigate the role of Yap and Taz in NCCs. To study Yap and Taz function in NCC proliferation and differentiation, Yap and Taz knockdown cells were generated in O9-1 cells by using siRNA, and Yap knockout cells were generated by using CRISPR-Cas9 genome editing. The same gene loss-of-function strategies can be applied to different target genes in other pathways. In addition, gain-of-function studies and transfection assays can also be applied to O9-1 cells to study gene function and regulation. The protocols described here are intended to be used by investigators as guides for culturing O9-1 cells to maintain multipotent stemness, for differentiating O9-1 cells into other cell types under different culture conditions, and for studying gene function and the molecular mechanisms of NCCs.
Protocol
1. Preparation Before O9-1 Cell Culture
NOTE: Basal media used for O9-1 cell culture must have been conditioned by Sandos inbred mice thioguanine/ouabain-resistant (STO) mouse fibroblast cells; therefore, STO cells need to be obtained and prepared as described below before starting O9-1 cell culture.
- Active STO cell culture
- Prepare media for culturing active STO cells by making complete Dulbecco's modified Eagle's media (DMEM) with 7% fetal bovine serum (FBS) (embryonic stem cell culture grade), 100 U/mL penicillin, 100 µg/mL streptomycin, and 2 mM L-glutamine (final concentrations are indicated). NOTE: STO medium is used for the cultivation of active STO feeder cells. Penicillin, streptomycin, and L-glutamine may form precipitation in storage; dissolve completely by pipetting before use.
- Coat a standard 100 mm cell culture plate with 0.1% gelatin for 30 min at room temperature. After the incubation period, aspirate the extra gelatin. Use the plate immediately after coating. NOTE: Alternatively, cover the plate with STO media and leave the plate in a humidified incubator to avoid the drying of the plate (this is meant only for short-term storage and not for storing precoated plates).
- Thaw the STO cells rapidly in a 37 °C water bath; wipe the cryovial with 70% ethanol before opening, and immediately transfer the whole vial of cells to a centrifuge tube.
- Add the STO media dropwise to the centrifuge tube; the ratio by volume of the cells to media is 1:10.
- Centrifuge the cells at 180 x g for 3 min at RT.
- Mix by gentle pipetting and transfer cells to the gelatin-coated plate.
- Allow STO cells to grow for 24 h in a standard incubator (37 °C, 5% CO2).
- Change the medium (only after the cells adhere) 24 h after seeding and discard the old medium.
- Check STO cells every day under a microscope and passage at 80% confluency.
- Before starting STO cell passaging, coat plates with 0.1% gelatin (gelatin volume equals half of growth media volume for that vessel) for 30 min at room temperature.
- To passage STO cells, aspirate growth media and wash cells with phosphate-buffered saline (PBS) twice (add PBS in an equal volume of growth media).
- Aspirate PBS and add 0.25% trypsin-EDTA (adjust volume according to the vessel size); then, incubate at 37 °C for 5 min. NOTE: For a 100 mm plate, use 10 mL of growth media and 3 mL of trypsin solution. For a 150 mm plate, use 20 mL of growth media and 8 mL of trypsin solution. The volume of wash buffer used is equal to the volume of growth media.
- Inactivate trypsin with an equal volume of STO media. Seed STO cells to new plates with a ratio from 1:3 to 1:10. NOTE: A common expanding scheme is to expand from one cryovial into one 100 mm plate, then to one 150 mm plate, then to four 150 mm plates, and finally to 16 150 mm plates.
- Inactivation and freezing of STO cells
- For preparing 2x freezing media, add the following to STO media (final concentrations are indicated): 20% FBS, 20% dimethyl sulfoxide (DMSO).
- After mixing, filter sterilize the 2x freezing media (pore size 0.22 µm) and store it at 4 °C until use. Make 2x freezing media the same day of use and discard unused freezing media.
- Prepare mitomycin C solution in PBS at a concentration of 0.5 mg/mL. To dissolve mitomycin C in PBS, tape the bottle on a vortexer and vortex at a low setting for approximately 45 min to dissolve the particles completely. NOTE: Mitomycin C is light sensitive and hard to dissolve. Caution: Mitomycin C is acutely toxic and may cause cancer. Handle only with proper personal protective equipment.
- Inactivate STO cells by adding a final concentration of 0.01 mg/mL mitomycin C to the existing media. Incubate for 2 h inside a standard incubator (37 °C, 5% CO2).
- Wash plates (150 mm) with 20 mL PBS twice after inactivation with mitomycin C.
- Aspirate the PBS solution, dissociate cells by using 8 mL of 0.25% trypsin-EDTA, and incubate for 5 min inside a standard humidified incubator (37 °C, 5% CO2).
- Inactivate trypsin with 8 mL of STO media.
- Transfer the cell solution to a centrifuge tube. Combine more than one plate into one centrifuge tube to save time. Centrifuge for 5 min at 180 x g.
- Aspirate the supernatant and resuspend the cell pellet in 5 mL PBS.
- Use 10 µL of the cell solution to count cells by using a hemocytometer. Count the cells in the central gridded square and multiply the number obtained by 104 to determine the number of cells per 1 mL. Count twice and average the two numbers to obtain the final estimation of the number of cells per milliliter.
- Centrifuge cells for 5 min at 180 x g.
- Aspirate PBS and add 1:1 (by volume) STO media and 2x freezing media to the cell pellet. Adjust the volume for a final cell concentration of 4 x 106 cell/mL (this amount is good for one 100 mm plate). NOTE: For example, from four 150-mm plates yielding a total of 60 x 106 cells, freeze 4 x 106 cells per cryovial, with each cryovial containing 1 mL of cell solution. Four plates yield approximately 15 cryovials (60/4=15). Add 7.5 mL of STO media and 7.5 mL of 2x freezing media to the cell pellet, resuspend, and aliquot 1 mL into each cryovial.
- Resuspend by slowly pipetting freezing media with cell pellet. Transfer 1 mL of cell solution to each cryovial and label accordingly.
- Place cryovials inside a lidded polystyrene box (this provides a slow cooling rate, which improves cell survival). Transfer the box to a -80 °C freezer for at least 24 h before transferring the cryovial to liquid nitrogen. NOTE: For best results, minimize the cell counting time, as well as the time between adding 2x freezing media to cells and transferring vials to -80 °C.
2. O9-1 Cell Culture
Prepare basal media for O9-1 cell culture by adding the following in DMEM (final concentrations are indicated): 15% FBS, 0.1 mM minimum essential media (MEM) nonessential amino acids, 1 mM sodium pyruvate, 55 μM beta-mercaptoethanol, 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine, 103 units/mL leukemia inhibitory factor (LIF; added immediately before use, do not add to stock bottle), and 25 ng/mL fibroblast growth factor-basic (bFGF; added immediately before use, do not add to stock bottle).
Filter sterilize the basal media (pore size 0.22 µm) and wrap in foil to protect from light.
- Collect conditioned basal media from inactivated STO cells. NOTE: Proceed only after obtaining inactivated STO cells. O9-1 basal media collected from STO cell plates is hereafter referred to as conditioned basal media.
- Thaw inactivated STO cells rapidly as described above (one cryovial containing 4 x 106 cells is good for one 100 mm plate and yields approximately 100 mL conditioned basal media after 10 days of collection). Seed inactivated STO cells to the gelatin-coated plate by using STO media. Allow cells to attach overnight in a standard humidified incubator (37 °C, 5% CO2).
- 24 h after thawing STO cells, discard the existing STO media and replace with the basal media. NOTE: Do not add LIF and bFGF to inactivated STO cell culture dishes; only add to O9-1 cell culture dishes.
- Every 24 h, change media by using O9-1 basal media and collect the basal media from STO cell plates into a bottle. Wrap the bottle containing conditioned basal media in foil to protect from light. Store all the collected media at 4 °C. NOTE: Because the cells were inactivated, they may not appear as healthy as they did after several days of collection; this is to be expected.
- Optionally to check for contamination, collect the basal media for each collection day in different tubes (instead of one bottle) wrapped in foil. Using a 24-well plate, place 1 mL of media from each day into each well and incubate overnight in a standard incubator to check for bacterial contamination under the microscope. NOTE: The media remains a red color if there is no contamination but changes to yellow if bacterial contamination is present.
- After collecting conditioned basal media for 10 days, discard the STO cells. Combine (if applicable) all collected conditional basal media and filter sterilize (pore size 0.22 µm) into a sterile bottle. Wrap the bottle in foil to protect from light. Keep the filtered conditioned basal media at 4 °C for a maximum of one month from the filter date.
3. Maintaining O9-1 Cells
NOTE: Working O9-1 basal media is filter sterilized conditioned basal media, to which LIF (final concentration 103 units/mL) and bFGF (final concentration 25 ng/mL) are added immediately to the cell culture dish before use. This media needs to be protected from light and stored at 4 °C.
- Recovery of O9-1 cells
- Slowly thaw the stock bottle of the basement membrane matrix (e.g., Matrigel) overnight at 4 °C to prepare aliquots.
- Add 5 mL of DMEM with 10% FBS to 5 mL of the stock basement membrane matrix membrane. Mix well by pipetting with cold pipette tips stored at -20 °C. Keep the original bottle and aliquot tubes on ice. Freeze aliquots in different volumes (0.5 mL or 1 mL aliquots) depending on the experiment needs. Avoid freeze-thawing basement membrane matrix multiple times. NOTE: Basement membrane matrix polymerizes rapidly at room temperature; use cold pipette tips and keep tubes cold when working to avoid unintentional polymerization.
- Thaw an aliquot of the basement membrane matrix at 4 °C or on ice 2 h before starting the experiment. Do not store the matrix at 4 °C for an extended time.
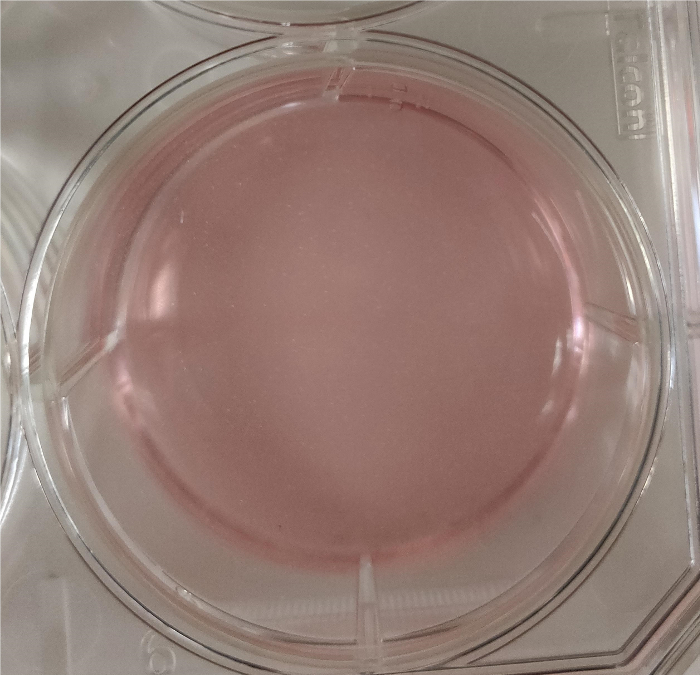
- Use filter-sterilized DMEM with 10% FBS to dilute the basement membrane matrix to a final concentration of 0.5 mg/mL to coat the plates.Coat plates with the basement membrane matrix (0.5 mg/mL) at room temperature for 1 h. Ensure that it fully covers the plate (as shown in Figure 1).
- Tilt the plate when aspirating the basement membrane matrix to avoid touching the coated surface; dry plates at 37 °C for 30 min. Do not over-dry coated plates. NOTE: Proceed only when all media and reagents are ready to use (conditioned basal media, sterile-filtered 10% FBS in DMEM, LIF, and bFGF).
- Recover O9-1 cells rapidly by placing the cryovial in a 37 °C water bath; slowly swirl the vial until the solution completely turns to liquid and proceed immediately to the next step.
- Transfer all the cell solution from the cryovial (approximately 1 mL) to a 15 mL centrifuge tube. Add 5 volumes of DMEM with 10% FBS (filter sterilized with a filter pore size of 0.22 µm) and resuspend.
- Centrifuge for 3 min at 180 x g. Aspirate the supernatant without disturbing the cell pellet. Resuspend the cell pellet (by tapping) in conditioned basal media supplemented with LIF and bFGF. Count the cells by using a hemocytometer. NOTE: Conditioned basal media supplemented with LIF and bFGF is critical to maintain the multipotency of the O9-1 cell line. Use care to add the correct media to avoid unintentional cell differentiation.
- Seed cells from 10,000 to 15,000 cell/cm2 to a 6-well plate. Allow cells to attach and grow in a standard cell culture incubator (37 °C, 5% CO2) overnight before changing media.
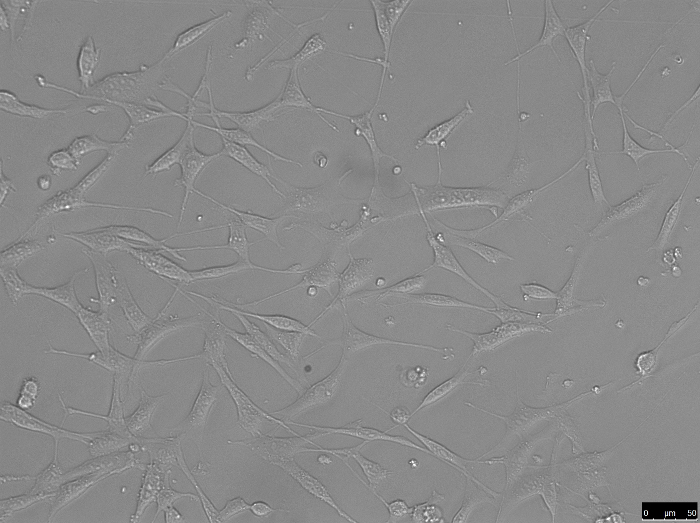
- Ensure that the cells are attached before proceeding to change media (healthy attached cells look like those shown in Figure 2).
- Always change culture media the next day after the recovery of O9-1 cells (use conditioned basal media supplemented with LIF and bFGF).
- Passage of O9-1 cells
- When O9-1 cells reach 80% confluency, thaw the basement membrane matrix and prepare a basement membrane matrix-coated plate as described above. Add conditioned basal media supplemented with LIF and bFGF to the vessel. Alternatively, add DMEM without FBS to keep the basement membrane matrix from drying and store inside a standard humidified incubator for no more than 3 days.
- Rinse the O9-1-containing wells (6-well plate) with 2 mL of Dulbecco's phosphate-buffered saline (DPBS) twice and gently pipette to avoid losing cells.
- Dissociate cells with 1 mL of 0.05% trypsin-EDTA at 37 °C for 3 min. Neutralize trypsin with an equal amount of DMEM with 10% FBS. Repeatedly pipette the liquid over the whole surface of the well to dissociate as many cells from the plate as possible. NOTE: Trypsin concentration is 0.05% instead of 0.25%. Use DPBS to dilute from the stock bottle.
- Avoid generating bubbles when dissociating cells from the plate.
- Transfer all cell solution to a centrifuge tube and centrifuge for 3 min at 180 x g. Aspirate the supernatant without disturbing the cell pellet. Resuspend the cell pellet in the centrifuge tube by gentle pipetting in conditioned basal media supplemented with LIF and bFGF.
- Use a hemocytometer to count the total cell number as described above. Seed cells (10,000 to 15,000 cells/cm2) to the coated plate prepared as described in steps 3.1.4 to 3.1.8. One well of a 6-well plate would require 100,000 cells to seed.
- Allow cells to attach and grow in a standard cell culture incubator (37 °C, 5% CO2). Always change culture media the next day after passaging O9-1 cells.
- Freezing of O9-1 cells
- To prepare 2x freezing media for O9-1 cells, dilute the following in DMEM (final concentrations are indicated): 40% FBS and 20% DMSO.
- Filter sterilize 2x freezing media and keep it at 4 °C. The 2x freezing media must be made on the day it is used. Discard all unused freezing media. NOTE: Freeze O9-1 cells at 80% confluency.
- Rinse wells with DPBS twice; pipette gently to avoid losing cells.
- Dissociate cells with 0.05% trypsin-EDTA at 37 °C for 3 min, then neutralize trypsin with an equal amount of 10% FBS in DMEM. Repeatedly pipette the liquid over the whole surface of the well to dissociate as many cells from the plate as possible.
- Transfer all plate contents to a centrifuge tube and centrifuge for 3 min at 180 x g. Aspirate the supernatant and add 2 mL of PBS and resuspend by tapping.
- Use 10 µL of cell solution to count cells by using a hemocytometer as described above.
- Centrifuge cell solution for 3 min at 180 x g. Aspirate the supernatant and adjust the amount of basal media needed; then, add an equal amount of 2x freezing media. NOTE: Cells are frozen at a concentration of 1 x 106 cells/mL (1 mL per cryovial).
- Transfer cells to cryovials and label accordingly. Place cryovials inside a lidded polystyrene box for a slow cooling rate at -80 °C for at least 24 h before transferring to liquid nitrogen. NOTE: For best results, minimize the cell counting time and the time between adding 2x freezing media and transferring vials to -80 °C.
4. Manipulation of O9-1 cells
- Performing siRNA knockdown in O9-1 cells
- Thaw and coat a 24-well plate with basement membrane matrix as described above (steps 3.1.4–3.1.8) before starting the experiment. Recover and seed O9-1 cells, allowing them to grow to 60% to 80% confluency.
- Dilute liposomes in serum-free media according to the appropriate well volume. Dilute siRNA in serum-free media to the final. Add diluted siRNA to diluted liposomes in a ratio recommended by the manufacturer. Mix well by pipetting and incubate. NOTE: Follow the manufacturer's guide on the volume used, time and temperature of incubation.
- Add an appropriate volume of siRNA-lipid complex to cells according to the manufacturer's recommendations.
- Incubate cells for 24 h in a standard cell culture incubator (37 °C, 5% CO2). Perform downstream steps as appropriate (extract RNA, extract proteins, staining, etc.) NOTE: siRNA knockdown times and concentrations can vary depending on the individual experiment.
- Performing gene knockout in O9-1 cells by using CRISPR-Cas9 genome editing NOTE: Refer to the previously published protocol for using CRISPR-Cas9 in mammalian cell lines29. Provided here is a simplified description of CRISPR-Cas9 genome editing and related steps.
- Obtain sgRNA sequences by using a sgRNA design tool.
- Ligate the sgRNA to pSpCas9(bb)-2a-GFP vector30.
- Transfect O9-1 cells with the vector by using a standard lipofection protocol according to the manufacturer's recommendations.
- Incubate cells in a standard cell culture incubator at 37 °C with 5% CO2 for 24 h.
- Perform fluorescence-activated cell sorting (FACS) based on GFP/7-AAD. Seed GFP+ single cells into 96-well plates.
- Grow the cells in the incubator for an additional 4 days. Discard all wells that have more than one colony.
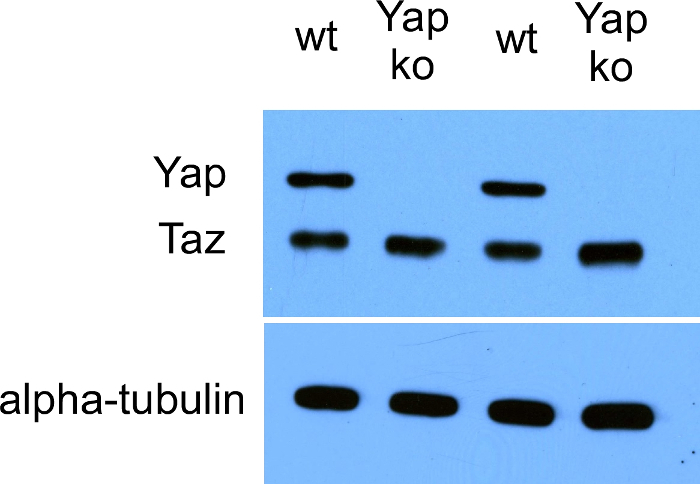
- Perform PCR with primers designed for detecting the deletion. After PCR, run PCR products on an agarose gel to evaluate the band size. Validate the knockout efficiency achieved by using CRISPR-Cas9 editingby performing Western blotting (an example of Yap knockout results is shown in Figure 3).
5. O9-1 cell differentiation
- Differentiation of O9-1 cells into osteoblasts
- To prepare osteogenic differentiation media, dilute the following in alpha-MEM (final concentrations are indicated): 0.1 μM dexamethasone, 100 ng/mL bone morphogenetic protein 2 (BMP2), 50 µg/mL ascorbic acid, 10 mM b-glycerophosphate, 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin.
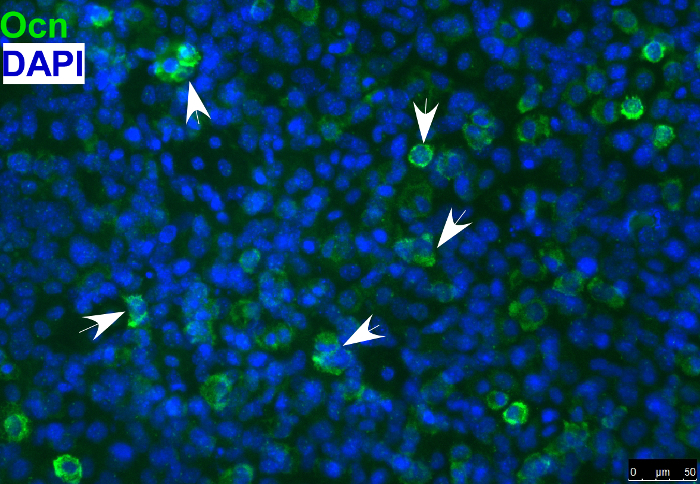
- Detect the osteoblast marker, osteocalcin, in differentiated osteoblasts by immunostaining as shown in Figure 4. NOTE: Osteogenic differentiation can also be evaluated by using other markers of osteoblasts or with Alizarin red staining or alkaline phosphatase staining.
- Differentiation of O9-1 cells into chondrocytes
- To prepare chondrocyte differentiation media, dilute the following in alpha-MEM (final concentrations are indicated): 5% fetal calf serum (FCS), 1% insulin-transferrin-selenium (ITS), 100 U/mL penicillin, 100 μg/mL streptomycin, 10 ng/mL transforming growth factor beta (TGF-b3), 50 μg/mL ascorbic acid, 10 ng/mL BMP2, 0.1 μM dexamethasone, and 1 mM sodium pyruvate.
- First culture a monolayer of O9-1 cells with osteogenic media for 3 days and then trypsinize and culture as a micromass format in chondrocyte differentiation media for an additional 7 days.
- Asses chondrogenic differentiation with Alcian blue staining or markers of chondrocytes.
- Differentiation of O9-1 cells into smooth muscle cells
- To prepare smooth muscle cell differentiation media, dilute the following in DMEM (final concentrations are indicated): 100 U/mL penicillin, 100 µg/mL streptomycin, and 10% FBS.
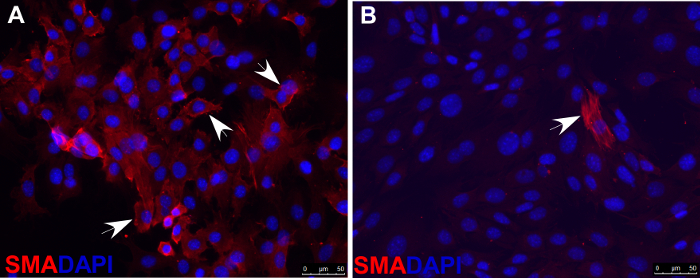
- Asses smooth muscle cell differentiation with immunofluorescence staining by using antibodies against markers of smooth muscle cells, such as smooth muscle actin (SMA), shown as an example in Figure 5.
- Differentiation of O9-1 cells into glial cells
- To prepare glial cell differentiation media, dilute the following in DMEM/F12 (final concentrations are indicated): 1x B-27 supplement, 2 mM L-glutamine, 50 ng/mL BMP2, 100 U/mL penicillin, 100 μg/mL streptomycin, 50 ng/mL LIF, and 1% heat-inactivated FBS.
- Evaluate glial cell differentiation by performing immunofluorescence staining with antibodies against glial cell markers such as fatty acid binding protein 7 (FABP7).
Representative Results
The goal of our knockdown and knockout experiments was to study the effects of Yap and Taz loss-of-function in O9-1 cells. Before the knockdown and knockout experiments, we have to make sure that prepare for basal media and culture O9-1 cells as described above (for example, basement membrane matrix needs to cover the whole plate as shown in Figure 1, and O9-1 cells recovered from liquid nitrogen as shown in Figure 2). We performed knockdown experiments as described above in which Yap and Taz were simultaneously knocked down. Equal amounts of Yap siRNA and Taz siRNA were added to the final volume recommended by the manufacturer. To the control plate, an equal volume of nontargeting siRNA was added.
A Yap-null O9-1 cell line was generated by deleting exon 3 of Yap by using CRISPR/Cas9 genome editing, as described by Wang et al.20. We performed knockout experiments by using CRISPR-Cas9 genome editing as described above and in Wang et al.20. We used the following sgRNA sequences, which flanked exon 3 of Yap: 5′-caccgtggattacgtgggtatgtt-3′ (sgRNA1-forward), 5′- aaacaacatacccacgtaatccac-3′ (sgRNA1-reverse), 5′-caccgagatggtctaatgtagtga-3′ (sgRNA2-forward), 5′-aaactcactacattagaccatctc-3′ (sgRNA2-reverse)
CACC was added to the forward strand, and AAAC was added to the reverse strand. G was added at the 5' end of the forward strand because the oligo did not begin with G, and C was added to the 3' end of the reverse strand. During the CRISPR-Cas9 genome editing steps described above and in Wang et al.20, cells were in conditioned basal media for O9-1 cells supplemented with LIF and bFGF to avoid unwanted differentiation. The following PCR primers were used for detecting the Yap deletion: 5′-AAAACAGTCTCCACTACCCCTT-3′ (forward) and 5′-GGCCATCATAGATCCTGGACG-3′ (reverse). The position of the sgRNAs is from base #7973399 to #7974433 on mouse chromosome 9. The PCR primer position on the chromosome is from base #7973306 to #7974478. After PCR, the PCR band for the Yap knockout with exon 3 deleted appeared as a 138-bp band on an agarose gel; wild-type Yap appeared as an 1173-bp band. The efficiency of Yap knockout by using CRISPR-Cas9 genome editing was validated with Western blotting, as shown in Figure 3.
As described above, O9-1 cells can be cultured in specific differentiation-inducing media to promote differentiation into particular cell types. Evaluation of differentiation can be done by using various approaches, such as quantitative PCR or immunostaining of specific cell markers. As an example, Figure 4 shows O9-1 cells that were cultured in osteoblast-inducing media and differentiated into osteoblasts, which were evaluated by immunostaining cells with the antibody against osteocalcin (Ocn, an osteoblast marker). Combined with the gene knockdown and knockout experiments described above, O9-1 cells can be broadly used for gene function studies and phenotypic analyses. As an example, both wild-type and Yap-null O9-1 cells were cultured in smooth muscle cell differentiation media and evaluated by immunostaining cells with the antibody against smooth muscle actin (SMA, a smooth muscle cell marker). Most wild-type O9-1 cells gave rise to SMA-positive smooth muscle cells (Figure 5A), whereas, Yap-null O9-1 cells failed to differentiate into SMA-positive smooth muscle cells (Figure 5B), which indicated that Yap plays a critical role in the differentiation of NCCs into smooth muscle cells.
Figure 1: Matrigel fully covering a 35 mm plate. Please click here to view a larger version of this figure.
Figure 2: O9-1 cells 24 h after recovery from liquid nitrogen. Please click here to view a larger version of this figure.
Figure 3: Western blot data showing the efficient knockout (ko) of Yap in O9-1 cells by using CRISPR-Cas9 genome editing. wt: wild-type. Please click here to view a larger version of this figure.
Figure 4: Immunofluorescence staining of osteoblast marker osteocalcin (Ocn) indicating that O9-1 cells gave rise to osteoblast cells under osteogenic differentiation conditions. Cells were stained with osteoblast marker Ocn antibody (green), and nuclei were stained with DAPI (blue). Arrows indicate Ocn-positive cells. Osteocalcin (Ocn, an osteoblast marker); DAPI (′,6-diamidino-2-phenylindole). Please click here to view a larger version of this figure.
Figure 5. Immunofluorescence staining of smooth muscle actin (SMA) indicating that, under smooth muscle cell differentiation conditions, most wild-type O9-1 cells gave rise to smooth muscle cells (A), whereas Yap-null O9-1 cells failed to differentiate into smooth muscle cells (B). Cells were stained with SMA antibody (red), and nuclei were stained with DAPI (blue). Arrows indicate SMA-positive cells. Please click here to view a larger version of this figure.
Discussion
The NCC is a versatile and key contributor to different tissues and organs during embryonic morphogenesis.The O9-1 cell line maintains its potential to differentiate into many different cell types and mimics the in vivo characteristics of NCCs, making it a useful in vitro tool for studying gene function and molecular regulation in NCCs. The different status of O9-1 cells may correspond to different neural crest progeny in vivo, depending on the culture conditions of O9-1 cells. O9-1 cells can be maintained in the multipotent state when cultured under non-differentiating culture conditions, similar to regular stem cell culture. The non-differentiating O9-1 cells may correspond to pre-migratory and migratory multipotent stem-like NCCs in vivo. Furthermore, O9-1 cells can differentiate into many different cell types under specific differentiation culture conditions, which is an important advantage for studying gene function and regulation during the differentiation of NCCs from multipotent stem cells into a specific differentiated cell type. The differentiating O9-1 cells may correspond to post-migratory neural crest progeny in vivo, which have a different cell fate to differentiate into various cell types. Depending on the research interest, O9-1 cells can be manipulated and cultured differently to complement in vivo NCC studies with animal models.
There are various ways to manipulate O9-1 cells to study NCC events, including gene loss-of-function and gain-of-function. Here, examples are presented in which siRNA knockdown and CRISPR-Cas 9 gene editing experiments were performed in O9-1 cells for Hippo pathway effector Yap loss-of-function studies20. With the combined use of a mouse model and O9-1 cells, our study indicated that Yap plays a critical role in regulating NCC proliferation and differentiation into smooth muscle cells20. Other studies in different models have also indicated an important role for Yap in NCCs. Yap was shown to be required for mouse neural crest smooth muscle differentiation31 and to enhance human NCC fate and migration32. Furthermore, Yap is expressed during early Xenopus development33. The efficiency of gene knockdown and knockout in O9-1 cells, combined with the convenience of performing other downstream molecular techniques such as quantitative PCR (qPCR), Western blotting, and immunostaining supports that the O9-1 cell line is an excellent NCC model that can be easily manipulated in vitro. In addition to the loss-of-function studies we have described above and previously20, various other applications of O9-1 cells have been described for studying NCCs. O9-1 cells can be used as an efficient alternative for experiments that require a relatively large amount of tissue, such as chromatin immunoprecipitation sequencing (ChIP-seq). However, many in vivo cell events such as NCC migration rely on complex tissue-to-tissue interactions, so it remains unclear how well O9-1 cells can accurately mimic in vivo NCC migration. Because of the limitations of in vitro studies with O9-1 cells, it is recommended to validate observations made with O9-1 cells in vivo by using animal models.
When using the O9-1 cell line to study NCCs in vitro, maintaining the multipotency of the cells during routine culture is critical. The multipotency of O9-1 cells can be tested at the beginning of culture by evaluating the expression of NCC marker genes such as AP-2a and Sox9. Similarly, the differentiation ability of O9-1 cells can be tested by differentiating them into specific cell types. To avoid unwanted differentiation during the routine culturing of O9-1 cells, the experimental procedures must be carried out in a manner consistent with the established protocol described here. Careful preparation of the conditioned basal media and the fresh addition of the correct concentrations of LIF and bFGF are critical for maintaining the multipotency of O9-1 cells. In addition, the experimental timeline needs to be carefully planned, given that culturing O9-1 cells requires first preparing inactive feeder STO cells and collecting conditioned basal media, which is good for only about one month. Previous studies have shown that Basement membrane matrix is important for maintaining the differentiation potential of muscle and neural precursor cells, as well as mesenchymal stem cells34,35,36. In the context of the protocol described here, Basement membrane matrix provides a platform for O9-1 cells to attach and helps maintain differentiation potential. Therefore, Basement membrane matrix is an important factor that may affect experimental results and their consistency when repeated. We recommend preparing Basement membrane matrix strictly according to the protocol described above and avoiding multiple freeze-thaw cycles of the Basement membrane matrix. Also, when recovering O9-1 cells from the frozen state to culture, the process of moving the cells from liquid nitrogen to a 37 °C water bath needs to be done quickly, avoiding any vortexing during the entire process. In addition, the overgrowth of O9-1 cells also needs to be avoided to maintain the multipotency of O9-1 cells. When passaging O9-1 cells, ensure that the trypsin concentration is diluted to 0.05% instead of 0.25% and that cells appear properly dissociated. Repeatedly pipetting very gently to achieve a single-cell suspension is necessary for avoiding the formation of aggregated cells. In general, the entire process of handling the O9-1 cell line needs to be done gently and carefully.
In summary, the O9-1 cell line is a very useful tool for studying NCCs in vitro, especially when used as a method complementary to in vivo studies of NCCs. O9-1 cells have obvious advantages such as the ease with which they can be accessed and the ease with which sufficient cell numbers can be obtained for experiments. Given the differentiation capabilities of O9-1 cells, they have great potential for use in a wide range of applications in various fields of research. O9-1 cells can be conveniently used for applications such as quick drug testing and screening, ChIP-seq, transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) and RNA sequencing (RNA-Seq), gene gain-of-function and loss-of-function studies, as well as signaling regulation studies. The use of a standardized and complete protocol for the handling of O9-1 cells will facilitate the reproducibility of studies in which they are used.
Disclosures
The authors have nothing to disclose.
Acknowledgments
Nicole Stancel, Ph.D., ELS, of Scientific Publications at the Texas Heart Institute, provided editorial support. We also thank the following funding sources: the American Heart Association's National Center Scientist Development Grant (14SDG19840000 to J. Wang), the 2014 Lawrence Research Award from the Rolanette and Berdon Lawrence Bone Disease Program of Texas (to J. Wang), and the National Institutes of Health (DE026561 and DE025873 to J. Wang, DE016320 and DE019650 to R. Maxson).
References
- Achilleos A, Trainor PA. Neural crest stem cells: discovery, properties and potential for therapy. Cell Research. 2012;22(2):288–304. doi: 10.1038/cr.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Douarin NM, Dupin E. Multipotentiality of the neural crest. Current Opinion in Genetics and Development. 2003;13(5):529–536. doi: 10.1016/j.gde.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Santagati F, Rijli FM. Cranial neural crest and the building of the vertebrate head. Nature Reviews. Neuroscience. 2003;4(10):806–818. doi: 10.1038/nrn1221. [DOI] [PubMed] [Google Scholar]
- Cordero DR, et al. Cranial neural crest cells on the move: their roles in craniofacial development. American Journal of Medical Genetics. Part A. 2011;155(2):270–279. doi: 10.1002/ajmg.a.33702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trainor PA. Craniofacial birth defects: The role of neural crest cells in the etiology and pathogenesis of Treacher Collins syndrome and the potential for prevention. American Journal of Medical Genetics. Part A. 2010;152(12):2984–2994. doi: 10.1002/ajmg.a.33454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner ME, Simoes-Costa M. The neural crest migrating into the twenty-first century. Current Topics in Developmental Biology. 2016. pp. 115–134. [DOI] [PMC free article] [PubMed]
- Zurkirchen L, Sommer L. Quo vadis: tracing the fate of neural crest cells. Current Opinion in Neurobiology. 2017;47:16–23. doi: 10.1016/j.conb.2017.07.001. [DOI] [PubMed] [Google Scholar]
- Sauka-Spengler T, Bronner-Fraser M. A gene regulatory network orchestrates neural crest formation. Nature Reviews. Molecular Cell Biology. 2008;9(7):557–568. doi: 10.1038/nrm2428. [DOI] [PubMed] [Google Scholar]
- Knecht AK, Bronner-Fraser M. Induction of the neural crest: a multigene process. Nature Reviews. Genetics. 2002;3(6):453–461. doi: 10.1038/nrg819. [DOI] [PubMed] [Google Scholar]
- Steventon B, Carmona-Fontaine C, Mayor R. Genetic network during neural crest induction: from cell specification to cell survival. Seminars in Cell and Developmental Biology. 2005;16(6):647–654. doi: 10.1016/j.semcdb.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Raible DW. Development of the neural crest: achieving specificity in regulatory pathways. Current Opinion in Cell Biology. 2006;18(6):698–703. doi: 10.1016/j.ceb.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Dupin E, Sommer L. Neural crest progenitors and stem cells: from early development to adulthood. Developmental Biology. 2012;366(1):83–95. doi: 10.1016/j.ydbio.2012.02.035. [DOI] [PubMed] [Google Scholar]
- Henion PD, Weston JA. Timing and pattern of cell fate restrictions in the neural crest lineage. Development. 1997;124(21):4351–4359. doi: 10.1242/dev.124.21.4351. [DOI] [PubMed] [Google Scholar]
- Harris ML, Erickson CA. Lineage specification in neural crest cell pathfinding. Developmental Dynamics. 2007;236(1):1–19. doi: 10.1002/dvdy.20919. [DOI] [PubMed] [Google Scholar]
- Luo R, Gao J, Wehrle-Haller B, Henion PD. Molecular identification of distinct neurogenic and melanogenic neural crest sublineages. Development. 2003;130(2):321–330. doi: 10.1242/dev.00213. [DOI] [PubMed] [Google Scholar]
- Betters E, Liu Y, Kjaeldgaard A, Sundstrom E, Garcia-Castro MI. Analysis of early human neural crest development. Developmental Biology. 2010;344(2):578–592. doi: 10.1016/j.ydbio.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SY, Beavan M, Chau KY, Taanman JW, Schapira AHV. A human neural crest stem cell-derived dopaminergic neuronal model recapitulates biochemical abnormalities in GBA1 mutation carriers. Stem Cell Reports. 2017;8(3):728–742. doi: 10.1016/j.stemcr.2017.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii M, et al. A stable cranial neural crest cell line from mouse. Stem Cells and Development. 2012;21(17):3069–3080. doi: 10.1089/scd.2012.0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yumoto K, et al. TGF-beta-activated kinase 1 (Tak1) mediates agonist-induced Smad activation and linker region phosphorylation in embryonic craniofacial neural crest-derived cells. Journal of Biological Chemistry. 2013;288(19):13467–13480. doi: 10.1074/jbc.M112.431775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, et al. Yap and Taz play a crucial role in neural crest-derived craniofacial development. Development. 2016;143(3):504–515. doi: 10.1242/dev.126920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heallen T, et al. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332(6028):458–461. doi: 10.1126/science.1199010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Martin JF. Hippo pathway: An emerging regulator of craniofacial and dental development. Journal of Dental Research. 2017;96(11):1229–1237. doi: 10.1177/0022034517719886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D. The hippo signaling pathway in development and cancer. Developmental Cell. 2010;19(4):491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Leach J, Wang J, Martin JF. Hippo/Yap signaling in cardiac development and regeneration. Current Treatment Options in Cardiovascular Medicine. 2016;18(6):38. doi: 10.1007/s11936-016-0461-y. [DOI] [PubMed] [Google Scholar]
- Yu FX, Meng Z, Plouffe SW, Guan KL. Hippo pathway regulation of gastrointestinal tissues. Annual Review of Physiology. 2015;77:201–227. doi: 10.1146/annurev-physiol-021014-071733. [DOI] [PubMed] [Google Scholar]
- Fu V, Plouffe SW, Guan KL. The Hippo pathway in organ development, homeostasis, and regeneration. Current Opinion in Cell Biology. 2017;49:99–107. doi: 10.1016/j.ceb.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya IM, Halder G. The Hippo pathway in cellular reprogramming and regeneration of different organs. Current Opinion in Cell Biology. 2016;43:62–68. doi: 10.1016/j.ceb.2016.08.004. [DOI] [PubMed] [Google Scholar]
- Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiological Reviews. 2014;94(4):1287–1312. doi: 10.1152/physrev.00005.2014. [DOI] [PubMed] [Google Scholar]
- Bauer DE, Canver MC, Orkin SH. Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. Journal of Visual Experiments : JoVE. 2015. p. e52118. [DOI] [PMC free article] [PubMed]
- Ran FA, et al. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 2013;8(11):2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manderfield LJ, et al. Hippo signaling is required for Notch-dependent smooth muscle differentiation of neural crest. Development. 2015;142(17):2962–2971. doi: 10.1242/dev.125807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindley CJ, et al. The Hippo pathway member YAP enhances human neural crest cell fate and migration. Scientific Reports. 2016;6:23208. doi: 10.1038/srep23208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nejigane S, Haramoto Y, Okuno M, Takahashi S, Asashima M. The transcriptional coactivators Yap and TAZ are expressed during early Xenopus development. International Journal of Developmental Biology. 2011;55(1):121–126. doi: 10.1387/ijdb.103130sn. [DOI] [PubMed] [Google Scholar]
- Grefte S, Vullinghs S, Kuijpers-Jagtman AM, Torensma R, Von den Hoff JW. Matrigel, but not collagen I, maintains the differentiation capacity of muscle derived cells in vitro. Biomedical Materials. 2012;7(5):055004. doi: 10.1088/1748-6041/7/5/055004. [DOI] [PubMed] [Google Scholar]
- Kang BJ, et al. Effect of matrigel on the osteogenic potential of canine adipose tissue-derived mesenchymal stem cells. Journal of Veterinary Medical Science. 2012;74(7):827–836. doi: 10.1292/jvms.11-0484. [DOI] [PubMed] [Google Scholar]
- Uemura M, et al. Matrigel supports survival and neuronal differentiation of grafted embryonic stem cell-derived neural precursor cells. Journal of Neuroscience Research. 2010;88(3):542–551. doi: 10.1002/jnr.22223. [DOI] [PubMed] [Google Scholar]