

Abstract
The biological small-angle neutron scattering instrument at the High-Flux Isotope Reactor of Oak Ridge National Laboratory is dedicated to the investigation of biological materials, biofuel processing, and bio-inspired materials covering nanometer to micrometer length scales. The methods presented here for investigating physical properties (i.e., size and shape) of membrane proteins (here, MmIAP, an intramembrane aspartyl protease from Methanoculleus marisnigri) in solutions of micelle-forming detergents are well-suited for this small-angle neutron scattering instrument, among others. Other biophysical characterization techniques are hindered by their inability to address the detergent contributions in a protein-detergent complex structure. Additionally, access to the Bio-Deuteration Lab provides unique capabilities for preparing large-scale cultivations and expressing deuterium-labeled proteins for enhanced scattering signal from the protein. While this technique does not provide structural details at high-resolution, the structural knowledge gap for membrane proteins contains many addressable areas of research without requiring near-atomic resolution. For example, these areas include determination of oligomeric states, complex formation, conformational changes during perturbation, and folding/unfolding events. These investigations can be readily accomplished through applications of this method.
Keywords: Biochemistry, Issue 140, Small angle scattering, neutron scattering, biophysics, membrane protein, contrast variation, surfactant, micelle, intramembrane aspartyl protease, presenilin, signal peptide peptidase
Introduction
Membrane proteins are encoded by an estimated 30% of all genes1 and represent a strong majority of targets for modern medicinal drugs.2 These proteins perform a wide array of vital cellular functions,3 but despite their abundance and importance — only represent about 1% of total structures deposited in the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank.4 Due to their partially hydrophobic nature, structural determination of membrane-bound proteins has been exceedingly challenging.5,6,7
As many biophysical techniques require monodisperse particles in solution for measurement, isolating membrane proteins from native membranes and stabilizing these proteins in a soluble mimic of the native membranes has been an active area of research in recent decades.8,9,10 These investigations have led to the development of many novel amphiphilic assemblies to solubilize membrane proteins, such as nanodiscs,11,12,13 bicelles,14,15 and amphipols.16,17 However, the use of detergent micelles remains one of the most common and straightforward approaches for satisfying the solubility requirements of a given protein.18,19,20,21,22,23,24,25 Unfortunately, no single detergent or magic mixture of detergents currently exists that satisfies all membrane proteins; thus, these conditions must be empirically screened for the unique requirements of each protein.26,27
Detergents self-assemble in solution above their critical micelle concentration to form aggregate structures called micelles. Micelles are composed of many detergent monomers (typically ranging from 20-200) with hydrophobic alkyl chains forming a micelle core and hydrophilic head groups arranged in a micelle shell layer facing the aqueous solvent. The behavior of detergents and micelle formation has been classically described by Charles Tanford in The Hydrophobic Effect,28 and sizes and shapes of micelles from commonly used detergents in membrane protein studies have been characterized using small-angle scattering.29,30 Detergent organization about membrane proteins has also been studied, and the formation of protein-detergent complexes (PDCs) is expected with detergent molecules surrounding the protein in an arrangement that resembles the neat detergent micelles.31
One added advantage in using detergents is that the resulting micelle properties can be manipulated by incorporating other detergents. Many detergents exhibit ideal mixing, and select properties of mixed micelles may even be predicted from the components and ratio of mixing.22 However, the presence of detergent can still present challenges for biophysical characterizations by contributing to the overall signal. For example, with X-ray and light scattering techniques, signal from detergent in the PDC is practically indistinguishable from protein.32 Investigations with single-particle cryo-electron microscopy (cryo-EM) typically rely on trapped (frozen) particles; structural details of the protein are still obscured by certain detergents or a high concentration of detergent which adds to the background.33 Alternate approaches toward interpreting the full PDC structure (including the detergent) have been made through computational methods which seek to reconstruct the detergent around a given membrane protein.34
For the case of neutron scattering, the core-shell arrangement of detergent in the micelle produces a form factor which contributes to the observed scattering. Fortunately, solution components can be altered such that they do not contribute to the net observed scattering. This "contrast matching" process is achieved by substituting deuterium for hydrogen to achieve a scattering length density that matches that of the background (buffer). A judicious choice of detergent (with available deuterated counterparts) and their ratio of mixing must be considered. For detergent micelles, this substitution can be performed using a detergent with the same head group but having a deuterated alkyl chain (d-tail instead of h-tail). Since the detergents are well-mixed,35 their aggregates will have a scattering length density that is the mole-fraction weighted average of the two components (h-tails and d-tails). When this average contrast is consistent with that of the head group, the uniform aggregate structures can be fully matched to remove all contributions to observed scattering.
We present here a protocol to manipulate the neutron contrast of detergent micelles by incorporating chemically identical detergent molecules with deuterium-labeled alkyl chains.19,36,37 This permits complete simultaneous contrast matching of micelle core and shell, which is a unique capability of neutron scattering.35,38 With this significantly refined level of detail, contrast matching can enable otherwise unfeasible studies of membrane protein structures. Additionally, this contrast-matching approach could be extended to other systems involving detergent, such as polymer exchange reactions39 and oil-water dispersants,40 or even other solubilizing agents, such as bicelles,41 nanodiscs,42 or block copolymers.43 A similar approach as outlined in this manuscript, but employing a single detergent species with partial deuterium substitutions on the alkyl chain and/or head group, was recently published.37 While this can be expected to improve the random distribution of hydrogen and deuterium throughout the detergent compared to the approach presented here, the limited number of available positions on the detergent for substitution and two-step detergent synthesis required poses additional challenges for consideration.
Steps 1 and 2 of the protocol detailed below often overlap since initial experiment planning must be done to submit a quality proposal. However, proposal submission is considered here as the first step to emphasize that this process should be started well in advance of a neutron experiment. It should also be noted that a prerequisite step, which should be demonstrated by the proposal, is to have biochemical and physical characterization (including purity and stability) of the sample supporting the need for neutron studies. A general discussion of small-angle neutron scattering (SANS) is beyond the scope of this article. A brief but thorough introduction is available in the reference work Characterization of Materials by Kaufmann,44 and a comprehensive textbook focused on biological small-angle solution scattering has recently been published.45 Further recommended reading is given in the Discussion section. Small angle scattering uses the so called scattering vector Q as the central quantity that describes the scattering process. This article uses the widely accepted definition Q = 4π sin(θ)/λ, where θ is half the angle between incoming and scattered beam and λ is the wavelength of the neutron radiation in Angstroms. Other definitions exist that use different symbols such as 's' for the scattering vector, and that may differ by a factor 2π or by using nanometers in place of Angstrom (see also discussion of Figure 10).
Protocol
1. Prepare and Submit a Neutron Facility Beam Time and Instrument Proposal
Consult online resources for identifying neutron scattering facilities that provide general user neutron beam time access, such as Oak Ridge National Laboratory (ORNL). A map of neutron facilities and information about neutron research worldwide is available online.46 Be aware that these facilities typically have regular calls for proposals; this determines when the next beam time will be available. Neutrons are used for a variety of applications; search for small-angle neutron scattering (SANS) instruments, particularly those with capabilities for biological samples.
Use resources provided by the neutron facilities to help navigate the neutron beam time proposal submission process. Consult with a Neutron Scattering Scientist (NSS) that is associated with the instrument for which you will apply. Review the neutron facility’s current information regarding proposal calls, deadlines, and advice for submission; for Oak Ridge this is available online.47 Submit the beam time proposal following all guidelines for a successful submission.
If deuterated protein is required (see 2.2), neutron scattering facilities are often supported by laboratories and expertise dedicated to the production of deuterated materials. To request access to the Bio-Deuteration Lab (BDL) at ORNL, select the BDL as the second instrument in the proposal system. While the SANS proposal is reviewed for feasibility and by a scientific review committee, BDL requests are only screened for feasibility. To help with the BDL feasibility review, submit a completed information request form48 that describes the protein expression protocol and estimated yield (available online).
After successful peer-review of the beam time proposal, confirm the awarded date of beam time in advance of the experiment. Ensure that all sample and buffer details and formulas are listed correctly in the proposal for proper review. Any changes to the proposed experimental plan, including changes in sample and buffer composition, require notification of the facility. NOTE: Changes should be discussed as soon as possible with the assigned NSS.
2. Determine Neutron Contrast Match Points and Necessary Contrast for Protein Measurement
Gather information (from supplier’s product information, online databases, published values, etc.) relating to atomic compositions and volumes for the detergent system components to be contrast-matched. This information is used to determine neutron scattering length densities (SLDs) and thus contrast match points (CMPs) in solution, which is critical to obtaining quality SANS data and structural information for the membrane protein of interest in the context of a PDC. A table summarizing the physical properties and contrast match points for detergents commonly used in biophysical studies of membrane proteins is included (Table 1).
- Determine neutron SLDs using the web application MULCh: ModULes For The Analysis Of Contrast Variation Data49 (available online50 free-of-charge). A link to the user manual is located on the home page. Access the website and read the accompanying documentation. Figure 1 and Figure 2 provide an overview of Contrast module input and output for two related examples. NOTE: Other websites for calculating SLDs are available, such as the one maintained by the National Institute of Standards and Technology (NIST) Center for Neutron Research.51
- Open the Contrast module by clicking ‘Contrast’ located in the left navigation pane. Provide text for a project title and enter details below for two components (e.g., detergent head group and tail) as ‘subunit 1’ and ‘subunit 2’.
- Enter the molecular formula for each component in the ‘Formula’ text box. NOTE: The radio button ‘M’ must be selected under ‘Substance Type’ to enter a molecular formula. Additionally, use the letter ‘X’ in the formula to indicate readily exchangeable hydrogen atoms. Protein, RNA, or DNA sequences can be entered if the corresponding ‘P’, ‘R’, or ‘D’ radio buttons are selected. If multiple copies of a component exist within the subunit, the value for ‘Nmolecules’ can be changed accordingly. A practical example here relates to lipid-like detergents containing two identical tails, allowing the formula for one tail to be entered with Nmolecules equal to 2.
- Enter the volume in cubic Angstroms for each component in the box below ‘Volume (Å3)’. Use Tanford’s formula28 for alkyl chains of length n, Vtail = 27.4 + n * 26.9, to calculate approximate tail volume, or obtain molecular volumes from product information provided or published values in the literature.
- Enter details for buffer components. Change the ‘Number dissolved species in the solvent’ using the dropdown menu to add or remove rows for each buffer component. In the case of micelle-forming detergents, include the free detergent monomers as separate buffer components at the detergent’s critical micelle concentration (CMC).
- Click ‘Submit’ to perform the neutron contrast calculations and generate a results page that provides a table of scattering length density and contrast match points as well as formulae for determining these parameters at any given percentage of D2O in the buffer. Review the scattering parameters with particular attention to the component CMPs. If the match points are similar (within 10% D2O) and the free micelle concentration is low, then the average CMP can be used for contrast-matching the detergent contributions. In most cases, the CMP of the detergent head group and tail will differ by more than 10-15%, producing a core-shell form factor in the SANS data which obfuscates direct collection of the scattering signal from the protein alone.
- Assess the contrast between detergent head group and tail. When the detergent head group and alkyl chain tail CMPs are not well-matched, the availability and incorporation of a deuterium-substituted detergent can permit scattering length densities of select components to be manipulated. In most cases, this will require forming a mixed micelle through the incorporation of an identical detergent with deuterium-substituted alkyl chains. The addition of deuterium raises the scattering length density of the core formed by the alkyl chain tails. The desired endpoint, in this case, is such that the head group shell CMP and alkyl chain core CMP have approximately equal values.
- Raise the CMP of the detergent micelle core to be approximately equal to the CMP of the head group shell by determining the appropriate ratio of mixing between h-tails and d-tails that achieves complete contrast matching with the head groups. A prerequisite for this determination is knowledge of the CMP for a deuterium-substituted alkyl chain tail. A commercially-available counterpart to n-Dodecyl β-D-maltoside (DDM) is available with all alkyl chain hydrogen replaced by deuterium (d25-DDM). Repeat the contrast calculations outlined in step 2.1 for a ‘d-tail’ in place of the ‘h-tail’.
- Calculate the mole ratio of h-tail to d-tail required for contrast-matching with the head group CMP. The following formula can assist with this determination: CMPhead = CMPh-tail * χh-tail + CMPd-tail * χd-tail where CMP is the scattering length density and χ is the volume fraction for the detergent head, h-tail, or d-tail component. For buffered solutions of DDM, incorporation of 44% by weight (43% by mole) of the total detergent as d25-DDM with deuterium-substituted alkyl chains produces a single CMP in 48.5% D2O for both head and tail components.
Consider the degree of deuterium substitution for the membrane protein. To measure sufficient scattering signal from the protein, the CMP for the protein should be at least 15% D2O in solution away from the detergent CMP and measurement conditions. The signal increases with the square of this% difference. Since the CMP of an unlabeled protein typically occurs with ~42% D2O in solution (a CMP similar to many detergent head groups), deuterium labeling of the protein is almost always a necessity.52
3. Express and Purify the Membrane Protein of Interest
- Prepare LB (lysogeny broth) medium and grow Escherichia coli (E. coli) cells harboring an inducible expression vector for the target protein sequence.
- Prepare minimal media. In either H2O or D2O, dissolve 7.0 g/L (NH4)2SO4, 5.25 g/L Na2HPO4, 1.6 g/L KH2PO4, 0.50 g/L diammonium hydrogen citrate, 5.0 g/L glycerol, 1.0 mL/L of 20% w/v MgSO4·7H2O, and 1.0 mL/L of Holme trace metals (0.50 g/L CaCl2·2H2O, 0.098 g/L CoCl2, 0.102 g/L CuSO4, 16.7 g/L FeCl3·6H2O, 0.114 g/L MnSO4·H2O, 22.3 g/L Na2EDTA·2H2O, and 0.112 g/L ZnSO4·H2O).53,54
- Prepare appropriate antibiotic stock solutions using H2O or D2O.
- Prepare an isopropyl-β-D-1-thiogalactopyranoside stock solution using H2O or D2O.
- Sterile-filter all solutions into dry, sterile containers using bottle-top vacuum and syringe filters with a 0.22 micron pore size.
- Adapt E. coli cells to deuterium-labeled medium prior to scale-up for overexpression of a deuterated protein.
- Inoculate 3 mL of LB medium with an isolated colony from a Lysogeny Broth (LB) agar plate or cells from a frozen glycerol stock. Grow cells at 37 °C in a shaking incubator at 250 RPM or reduce the temperature to 30 °C or below to avoid overgrowth of the culture when incubating overnight. NOTE: The adaptation procedure can be varied.55,56,57
- Once the LB culture has grown to an optical density at 600 nm (OD600) of ~1, dilute it 1:20 in 3 mL of H2O minimal medium and grow to an OD600 of ~1. Repeat the 1:20 dilutions using minimal medium containing 50, 75 and 100% D2O (or up to the desired D2O percentage). NOTE: As the D2O content is increased, growth rates will decrease. The relationship between the D2O percentage in the growth media and deuterium substitution in the protein has been reported in the literature.58,59,60
- Harvest and lyse E. coli cells for membrane extraction and protein purification
- Pellet the cells by centrifugation at ~6,000 x g for ~30-45 min at 4 °C.
- Soften the cell pellet by soaking in buffer on ice for ~30 min. Then, gently resuspend the cell pellet in buffer by gently pipetting with a serological pipet, or gentle rocking on a 2D rocker, to a final concentration of ~1 g wet cell mass/10 mL buffer. NOTE: An example buffer is 50 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), pH 7.5, 200 mM NaCl supplemented with a protease inhibitor tablet.
- Lyse resuspended cells with three passes through a high-pressure homogenizer at 10,000 psi while chilling the outflow. NOTE: Depending on the scale required, other lysis methods may be used (e.g., by French press or sonication).
- Pellet cell debris by centrifugation at ~4,000 - 5,000 x g for 15 min at 4 °C. Repeat this step until the supernatant is clarified.
- Ultracentrifuge (~150,000 x g for 30-45 min) the supernatant from the prior step, which contains the soluble and membrane fractions. The pellet after ultracentrifugation contains the membrane fraction.
- Resuspend the membrane pellet in a large Dounce homogenizer and isolate the membrane fraction again by ultracentrifugation (step 3.3.5). Repeat this step at least once to remove loosely bound proteins.
- Solubilize membranes by gentle rocking for ~30 min at 4 °C in a buffer containing detergent suitable for purification. Solubilization is apparent when the suspension turns from cloudy to transparent. Remove insoluble material by ultracentrifugation (~150,000 x g for 30-45 min). NOTE: An example buffer is 50 mM HEPES, pH 7.5, 500 mM NaCl, 20 mM imidazole and 4% (w/v) DDM for purification by Ni2+ affinity chromatography.
Purify the protein following a protocol previously developed for protonated forms. NOTE: See Naing et al. for an example purification protocol for a hexahistidine tagged M. marisnigri JR1 intramembrane aspartyl protease (d-MmIAP).65 The final yield was ~ 1 mg of deuterated from 5 L deuterated cell culture growth, all of which was used in the SANS experiment (Figure 4).
- Exchange the purified protein into the “Final Exchange Buffer” for contrast matching.
- Prepare 200 mL of “Final Exchange Buffer” containing the correct mixture for contrast matching, e.g.,20 mM HEPES, pH 7.5, 250 mM NaCl, and 0.05% total DDM (0.028% DDM and 0.022% d25-DDM) in 49% D2O.
- Equilibrate a size exclusion column with >2 column volumes (CV) of “Final Exchange Buffer” in 3.5.1 using a Fast Protein Liquid Chromatography (FPLC) system.
- After injecting the sample, run the column and isolate fractions corresponding to the protein of interest.
- Concentrate the protein to the desired final concentration (2 - 5 mg/mL). NOTE: A volume of ~300 μL is required to fill a 1 mm cylindrical quartz cuvette used for SANS.
- Reserve an aliquot of matched buffer for SANS background subtraction. NOTE: The aliquot can be taken directly from the prepared buffer, or ideally from a clean elution fraction at the end of the FPLC run.
4. Make Final Preparations for Beam Time and Collect SANS Data
After the proposal has been accepted and site access has been approved, complete all required training in advance of assigned beam time. This includes prearrival, web-based training as well as on-site radiological, laboratory, and instrument-specific training. NOTE: Training requirements may dictate advance arrival for first time users. More information is available online.66
- Load samples and buffers for data collection. NOTE: An overview of the biological small-angle neutron scattering (Bio-SANS) instrument sample environment area is shown in Figure 5.
- Load samples and buffers into quartz cells. Quartz cells are available from the instrument scientist or facility. Use gel-loading tips to access the narrow opening of most sample cells.
- Ensure that the beam shutter is closed, then approach the sample environment area and place the quartz cells in the sample changer. Record the sample changer position for each sample cell.
- Check the area and ensure the beam path is free from obstruction, then leave the sample environment area and open the beam shutter. NOTE: Carefully follow all facility rules and regulations, obey all postings, and heed advice of the instrument scientist. Samples may not be removed from the beam and manipulated after exposure to the beam without following special precautions. Consult with a Radiological Control Technician (RCT) prior to these procedures.
- Execute table scan for automated data collection
- Operate the instrument through custom control LabVIEW-based software, Spectrometer Instrument Control Environment (SpICE). Follow instructions for instrument operation provided by the Neutron Scattering Scientist. An overview of the table scan operation windows is provided in Figure 6.
- After entering the information required in the appropriate areas, execute the table scan and an automated data collection process will begin. Monitor progress via the ‘Table Scan Status’ tab.

5. Reduce SANS Data from 2D Image to 1D Plot
Identify each sample and buffer data file from the recorded scan numbers. Each measurement will be assigned a unique scan number. This information is helpful during data reduction to identify each corresponding sample and buffer pair.
- Use MantidPlot software and Python script for reduction
- Obtain a User Reduction Script from the Neutron Scattering Scientist (this will usually happen during the experiment); this script is Python-based and will contain all the necessary calibration and scaling adjustments to reduce the 2D scattering image into a 1D plot of scattered intensities as a function of scattering angle, I(Q).
- Open the provided User Reduction Script in MantidPlot and place the corresponding scan numbers or identification for the sample and buffer pairs in the appropriate list.
- Execute the script to generate reduced data as a four-column text file (column order is Q, I(Q), I(Q) error, Q error) in the specified location. Right click the appropriate workspace in MantidPlot and select “Plot with errors…” for an initial examination of the scattering profiles.
6. Analyze Data for Structural Parameters of the Scattering Particle
Transfer the reduced data file from the analysis server to a local computer using secure FTP file transfer. This can be performed with the use of software such as FileZilla or CyberDuck. Additional instructions and details for connecting to the analysis server file system are provided on the analysis.sns.gov login page.
Download the ATSAS software suite65,68 (whole ATSAS suite).69 Individual programs70 are also available online for analyzing the SANS data, namely determining structural parameters and ab initio models. NOTE: Many options are available for SANS data analysis of biomacromolecules.45
- Use PRIMUS71 for data plotting, buffer scaling and subtraction, and Guinier analysis.
- Launch the ATSAS ‘SAS Data Analysis’ application and load the reduced data files corresponding to the sample and buffer pair.
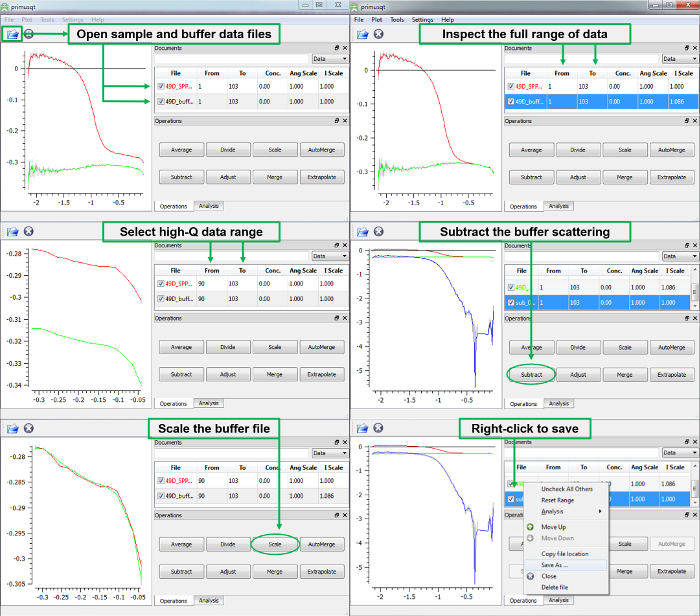
- To scale the buffer properly, select a data range at high Q (Q > 0.5 Å-1) where both profiles are similar and flat, and click the ‘Scale’ button located under the ‘Operations’ tab. A scale factor will be applied to the buffer such that these two flat regions should overlap. Use caution: there should be a plausible physical reason that justifies scaling the buffer intensity to match the sample. If the discrepancy is large, consult an instrument scientist for valid approaches to background subtraction.44,45,72
- Increase the data range to view all points. Click ‘Subtract’ to perform this operation. Right click the buffer-subtracted datafile in the legend and save this file for subsequent analysis. Figure 9 outlines the use of the ‘Operations’ tab.
- Perform a Guinier analysis of the buffer-subtracted sample data using the ‘Analysis’ tab of the ‘SAS Data Analysis’ application.
- Be sure the proper file is selected in the list and click ‘Radius of Gyration’. An automated attempt to perform a Guinier fit will be provided by clicking on the ‘Autorg’ button.
- Expand the range of data used to include all of the low-Q data and begin narrowing the data range by taking away high-Q points until the Qmax*Rg limit is below 1.3. Use the plot of residuals to verify that data are linear in the fit range. The Guinier fit yields an approximate Rg and I0 for the scattering particles.
- Make small adjustments to the fit region and monitor the sensitivity of these values to the range of data used in the fit. Figure 10A demonstrates the use of the ‘Radius of Gyration’ tool.
- Obtain the probability distribution function (P(r)) in GNOM.73 The output file generated here will be used as an input for the ab initio modeling process.
- Start the Distance Distribution Wizard within the ‘Analysis’ tab. Obtaining a good fit to the data is essential to obtaining a quality model. For more information on obtaining accurate fits, please see the review74 by Putnam, et al. NOTE: The GNOM program provides additional information about the fit beyond the Distance Distribution Wizard. Figure 10B demonstrates proper use of the ‘Distance Distribution’ tool, and Figure 11 illustrates some of the common errors encountered.
- Determine Dmax, the maximum interatomic distance within the molecule.
- Estimate a value for Dmax by unchecking the box to force Rmax=0 and entering a large value for Dmax (150 Å, for example). The first x-intercept in the plot of P(r) yields this estimate.
- Make incremental changes to this Dmax value and the range of data used or number of points used in the fit to optimize the GNOM fit to the data and the resulting P(r) curve.
- Continue to refine the GNOM fit and save the GNOM output files with good fit parameters for subsequent ab initio modeling steps.
- Simulate SANS profiles from high resolution PDB models using the program CRYSON.75
- Open the program and select option ‘0’. Select the PDB file name in the popup file browser. Press enter to accept the default settings, except for specifying chain deuteration fractions and the fraction of D2O in the solvent to achieve the proper contrast parameters.
- Fit the model to an experimental data set by entering ‘Y’ and selecting the data file in the popup file browser. Accept the remaining default values, and output files will be written in the PDB file location. The ‘.fit’ file contains information for the predicted scattering profile from the high-resolution 3D model.
7. Create Ab Initio Models from the SANS Data.
NOTE: DAMMIF76 and DAMMIN77 within the ATSAS software suite is used to reconstruct dummy atom models (DAMs) using a simulated annealing process from the GNOM output, which contains P(r) data, or information about the probability or frequency of interatomic distances within the scattering particle. These programs may be run in batch mode or on the ATSAS-Online web server.
- Start the PRIMUS Shape Wizard from the ‘Dammif’ button on the ‘Analysis’ tab of ‘SAS Data Analysis’, and use a manual selection of parameters. The input for the wizard is illustrated in Figure 12.
- Define the Guinier range from the fit (step 6.3.2) and proceed to the next step using the navigation buttons. Define fit values from the P(r) plot (step 6.4) and proceed to the next step.
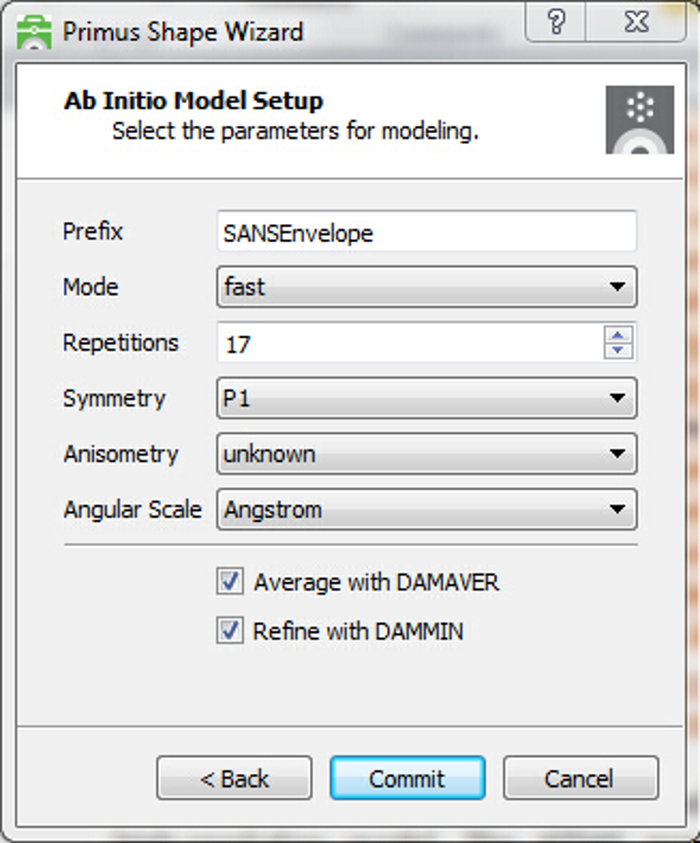
- Provide parameters for the ab initio modeling process. Enter a prefix for the model output filenames (e.g., SANSEnvelope), choose either ‘fast’ or ‘slow’ mode modeling, enter a value for the number of models (17 recommended for statistical significance), select from available options for particle symmetry, anisometry, and angular scale (if known). Ensure that the boxes are checked to average models with DAMAVER and refine the final model with DAMMIN.
- Initiate the process using the ‘commit’ button. Once the process is complete, view and save the working directory. NOTE: The typical modeling process for a single, small protein can take up to a few hours with a typical personal computer. Files are stored in temporary directories until saved.
Overlay and superimpose the final SANS envelope with a related high-resolution model using SUPCOMB within ATSAS. Place a copy of the high-resolution PDB model to be fit to the SANS envelope in the working directory. Execute SUPCOMB from the command line using the PDB filenames as two arguments with the template structure listed first: $ supcomb HiRes.pdb SANSEnvelope.pdb NOTE: The second filename listed will be rewritten as a new file with an “r” appended to the end and having new coordinates for superimposition on the template structure.
- Visualize the ab initio model results using PyMOL (pymol.org), a 3D molecular graphics viewing program. Figure 13 provides an overview of the PyMOL operations. NOTE: There are publication guidelines for structural modeling of small-angle scattering data from biomolecules in solution.78
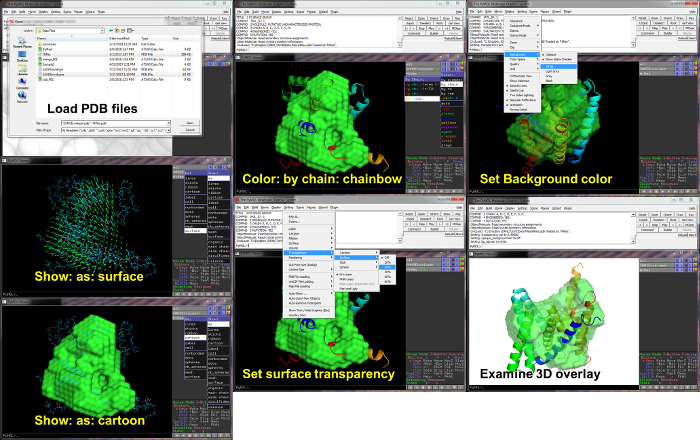
- Launch the PyMOL application and open the .pdb files corresponding to the 3D SANS envelope and the SUPCOMB-aligned high-resolution structure to be viewed.
- Visualize the SANS envelope by representing this model as a surface. Click the ‘S’ button next to the model and select ‘Show: As: Surface’.
- Visualize the high-resolution protein backbone structure by representing this model as a cartoon. Select the ‘S’ button next to this model and select ‘Show: As: Cartoon’. Display the chain as a rainbow from N- to C- terminus by selecting the ‘C’ button and ‘By Chain: Chainbows’.
- Modify the transparency of the surface representation for clarity. From the ‘Setting’ menu bar, select ‘Transparency » Surface » 40%’.
- Make the background white and non-opaque. From the ‘Display’ menu bar, select ‘Background »’ and select ‘Opaque’ to uncheck this option and ‘White’ to mark this color for background. NOTE: Additional operations and uses for PyMOL can be found at PyMolWiki.org.
Representative Results
A beam time and instrument proposal should clearly convey all information needed to the review committee so that a valid assessment of the proposed experiment can be made. Communication with an NSS is highly suggested for inexperienced users. The NSS can assess initial feasibility and guide proposal submission to emphasize feasibility, safety, and the potential for high-impact science. The information provided in the proposal should include background information and context for the significance of the research; knowledge that is expected to be gained and how this impacts the current understanding in the related field of science; a description of the work, samples, methods, and procedures that will be employed; and, if applicable, previous productivity of the team at the facility, including relevant publications and results. Useful resources, such as proposal templates and tips for preparing the proposal, are available to Users via the Neutron Science User Portal (neutrons.ornl.gov/users).
Experiment planning is a dynamic process that often begins during the initial stages of proposal submission, but may not be fully conceived until just prior to the experiment. However, keep in mind that any changes that deviate significantly from the description in the proposal (including changes to buffer conditions or sample composition) must be approved by the facility prior to the start of the experiment.
This protocol assumes that a method for expressing and purifying the membrane protein of interest into detergent micelles in solution has been successfully demonstrated. In this case, the membrane protein of interest is an intramembrane aspartyl protease (IAP), which has an established purification protocol and has been previously determined to be soluble and active in buffer containing DDM micelles.
Here, we demonstrate neutron contrast calculations using the MULCh: Contrast module for a solution of IAP protein in contrast-matched mixed DDM / d25-DDM micelles. The strategy outlined herein was to first determine the degree of mixing between the two detergents necessary to achieve a complete contrast match of the micelle. The end result was to determine the relative contrast of each component and the background (H2O/D2O ratio) necessary to contrast-match the scattering from detergent in order to observe only the protein scattering.
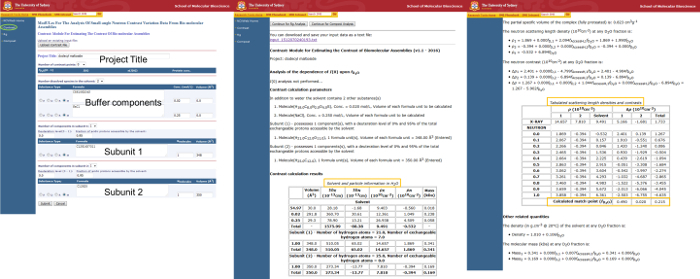
Figure 1 demonstrates a proper input for the MULCh Contrast module and the resulting output for contrast calculations of the DDM detergent head group and tail as subunits 1 and 2, respectively. The formula used for the head group is C12H14X7O11 with a volume of 348 Å3, and the alkyl chain tail formula is given as C12H25 with a volume of 350 Å3.
It is apparent from the Contrast module output that the DDM head group and tail have different scattering length densities, and thus contrast between the two components will be observed. For DDM, the head groups have a CMP in 49% D2O while the alkyl chain tails have a CMP in 2% D2O, with their average CMP occurring in 22% D2O. Therefore, the aim of the next step will be to design a mixed micelle that incorporates deuterium-substituted alkyl chains to increase the average CMP of the detergent tails to match the CMP of the head groups. The contrast calculation was repeated for a substituted detergent, DDM with a fully-deuterated tail (d25-DDM), which similarly results in contrast between the head group and tail. However, contrast values of the h-tails and d-tails are significantly different. Recall that detergent mixtures are known to produce well-mixed micelles in solution,22 thus a blend of these h- and d-tails in the micelle core should produce an average SLD equal to that of the head group shell, yielding a micelle with single CMP. Since the head group is common to both DDM and d25-DDM, the strategy is to find a mixture of these detergents that produces an average contrast value from the mixed tails that matches the contrast of the head groups.
The target average CMP for the detergent tails is that of the maltoside head groups, or 49% D2O. To estimate the ratio of mixing the average CMP of each h-tail and d-tail component must be known. These values for some common detergents and commercially-available deuterium labelled counterparts are provided in Table 1. Using these CMP values for DDM and d25-DDM, the mole fraction of h-tails and d-tails that satisfies the equation provided in step 2.2 is χd-tails = 0.43.
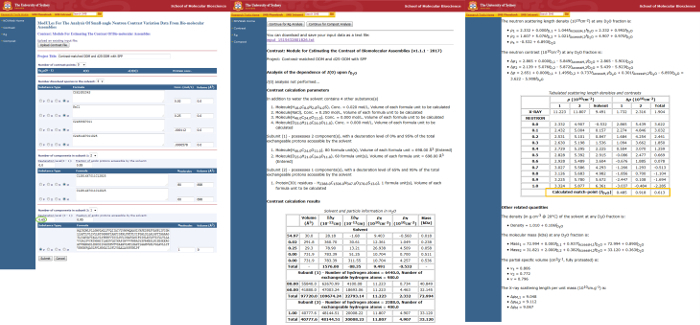
Figure 2 demonstrates a more advanced input to the MULCh Contrast module that can be used to confirm the final mixed-micelle contrast match conditions and determine the degree of deuteration necessary on the protein. Here, subunit 1 refers to the contrast-matched mixed micelle with 2 components, DDM and d25-DDM, while subunit 2 refers to the M. marisnigri JR1 intramembrane aspartyl protease (MmIAP) given by its amino acid sequence. The SLD of the protein has been elevated by expression in deuterated growth media to yield a CMP for the protein in buffer containing ~92% D2O. The degree of separation between the protein's match point in 92% D2O and the measurement condition (48.5% D2O) suggests that sufficient scattering signal will be obtained from the protein.
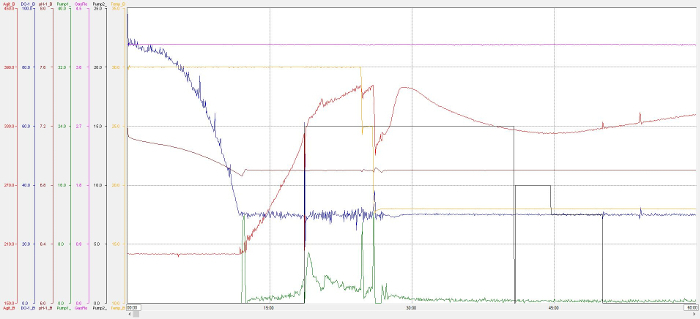
Production of d-MmIAP was carried out with Rosetta 2 E. coli cells harboring the pET22b-MmIAP vector. Minimal medium with unlabeled glycerol in 90% D2O was selected as the growth medium. After adaptation to 90% D2O minimal medium, the culture volume was scaled up to 400 mL and used to inoculate 3.6 L of fresh 90% D2O minimal medium in a 5.5 L bioreactor vessel.
Figure 3 provides a trace of the process values during the fed-batch cultivation. At the time of inoculation, the temperature set point was 30 °C, the dissolved oxygen (DO) set point was 100%, the agitation set point was 200 rpm, and the flow rate of compressed air was 4 L/min. The pH was held above a set point of 6.9 using a 10% w/v solution of sodium hydroxide in 90% D2O. Once the DO spike signaled depletion of the initial 5 g/L glycerol, feeding(30% w/v glycerol, 0.2% MgSO4 in 90% D2O) was initiated, which continued throughout the cultivation. After around 7 hours of feeding, the culture temperature was reduced to 18 °C and isopropyl-β-D-1-thiogalactopyranoside was added to a final concentration of 1 mM. Upon harvesting the culture, approximately 145 g wet weight of cell paste was collected via centrifugation (6,000 x g for 45 min)
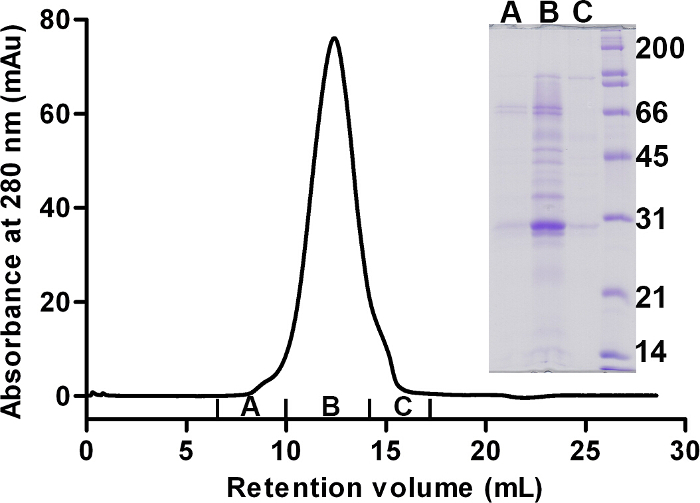
Purification of d-MmIAP proceeded as for the protonated enzyme except that enzyme yield was significantly lower and final purity was somewhat lower than typical. From 5 L, ~20 g of wet membranes was isolated, all of which was solubilized in DDM for purification and loaded onto the first Ni2+ affinity column. To maximize purification yield, the flow-through fraction was diluted and re-rerun on Ni2+ affinity column. The procedure was repeated a third time before polishing the concentrated d-MmIAP sample by size exclusion chromatography (Figure 4).
Figure 5 depicts a quartz sample cell and its related sample changer setup. Sample environments are managed by the Bio-SANS operations team and NSS. A variety of different environments can be arranged to perform measurements with temperature control, humidity control, mechanical tumbling, high temperature, and sustained pressure, among others.
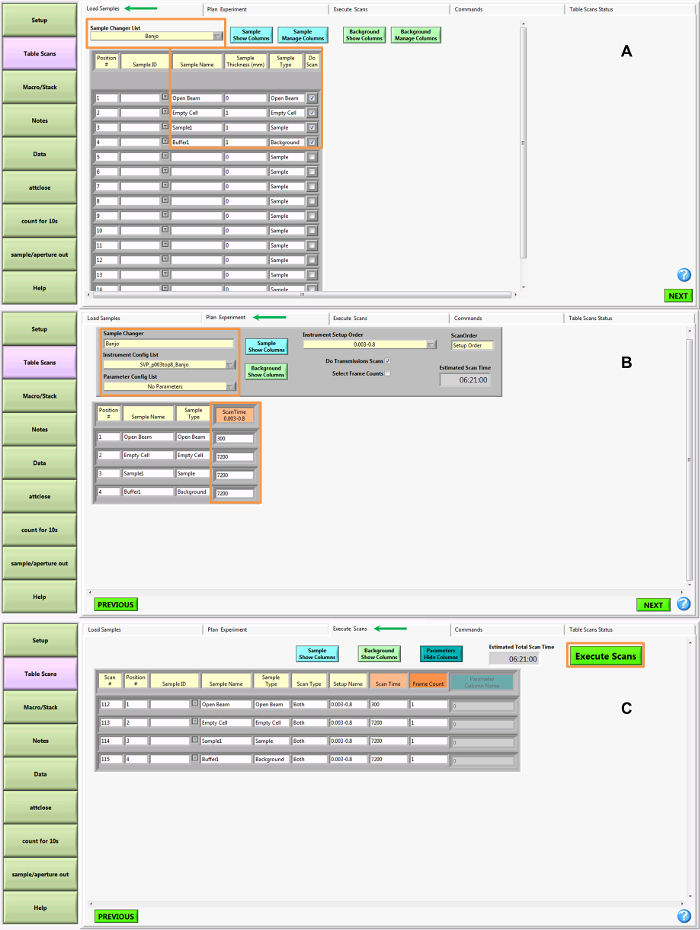
Figure 6 demonstrates Bio-SANS operations and execution of the automated table scans. The table scan is configured to be intuitive and user-friendly. On the first tab (Load Samples), information is provided about the sample changer and setup, such as identification of samples at each position in the sample changer, cell thicknesses, and sample type (open beam, sample, background, etc.). Additional metadata tags can be applied here also. On the next tab (Plan Experiment), the instrument configuration and any associated parameters (such as temperature control) are defined. This step is arranged by the NSS at the start of the experiment. The user must simply input the measurement times for each sample (in seconds). The final tab (Execute Scans) provides a summary of the table scan data and allows the automated scan to be executed.
After the 2D scattering images are recorded, this data must be reduced to a 1D plot of the intensity versus Q - a function of the scattering angle. During data reduction, image corrections such as pixel sensitivity masks and dark background subtractions are also applied. MantidPlot software was designed to interpret the raw Bio-SANS instrument data. The NSS will generally assist in reducing and retrieving the final data at the end of the experiment.
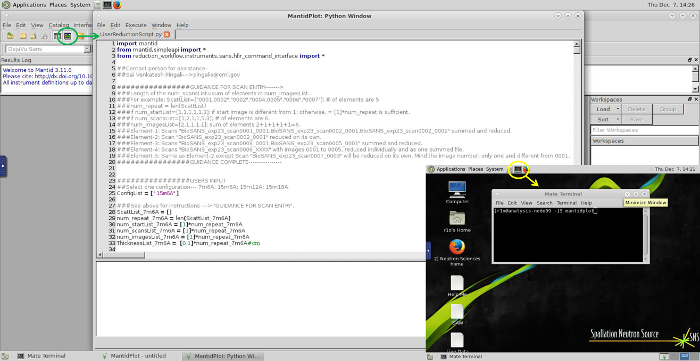
Figure 7 provides an overview of the remote analysis cluster access to MantidPlot and the Script Window used for data reduction. Raw data are accessible at the remote analysis cluster (analysis.sns.gov) via UCAMS/XCAMS user authentication. After logging into the remote desktop, the MantidPlot software can be launched from a terminal window by simply executing 'MantidPlot' under any path. Open the Script Window in MantidPlot and edit the User Reduction Script as directed by the NSS. Executing this script will generate the reduced data files, which can then be transferred to a local machine for analysis using secure FTP.
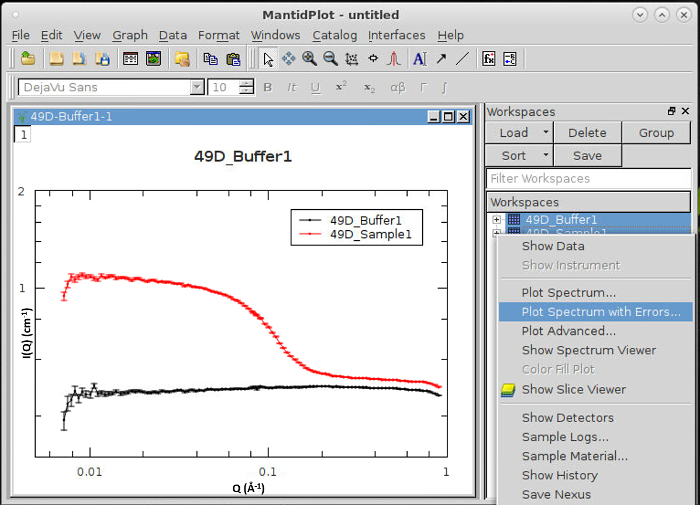
Figure 8 demonstrates plotting and viewing the data after executing the User Reduction Script. Reduced data will appear in the Workspaces window. By default, final merged data will have the suffix _f appended. Right-click will allow Plot Spectrum with Errors to be selected and data to be plotted. Display preferences are accessed by selecting Preferences from the 'View' menu bar. Formatting of axes and plotting range is easily accessible by double-clicking the axes. Scattering profiles of buffers, which contain no scattering particles of significant size should appear as a relatively flat line (constant intensity). For samples, intensity should be observed at lower scattering angles (Q) with exponential decay to a flat incoherent background.
Figure 9 provides an overview of proper buffer subtraction using the SAS Data Analysis (or primusqt) software package following data reduction. Quite often the incoherent background (intensity at high-Q) is slightly mismatched between the sample and buffer. For proper subtraction, data in this region should overlap. The scale correction applies an intensity scale factor to the data which results in overlapping data, and the subtraction can be performed. If the scale factor results in buffer data points with larger intensity than corresponding points in the sample, then these points will be negative and not rendered in a log plot. If negative values in the buffer-subtracted file are abundant, make a minor reduction in the buffer scale factor and perform the subtraction again.
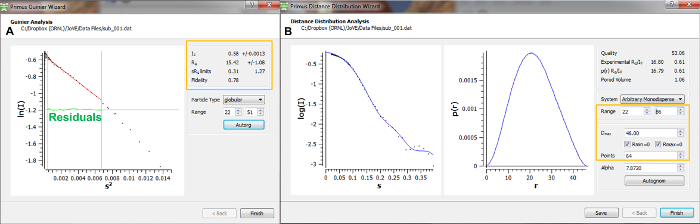
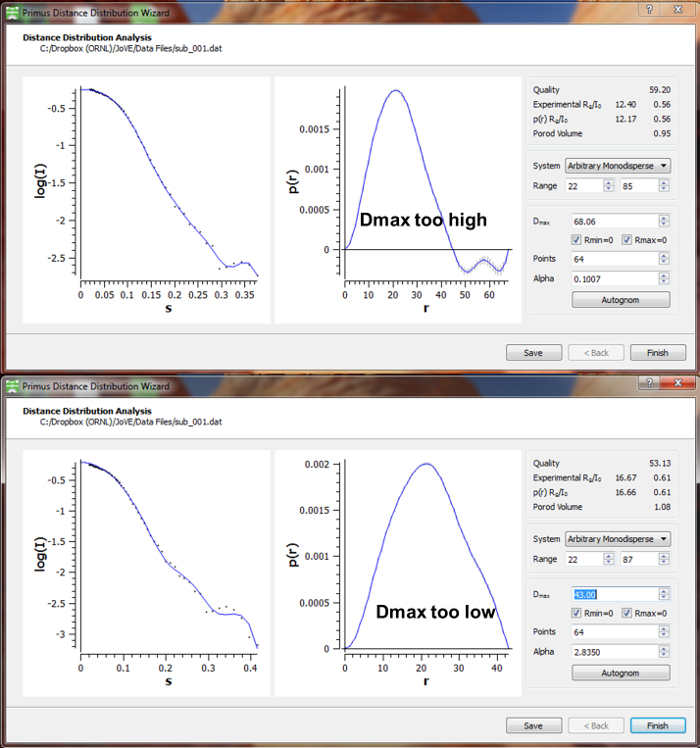
Figure 10 provides example usages of the Primus Guinier and Distance Distribution Wizards. A Guinier analysis provides a preliminary estimate of the particles radius of gyration from the low-angle scattering data. As data approach zero, a linear region should be present in the data. Fitting of a line in this region allows the Rg to be determined from the slope and an I0 to be extrapolated from the intensity intercept at Q=0. An upward trend in the data as Q approaches zero indicates aggregation in the sample, while downward trends are usually indicative of interparticle repulsions. These trends are most apparent when the distribution of residuals is non-stochastic. The upper limit of the Guinier fit for globular particles is constrained by Q*Rg < 1.3 ('s' is used in place of Q in the ATSAS software).
The Rg determined for d-MmIAP from this Guinier fit was 15.4 ± 1.1 Å with an estimated I0 given as 0.580 ± 0.001.
The distance distribution wizard allows systematic changes to be made to parameters of the P(r) fit. The P(r) curve is an indirect Fourier transform of the curve fit to the experimental data. Therefore, it is essential to verify that the fit in the left panel accurately reflects the experimental data. For the fit shown in this example, a Dmax of 46 Å was obtained.
Figure 11 illustrates common errors in Dmax selection. A Dmax that is artificially large often causes P(r) values to become negative as the P(r) function oscillates about the x-axis. A Dmax that is artificially small leads to a truncated P(r) curve with an abrupt transition to Rmax=0.
The first steps in the Primus Shape Wizard are identical to the Guinier and Distance Distribution Wizards. Once this information has been provided to obtain good fits to the experimental data, the ab initio model setup is configured.
Figure 12 illustrates a proper input for the Primus Shape Wizard. Here, experimental SANS data for the IAP was provided with a scale in Angstroms, and the expected file prefix of "SANSEnvelope" was given. A set of 17 initial dummy atom models were requested to be generated using "fast" model mode with no symmetry (P1) applied and no anisometry selected. The set of 17 models will be aligned, averaged, and filtered with DAMAVER, and the average model refined using DAMMIN. Inspection of the prefix-damsel.log text document provides a summary of the selection criterion and any models discarded from the set. The final refined model was written as SANSEnvelope-1.pdb.
The program SUPCOMB is used to perform a superimposition of the SANS envelope model with the high-resolution model, with only the two PDB structure files as input. By default, "r" is appended to the reoriented and superimposed file name.
Once the SANS envelope and high-resolution structure have been superimposed, these models can be visualized using any molecular graphics viewing program. The results from PyMOL are suitable for publication quality images and can be used to emphasize biophysical results relating to the structural investigation. Here, we demonstrate a few basic PyMOL operations used to provide 3D representations and views of the resulting superimposed structures.
Figure 13 provides an overview of the PyMOL visualization process. Once the PDB structure files have been opened in PyMOL, the models should be visible in the PyMOL Viewer window. Change the representations of each model using the 'S' button next to each file name. Use a surface representation for the SANS envelope and a cartoon representation of the protein backbone from the high-resolution model. Select a suitable color scheme from options available under the 'C' button. For protein chains, a "chainbow" coloring provides a color gradient from N- to C- terminus to aid interpretation. Transparency is applied to the surface representation to allow better visualization of the protein structure within the envelope. For publication images, a white background is recommended. Once these visualization queues have been applied, the 3D structures can be examined. Manipulations of the perspective and molecule rotation/translation can be performed by clicking and dragging the structure.
Figure 1. Usage of the MULCh Contrast module to determine contrast parameters for components of dodecyl maltoside (DDM). Input values are shown in the screen on the left with the subsequent output to the right. The Contrast module input page is accessed by clicking 'Contrast' (green circle) from the navigation menu on the left side of each screen. Input areas for the project title, buffer components, and subunits 1 and 2 are labelled as such. Output pages contain important particle information and contrast properties for the described system. Relevant tables and calculated match points have been boxed in orange for clarity. Please click here to view a larger version of this figure.
Figure 2. Usage of the MULCh Contrast module to determine contrast parameters for components of the membrane protein-detergent complex. In this instance, input has been given to describe the overall PDC system in buffered solution. Subunit 1 describes the contrast-matched mixed micelle composed of DDM with 43% by mole as d25-DDM. Subunit 2 describes the membrane protein, with the amino acid sequence for MmIAP input as the formula. The deuteration level (green circle) describes the protein's degree of deuterium substitution, and has a direct impact on the SLD and calculated match-point that will be estimated for the protein. Results (orange box) indicate that match-point for the mixed detergent will occur in 48.5% D2O, while the protein's match-point occurs at ~90% D2O. Please click here to view a larger version of this figure.
Figure 3. Sample preparation – Bioreactor trace. The process values displayed are temperature set point (orange line), dissolved oxygen (DO) set point (blue line), agitation set point (red line), and compressed air flow rate (pink line). Pump 1 (green line) was used for pH control. The DO spike at 18:40 signaled depletion of the intial glycerol and was used to initiate feeding of glycerol solution via pump 2 (black line). X-axis denotes hours. Please click here to view a larger version of this figure.
Figure 4. Sample preparation – FPLC and SDS-PAGE characterization of the sample. Final size exclusion chromatogram for d-MmIAP equilibrated with 20 mM HEPES, pH 7.5, 250 mM NaCl, 48.5% D2O and 0.05% total DDM, of which 44% (w/v) is tail-deuterated d25-DDM. Inset: SDS-PAGE analysis with Coomassie staining. Pooled fractions are labeled A, B, and C. Region "B" was used in the SANS experiment. Annotated molecular weight marker is in kDa. Image reproduced with permission from Naing et al.65 Please click here to view a larger version of this figure.
Figure 5. Data collection – Quartz banjo sample cell and autosampler setup. (A) An empty quartz banjo cell is shown resting against the autosampler block. Cells are filled with sample and inserted into one of the 15 available positions. (B) The sample block is mounted behind an aperture selector (not shown) between the Beam Tube Extender and Silicon window at the entrance to the detector tank. Hoses connecting to a temperature-controlled water bath and channels throughout the sample block provide temperature regulation at the sample positions. The arrow indicates the direction of the neutron beam. Please click here to view a larger version of this figure.
Figure 6. Data Collection – Overview of SPICE operations and table scan. The table scan operations are accessible from the 'Table Scan' button on the far left menu of the SPICE instrument control software. Green arrows denote the active tab at the top of the screen and orange boxes highlight areas in the table for active user inputs. (A) The first tab of the table scan is used to provide information about the sample changer and sample cell positions. Provide labels, sample thicknesses, and sample types for each position used in the sample changer. Checking the 'Do Scan' box will add the corresponding row to the table scan queue. (B) On the second tab, details about the instrument configuration and measurement time for each sample (in seconds) are recorded. Instrument configurations are preconfigured by the Neutron Scattering Scientist and provided to users based on details provided in the beam time and instrument proposal. Additional parameters can be added to the instrument control, such as the ability to define temperature setpoints and hold times, throughout the measurement queue. (C) The final tab provides an overview of the measurements to be made for each row in the table. If no changes are needed, the automated scan is initiated by clicking the 'Execute Scans' button. Please click here to view a larger version of this figure.
Figure 7. Data Reduction – Overview of reduction script and MantidPlot operations. MantidPlot software is accessible on the analysis cluster by typing "mantidplot" in a terminal command prompt at any active directory (the yellow circle and arrow are added for emphasis). Once the software is open, access the script window (toggled by button marked in green) and open the script provided by the Neutron Scattering Scientist. Follow the instructions provided in the script, which should only require the scan numbers for each sample and buffer to be entered in a list for automated reduction, as well as scan numbers for the empty cell for subtraction and empty beam for transmission and beam center determination. Please click here to view a larger version of this figure.
Figure 8. Data Analysis – Plotting of data using MantidPlot. Data are referenced as 'Workspaces' in MantidPlot. The workspace area will be populated with filenames as produced by the user reduction script. Workspace data for 1D data sets can be plotted by right-clicking the workspace and selecting "Plot Spectrum with Errors…". Additional data can be added to the current plot by simply dragging and dropping workspaces. Formatting of the plot window (such as for log-log plots) can be performed by selecting 'Preferences' from the 'View' menu bar, or double-clicking on the axis labels. Please click here to view a larger version of this figure.
Figure 9. Data Analysis – ATSAS software: basic operations and buffer subtraction. The primusqt application (SAS Data Analysis) provides visualization for proper background (buffer) subtraction. The buffer file should be scaled for subtraction such that data overlap at high-Q (where scattering is flat as a result of remaining signal from incoherent background). When this high-Q range of data is selected, the Scale operation will apply a scale factor to the lower data file in the table that produces overlap in the defined region. After scaling, the buffer file can be subtracted to yield the net scattering profile of the particle. Please click here to view a larger version of this figure.
Figure 10. Data Analysis – Guinier and P(r) (distance distribution) determination. (A) The Primus Guinier Wizard is used to perform a Guinier analysis, providing an initial estimate of I0 and Rg. The particle type and range of data to fit are used to define the fit. A plot of the fit and corresponding residuals are shown to the left, while Guinier terms (I0 and Rg), Q*Rg limits, and quality of linear fit are provided as output in the area marked by the orange box. (B) The Primus Distance Distribution Wizard is used to perform the distance distribution analysis, providing the P(r) curve which defines the probability of interatomic distances within the scattering particle and includes the Dmax or maximum interatomic distance. Parameters indicated by the orange box can be systematically investigated to determine a proper distance distribution function. Please click here to view a larger version of this figure.
Figure 11. Improper results from the distance distribution analysis. A properly selected Dmax should produce a P(r) curve that peaks with a gradual decay to zero. Dmax values that are too large will likely produce negative P(r) values or oscillations near the x-axis at high values of r. Dmax values that are artificially small often result in P(r) curves where the upper bound appears truncated. Please click here to view a larger version of this figure.
Figure 12. Modeling – Ab initio model setup using the Primus Shape Wizard.Ab initio modeling parameters are provided in a single input window. A prefix is supplied for output file names with other options for processing available. Annealing procedure can be fast (bigger beads with faster cooling) or slow (smaller beads with slower cooling). Number of repetitions should be of sufficient size to examine reproducibility of model features. Particle symmetry and anisometry can be provided, if known. Angular scale should correspond to the units of the measured data. DAMAVER averaging will align and average all dummy atom models, and then apply a selection criterion which excludes any outlier models. A core of fixed atoms from the averaged model will be further refined using DAMMIN if this option is selected. This refined model should represent the 3D low-resolution envelope of the experimental SANS profile. Please click here to view a larger version of this figure.
Figure 13. Visualization – Experimental SANS envelope and overlay with high-resolution model using PyMOL. After opening the PDB structure files in PyMOL, models appear in the PyMOL Viewer. Active models are listed in the table to the right, along with action buttons which can be used to manipulate the model and its representation. Basic operations are provided for visualizing these models which allow regions of agreement (or mismatch) between the SANS envelope and high-resolution structure to be identified. Please click here to view a larger version of this figure.
Table 1. Physical properties of detergents commonly used in membrane protein investigations. Please click here to download this file.
Discussion
Structural biology researchers take advantage of complementary structural techniques like solution scattering to obtain biochemical and structural details (such as overall size and shape) from biomolecules in solution. SANS is a particularly attractive technique for determining low resolution structures of membrane proteins, a core focus of modern structural biology and biochemistry. SANS requires quantities of purified proteins comparable to those of crystallographic trials (1 mg/sample). The recently expanding commercial availability of high-purity deuterated detergents relevant to membrane protein studies makes accessible a means to manipulate this hydrogen/deuterium content for SANS experiments of membrane protein-detergent complexes, allowing the protein signal to be recorded directly. When successful, an ab initio model molecular envelope can be calculated from data collected over just several hours. Thus, membrane protein biochemists, biophysicists, and structural biologists can readily take advantage of SANS to obtain coveted initial 3D models of membrane proteins in solution, structures in complex with binding partners or substrates, and models obtained by SANS can be used for phasing diffraction data. Routine usage of SANS to characterize membrane proteins would be transformative for the membrane protein biochemistry field and have ripple effects from basic structure function to drug discovery and development. The added advantage of neutron contrast matching with deuterium substitution makes SANS a valuable technique for studying protein-protein, protein-DNA, or other biomolecular complexes, which can be readily manipulated in their hydrogen/deuterium content.
For additional details about small-angle scattering instrument design and theory, the following reviews are recommended: The Bio-SANS instrument at the High Flux Isotope Reactor of Oak Ridge National Laboratory,79 Small-angle scattering for structural biology – expanding the frontier while avoiding the pitfalls,72 and Small-angle scattering studies of biological macromolecules in solution.80 The Neutron Scattering Scientist can also discuss current instrument configurations and the optimal parameters for a given system. For instance, data at lower Q (which is a function of the scattering angle and neutron wavelength) is generally desired for larger systems. The most common instrument configuration at Bio-SANS provides a Qmin of 0.003 Å-1 and is suitable for larger protein complexes up to hundreds of kDa. A solution containing ~3 mg mL-1 of membrane protein of approximate size of 30 kDa (monomer) in contrast-matched micelles with 48.5% D2O in solution requires a typical measurement time around 8 hours each for sample, buffer, and empty cell (24 hours = 1day total). Instrument measurement times at a given contrast condition can be approximately scaled according to the product of the scattering particle's molecular mass and concentration. However, scattering particles must be measured under non-interacting conditions, including any non-specific protein aggregation, which places a practical upper limit on the concentration of the sample. Hour-long measurements place limits on the stability of certain proteins. Fortunately, SANS measurements can be done routinely at refrigerated temperatures to improve protein life time, and the employed beam of low-energy neutrons does not cause any radiation damage during the measurement.
An overview of this process and determination of many key factors toward the successful recording of the neutron scattering from a sample measured at the contrast match point of the detergent are presented in this manuscript. This includes the critical step toward obtaining a complete match of the aggregate detergent by designing a detergent mixture with a uniform neutron contrast between the detergent head group and alkyl chain components. Measurements at this single detergent match point provide a scattering profile attributed to only the membrane protein of interest and allowed an ab initio envelope representing the membrane protein to be reconstructed from the data. The data analysis and ab initio modeling protocols also demonstrate the potential information obtained from such investigations, which could aim to address overall structure, conformational changes, oligomeric states, among others. One limitation is that to date only very few detergents are commercially available with deuterium substitutions.19
While perdeuteration may not be necessary, the aim in this case should be to achieve a protein with >65% deuterium labeling in the protein. Alternatively, if detergent with selectively-deuterated head groups and tails is available, and the CMP of the detergent micelle occurs near 100% D2O, then sufficient contrast from the protein can be achieved without the need for deuterium labeling. Determine the appropriate deuteration level of the protein to achieve sufficient scattering when measured at the contrast-match point of the detergent.There are numerous challenges associated with membrane protein expression and purification,81 which remain beyond the scope of this article. Unfortunately, high levels of deuteration are currently not (yet) possible for eukaryotic systems. While we realize this is a limitation for some eukaryotic proteins, most membrane proteins have bacterial orthologs for which this method is suitable.
Disclosures
The authors Volker S. Urban, Sai Venkatesh Pingali, Kevin L. Weiss, and Ryan C. Oliver are contracted through UT-Battelle with US DOE to support the Neutron Science User Program and Bio-SANS instrument at Oak Ridge National Laboratory used in this Article.
This manuscript has been co-authored by UT-Battelle, LLC, under contract DE-AC05-00OR22725 with the US Department of Energy (DOE). The US government retains and the publisher, by accepting the article for publication, acknowledges that the US government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for US government purposes. DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan.82
Acknowledgments
The Office of Biological and Environmental Research supported research at ORNL's Center for Structural Molecular Biology (CSMB) and Bio-SANS using facilities supported by the Scientific User Facilities Division, Office of Basic Energy Sciences, US Department of Energy. Structural work on membrane proteins in the Lieberman lab has been supported by NIH (DK091357, GM095638) and NSF (0845445).
References
- Wallin E, Heijne GV. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Science. 1998;7(4):1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautermann CS. GPCR structures in drug design, emerging opportunities with new structures. Bioorganic & Medicinal Chemistry Letters. 2014;24(17):4073–4079. doi: 10.1016/j.bmcl.2014.07.009. [DOI] [PubMed] [Google Scholar]
- Cournia Z, et al. Membrane protein structure, function, and dynamics: A perspective from experiments and theory. The Journal of Membrane Biology. 2015;248(4):611–640. doi: 10.1007/s00232-015-9802-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerr A. Membrane protein structures. Nature Methods. 2008;6:35. [Google Scholar]
- Bill RM, et al. Overcoming barriers to membrane protein structure determination. Nature Biotechnology. 2011;29:335. doi: 10.1038/nbt.1833. [DOI] [PubMed] [Google Scholar]
- Carpenter EP, Beis K, Cameron AD, Iwata S. Overcoming the challenges of membrane protein crystallography. Current Opinion in Structural Biology. 2008;18(5):581–586. doi: 10.1016/j.sbi.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders CR, Sönnichsen F. Solution NMR of membrane proteins: Practice and challenges. Magnetic Resonance in Chemistry. 2006;44(S1):S24–S40. doi: 10.1002/mrc.1816. [DOI] [PubMed] [Google Scholar]
- Garavito RM, Ferguson-Miller S. Detergents as tools in membrane biochemistry. Journal of Biological Chemistry. 2001;276(35):32403–32406. doi: 10.1074/jbc.R100031200. [DOI] [PubMed] [Google Scholar]
- Privé GG. Detergents for the stabilization and crystallization of membrane proteins. Methods. 2007;41(4):388–397. doi: 10.1016/j.ymeth.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Wessel D, Flügge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Analytical Biochemistry. 1984;138(1):141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- Bayburt TH, Grinkova YV, Sligar SG. Self-assembly of discoidal phospholipid bilayer nanoparticles with membrane scaffold proteins. Nano Letters. 2002;2(8):853–856. [Google Scholar]
- Bayburt TH, Sligar SG. Membrane protein assembly into Nanodiscs. FEBS Letters. 2010;584(9):1721–1727. doi: 10.1016/j.febslet.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skar-Gislinge N, et al. Elliptical structure of phospholipid bilayer nanodiscs encapsulated by scaffold proteins: Casting the roles of the lipids and the protein. Journal of the American Chemical Society. 2010;132(39):13713–13722. doi: 10.1021/ja1030613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders CR, Schwonek JP. Characterization of magnetically orientable bilayers in mixtures of dihexanoylphosphatidylcholine and dimyristoylphosphatidylcholine by solid-state NMR. Biochemistry. 1992;31(37):8898–8905. doi: 10.1021/bi00152a029. [DOI] [PubMed] [Google Scholar]
- Vestergaard M, Kraft JF, Vosegaard T, Thøgersen L, Schiøtt B. Bicelles and other membrane mimics: comparison of structure, properties, and dynamics from MD simulations. The Journal of Physical Chemistry B. 2015;119(52):15831–15843. doi: 10.1021/acs.jpcb.5b08463. [DOI] [PubMed] [Google Scholar]
- Popot J-L, et al. Amphipols From A to Z. Annual Review of Biophysics. 2011;40(1):379–408. doi: 10.1146/annurev-biophys-042910-155219. [DOI] [PubMed] [Google Scholar]
- Tribet C, Audebert R, Popot J-L. Amphipols: Polymers that keep membrane proteins soluble in aqueous solutions. Proceedings of the National Academy of Sciences. 1996;93(26):15047. doi: 10.1073/pnas.93.26.15047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández C, Wüthrich K. NMR solution structure determination of membrane proteins reconstituted in detergent micelles. FEBS Letters. 2003;555(1):144–150. doi: 10.1016/s0014-5793(03)01155-4. [DOI] [PubMed] [Google Scholar]
- Hiruma-Shimizu K, Shimizu H, Thompson GS, Kalverda AP, Patching SG. Deuterated detergents for structural and functional studies of membrane proteins: Properties, chemical synthesis and applications. Molecular Membrane Biology. 2015;32(5-8):139–155. doi: 10.3109/09687688.2015.1125536. [DOI] [PubMed] [Google Scholar]
- Krueger-Koplin RD, et al. An evaluation of detergents for NMR structural studies of membrane proteins. Journal of Biomolecular NMR. 2004;28(1):43–57. doi: 10.1023/B:JNMR.0000012875.80898.8f. [DOI] [PubMed] [Google Scholar]
- Linke D. In: Methods in Enzymology. Burgess RR, Deutscher MP, editors. Vol. 463. Academic Press; 2009. pp. 603–617. [Google Scholar]
- Oliver RC, et al. Tuning micelle dimensions and properties with binary surfactant mixtures. Langmuir. 2014;30(44):13353–13361. doi: 10.1021/la503458n. [DOI] [PubMed] [Google Scholar]
- Orwick-Rydmark M, Arnold T, Linke D. The use of detergents to purify membrane proteins. Current Protocols in Protein Science. 2016;84(1) doi: 10.1002/0471140864.ps0408s84. [DOI] [PubMed] [Google Scholar]
- Tanford C, Reynolds JA. Characterization of membrane proteins in detergent solutions. Biochimica et Biophysica Acta (BBA) - Reviews on Biomembranes. 1976;457(2):133–170. doi: 10.1016/0304-4157(76)90009-5. [DOI] [PubMed] [Google Scholar]
- Tulumello DV, Deber CM. Efficiency of detergents at maintaining membrane protein structures in their biologically relevant forms. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2012;1818(5):1351–1358. doi: 10.1016/j.bbamem.2012.01.013. [DOI] [PubMed] [Google Scholar]
- Arachea BT, et al. Detergent selection for enhanced extraction of membrane proteins. Protein Expression and Purification. 2012;86(1):12–20. doi: 10.1016/j.pep.2012.08.016. [DOI] [PubMed] [Google Scholar]
- Seddon AM, Curnow P, Booth PJ. Membrane proteins, lipids and detergents: Not just a soap opera. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2004;1666(1):105–117. doi: 10.1016/j.bbamem.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Tanford C. The Hydrophobic Effect: Formation of Micelles and Biological Membranes. 2nd edn. John Wiley; 1980. [Google Scholar]
- Littrell K, Urban V, Tiede D, Thiyagarajan P. Solution structure of detergent micelles at conditions relevant to membrane protein crystallization. Journal of Applied Crystallography. 2000;33(3 Part 1):577–581. [Google Scholar]
- Oliver RC, et al. Dependence of micelle size and shape on detergent alkyl chain length and head group. PLOS ONE. 2013;8(5):e62488. doi: 10.1371/journal.pone.0062488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maire M, Champeil P, Møller JV. Interaction of membrane proteins and lipids with solubilizing detergents. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2000;1508(1):86–111. doi: 10.1016/s0304-4157(00)00010-1. [DOI] [PubMed] [Google Scholar]
- Hong X, Weng Y-X, Li M. Determination of the topological shape of integral membrane protein light-harvesting complex LH2 from photosynthetic bacteria in the detergent solution by small-angle X-ray scattering. Biophysical Journal. 2004;86(2):1082–1088. doi: 10.1016/S0006-3495(04)74183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinothkumar KR. Membrane protein structures without crystals, by single particle electron cryomicroscopy. Current Opinion in Structural Biology. 2015;33:103–114. doi: 10.1016/j.sbi.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez J, Koutsioubas A. Memprot: A program to model the detergent corona around a membrane protein based on S-SAXS data. Acta Crystallographica Section D. 2015;71(1):86–93. doi: 10.1107/S1399004714016678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver RC, Pingali SV, Urban VS. Designing mixed detergent micelles for uniform neutron contrast. The Journal of Physical Chemistry Letters. 2017;8(20):5041–5046. doi: 10.1021/acs.jpclett.7b02149. [DOI] [PubMed] [Google Scholar]
- Hiruma-Shimizu K, Kalverda AP, Henderson PJF, Homans SW, Patching SG. Synthesis of uniformly deuterated n-dodecyl-β-D-maltoside (d39-DDM) for solubilization of membrane proteins in TROSY NMR experiments. Journal of Labelled Compounds & Radiopharmaceuticals. 2014;57(14):737–743. doi: 10.1002/jlcr.3249. [DOI] [PubMed] [Google Scholar]
- Midtgaard SR, et al. Invisible detergents for structure determination of membrane proteins by small-angle neutron scattering. The FEBS Journal. 2018;285(2):357–371. doi: 10.1111/febs.14345. [DOI] [PubMed] [Google Scholar]
- Gabel F. In: Biological Small Angle Scattering: Techniques, Strategies and Tips. Chaudhuri B, Muñoz IG, Qian S, Urban VS, editors. Singapore: Springer; 2017. pp. 201–214. [Google Scholar]
- Schantz AB, et al. PEE-PEO Block copolymer exchange rate between mixed micelles is detergent and temperature activated. Macromolecules. 2017;50(6):2484–2494. [Google Scholar]
- Liyana-Arachchi TP, et al. Bubble bursting as an aerosol generation mechanism during an oil spill in the deep-sea environment: Molecular dynamics simulations of oil alkanes and dispersants in atmospheric air/salt water interfaces. Environmental Science: Processes & Impacts. 2014;16(1):53–64. doi: 10.1039/c3em00391d. [DOI] [PubMed] [Google Scholar]
- Dos Santos Morais R, et al. Contrast-matched isotropic bicelles: A versatile tool to specifically probe the solution structure of peripheral membrane proteins using SANS. Langmuir. 2017;33(26):6572–6580. doi: 10.1021/acs.langmuir.7b01369. [DOI] [PubMed] [Google Scholar]
- Maric S, et al. Stealth carriers for low-resolution structure determination of membrane proteins in solution. Acta Crystallographica Section D. 2014;70(2):317–328. doi: 10.1107/S1399004713027466. [DOI] [PubMed] [Google Scholar]
- Pedersen JS, Svaneborg C, Almdal K, Hamley IW, Young RN. A small-angle neutron and x-ray contrast variation scattering study of the structure of block copolymer micelles: Corona shape and excluded volume interactions. Macromolecules. 2003;36(2):416–433. [Google Scholar]
- Urban VS. In: Characterization of Materials. Kaufmann EN, editor. John Wiley and Sons; 2012. [Google Scholar]
- Chaudhuri B, Muñoz IG, Qian S, Urban VS, et al. In: Advances in Experimental Medicine and Biology. Cohen IR, editor. Vol. 1009. Singapore: Springer Singapore; 2017. pp. 1–268. [Google Scholar]
- Neutron Facilities WorldWide. 2018. Available from: http://neutronsources.org.
- Submitting a Research Proposal. 2018. Available from: https://neutrons.ornl.gov/users/proposals.
- Accompanying Bio-Deuteration Laboratory Proposal. 2014. Available from: https://www.ornl.gov/sites/default/files/BDL_info_request.docx.
- Whitten AE, Cai S, Trewhella J. MULCh: Modules for the analysis of small-angle neutron contrast variation data from biomolecular assemblies. Journal of Applied Crystallography. 2008;41(1):222–226. [Google Scholar]
- MULCh: ModULes for the Analysis of Contrast Variation Data. 2018. Available from: http://smb-research.smb.usyd.edu.au/NCVWeb/
- Scattering Length Density Calculator by National Institute of Standards and Technology (NIST) Center for Neutron Research. 2018. Available from: https://www.ncnr.nist.gov/resources/activation/
- Ibel K, Stuhrmann HB. Comparison of neutron and X-ray scattering of dilute myoglobin solutions. Journal of Molecular Biology. 1975;93(2):255–265. doi: 10.1016/0022-2836(75)90131-x. [DOI] [PubMed] [Google Scholar]
- Holme T, Arvidson S, Lindholm B, Pavlu B. Enzymes: Laboratory-scale production. Process Biochemistry. 1970;5(9):62–66. [Google Scholar]
- Larsson G, Enfors S-O. Protein release and foaming in Escherichia coli cultures grown in minimal medium. Bioprocess Engineering. 1996;15(5):231–237. [Google Scholar]
- Artero J-B, Hartlein M, McSweeney S, Timmins P. A comparison of refined X-ray structures of hydrogenated and perdeuterated rat [gamma]E-crystallin in H2O and D2O. Acta Crystallographica Section D. 2005;61(11):1541–1549. doi: 10.1107/S0907444905028532. [DOI] [PubMed] [Google Scholar]
- Paliy O, Bloor D, Brockwell D, Gilbert P, Barber J. Improved methods of cultivation and production of deuteriated proteins from E. coli strains grown on fully deuteriated minimal medium. Journal of applied microbiology. 2003;94(4):580–586. doi: 10.1046/j.1365-2672.2003.01866.x. [DOI] [PubMed] [Google Scholar]
- Sivashanmugam A, et al. Practical protocols for production of very high yields of recombinant proteins using Escherichia coli. Protein Science. 2009;18(5):936–948. doi: 10.1002/pro.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopes JT, Elberson MA, Preston RJ, Reddy PT, Kelman Z. In: Methods in Enzymology. Kelman Z, editor. Vol. 565. Academic Press; 2015. pp. 27–44. [DOI] [PubMed] [Google Scholar]
- Leiting B, Marsilio F, O'Connell JF. Predictable deuteration of recombinant proteins expressed in Escherichia coli. Analytical Biochemistry. 1998;265(2):351–355. doi: 10.1006/abio.1998.2904. [DOI] [PubMed] [Google Scholar]
- Perkins SJ. Estimation of deuteration levels in whole cells and cellular proteins by 1H n.m.r. spectroscopy and neutron scattering. Biochemical Journal. 1981;199(1):163–170. doi: 10.1042/bj1990163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obom KM, Magno A, Cummings PJ. Operation of a benchtop bioreactor. Journal of Visualized Experiments. 2013. p. e50582. [DOI] [PMC free article] [PubMed]
- Duff AP, Wilde KL, Rekas A, Lake V, Holden PJ. In: Methods in Enzymology. Kelman Z, editor. Vol. 565. Academic Press; 2015. pp. 3–25. [DOI] [PubMed] [Google Scholar]
- Haertlein M, et al. In: Methods in Enzymology. Kelman Z, editor. Vol. 566. Academic Press; 2016. pp. 113–157. [DOI] [PubMed] [Google Scholar]
- Meilleur F, Weiss KL, Myles DAA. T Micro and Nano Technologies in Bioanalysis. Methods in Molecular Biology. 2009;544:281–292. doi: 10.1007/978-1-59745-483-4_18. [DOI] [PubMed] [Google Scholar]
- Naing S-H, Oliver RC, Weiss KL, Urban VS, Lieberman RL. Solution structure of an intramembrane aspartyl protease via small angle neutron scattering. Biophysical Journal. 2018;114(3):602–608. doi: 10.1016/j.bpj.2017.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Training Requirements for First Time and Repeat Users. 2018. Available from: https://neutrons.ornl.gov/users.
- Remote Analysis Cluster. 2018. Available from: https://analysis.sns.gov.
- Franke D, et al. ATSAS 2.8: A comprehensive data analysis suite for small-angle scattering from macromolecular solutions. Journal of Applied Crystallography. 2017;50(4):1212–1225. doi: 10.1107/S1600576717007786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Software Suite. 2018. Available from: https://www.embl-hamburg.de/biosaxs/download.html.
- Software Individual Programs. 2018. Available from: https://www.embl-hamburg.de/biosaxs/software.html.
- Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: A Windows PC-based system for small-angle scattering data analysis. Journal of Applied Crystallography. 2003;36(5):1277–1282. [Google Scholar]
- Jacques DA, Trewhella J. Small-angle scattering for structural biology-Expanding the frontier while avoiding the pitfalls. Protein Science. 2010;19(4):642–657. doi: 10.1002/pro.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. Journal of Applied Crystallography. 1992;25(4):495–503. [Google Scholar]
- Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Quarterly Reviews of Biophysics. 2007;40(3):191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- Svergun DI, et al. Protein hydration in solution: Experimental observation by x-ray and neutron scattering. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(5):2267–2272. doi: 10.1073/pnas.95.5.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke D, Svergun DI. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. Journal of Applied Crystallography. 2009;42(2):342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun DI. Restoring Low Resolution Structure of Biological Macromolecules from Solution Scattering Using Simulated Annealing. Biophysical Journal. 1999;76(6):2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques DA, Guss JM, Svergun DI, Trewhella J. Publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution. Acta Crystallographica Section D. 2012;68(6):620–626. doi: 10.1107/S0907444912012073. [DOI] [PubMed] [Google Scholar]
- Heller WT, et al. The Bio-SANS instrument at the High Flux Isotope Reactor of Oak Ridge National Laboratory. Journal of Applied Crystallography. 2014;47(4):1238–1246. [Google Scholar]
- Svergun DI, Koch MHJ. Small-angle scattering studies of biological macromolecules in solution. Reports on Progress in Physics. 2003;66(10):1735. [Google Scholar]
- Johnson JL, Kalyoncu S, Lieberman RL. In: Heterologous Expression of Membrane Proteins: Methods and Protocols. Mus-Veteau I, editor. New York: Springer; 2016. pp. 281–301. [Google Scholar]
- Department of Energy Public Access Plan. 2018. Available from: http://energy.gov/downloads/doe-public-access-plan.