

Abstract
Probing an individual cell's gene expression enables the identification of cell type and cell state. Single-cell RNA sequencing has emerged as a powerful tool for studying transcriptional profiles of cells, particularly in heterogeneous tissues such as the central nervous system. However, dissociation methods required for single cell sequencing can lead to experimental changes in the gene expression and cell death. Furthermore, these methods are generally restricted to fresh tissue, thus limiting studies on archival and bio-bank material. Single nucleus RNA sequencing (snRNA-Seq) is an appealing alternative for transcriptional studies, given that it accurately identifies cell types, permits the study of tissue that is frozen or difficult to dissociate, and reduces dissociation-induced transcription. Here, we present a high-throughput protocol for rapid isolation of nuclei for downstream snRNA-Seq. This method enables isolation of nuclei from fresh or frozen spinal cord samples and can be combined with two massively parallel droplet encapsulation platforms.
Keywords: Neuroscience, Issue 140, Nuclei, RNA sequencing, snRNA-Seq, massively parallel, spinal cord, sucrose gradient
Introduction
The nervous system is comprised of heterogenous groups of cells that display a diverse array of morphological, biochemical, and electrophysiological properties. While the bulk RNA sequencing has been useful for determining tissue-wide changes in the gene expression under different conditions, it precludes the detection of transcriptional changes at the single-cell level. Recent advances in the single-cell transcriptional analysis have enabled the classification of heterogenous cells into functional groups based on their molecular repertoire and can even be leveraged to detect sets of neurons that had been recently active.1,2,3,4 Over the last ten years, the development of single cell RNA sequencing (scRNA-Seq) has enabled the study of gene expression in individual cells, providing a view into cell-type diversity.5
The emergence of scalable approaches such as massively parallel scRNA-Seq, has provided platforms to sequence heterogeneous tissues, including many regions of the central nervous system.6,7,8,9,10,11,12,13,14,15 However, single cell dissociation methods can lead to the cell death as well as experimental changes in gene expression.16 Recent work has adapted single cell sequencing methods to enable preservation of endogenous transcriptional profiles.1,3,4,17,18,19 These strategies have been particularly suitable for detecting immediate early gene (IEG) expression following sensory stimulus or behavior.3,4 In the future, this strategy could also be used to study dynamic changes in tissues in disease states or in response to stress. Of these methods, single nucleus RNA sequencing (snRNA-Seq) is a promising approach that does not involve stress-inducing cell dissociation and can be used on difficult to dissociate tissue (such as the spinal cord), as well as frozen tissue.4,17,18,19 Adapted from previous nuclei isolation methods,20,21,22,23,25 snRNA-Seq typically utilizes rapid tissue disruption and cell lysis under cold conditions, centrifugation, and separation of nuclei from cellular debris.4 Nuclei can be isolated for the downstream next-generation sequencing on multiple microfluidic droplet encapsulation platforms.4,7,24,25 This method allows for a snapshot of the transcriptional activity of thousands of cells at a moment in time.
There are multiple strategies for releasing nuclei from cells before isolation and sequencing, each with their own advantages and disadvantages. Here, we describe and compare two protocols to enable isolation of nuclei from the adult spinal cord for the downstream massively parallel snRNA-Seq: detergent-mechanical lysis and hypotonic-mechanical lysis. Detergent-mechanical lysis provides complete tissue disruption and a higher final yield of nuclei. Hypotonic mechanical-lysis includes a controllable degree of tissue disruption, providing an opportunity for selecting a balance between the quantity and purity of the final nuclear yield. These approaches provide comparable RNA yield, detected numbers of genes per nucleus, and cell-type profiling and also can both be used successfully for snRNA-Seq.
Protocol
All animal work was performed in accordance with a protocol approved by the National Institute of Neurological Disorders and Stroke Animal Care and Use Committee. Balanced samples of male and female ICR/CD-1 wild-type mice, between 8 and 12 weeks old, were used for all experiments. Mice should be handled in accordance with local Institutional Animal Care and Use Committee guidelines.
1. Preparation of Materials and Buffers
- Prepare all buffers the day of use and pre-chill on ice (see Table 1).
- If using detergent-mechanical lysis, prepare the detergent lysis buffer (> 500 μL per sample), low sucrose buffer (> 6 mL per sample), sucrose density buffer (> 12.5 mL per sample), and the resuspension solution (> 1 mL).
- If using hypotonic-mechanical lysis, prepare the hypotonic lysis buffer (> 5 mL per sample), HEB medium (> 5 mL per sample), low sucrose buffer (> 3 mL per sample), sucrose density buffer (>12.5 mL per sample), and the resuspension solution (> 1 mL).
- Add 25 μL of dithiothreitol (DTT) to 25 mL of the low sucrose buffer and another 25 μL of DTT to 25 mL of the sucrose density gradient buffer just before starting the protocol.
Cover the dissecting surface with aluminum-foil to minimize contamination of the sample with fibers from paper towels or bench protectors, which can clog microfluidic channels used for capturing single nuclei.
Spray dissecting tools and bench space with an RNase decontamination solution. Additionally, spray the inside of the Dounce homogenizer tube (if using detergent-mechanical cell lysis) and Oak Ridge tube with an RNase decontamination solution. Rinse out the Dounce and Oak Ridge tube with ultrapure, RNase-free water.
Pre-chill all collection tubes (50 mL conical, Oak Ridge) and Dounce homogenizer tubes on ice.
Fire polish a series of Pasteur pipettes (if using hypotonic-mechanical cell lysis).
2. Preparation of the Spinal Cord
If using fresh tissue, euthanize the mouse by CO2 inhalation. Following euthanasia, spray the coat of the mouse with 70% ethanol to minimize hair contamination in the sample.
Decapitate the mouse with sharp, RNase-free surgical scissors. Next, gently lifting the abdominal skin with forceps and make an incision along the length of the body to expose the inner organs.
- Eviscerate the mouse by pulling the inner organs from the body cavity using forceps. Do not use paper towels to clean the area or to remove organs as this may introduce contaminants. Using scissors, cut the vertebral column between the L2 and L3 spinal vertebrae. NOTE: With practice, this step can be achieved in less than 30 seconds.
- To eject the spinal cord, fit a 3 mL syringe containing ice-cold PBS with a 25 G ¼ inch needle. Place the tip of the needle into the sacral end of the vertebral column. Use two fingers to pinch the vertebrae to create a tight seal around the tip of the needle and press down on the plunger to eject the spinal cord rostrally. Place the spinal cord in a petri dish with ice-cold PBS.
- At this point, freeze the tissue and store at -80 °C or use immediately for either detergent-mechanical (Step 3) or hypotonic-mechanical (Step 4) lysis.
If using frozen tissue, maintain the tissue on dry ice, proceed to detergent-mechanical (Step 3) or hypotonic-mechanical (Step 4) lysis.
3. Detergent-Mechanical Cell Lysis
Place the lumbar spinal cord in a pre-chilled Dounce homogenizer and add 500 μL pre-chilled detergent lysis buffer. NOTE: A mouse lumbar spinal cord is 325.5 mg ± 63.9 mg standard error of the mean (SEM, N = 4). 50 mg–1.5 g of tissue can be successfully used.
Dounce with 5 strokes of pestle A (‘loose’ pestle), then 5-10 strokes of pestle B (‘tight’ pestle). Avoid lifting the homogenizer out of the lysis solution in between strokes and avoid introducing bubbles.
Place a 40 mm strainer over a pre-chilled 50 mL conical tube and prewet with 1 mL of low sucrose buffer.
Add 1 mL of low sucrose buffer to the Dounce homogenizer containing the crude nuclei in the lysis buffer and mix gently by pipetting 2–3 times.
Pass the crude nuclei prep over the 40 mm strainer into the pre-chilled 50 mL conical tube.
Pass an additional 1 mL low sucrose buffer over the 40 mm strainer, bringing the final volume to 3 mL of the low sucrose buffer and 500 μL of the lysis buffer.
Repeat steps 3.1–3.6 if combining multiple cords, pooling in the same conical tube.
Centrifuge the sample at 3,200 x g for 10 min at 4 °C. Once the centrifugation is complete, decant the supernatant. Proceed to Step 5.
4. Hypotonic-mechanical Cell Lysis
Place the lumbar spinal cord in 5 mL of the hypotonic lysis buffer in a tissue culture dish. Use the blunt end of spring scissors to bisect the spinal cord, then use spring scissors to cut the cord into 3–4 mm pieces, but do not mince. Note: 50 mg–1.5 g of tissue can be successfully used.
Incubate on the ice for 15 min, swirling 2–3 times.
Add 5 mL of HEB medium to dilute the hypotonic lysis buffer.
Triturate the tissue 10 times with a 5 mL serological pipette, or until all of the pieces of the tissue move smoothly through the opening of the pipette.
- Triturate with a series of three fire-polished Pasteur pipettes with progressively narrower diameters (~900–600 mm).
- For each pipette, triturate 5-15 times, allow tissue to settle, remove 1–2 mL of supernatant containing dissociated nuclei and pass over a 40 mm strainer into a pre-chilled 50 mL conical tube.
- After trituration with the smallest-sized Pasteur pipette, ensure that the homogenate flows smoothly through the pipette tip. Pass the remaining solution over the 40 mm strainer into the 50 mL conical tube. NOTE: The total number of triturations can be adjusted as desired. The meninges of the mouse spinal cord will remain, but it is important to triturate any visible chunks of spinal cord. Pass the remaining homogenate over the 40 m strainer. Avoid introducing bubbles during trituration.
Centrifuge the filtered sample at 1,000 x g for 10 min at 4 °C. Once the centrifugation is complete, decant and discard the supernatant. Proceed to Step 5.
5. Homogenization and Sucrose Density Gradient
After either Step 3 or 4, resuspend the pellet using 3 mL of low sucrose buffer. Gently swirl to remove the pellet from the wall to facilitate the resuspension. Let the sample sit on ice for 2 min and transfer the suspension to an Oak Ridge tube.
Using the homogenizer at setting 1, homogenize the nuclei in low sucrose buffer for 15–30 s, keeping the sample on ice. NOTE: Use 15 s if using one lumbar spinal cord or 30 s if using pooled samples or a whole spinal cord.
Using a serological pipette, layer 12.5 mL of density sucrose buffer underneath the low sucrose buffer homogenate, taking care not to create a bubble that disrupts the density layers.
Centrifuge the tubes at 3,200 x g for 20 min at 4 °C.
Once the centrifugation is complete, immediately decant the supernatant in a flicking motion. NOTE: A residual volume (less than 400 μL) of sucrose buffer can be discarded if desired to produce a lower volume and cleaner final sample, but this residual volume does contain nuclei and can be preserved to maximize nuclei yield.
Using 100 μL - 1 mL of resuspension solution, resuspend the nuclei remaining on the wall. Avoid the myelin ‘frown’ that remains with the detergent-based preparation.
Filter the nuclei through a 30–35 mm pore-size strainer and collect in a pre-chilled tube.
Determine the nuclei yield using a hemocytometer to count nuclei under a 10X objective. NOTE: Trypan blue can be added to visualize nuclei, which should appear blue. Note the amount of cellular debris.
Proceed to either Step 6 or 7.
6. Massively Parallel snRNA-Sequencing: Academic Platform7
- Perform the massively parallel snRNA sequencing (e.g., Drop-Seq) method as previously described7 with the following modifications:4
- Adjust nuclei to a final concentration of 225 nuclei per μL.
- Prepare barcoded beads at a concentration of 250 beads per μL.
- Prepare the lysis buffer with 0.7% sarkosyl.
- Adjust the flow rates to 35 μL per min for beads, 35 μL per min for nuclei, and 200 μL per min for oil.
7. Massively parallel snRNA-sequencing: Commercial Platform26
- Perform massively parallel snRNA-sequencing using the commercial platform (e.g., Chromium Single Cell Gene Expression Solution) products according to the manufacturer’s instructions26 with the following modification:
- Following reverse-transcription, add an additional PCR cycle to the calculated number of cycles for cDNA amplification based on the targeted cell recovery to compensate for decreased cDNA from nuclei compared to cells.
Representative Results
Here, we performed isolation of nuclei from the adult mouse lumbar spinal cord for downstream massively parallel RNA sequencing. The protocol involved three main components: tissue disruption and cellular lysis, homogenization, and sucrose density centrifugation (Figure 1). Within seconds, the detergent-mechanical lysis yielded a crude nuclei preparation with a large number of nuclei as well as cellular and tissue debris (Figure 2A, Table 2). After fifteen minutes, the hypotonic-mechanical lysis yielded a crude nuclei preparation that had less debris, but also fewer nuclei (Figure 2B, Table 2). Both preparations underwent homogenization (Figure 2Cand D) and sucrose density gradient centrifugation before resuspension in PBS with 0.04% BSA (Figure 2Eand F). On an average, a mouse lumbar spinal cord (325.5 mg ± 63.9 mg standard error of the mean, SEM, N = 4) yielded 5.1 x 105 nuclei (± 6.3 x 104 SEM, N = 3) following the detergent-mechanical lysis and 2.0 x 105 nuclei (± 5.9 x 104 SEM, N = 3) following the hypotonic-mechanical lysis. The number of nuclei in the lumbar spinal cord was estimated from the initial crude preparation after Dounce homogenization in the detergent-mechanical lysis protocol (2.6 x 106 nuclei ± 4.0 x 105 SEM, N = 3, Table 2). The final sample from the detergent-mechanical lysis protocol consists of 20% of the initial nuclei (± 2% SEM, N = 3, Table 2). The crude nuclei preparation from the hypotonic-mechanical lysis following trituration contains 62% of the initial nuclei (± 2% SEM, N = 3, Table 2). The final hypotonic-mechanical lysis sample contains only 8% of initial nuclei (± 1% SEM, N = 3, Table 2). We did not detect any difference in the total RNA yield or the cDNA yield for a housekeeping gene (Gapdh) between the two preparation methods. Using qPCR, the detergent method yielded 463.7 ng (± 98.9 SEM, N= 6) of total RNA and an average detection threshold cycle of 25.2 (± 1.3 SEM) for Gapdh cDNA by qPCR and the hypotonic method yielded 419.2 ng (± 85.3 SEM, N = 6) of total RNA and an average detection threshold cycle of 26.1 for Gapdh cDNA (± 0.8 SEM). The two lysis options both isolate nuclei from difficult-to-dissociate tissues and provide the high-quality material for the downstream single-nucleus RNA sequencing.

Given the size of microfluidic channels for downstream massively parallel single nucleus sequencing platforms, it is critical to input a nuclei suspension free of large particles or cellular debris to prevent clogging. Following the protocol presented here, there were no instances of clogging on the platform adapted from Macosko et al. 2015 (N = 17) and one partial clog on the commercial platform (N = 16).
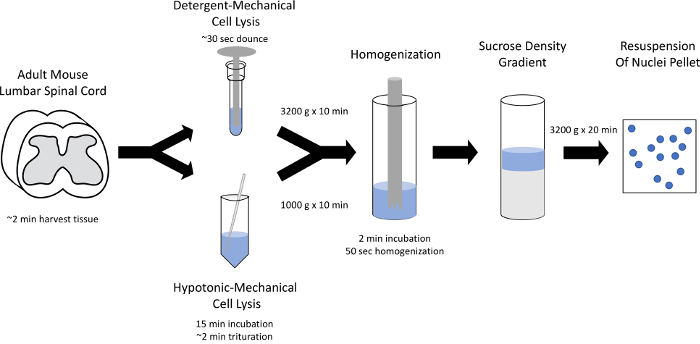
The detergent-mechanical and hypotonic-mechanical procedures were used to isolate nuclei successfully for two massively parallel droplet encapsulation platforms and representative results are shown in Figure 3. Both of these approaches enabled transcriptional profiling of thousands of nuclei, and classification of cell types in the adult mouse lumbar spinal cord (Figure 3).4 These approaches resulted in comparable genes per nucleus for each cell type (Figure 3Cand D). The rates of recovery of input nuclei between the two platforms differ. The platform adapted from Macosko et al. 2015 with modifications from Sathyamurthy et al. 2018 recovered an estimated 0.59% of nuclei (± 0.05% SEM, N = 17), while the commercial platform recovered an estimated 53.7% nuclei (N = 2).
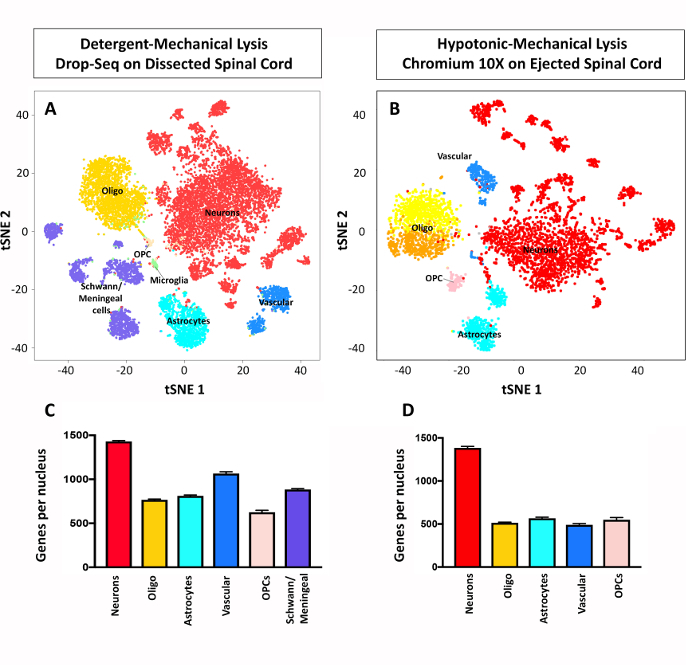
This protocol slightly enriches for neuronal nuclei in the final preparation. In lumbar spinal cord tissue sections, we found that 27% of nuclei were positive for the neuronal marker NeuN (N = 7,368 nuclei from 2 animals), while detergent-mechanical nuclei preparation of the lumbar spinal cord resulted in 31.9% of total nuclei expressing NeuN, as determined by fluorescence-activated cell sorting (FACS, ± 2.0% SEM, N = 13 independent nuclei preparations using pooled samples from multiple animals in each preparation, Figure 4). This is similar to what has been observed previously for the percent of NeuN-positive nuclei in the entire spinal cord (20% to 24% depending on age),27 including the cervical and thoracic regions that have more white matter and oligodendrocytes. Of note, NeuN/Rbfox3 is not expressed in all neurons and, accordingly, these numbers are likely modest underestimates. It is possible that smaller non-neuronal cells are slightly depleted during the sucrose gradient purification. In addition, downstream filtering and analysis parameters following sequencing may alter the final cell-type distribution because neurons have more genes per nucleus (Figure 3Cand D) and, therefore, are less likely to be removed during the filtering process.
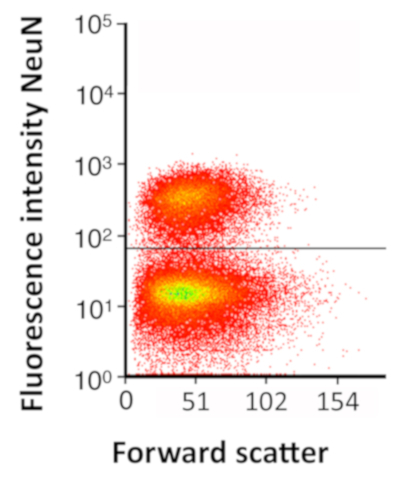
There are several key steps in this protocol that require care. First, excessive douncing or trituration (in steps 3 or 4, respectively) can lead to an increase in cellular debris and particle formation. Although filtration and sucrose density centrifugation can separate large particles, once small particles are generated during cellular lysis, they are difficult to remove. Secondly, during homogenization, do not place the homogenizer directly onto the bottom of the Oak Ridge tube. Instead, submerge the end of the homogenizer into the low sucrose solution containing resuspended nuclei, without touching the bottom of the tube. Homogenization improves nuclear isolation by removing cellular debris and reducing clumps and multiplets (Figure 5). Following sucrose density centrifugation, it is critical to immediately remove the Oak Ridge tube from the centrifuge, and quickly decant the supernatant in a rapid 'flicking' motion. When resuspending nuclei from the wall of the Oak Ridge tube, resuspend the 'salty' pellet from halfway between the myelin band and the bottom of the tube. Note that the pellet may not be visible. Resuspending nuclei higher along the tube may result in myelin contamination in the nuclei preparation. The cellular lysis and sucrose density centrifugation steps are the most critical to reducing particulates that may clog microfluidic channels for downstream application.
| Name of Material/ Equipment | Stock Concentration | Final Concentration | Volume / Amount |
| Detergent Lysis Buffer | |||
| Low sucrose buffer | - | - | 600 μL |
| Triton-X | 20% | 0.10% | 3 μL |
| Hypotonic Lysis buffer | |||
| Tris-HCl (pH = 7.4) | 1 M | 10 mM | 100 μL |
| NaCl | 5 M | 10 mM | 20 μL |
| MgCl2 | 1 M | 3 mM | 30 μL |
| Nonidet P40 | - | 0.01% | 1 μL |
| Nuclease-free water | up to 10 mL | ||
| HEB Medium | |||
| Hibernate-A | - | - | 10 mL |
| Glutamax | - | - | 100 μL |
| B27 | - | - | 200 μL |
| Low sucrose buffer | |||
| Sucrose | - | 0.32 M | 2.75 g |
| HEPES (pH = 8.0) | 1 M | 10 mM | 250 μL |
| CaCl2 | 1 M | 5 mM | 125 μL |
| MgAc | 1 M | 3 mM | 75 μL |
| EDTA | 0.5 M | 0.1 mM | 5 μL |
| DTT | 1 M | 1 mM | 25 μL |
| Nuclease-free water | up to 25 mL | ||
| Sucrose density buffer | |||
| Sucrose | - | 1 M | 8.6 g |
| HEPES (pH = 8.0) | 1 M | 10 mM | 250 μL |
| MgAc | 1 M | 3 mM | 75 μL |
| DTT | 1 M | 1 mM | 25 μL |
| Nuclease-free water | up to 25 mL | ||
| Resuspension Solution | |||
| 1X PBS | - | - | 1 mL |
| BSA | 20 mg/mL | 0.4 mg/mL | 20 μL |
| RNAse Inhibitor | 40 U/μL | 0.2 U/μL | 5 μL |
Table 1: Table of Solutions.
| Crude | Homogenized | Final | |
| Detergent-Mechanical | 100 ± 15% | 87 ± 9% | 20 ± 2% |
| Hypotonic-Mechanical | 62 ± 12% | 35 ± 4% | 8 ± 1% |
Table 2: Yield of nuclei at each step in the protocol. The number of nuclei in the initial crude preparation after dounce homogenization in the detergent-mechanical lysis protocol was used to estimate the number of nuclei in the lumbar spinal cord. The initial nuclei yield (2.6 x 106 nuclei ± 4.0 x 105 SEM, N = 3) was used to calculate the nuclei yield at each downstream step for both detergent- and hypotonic-mechanical lysis protocols. The number of nuclei isolated by the hypotonic-mechanical preparation was normalized to the estimated initial nuclei. Values in the table are mean ± SEM, N = 3.
Figure 1: Schematic of nuclear isolation. Nuclei from the adult spinal cord can be isolated using detergent-mechanical or hypotonic-mechanical cell lysis, followed by homogenization, and sucrose density gradient centrifugation. Please click here to view a larger version of this figure.
Figure 2: Representative brightfield and DAPI-stained nuclei at key steps in the protocol. (A,B) Crude nuclei following detergent-mechanical or hypotonic-mechanical lysis. (C,D) Nuclei following homogenization. (E,F) Nuclei resuspended in PBS with 0.04% BSA following sucrose density centrifugation. Nuclei were fixed with 2% paraformaldehyde and subsequently stained using Trypan Blue or DAPI. Images were taken at 10X (Scale bar = 100 µm) using brightfield and epi-fluorescence. Please click here to view a larger version of this figure.
Figure 3: Representative tSNE plot of sequenced nuclei: using detergent-mechanical and hypotonic-mechanical lysis. (A) Results obtained from sequencing over 17,000 nuclei from the dissected adult mouse lumbar spinal cord following detergent-mechanical lysis and according to Macosko et al. 2015 with modifications from Sathyamurthy et al. 2018. This figure has been modified with permission from Sathyamurthy et al. 2018.4 (B) Results obtained from sequencing 5,000 nuclei from the ejected adult lumbar spinal cord following hypotonic-mechanical lysis and a commercial microfluidic single cell encapsulation platform.26 (C,D) Average genes per nucleus results following clustering of major cell types in the adult mouse spinal cord ± SEM. Of note, the detergent-mechanical lysis procedure followed by the Macosko et al. 2015 platform was performed using dissected lumbar spinal cord, while the hypotonic-mechanical lysis followed by the commercial platform was performed using ejected lumbar spinal cord (as described in this protocol). Given that ejecting the cord removes the dura and dorsal root ganglia, the meningeal/Schwann cell cluster is absent from Figure 3Band D. Please click here to view a larger version of this figure.
Figure 4: FACS plot of NeuN+ nuclei following detergent-mechanical lysis. FACS plot showing fixed nuclei stained for NeuN (average 31.9% of total nuclei ± 2.0% SEM, N = 13), isolated using the detergent-mechanical lysis protocol. For the immediate fixation, nuclei for FACS validation, a crude nuclei preparation was obtained by dounce homogenization of spinal cords using the detergent-mechanical preparation, followed by immediate fixation with 1% PFA with a 5 min incubation period. Fixation was quenched with 250 mM glycine, and nuclei were collected. Staining with anti-NeuN antibody was performed in solution. FACS was performed on fixed, NeuN stained nuclei using a cell sorter. Please click here to view a larger version of this figure.
Figure 5: Nuclei preparation without homogenization. Nuclei were resuspended prior to sucrose density centrifugation following A detergent-mechanical or B hypotonic-mechanical lysis, without homogenization. * Denotes cellular debris attached to nuclei (A) and a multiplet of nuclei attached by cellular debris (B). Nuclei resuspended in PBS with 0.04% BSA following sucrose density centrifugation. Nuclei were fixed with 2% paraformaldehyde and subsequently stained using Trypan Blue or DAPI. Images were taken at 10X (scale bar 100 µm) using brightfield and epi-fluorescence. Please click here to view a larger version of this figure.
Discussion
The ultimate goal of this protocol is to isolate nuclei containing high-quality RNA for downstream transcriptional analysis. We adapted snRNA-Seq methods in order to profile all of the cell types in the spinal cord. Initially, we found that typical cell dissociation methods were ineffective for single cell RNA sequencing, as spinal cord neurons are particularly vulnerable to cell death. Furthermore, cell dissociation methods induce expression of various activity- and stress-response genes by up to several hundred-fold.3,4,16 Given the drawbacks associated with single cell preparations, we and others have used nuclei as an alternative.16,17,18,24 This method can also be used on human tissue, including frozen spinal cord tissue.4,19,24 Here, we will describe the strengths and limitations of this approach.
Strengths of this method include the avoidance of experimentally-induced IEGs as well as the ability to use both fresh and frozen tissue.4 Thus, this approach can be useful for probing endogenous IEGs following a behavior or stimulus.1,3,4 One of the benefits of this method is that it does not require specialized devices for utilization of nuclei for massively parallel single nucleus sequencing, but can use the platform developed by Macosko et al. 2015, with minor adjustments of lysis buffer and flow rate, or use commercially available systems. Moreover, single nucleus sequencing is proven to be a comparable method to that of single cell sequencing for the identification of cell types, lending to the strength of this approach.28,29 However, there are several important limitations of this approach. Nuclei contain approximately 20-50% of cellular mRNA,29 and this is reflected in a lower number of transcripts per nucleus compared to single cell sequencing.18,29 Including intronic reads from snRNA-Seq is one approach to increase the number of detected genes.
There are several available protocols that enable isolation of nuclei from tissue.2,16,17,18,24,30 In comparison with most other methods, the protocols presented here do not require myelin removal, ultracentrifugation, or many centrifugation steps or washes that can lead to lower final numbers of nuclei. Furthermore, this protocol takes 45 min (detergent-mechanical) or 1 hour (hypotonic-mechanical) to complete. Commercial protocols supported on microfluidic platforms are more than double the time, and require many more centrifugation steps, increasing the risk of losing nuclei. In contrast with nuclei isolation protocols that involve only lysis and filtering, the methods presented here include a sucrose gradient to increase the purity of the final nuclei. This step is required for adult spinal cord tissue due to the large percentage of white matter and the resulting myelin debris.
The detergent-mechanical lysis protocol can be used for the complete tissue dissociation and lysis, and the hypotonic-mechanical lysis protocol can be used to control the amount of tissue dissociation and cellular debris allowed in the downstream application. These protocols can be used for bio-bank material, difficult to dissociate tissues and for the investigation of activity-dependent transcriptional changes through the isolation of nuclei for downstream massively parallel snRNA-Seq. In addition to massively parallel single nucleus RNA sequencing, this protocol may be used to isolate nuclei for alternative applications, including immunofluorescence and FACS and epigenetic analysis such as DNA methylation studies and ChIP-Seq (Figure 4).23
Disclosures
We have no conflicts of interest to disclose.
Acknowledgments
This work was supported by the intramural program of NINDS (1 ZIA NS003153 02) and NIDCD (1 ZIA DC000059 18). We thank L. Li and C.I. Dobrott for their technical support and helpful discussions, and C. Kathe for reviewing the manuscript.
References
- Hrvatin S, et al. Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex. Nature Neuroscience. 2018;21(1):120–129. doi: 10.1038/s41593-017-0029-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P, et al. Dissecting Cell-Type Composition and Activity-Dependent Transcriptional State in Mammalian Brains by Massively Parallel Single-Nucleus RNA-Seq. Molecular Cell. 2017;68(5):1006–1015. doi: 10.1016/j.molcel.2017.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YE, Pan L, Zuo Y, Li X, Hong W. Detecting Activated Cell Populations Using Single-Cell RNA-Seq. Neuron. 2017;96(2):313–329. doi: 10.1016/j.neuron.2017.09.026. [DOI] [PubMed] [Google Scholar]
- Sathyamurthy A, et al. Massively Parallel Single Nucleus Transcriptional Profiling Defines Spinal Cord Neurons and Their Activity during Behavior. Cell Reports. 2018;22(8):2216–2225. doi: 10.1016/j.celrep.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nature Methods. 2009;6(5):377–382. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- Campbell JN, et al. A molecular census of arcuate hypothalamus and median eminence cell types. Nature Neuroscience. 2017;20(3):484–496. doi: 10.1038/nn.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161(5):1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Wu X, Jiang L, Zhang Y. Single-Cell RNA-Seq Reveals Hypothalamic Cell Diversity. Cell Reports. 2017;18(13):3227–3241. doi: 10.1016/j.celrep.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin DA, et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 2014;343(6172):776–779. doi: 10.1126/science.1247651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CL, et al. Somatosensory neuron types identified by high-coverage single-cell RNA-sequencing and functional heterogeneity. Cell Research. 2016;26(8):967. doi: 10.1038/cr.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, et al. Single-Cell RNA-Seq with Waterfall Reveals Molecular Cascades underlying Adult Neurogenesis. Cell Stem Cell. 2015;17(3):360–372. doi: 10.1016/j.stem.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nature Neuroscience. 2016;19(2):335–346. doi: 10.1038/nn.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usoskin D, et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nature Neuroscience. 2015;18(1):145–153. doi: 10.1038/nn.3881. [DOI] [PubMed] [Google Scholar]
- Villani AC, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. 2017;356(6335) doi: 10.1126/science.aah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347(6226):1138–1142. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
- Lacar B, et al. Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nature Communications. 2016;7:11022. doi: 10.1038/ncomms11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake BB, et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science. 2016;352(6293):1586–1590. doi: 10.1126/science.aaf1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grindberg RV, et al. RNA-sequencing from single nuclei. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(49):19802–19807. doi: 10.1073/pnas.1319700110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnaswami SR, et al. Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nature Protocols. 2016;11(3):499–524. doi: 10.1038/nprot.2016.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matevossian A, Akbarian S. Neuronal nuclei isolation from human postmortem brain tissue. Journal of Visualized Experiments. 2008. [DOI] [PMC free article] [PubMed]
- Bergmann O, Jovinge S. Isolation of cardiomyocyte nuclei from post-mortem tissue. Journal of Visualized Experiments. 2012. [DOI] [PMC free article] [PubMed]
- Nohara K, Chen Z, Yoo SH. A Filtration-based Method of Preparing High-quality Nuclei from Cross-linked Skeletal Muscle for Chromatin Immunoprecipitation. Journal of Visualized Experiments. 2017. [DOI] [PMC free article] [PubMed]
- Halder R, et al. DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nature Neuroscience. 2016;19(1):102–110. doi: 10.1038/nn.4194. [DOI] [PubMed] [Google Scholar]
- Habib N, et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nature Methods. 2017;14(10):955–958. doi: 10.1038/nmeth.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10X Genomics. Sample Preparation Demonstrated Protocols: Isolation of Nuclei for Single Cell RNA Sequencing. 2018. Available from: https://support.10xgenomics.com/single-cell-gene-expression/sample-prep/doc/demonstrated-protocol-isolation-of-nuclei-for-single-cell-rna-sequencing.
- 10X Genomics. Single Cell 3' Reagent Kits v2 User Guide. 2018.
- Fu Y, Rusznak Z, Herculano-Houzel S, Watson C, Paxinos G. Cellular composition characterizing postnatal development and maturation of the mouse brain and spinal cord. Brain Structure and Function. 2013;218(5):1337–1354. doi: 10.1007/s00429-012-0462-x. [DOI] [PubMed] [Google Scholar]
- Lake BB, et al. A comparative strategy for single-nucleus and single-cell transcriptomes confirms accuracy in predicted cell-type expression from nuclear RNA. Scientific Reports. 2017;7(1):6031. doi: 10.1038/s41598-017-04426-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakken TE. Equivalent high-resolution identification of neuronal cell types with single-nucleus and single-cell RNA-sequencing. bioRxiv. 2018.
- Habib N, et al. Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science. 2016;353(6302):925–928. doi: 10.1126/science.aad7038. [DOI] [PMC free article] [PubMed] [Google Scholar]