

Abstract
A comprehensive identification of RNA-binding proteins (RBPs) is key to understanding the posttranscriptional regulatory network in cells. A widely used strategy for RBP capture exploits the polyadenylation [poly(A)] of target RNAs, which mostly occurs on eukaryotic mature mRNAs, leaving most binding proteins of non-poly(A) RNAs unidentified. Here we describe the detailed procedures of a recently reported method termed click chemistry-assisted RNA-interactome capture (CARIC), which enables the transcriptome-wide capture of both poly(A) and non-poly(A) RBPs by combining the metabolic labeling of RNAs, in vivo UV cross-linking, and bioorthogonal tagging.
Keywords: Biochemistry, Issue 140, RNA, RNA-protein interactions, proteomics, bioorthogonal chemistry, noncoding RNA
Introduction
The human genome is transcribed into various types of coding and noncoding RNAs (ncRNAs), including mRNAs, rRNAs, tRNAs, small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs), and long non-coding RNAs (lncRNAs)1. Most of these RNAs possess clothing of RBPs and function as ribonucleoprotein particles (RNPs)2. Therefore, a comprehensive identification of RBPs is a prerequisite for understanding the regulatory network between RNAs and RBPs, which is implicated in various human diseases3,4,5.
The past few years have witnessed a great boost of RBPs discovered in various eukaryotic systems2,6, including human7,8,9,10,11, mouse12,13,14, yeast9,15,16, zebrafish17, Drosophila melanogaster18,19, Caenorhabditis elegans16, Arabidopsis thaliana20,21,22, and human parasites23,24,25. These advances have been facilitated by an RBP capture strategy developed by Castello et al.7 and Baltz et al.8 in 2012, which combines in vivo UV cross-linking of RNA and its interacting proteins, oligo(dT) capture of poly(A) RNAs, and mass spectrometry (MS)-based proteomic profiling. However, given the fact that poly(A) mostly exists on mature mRNAs, which account for only ~3% - 5% of the eukaryotic transcriptome26, this widely used strategy is not capable of capturing RBPs interacting with non-poly(A) RNAs, including most ncRNAs and pre-mRNAs.
Here, we report the detailed procedures of a recently developed strategy for the transcriptome-wide capture of both poly(A) and non-poly(A) RBPs27. Termed CARIC, this strategy combines in vivo UV cross-linking and metabolic labeling of RNAs with photoactivatable and "clickable" nucleoside analogs (which contain a bioorthogonal functional group that can participate in click reaction), 4-thiouridine (4SU), and 5-ethynyluridine (EU). Steps that are key to get ideal results with the CARIC strategy are efficient metabolic labeling, UV cross-linking and click reaction, and the maintenance of RNA integrity. Because Cu(I) used as the catalyst in click reaction can cause the fragmentation of RNAs, a Cu(I) ligand that can reduce RNA fragmentation is essential. We describe how to perform efficient click reactions in cell lysates without causing severe RNA degradation.
Although RBP capture and identification in HeLa cells only is described in this protocol, the CARIC strategy can be applied to various cell types and possibly to living organisms. Besides RBP capture, this protocol also provides streamlined step-by-step procedures for MS sample preparation and protein identification and quantification, which can be helpful for those who are not familiar with proteomic experiments.
Protocol
CAUTION: When applicable, the reagents used should be purchased in the form of RNase-free, or dissolved in RNase-free, solvents (for most cases, in diethyl pyrocarbonate (DEPC)-treated water). When handling RNA samples and RNase-free reagents, always wear gloves and masks, and change them frequently to avoid RNase contamination.
1. Preparation of Lysate of Metabolically Labeled and UV Cross-linked Cells
- Metabolic incorporation of EU and 4SU
- Culture HeLa cells in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C in a 5% CO2 atmosphere. Culture ~4 x 107 HeLa cells (in two 15-cm dishes) for preparing one experimental or control sample for one standard MS run.
- When the cultured HeLa cells reach ~80% confluence, remove the culture medium and add 15-mL of prewarmed fresh medium per 15-cm dish.
- Add 15 μL per dish of 100 mM EU (dissolved in phosphate-buffered saline (PBS)) to a final concentration of 1 mM, and 7.5 μL per dish of 100 mM 4SU (dissolved in PBS) to a final concentration of 0.5 mM for experimental and noUV-control samples. Add 15 μL per dish of 100 mM EU (dissolved in PBS) to a final concentration of 1 mM for no4SU-control samples. NOTE: 4SU is photo-activatable; thus, protection from light after adding 4SU is required.
- Cover the dishes with foil and culture the cells for 16 h. Add half of the amount of the EU and 4SU or only EU from step 1.1.3 to the experimental, noUV-, and no4SU-control samples, respectively, and continue culturing for another 2 h.
- In vivo UV cross-linking
- Remove the culture medium, wash the cells 3x with 5 mL of PBS per dish, and remove residual PBS as much as possible. NOTE: Residue liquid will significantly reduce cross-linking efficiency.
- For experimental and no4SU-control samples, place the dishes on ice with the lid removed and irradiate the cells with 365-nm UV light at 2 J/cm2 by a UV cross-linker.
- For noUV-control samples, place the dishes on ice and protect them from light. NOTE: All following steps for noUV-control samples should be performed in a darkened room.
- Cell lysis and homogenization
- Add 1 mL per dish of pre-lysis buffer (10 mM Tris∙HCl, pH 7.5, 50 mM LiCl, 0.02% Nonidet P-40, and ethylenediaminetetraacetic acid (EDTA)-free protease inhibitor cocktail) to the cells. Scrape the cells using a rubber cell lifter and collect the pre-lysis suspension in a 15-mL tube. NOTE: This step will break the cell membrane and release soluble cytoplasmic proteins and RNAs. DO NOT centrifuge the tube and remove the supernatant.
- For the suspension from two 15-cm dishes, adjust the volume to 6 mL by adding pre-lysis buffer. Add to the pre-lysis suspension an equal volume of R-lysis buffer (200 mM Tris∙HCl, pH 7.5, 500 mM LiCl, 2% lithium dodecyl sulfate [LDS]).
- Homogenize the cell lysate by passing it through a syringe with a narrow needle (27-G) several times till the lysate is clear and homogenous. Incubate the lysate at 4 °C with gentle rotation (~15 rpm) for 1 h. NOTE: This last step will allow the complete denaturing of proteins. The lysate can be safely stored at -70 °C for up to one month.
- Preparation for click reaction
- Dilute the lysate by adding 20 volumes of dilution buffer (50 mM Tris∙HCl, pH 7.5) and divide it into 15-mL fractions. NOTE: Solutions containing a high concentration of salt and detergent will compromise the efficiency of the Cu(I)-catalyzed click reaction; thus, the buffer of the lysate must be changed.
- Concentrate each fraction by using a 15-mL ultrafiltration tube (with a molecular weight cutoff of 10 kDa) till the volume is smaller than 1 mL. Use a swinging-bucket rotor to spin the ultrafiltration tube at 4,000 x g at 4 °C for ~15 min.
- Add 14 mL of dilution buffer to the concentrated lysate fraction and repeat step 1.4.2. Combine the fractions and concentrate them to a volume of 6 mL by ultrafiltration (4,000 x g at 4 °C for ~15 min). NOTE: Most of the salt and LDS will now be removed, so the lysate is ready for the click reaction. The lysate can be stored at -70 °C for up to one week. Avoid multiple freeze-thaw cycles, because they will result in significant RNA degradation. Aliquot the lysate if small-scale characterizations are required.
2. Preparation of Samples for RNA-interactome Capture
- Preclearing of the lysate
- Add 100-μL streptavidin magnetic beads per 6 mL of lysate, and gently rotate (~15 rpm) for 30 min at room temperature to eliminate naturally biotinylated proteins.
- Pellet the beads using a magnet (for ~20 min at 4 °C) and transfer the precleared lysate to a new tube.
- Performance of the click reaction
- Prepare the reaction mix: 6.5 μL of biotin stock (100 mM azide-biotin dissolved in dimethyl sulfoxide [DMSO] at a final concentration of 100 μM), 3.25 μL of copper stock (make it fresh; 1 M CuSO4 dissolved in water at a final concentration of 500 μM), 65 μL of ligand stock (200 mM THPTA dissolved in water at a final concentration of 2 mM), and 262.75 μL of H2O. NOTE: THPTA stands for Tris[(1-hydroxypropyl-1H-1,2,3-triazol-4-yl)methyl]amine.
- Add the reaction mix to 6 mL of precleared lysate and mix well. Then, add 162.5 μL of reducing reagent (make it fresh; 40 mg/mL sodium ascorbate at a final concentration 5 mM) to the lysate and mix well. The final volume should be 6.5 mL.
- Incubate the reaction mixture for 2 h at room temperature on an orbital shaker (800 rpm). Add 5 mM EDTA to the reaction mixture and incubate it for 5 min to quench the reaction.
- Small-scale characterizations
- Prepare the reaction mix as in step 2.2.1 with the biotin stock replaced by dye stock (e.g., 100 mM azide-Cy5 dissolved in DMSO). NOTE: The reagent amount should be adjusted according to the volume of lysate. Typically, a 20-μL aliquot of the lysate is enough for characterizations such as an in-gel fluorescence analysis.
- Add the reaction mix to the lysate and incubate it for 2 h at room temperature. Then, add one-third of the volume of the LDS sample buffer (4x), denature it at 55 °C for 5 min, and resolve the sample on a 10% bis-Tris gel. NOTE: To confirm the fluorescence signal is presented on RNAs, include controls with RNase A digestion after the click reaction.
- Cleaning up of the reaction mixture
- Add eight volumes of prechilled methanol (100%) to the quenched reaction mixture and incubate it for 30 min at -30 °C for precipitation. Perform the precipitation in 50-mL conical centrifuge tubes. NOTE: If the total volume is greater than 50 mL, divide the reaction mixture into two 50-mL conical centrifuge tubes.
- Prepare reconstitution buffer: combine one volume of buffer A (4% sodium dodecyl sulfate [SDS] and 10 mM EDTA) with eight volumes of buffer B (1% Brij-97, 150 mM NaCl, and 50 mM triethanolamine, pH 7.4).
- Centrifuge at 4,000 x g for 15 min at 4 °C and discard the supernatant. Add ~1 - 2 mL of prechilled methanol to the pellet. Pipette up and down to break the pellet and make sure the pellet is completely suspended with no visible chunks. Fill the tube with prechilled methanol. Repeat this step 2x.
- Centrifuge at 4,000 x g for 15 min at 4 °C and discard the supernatant. Put back the tubes and centrifuge again at 4,000 x g for 5 min. Carefully draw out the residual methanol as much as possible without disturbing the pellet.
- Add 10 mL of reconstitution buffer to the pellet. Pipette up and down to dissolve the pellet. Centrifuge at 4,000 x g for 10 min at 4 °C.
- Transfer the supernatant to a new tube. Collect 20 μL of the sample for quality control (see section 4). NOTE: Now, the sample is ready for RNA-interactome capture. The reconstituted sample can be stored at -70 °C for up to one week.
3. RNA-interactome Capture
- Preparation of the streptavidin-agarose beads
- Take 1,600 μL of streptavidin-agarose slurry (800 μL of settled beads) per 10 mL of reconstituted sample into a 15-mL conical centrifuge tube.
- Spin down the beads at 4000 x g for 5 min. Carefully remove the supernatant without disturbing the settled beads.
- Wash the beads with 10 mL of 50 mM Tris∙HCl (pH 7.5). Spin down the beads (4,000 x g for 5 min) and remove the supernatant. Repeat this step 2x.
- Affinity pulldown
- Transfer the cleaned-up and reconstituted sample from step 2.4.6 to the streptavidin-agarose beads (see step 3.1). Incubate overnight with gentle rotation at 4 °C.
- Washing of the streptavidin beads
- Spin down the beads (4,000 x g for 5 min) and transfer the supernatant to a new tube. Collect 20 μL of the sample for quality control.
- Wash the beads with 10 mL of wash buffer A (2% SDS in PBS, pH 7.4). Incubate for 10 min with gentle rotation (~12 rpm) at room temperature. Spin down the beads (4,000 x g for 5 min) and remove the supernatant. Repeat 1x.
- Repeat step 3.3.2 with wash buffer B (8 M urea and 250 mM NH4HCO3 dissolved in water). Repeat step 3.3.2 with wash buffer C (2.5 M NaCl in PBS, pH 7.4). Then, wash the beads with 10 mL of 50 mM Tris∙HCl (pH 7.5). Spin down the beads (4,000 x g for 5 min) and remove the supernatant.
- Split the beads evenly and transfer them to two 1.5-mL microcentrifuge tubes.
- Elution of the captured RNPs
- Prepare biotin elution buffer: 12.5 mM biotin, 75 mM NaCl, 7.5 mM Tris∙HCl (pH 7.5), 1.5 mM EDTA, 0.15% SDS, 0.075% sarkosyl, and 0.02% sodium deoxycholate. NOTE: Store the buffer at room temperature, for biotin may precipitate at 4 °C.
- To 400 μL of washed settled beads, add 400 μL of biotin elution buffer.
- Incubate them on an orbital shaker (1,500 rpm) at room temperature for 20 min. Then, incubate on an orbital shaker with a heat block (1,500 rpm, 65 °C) for 10 min. Spin down the beads (7,800 x g for 1 min) and collect the eluted RNP.
- To the beads, add 400 μL of fresh biotin elution buffer and repeat step 3.4.3. Combine the two elutes into one 15-mL tube.
- RNase digestion
- Add three volumes of dilution buffer to the eluted RNP to decrease the concentration of SDS. Concentrate the diluted sample by using a 0.5-mL ultrafiltration tube (with a molecular weight cutoff of 10 kDa; spin at 12,000 x g at 4 °C for ~30 min) to ~40 μL.
- Add 0.5 μg/μL RNase A and incubate it for 2 h at 37 °C to release RBPs from cross-linked RNAs. Collect 2 μL of RBPs for quality control (see section 4).
4. Quality Control
- Control of the efficiency of the affinity pulldown
- Take 10 μL of the “before-pulldown” sample from step 2.4.6 and 10 μL of the “after-pulldown” sample from step 3.3.1.
- Analyze the samples using standard western blot procedures (10% bis-Tris gel).
- Stain the polyvinylidene fluoride (PVDF) membrane with streptavidin-HRP conjugate to monitor the residue biotin signals of the “after-pulldown” sample. NOTE: If the biotin signal of the “after-pulldown” sample is greater than one-fifth of the signal of the “before-pulldown” sample, increase the amount of streptavidin-agarose beads used in step 3.1.1.
- Control of the total capture efficiency
- Take 2 μL of the released RBP sample from step 3.5.2 and 0.5 μL of the “before-pulldown” sample (as 0.1% input) from step 2.4.6.
- Analyze the samples using standard silver-staining procedures.
- Fix the gel with fixation buffer (40% ethanol, 10% acetic acid) for 20 min followed by sensitization (13 mM Na2S2O3, 83 mM sodium acetate, 30% ethanol) for 30 min.
- Wash the gel 3x with water for 5 min and, then, stain it with a 15 mM AgNO3 solution for 20 min. Wash the gel 2x with water for 1 min, develop it in 0.24 M Na2CO3 and 0.012% formaldehyde, and terminate with 45 mM EDTA when the staining is sufficient. NOTE: The silver-staining intensity of the captured RBPs should be similar to that of the 0.1% input.
5. Preparation of the Samples for MS
- In-gel trypsin digestion of captured RBPs 28
- Add one-fourth volume of SDS sample buffer (5x) to the released RBP samples from step 3.5.2. Denature the sample at 95 °C for 10 min.
- Resolve the RBPs on a 1.5-mm 10% SDS-polyacrylamide gel.
- Stain the gel with silver, following standard protocols.
- Excise the lane of experimental sample or control sample with stacking gel and the major band of RNase A (~15 kDa) removed.
- Cut the excised lane into small pieces (~1 - 1.5 mm x ~1 - 1.5 mm). NOTE: The shortest edge of the gel piece should be no shorter than 1 mm to prevent clogging in pipette tips.
- Transfer the gel pieces to a microcentrifuge tube and destain with destaining buffer (a mixture of equal volumes of 100 mM Na2S2O3 and 30 mM K3[Fe(CN)6]).
- Wash the gel pieces with 200 mM ammonium bicarbonate (ABC) till the gel pieces are totally colorless.
- Dehydrate the gel pieces in 1 mL of neat acetonitrile (ACN). Rehydrate with 200 μL of 10 mM dithiothreitol (dissolved in 50 mM ABC) and incubate at 56 °C for 45 min. NOTE: Completely dehydrated gel pieces should be very hard and opaque. If the gel pieces are still soft after dehydration, remove the ACN and add 1 mL of neat ACN to dehydrate again.
- Cool down the gel pieces to room temperature. Add 200 μL of 58 mM iodoacetamide (dissolved in 50 mM ABC) and incubate at room temperature for 45 min in the dark.
- After a brief wash with water, dehydrate the gel pieces in 1 mL of neat ACN. NOTE: The gel pieces must be completely dehydrated.
- Rehydrate the gel pieces with the appropriate amount of 10 ng/μL trypsin (dissolved in 50 mM ABC) and incubate at 37 °C for 12 - 16 h. NOTE: The gel pieces should be completely rehydrated with no opaque cores. Remove any excess liquid.
- Stable isotope dimethyl labeling of the digested peptides 29
- Extract the digested peptides from the gel pieces by adding 200 μL of extraction buffer (5% formic acid and 50% ACN in water) and incubate at 37 °C for 30 min with vortexing (at 1,200 rpm).
- Repeat step 5.2.1 2x. Combine the extracts into one microcentrifuge tube.
- Dry the extracted peptides by vacuum centrifugation.
- Reconstitute the peptides in 200 μL of 100 mM triethylammonium bicarbonate (TEAB, pH 8.5). CAUTION: Steps 5.2.4 - 5.2.6 should be performed on ice in a fume hood.
- Add 8 μL of 4% CH2O and 8 μL of 4% 13CD2O to the experimental and control samples, respectively. NOTE: To control the bias of stable isotopic labeling, swap the stable isotope for experimental and control samples of the other biologically independent replicate.
- Add 8 μL of 0.6 M NaBH3CN (make it fresh) and mix well.
- Incubate the samples at room temperature for 1 h with vortexing.
- Cool down the samples on ice. Quench the reaction by adding 32 μL of 1% ammonia aqueous solution. Then, further quench the reaction by adding 16 μL of formic acid.
- Combine the experimental sample with the corresponding control sample into one microcentrifuge tube. Dry the samples by vacuum centrifugation.
- Fractionation of dimethyl-labeled peptides
- Prepare the stop-and-go-extraction tips (StageTips)30.
- Insert a C18 membrane into an extended-length, 10-μL tip.
- Add 300 μg of high-pH C18 beads suspended in ACN to the tip.
- Place the tip upright in a microcentrifuge tube with a home-made rack which can stabilize the tip and lift the tip off the bottom.
- Spin the tip at 1,400 x g for 2 min. Discard the flow-through.
- Wash the tip with 50 μL of 80% ACN in 10 mM ABC (pH 10.0). Repeat 1x. NOTE: Adjust the pH of 10 mM ABC solution by adding 28% ammonium hydroxide.
- Wash the tip with 50 μL of 50% ACN in 10 mM ABC (pH 10.0). Repeat 1x.
- Wash the tip with 50 μL of 10 mM ABC (pH 10.0). Repeat 1x.
- Reconstitute the peptides in 50 μL of 10 mM ABC (pH 10.0).
- Add the reconstituted sample to the prepared tip. Reload the flow-through to the tip to ensure efficient peptide binding.
- Wash the tip with 50 μL of 10 mM ABC (pH 10.0). Repeat 1x.
- Elute the peptide stepwise for 12 fractions with 50 μL of 6%, 9%, 12%, 15%, 18%, 21%, 25%, 30%, 35%, 40%, 80%, and 6% ACN in 10 mM ABC (pH 10.0).
- Combine two fractions with an equal interval (fraction 1 with 7, 2 with 8, and so on) to get six combined fractions.
- Dry the samples by vacuum centrifugation. The dried peptides can be stored at -30 °C.
6. Performance of the MS and Data Analysis
- Peptide analysis by liquid chromatography-tandem mass spectrometry
- Reconstitute the dried peptide fractions from step 5.3.7 in 15 μL of water containing 0.1% formic acid. Check the pH of reconstituted peptides by spotting 1 μL of the solution on a pH strip (the pH should be under 3).
- Inject the reconstitute sample into the liquid chromatography (LC) column. Apply an appropriate gradient of solvent (solvent A is water containing 0.1% formic acid, solvent B is ACN containing 0.1% formic acid) in high-performance liquid chromatography (HPLC). A typical gradient of solvent B is as follows: 5% - 35% in 40 min; 35% - 70% in 4 min; and held at 75% for 10 min.
- Ionize the eluted peptides by electrospray and operate the mass spectrometer in data-dependent mode. Select 15 most abundant ions (multiply charged: 2+, 3+, or higher) in the initial MS scan for a tandem mass spectrometry (MS/MS) scan (collision-induced dissociation, CID). Set the dynamic exclusion size to 500 with a maximum duration time of 25 s.
- Protein identification and quantification using MaxQuant 31
- Set the false discovery rate (FDR) of protein identification to 0.01 and set the number of unique peptides to 2 in order to increase accuracy and reliability.
- Set the minimal required ratio counts (unique + razor) for protein quantification to 2, and enable the Re-quantify and Match between runs functions.
- Enrichment significance evaluation using the R/Bioconductor package limma 32
- Perform a moderated t-test implemented in limma to test the Log2-fold change against zero from at least three biological replicates. Use the read.table function to read the data table. Then, use the lmFit and eBayes functions for data fitting. Use the topTable function to export the calculation results (including the averaged Log2-fold change and P values).
- Correct the P values using the Benjamini–Hochberg method for controlling the FDR.
- Apply an FDR of 0.01 to generate a list of proteins significantly enriched in the experimental samples. Set a cutoff of two- or threefold change to further control the false positives.
Representative Results
The representative results of quality control steps are presented. The results include figures of the in-gel fluorescence analysis described in step 2.3.2 (Figure 1), the western blot analysis described in step 4.1.3 (Figure 2A), and the silver-staining analysis described in step 4.2.2 (Figure 2B). The quality control steps are critical for the optimization of CARIC protocols. Always include quality controls in the preparation of large-scale RBP identification experiments.
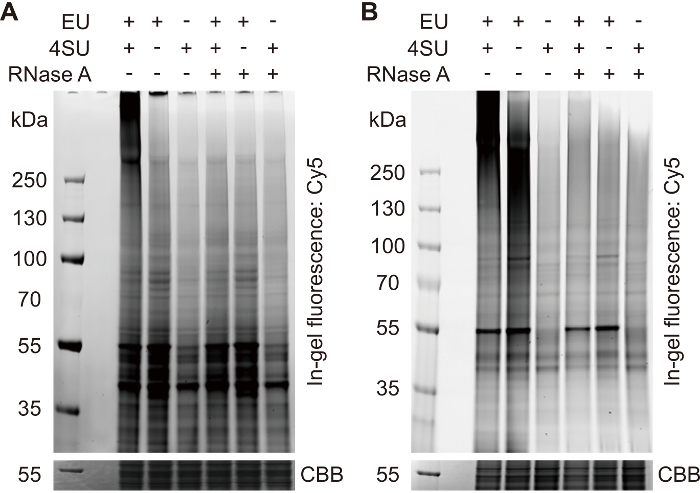
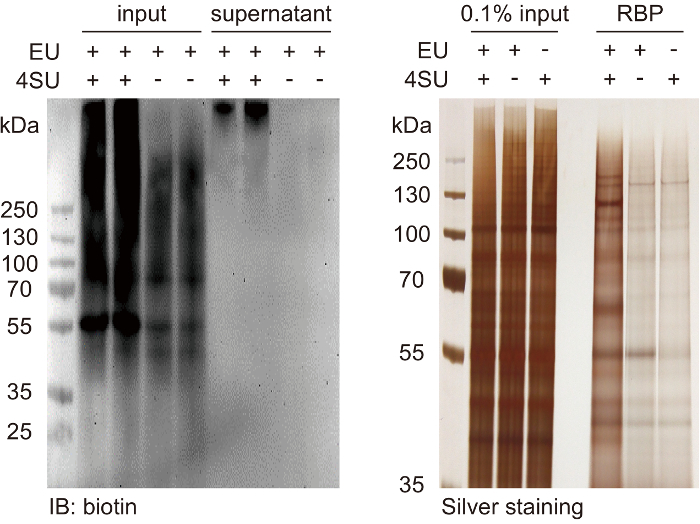
Figure 1: In-gel fluorescence analysis of the click-labeled samples described in step 2.3.2. (A) This panel shows a typical in-gel fluorescence pattern of click-labeled samples. Only the doubly labeled sample shows a strong smear band at a high molecular weight (> 250 kDa), which represents the signal of cross-linked RNPs. To abolish the RNP signal, omit either 4SU or EU or digest with RNase A. The background sharp bands at a lower molecular weight represent the signals of non-specific labeled proteins. (B) In some occasions, a strong smeared band (~130 - 250 kDa) can be observed in the no4SU-control sample. This band represents the signal of labeled uncross-linked RNAs, which will be degraded during the heat denaturation, for most cases. It will not interfere with the subsequent procedures. CBB = Coomassie brilliant blue. Please click here to view a larger version of this figure.
Figure 2: Quality control of affinity pulldown efficiency and the captured RBPs. (A) This panel shows a western blot analysis of the biotin signals in samples before pulldown (input) and in samples after pulldown (supernatant). Estimate the ratio of the remaining signals and optimize the bead amount used in step 3.1.1. (B) This panel shows a silver-staining analysis of captured RBPs compared to 0.1% input total proteins. For HeLa cells, the general total capture efficiency is ~0.05% - 0.1% of input proteins. This value can vary significantly due to the variance of the metabolic labeling efficiency of different cell types. Please click here to view a larger version of this figure.
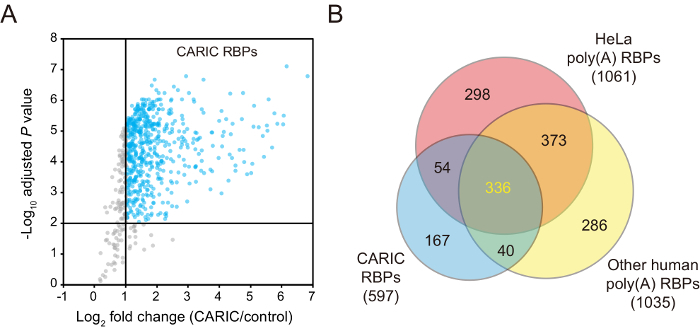
Figure 3: Representative MS results of CARIC. (A) This panel shows a volcano plot displaying the averaged Log2-fold change and adjusted P values of quantified proteins, calculated by the limma package. 597 of proteins with a Log2-fold change of > 2 and an adjusted P value of < 0.01 were classified as "CARIC RBPs". (B) This panel shows the overlap of the CARIC proteins with previously identified human poly(A) RBPs7,8,9,10,11. The overlapped proteins are mostly coding RBPs, while the rest of the CARIC RBPs are more likely to be non-coding RBPs. This figure is a reprinted from previously published work with permission from the National Academy of Sciences27. Please click here to view a larger version of this figure.
Discussion
The maintenance of fair RNA integrity is one of the keys to successful CARIC experiments. With appropriate ligands of Cu(I) and careful operation, RNA degradation can be significantly reduced, although partial degradation was observed. The substitution ratios of EU and 4SU in experimental samples are 1.18% and 0.46%, respectively (data not shown). For intact RNAs with a length of 2,000 nt, ~90% of RNAs contain at least one EU and one 4SU. For partially degraded RNAs with a length of 1,000 nt, ~70% of RNAs contain at least one EU and one 4SU. Therefore, partial degradation of RNAs does not dramatically decrease the efficiency of CARIC, while severe degradation is not acceptable.
Another critical step is step 1.4, the preparation for the click reaction. The Cu(I)-catalyzed click reaction on RNAs is sensitive to LDS concentration. A high concentration (> 0.1%) of LDS will lead to a decrease of labeling signals on EU-containing RNAs and an increase of background signals on proteins (data not shown).
In addition to EU, CARIC is also compatible with other clickable nucleosides, such as alkynyl and azido analogs of adenosine33,34,35,36. However, the application of CARIC is significantly limited by the metabolic efficiency of unnatural clickable nucleosides in a biological system of interest. Therefore, before performing CARIC using conditions other than those demonstrated in this protocol, always check the metabolic labeling efficiency (e.g., by fluorescent imaging).
Recently, a similar strategy called RICK (capture of the newly transcribed RNA interactome using click chemistry), which incorporates only EU to label total RNAs and uses 254-nm UV to cross-link RNAs and proteins, was reported37. Notably, 254-nm UV can activate all four natural nucleosides, as well as EU. Thus, 254-nm UV irradiation may cross-link free EU and its metabolites (e.g., EU phosphates) with corresponding binding proteins, which should be taken into consideration as possible false positives.
One intriguing application of CARIC is to identify RBPs in bacteria whose RNAs are mostly non-polyadenylated. The large-scale identification of RBPs will provide invaluable resources to understand the molecular basis of posttranscriptional regulations in bacteria38.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work is supported by the National Natural Science Foundation of China Grants 91753206, 21425204, and 21521003 and by the National Key Research and Development Project 2016YFA0501500.
References
- Djebali S, et al. Landscape of transcription in human cells. Nature. 2012;489(7414):101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nature Reviews Genetics. 2014;15(12):829–845. doi: 10.1038/nrg3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello A, Fischer B, Hentze MW, Preiss T. RNA-binding proteins in Mendelian disease. Trends in Genetics. 2013;29(5):318–327. doi: 10.1016/j.tig.2013.01.004. [DOI] [PubMed] [Google Scholar]
- Nussbacher JK, Batra R, Lagier-Tourenne C, Yeo GW. RNA-binding proteins in neurodegeneration: Seq and you shall receive. Trends in Neuroscience. 2015;38(4):226–236. doi: 10.1016/j.tins.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazurek M, Ciesiolka A, Starega-Roslan J, Bilinska K, Krzyzosiak WJ. Identifying proteins that bind to specific RNAs - focus on simple repeat expansion diseases. Nucleic Acids Research. 2016;44(19):9050–9070. doi: 10.1093/nar/gkw803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze MW, Castello A, Schwarzl T, Preiss T. A brave new world of RNA-binding proteins. Nature Reviews Molecular Cell Biology. 2018;19(5):327–341. doi: 10.1038/nrm.2017.130. [DOI] [PubMed] [Google Scholar]
- Castello A, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149(6):1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- Baltz AG, et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Molecular Cell. 2012;46(5):674–690. doi: 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- Beckmann BM, et al. The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nature Communications. 2015;6:10127–10135. doi: 10.1038/ncomms10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad T, et al. Serial interactome capture of the human cell nucleus. Nature Communications. 2016;7:11212–11222. doi: 10.1038/ncomms11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello A, et al. Comprehensive identification of RNA-binding domains in human cells. Molecular Cell. 2016;63(4):696–710. doi: 10.1016/j.molcel.2016.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SC, et al. The RNA-binding protein repertoire of embryonic stem cells. Nature Structural & Molecular Biology. 2013;20(9):1122–1130. doi: 10.1038/nsmb.2638. [DOI] [PubMed] [Google Scholar]
- Liepelt A, et al. Identification of RNA-binding proteins in macrophages by interactome capture. Molecular & Cellular Proteomics. 2016;15(8):2699–2714. doi: 10.1074/mcp.M115.056564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, et al. The cardiomyocyte RNA-binding proteome: Links to intermediary metabolism and heart disease. Cell Reports. 2016;16(5):1456–1469. doi: 10.1016/j.celrep.2016.06.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SF, Jain S, She MP, Parker R. Global analysis of yeast mRNPs. Nature Structural & Molecular Biology. 2013;20(1):127–133. doi: 10.1038/nsmb.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matia-González AM, Laing EE, Gerber AP. Conserved mRNA-binding proteomes in eukaryotic organisms. Nature Structural & Molecular Biology. 2015;22(12):1027–1033. doi: 10.1038/nsmb.3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despic V, et al. Dynamic RNA-protein interactions underlie the zebrafish maternal-to-zygotic transition. Genome Research. 2017;27(7):1184–1194. doi: 10.1101/gr.215954.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels HH, et al. The mRNA-bound proteome of the early fly embryo. Genome Research. 2016;26(7):1000–1009. doi: 10.1101/gr.200386.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sysoev VO, et al. Global changes of the RNA-bound proteome during the maternal-to-zygotic transition in Drosophila. Nature Communications. 2016;7:12128. doi: 10.1038/ncomms12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichel M, et al. In planta determination of the mRNA-binding proteome of Arabidopsis etiolated seedlings. Plant Cell. 2016;28(10):2435–2452. doi: 10.1105/tpc.16.00562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marondedze C, Thomas L, Serrano NL, Lilley KS, Gehring C. The RNA-binding protein repertoire of Arabidopsis thaliana. Scientific Reports. 2016;6:29766–29778. doi: 10.1038/srep29766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, et al. UV crosslinked mRNA-binding proteins captured from leaf mesophyll protoplasts. Plant Methods. 2016;12:42–53. doi: 10.1186/s13007-016-0142-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnik EM, et al. The mRNA-bound proteome of the human malaria parasite Plasmodium falciparum. Genome Biology. 2016;17:147–164. doi: 10.1186/s13059-016-1014-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lueong S, Merce C, Fischer B, Hoheisel JD, Erben ED. Gene expression regulatory networks in Trypanosoma brucei: insights into the role of the mRNA-binding proteome. Molecular Microbiology. 2016;100(3):457–471. doi: 10.1111/mmi.13328. [DOI] [PubMed] [Google Scholar]
- Nandan D, et al. Comprehensive identification of mRNA-binding proteins of Leishmania donovani by interactome capture. PLoS ONE. 2017;12(1):e0170068. doi: 10.1371/journal.pone.0170068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky E, Harris ME. Specificity and nonspecificity in RNA-protein interactions. Nature Reviews Molecular Cell Biology. 2015;16(9):533–544. doi: 10.1038/nrm4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R, Han M, Meng L, Chen X. Transcriptome-wide discovery of coding and noncoding RNA-binding proteins. Proceedings of the National Academy of Sciences of the United States of America. 2018;115(17):E3879–E3887. doi: 10.1073/pnas.1718406115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nature Protocols. 2006;1(6):2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, Heck AJR. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nature Protocols. 2009;4(4):484–494. doi: 10.1038/nprot.2009.21. [DOI] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nature Protocols. 2007;2(8):1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology. 2008;26(12):1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammel M, Hang H, Conrad NK. Chemical reporters for monitoring RNA synthesis and poly(A) tail dynamics. ChemBioChem. 2012;13(8):1112–1115. doi: 10.1002/cbic.201200091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curanovic D, et al. Global profiling of stimulus-induced polyadenylation in cells using a poly(A) trap. Nature Chemical Biology. 2013;9(11):671–673. doi: 10.1038/nchembio.1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YX, Beal PA. Synthesis and evaluation of an alkyne-modified ATP analog for enzymatic incorporation into RNA. Bioorganic & Medicinal Chemistry Letters. 2016;26(7):1799–1802. doi: 10.1016/j.bmcl.2016.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nainar S, et al. Metabolic incorporation of azide functionality into cellular RNA. ChemBioChem. 2016;17(22):2149–2152. doi: 10.1002/cbic.201600300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X, et al. Capturing the interactome of newly transcribed RNA. Nature Methods. 2018;15(3):213–220. doi: 10.1038/nmeth.4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmqvist E, Vogel J. RNA-binding proteins in bacteria. Nature Reviews Microbiology. 2018. Published online. [DOI] [PubMed]