

Abstract
Cell cycle analysis in eukaryotes frequently utilizes chromosome morphology, expression and/or localization of gene products required for various phases of the cell cycle, or the incorporation of nucleoside analogs. During S-phase, DNA polymerases incorporate thymidine analogs such as EdU or BrdU into chromosomal DNA, marking the cells for analysis. For C. elegans, the nucleoside analog EdU is fed to the worms during regular culture and is compatible with immunofluorescent techniques. The germline of C. elegans is a powerful model system for the studies of signaling pathways, stem cells, meiosis, and cell cycle because it is transparent, genetically facile, and meiotic prophase and cellular differentiation/gametogenesis occur in a linear assembly-like fashion. These features make EdU a great tool to study dynamic aspects of mitotically cycling cells and germline development. This protocol describes how to successfully prepare EdU bacteria, feed them to wild-type C. elegans hermaphrodites, dissect the hermaphrodite gonad, stain for EdU incorporation into DNA, stain with antibodies to detect various cell cycle and developmental markers, image the gonad and analyze the results. The protocol describes the variations in the method and analysis for the measurement of S-phase index, M-phase index, G2 duration, cell cycle duration, rate of meiotic entry, and rate of meiotic prophase progression. This method can be adapted to study the cell cycle or cell history in other tissues, stages, genetic backgrounds, and physiological conditions.
Keywords: Developmental Biology, Issue 140, C. elegans, EdU, S-phase, Cell cycle, Germline, meiotic S-phase, M-phase, G2-phase
Introduction
In animal development, hundreds, thousands, millions, billions, or even trillions of cell divisions are required to form the adult organism. The cell cycle, the set of cellular events composed of G1 (gap), S (synthesis), G2 (gap), and M (mitosis) define the series of events that are executed each cell division. The cell cycle is dynamic and best appreciated in real time, which can be technically difficult. The techniques presented in this protocol allow one to make the measurements of the phases and timing of the cell cycle from still images.
Labeling with nucleoside analogs such as 5-ethynyl-2'-deoxyuridine (EdU) or 5-bromo-2'-deoxyuridine (BrdU) is the gold standard to identify S-phase in the studies of cell cycle dynamics in the Caenorhabditis elegans (C. elegans) adult hermaphrodite germline1,2,3,4,5. Both EdU and BrdU can be used in nearly any genetic background, as they do not rely on any genetic construct. Visualizing BrdU requires harsh chemical treatment to expose the antigen for anti-BrdU antibody staining, which is often incompatible with the assessment of other cellular markers visualized by co-staining with additional antibodies. By contrast, visualizing EdU occurs by click chemistry under mild conditions and thus is compatible with antibody co-staining6,7.
The specificity of the label is clear, since nuclei only incorporate the thymidine (5-ethynyl-2'- deoxyuridine) analogs into DNA during S-phase. Visualization takes place in fixed tissue. The EdU label is invisible by itself until an azide-containing dye or fluorophore reacts covalently with the alkyne in EdU by copper-catalyzed click chemistry8. EdU labeling can provide immediate information on which nuclei are in S-phase, using a short pulse of labeling. EdU can also provide dynamic information, using pulse-chase or continuous labeling; for example, in a pulse-chase experiment, the label is diluted at each cell division or propagated as nondividing cells progress through development.
The C. elegans hermaphrodite germline is a powerful model system for the studies of signaling pathways, stem cells, meiosis, and cell cycle. The adult germline is a polarized assembly-line with stem cells found at the distal end followed by entry and progression through meiotic prophase, coordinated with the stages of gametogenesis more proximally (Figure 1). At the proximal end, oocytes mature, are ovulated and fertilized and begin embryogenesis in the uterus9,10,11. The ~20 cell-diameter long region near the distal tip cell, which includes the mitotically cycling germline stem, progenitor cells and meiotic S-phase cells but not the cells in meiotic prophase, is called the progenitor zone2,4,9,12. The cell membranes provide incomplete separation between the nuclei in the distal germline, but the progenitor zone cells undergo mitotic cell cycling largely independently. The median mitotic cell cycle duration of germline progenitor zone cells in young adult hermaphrodites is ~6.5 h; G1 phase is brief or absent, and quiescence is not observed1,2,13. Germline stem cell differentiation occurs through essentially direct differentiation and thus lacks transit-amplifying divisions4. During differentiation in the pachytene stage, approximately 4 out of 5 nuclei will not form oocytes but instead undergo apoptosis, acting as nurse cells by donating their cytoplasmic contents to the developing oocyte12,14,15.
In addition to labeling cells in S-phase with nucleoside analogs, one can identify the cells in mitosis and meiosis using antibody staining. Nuclei in mitosis are immunoreactive to anti-phospho-histone H3 (Ser10) antibody (called pH3)7,16. Nuclei in meiosis are immunoreactive to anti-HIM-3 antibody (a meiotic chromosome axis protein)17. Nuclei in the progenitor zone can be identified by the absence of HIM-3, the presence of nucleoplasmic REC-818, or the presence of WAPL-119. WAPL-1 intensity is highest in the somatic gonad, high in the progenitor zone, and low during early meiotic prophases19. Several cell cycle measurements are possible with a few variations in the protocol: I) identify nuclei in S-phase and measure S-phase index; II) Identify nuclei in M-phase and measure the M-phase index; III) determine whether nuclei were in mitotic or meiotic S-phase; IV) measure the duration of G2; V) measure the duartion of G2+M+G1 phases; VI) measure the rate of meiotic entry; VII) estimate the rate of meiotic progression.
One can make multiple cell cycle measurements from only a few types of wet-lab experiments. The protocol below describes a 30 min pulse labeling by feeding C. elegans adult hermaphrodites with EdU labeled bacteria and co-labeling M-phase cells by staining with anti-pH3 antibody and progenitor zone cells by staining with anti-WAPL-1 antibody. Only changes in the duration of EdU feed (Step 2.5), type of antibodies employed (Step 5), and analyses (Step 8.3) are required for the additional measurements.
Protocol
1. Preparation of EdU-labeled Bacteria
- Grow a starter culture of MG1693. Escherichia coli (E. coli) MG1693 carries a mutation in thyA.
- Streak out E. coli MG1693 from a frozen glycerol stock onto a 120 mm lysogeny broth (LB) agar Petri dish. Culture at 37 °C overnight.
- Inoculate from two individual E. coli MG1693 colonies into two duplicate 4 mL tubes of liquid LB. Culture at 37 °C for ~16 h. NOTE: MG1693 grows fine in LB without supplementing with thymine or thymidine.
- Grow E. coli MG1693 in minimal media supplemented with EdU.
- Autoclave a 500 mL conical flask.
- Use sterile technique to add 5 mL of 20% glucose, 50 μL of 10 mg/mL of thiamine, 120 μL of 5 mM thymidine, 100 μL of 1 M MgSO4, 200 μL of 10 mM EdU, 100 mL of M9 buffer, and 4 mL of freshly-grown overnight MG1693 culture. NOTE: The final concentration of EdU is 20 μM in this culture1,7. This concentration leads to DNA damage and cell cycle arrest if applied directly to mammalian cells in culture20. However, only a fraction of this EdU is incorporated into the E. coli and thus available to C. elegans. There is no evidence of cell cycle arrest and no change in the size of the progenitor zone or M-phase index after EdU treatment of young adult hermaphrodites.
- Culture overnight, but no longer than 24 h at 37 °C with shaking at 200 rpm.
- Concentrate EdU-labeled E. coli and apply to M9 agar Petri dishes.
- Pre-cool a tabletop centrifuge to 4 °C.
- Use sterile technique to transfer the culture into 2–4 sterile 50 mL conical tubes.
- Centrifuge the cultures at 3,000 x g at 4 °C for 30 min to pellet the EdU-labeled E. coli. NOTE: Dispose of EdU-containing supernatant according to local and institutional guidelines.
- Resuspend the pellets with 4 mL of fresh M9. Use a sterile 1 mL pipet tip or a sterile 5 mL serological pipet. Resuspending the pellets may take several minutes.
- Use the same pipet to apply and spread ~8 drops of resuspended EdU-labeled E. coli MG1693 to the center of room-temperature 60 mm M9 agar Petri dishes. One batch yields ~16 dishes.
- Allow the dishes to dry for several hours or overnight at room temperature, then seal each dish with a strip of laboratory film. Dishes can be stored at 15 °C for ~2 weeks and at 4 °C for ~2 months. Use the same batch of EdU dishes for each set of experiments.
2. Feeding EdU to C. elegans
Synchronize population by timed egg lay, alkaline hypochlorite treatment followed by hatching into S-medium with cholesterol, or by picking the appropriate stage21. Grow the animals to desired stage (here, 24 h post-L4) on Nematode Growth Medium seeded with E. coli OP50 at 20 °C. NOTE: Prepare 50–100 animals per experiment, as some may not dissect well, and others will be lost in the process of washing and transferring.
Allow the EdU dishes to warm to 20 °C (or the temperature required for the experiment).
Wash the animals from NGM dishes using M9 or phosphate-buffered saline (PBS) into a 1.5 mL tube. Allow the animals to settle briefly by gravity or a brief spin in a microcentrifuge.
Remove the supernatant and wash the animals 1–2 times with ~1 mL of M9 or PBS. Remove the supernatant.
Use a glass Pasteur pipette to transfer the animals in a tiny drop of M9 or PBS onto the center of the EdU lawn. Wait a few minutes for the liquid to be absorbed, then incubate at 20 °C for 30 min (for direct S-phase measurement) or longer (to measure history of S-phase), as needed. NOTE: EdU signal is detectable in germline nuclei after as little as 15 min of EdU feeding1.
Wash the worms off the EdU dish with ~2 mL of M9 or PBS into a glass dissecting dish.
3. Dissection and Fixation of C. elegans Germline
NOTE: This protocol for the dissection, fixation, and antibody staining of the C. elegans hermaphrodite germline is nearly identical to that published by Gervaise and Arur (2016)22, except that the 1 mL glass tubes can be centrifuged to speed up washing steps and a drawn-out glass Pasteur pipette can be used to remove the liquid from 1 mL glass tubes more effectively.
- Wash and dissect C. elegans germlines.
- Allow the animals to settle to the bottom of the dissecting dish, swirl to collect the animals in the center, and use a long Pasteur pipette to remove PBS. Wash 1–2 times with ~2 mL of PBS.
- Add 2 mL of PBS and 4 μL of 100 mM levamisole to immobilize the animals. Swirl the dish again to collect the animals in the center of the dish. NOTE: Immobilization can take between a few seconds and a few minutes. Complete immobilization is not necessary for successful dissection. Some people have better success when dissecting incompletely immobilized animals.
- Dissect the animals with a pair of 25G 5/8” needles by cutting at the head (approximately between the two pharyngeal bulbs) or the tail. Take care not to cut the loop of the germline. Intestine and germline should “pop out” of the body cavity due to internal pressure, but remain attached. This protocol is similar to previously published Gervaise and Arur (2016)22. NOTE: Keep the dissection time to ~5 min, certainly no more than 15 min. Longer dissection times may result in the loss of antibody staining signal (Sudhir Nayak, personal communication) and starvation in PBS may affect the animals’ physiology. Learning to dissect quickly and accurately may take some practice.
- If needed, swirl to collect the dissected animals in the center, and use a long Pasteur pipette to remove as much PBS as possible.
- Fix and dehydrate tissues
- Add 2 mL of 3% paraformaldehyde (PFA) in PBS solution. Cover the dish loosely with laboratory film and store on a bench or in a drawer for 10 min. NOTE: Thaw PFA solution in a 37 °C water bath and then cool to room temperature prior to dissecting germlines. CAUTION: PFA solution is moderately toxic and a probable carcinogen and teratogen. Vapors emitting from paraformaldehyde solutions are flammable. Wear nitrile gloves. Dilute PFA from 16% to 3% in a chemical fume hood. When working outside of fume hood, keep all containers covered.
- Transfer the gonads carefully to a clean 5 mL glass centrifuge tube.
- Add ~3 mL of PBSTw (PBS with 0.1% Tween-20) to the dish that contained the gonads to help retrieve remaining gonads and to dilute the PFA solution.
- Spin down in a clinical centrifuge at 870 x g for ~1 min. Younger or smaller animals require longer spin times than older or larger animals.
- Using a long glass pipette, transfer the supernatant to unwanted material beaker for eventual discard in unwanted material bottle in the chemical hood.
- Add 2 mL of high-grade methanol pre-chilled to -20 °C. Cover the centrifuge tube tightly with laboratory film. NOTE: Use of fresh high-grade “gold label” methanol is essential for proper morphology with certain antibodies. CAUTION: Methanol is a highly flammable liquid and vapor, that is toxic if swallowed, inhaled, or allowed to contact skin. Wear gloves and appropriate personal protective equipment. Use a freezer appropriate for small volumes of flammables.
- Store in -20 °C freezer for 1 h, even overnight or even several months. NOTE: The protocol can be paused here.
4. Rehydrate Germlines
Fill the glass centrifuge tube to top with PBSTw to dilute the methanol and rehydrate the gonads. Spin down in a clinical centrifuge at 870 x g for ~1 min.
Using a long glass Pasteur pipette, transfer the supernatant to unwanted material beaker for eventual discard in unwanted material bottle in the chemical hood.
Wash the gonads 3 times using ~5 mL of PBSTw, spinning down in a clinical centrifuge at 870 x g for ~1 min each time. Remove the supernatant.
Rinse a small 1 mL borosilicate glass tube and a long glass Pasteur pipette with PBSTw.
Add ~700 μL of PBSTw and use the long glass Pasteur pipette to transfer the gonads to the small tube. Use a few additional drops of PBSTw to ensure that all gonads are transferred.
Spin down in a clinical centrifuge at 870 x g for ~1 min. Using a drawn-out long glass Pasteur pipette, remove as much liquid as possible without disturbing the gonads. Leave no more than 50 μL. NOTE: If antibody detection is not necessary, skip to Step 6.
5. Detect Antigens with Antibodies
Dilute the primary antibodies in 30% serum in PBS. In the present example, anti-WAPL-1 antibody is diluted 1:2,000 and anti-pH3 antibody is diluted 1:500. Centrifuge freshly thawed serum for 10 min in a microfuge at 20,000 x g at 4 °C to remove the particulates. Use the supernatant, which can be stored at 4 °C for several days. Use the appropriate serum to match the host organism of secondary antibodies (goat serum is used here). An optional blocking step may be added prior to the addition of primary antibodies.
Apply 100 μL of diluted primary antibody to each small glass tube. Incubate at room temperature for 4 h. NOTE: Incubation times vary by antibody. For some antibodies, 2 h at room temperature is sufficient. Longer incubations (e.g., overnight) are possible, but may increase background.
Fill the tubes to top with PBSTw and spin down in a clinical centrifuge at 870 x g for ~1 min.
Wash the gonads 3 times using ~1 mL of PBSTw. Incubate for ~5 min per wash to allow excess primary antibody to diffuse into wash. Using a drawn-out long glass Pasteur pipette, remove as much liquid as possible without disturbing the gonads. Leave no more than 50 μL.
Dilute the secondary antibodies in 30% goat serum in PBS. In the present example, goat-anti-rabbit-594 and goat-anti-mouse-647 are each diluted at 1:400. NOTE: Select secondary antibodies carefully to make sure dyes are distinct from the dye in the EdU kit. In the present example, the EdU kit contained a 488 nm excitation dye.
Apply 100 μL of diluted secondary antibody to each small glass tube. Incubate in the dark at room temperature 2 h. NOTE: Incubation times vary by antibody. For some secondary antibodies, 1 h at room temperature is sufficient. Longer incubations (e.g., overnight) are possible, but may increase background.
Wash the gonads 3 times using ~1 mL of PBSTw. Incubate for ~5 min per wash to allow excess secondary antibody to diffuse into wash. Using a drawn-out long glass Pasteur pipette, remove as much liquid as possible without disturbing the gonads. Leave no more than 50 μL. NOTE: Gonads can be stored in 100 μL of PBS after this step, if necessary, although this may reduce signal. Remove PBS prior to proceeding.
6. Perform the EdU Click Reaction to Detect EdU
NOTE: Performing the EdU click reaction before the antibody staining steps (perform Step 6 before Step 5) is possible, depending on the antibodies used7. However, the click reagents may interfere with certain antigens (e.g., REC-8 antibody is sensitive to fixation and permeabilization). The order presented here yields bright antibody staining with the REC-8, WAPL-1, HIM-3, pH3, FLAG, and CYE-1 antibodies used, among others.
- Prepare the click EdU cocktail8 fresh by adding the following to a clean 1.5 mL tube. The order of additions is important. Protect from light and maintain all reagents on ice. This recipe yields enough for one sample (100 μL); multiply the recipe as needed.
- Add 2 mL of ultrapure water to the buffer additive. This makes 10x buffer additive, which must be diluted to 1x immediately prior to use.
- Add 8.5 μL of 10x buffer to 76.5 μL of ultrapure water. Mix well.
- Add 4 μL of 100 mM CuSO4 (may be labeled as Component E). Mix well.
- Add 0.25 μL of the 488 nm dye Azide. It must be thawed at room temperature, as its solvent, dimethyl sulfoxide, is solid at 4 °C. Mix well and protect from light.
- Mix 9 μL of ultrapure water with 1 μL of the buffer additive in the cap of the tube. Pipet from the cap to add to the remaining cocktail, and mix well by pipetting up and down.
- Perform the EdU click reaction.
- Add ~100 μL of click EdU cocktail to the gonads in the small tube. Cover with laboratory film and incubate for 30-60 min at room temperature.
- Wash once with 100 μL of reaction rinse buffer.
- Wash the gonads 4 times using ~1 mL of PBSTw. Incubate for ~15 min per wash to allow excess EdU cocktail components to diffuse into wash. Using a drawn-out long glass Pasteur pipette, remove as much liquid as possible without disturbing the gonads. Leave no more than 50 μL.
7. Stain DNA and Prepare Slides
Add 1 drop (~25 μL) of antifade mounting medium with 4’,6-diamidino-2-phenylindole (DAPI, used to visualize DNA) to the gonads. Wait a few minutes so it can settle and mix with the gonads. NOTE: Alternately, a 1:1,000 dilution of DAPI (from a 0.1 mg/mL stock) in PBS may be applied for 5 min, followed by 20 μL of 1,4-diazabicyclo[2.2.2]octane (DABCO) in 90% glycerol, or another antifade mounting medium.
Prepare a large 2.5% agarose pad on a standard glass microscope slide.
Use a new clean dust-free long glass Pasteur pipette to transfer the gonads to the agarose pad. Keep all liquid and gonads in the narrow bottom of the pipette to minimize the loss of gonads. NOTE: Gonads stuck to small glass tube or in long glass Pasteur pipette can be “rescued” by rinsing with PBSTw, collecting the liquid in a dissecting dish, and picking individual animals onto the slide with an eyelash.
Use an eyelash (or a loop of thin hair) glued to a toothpick to distribute the gonads over the agarose pad and remove the dust particles.
Apply a rectangular glass coverslip. Lower slowly from one side to avoid air bubbles. Use a tissue to remove excess solution thus preventing the coverslip from moving freely. NOTE: Choose coverslips that match to the microscope that will be used. #1 and #1.5 coverslips work well.
Allow the slides to settle and dry slightly overnight at room temperature or 4 °C. This helps to slightly flatten the gonads. Slides should be stored at 4 °C.
Optional: Seal the edges of the slide with nail polish, or another slide sealer. Sealing the corners first, then the sides, prevents the coverslip from shifting.
8. Confocal Imaging and Analysis
- Image the distal gonad with a spinning disc confocal fluorescent microscope equipped with a high energy light source, plan-apochromatic objectives, and a high efficiency microscope camera. Capture the images with 1 μm or tighter spacing between z-stacks. Take note of laser power, sensitivity, and exposure time for all channels.
- Use the following: 405 nm laser line excitation with a 485 nm (W60) emission filter for DAPI, 488 nm laser line excitation with a 527 nm (W55) emission filter for EdU, 561 nm laser line excitation with a 615 nm (W70) emission filter for WAPL-1, and a 640 nm laser line excitation with a 705 nm (W90) emission filter for pH3. NOTE: Signal intensity and background intensity will vary. Likewise, the required exposure times will vary, possibly up to 10-fold.
Use the Cell Counter plug-in23 in Fiji24,25 to manually count each nucleus. Label each individual nucleus according to the presence and absence of pH3, EdU, and WAPL-1. Use the classes of nuclei described in Table 1 and Figure 2, as these will facilitate all of the calculations outlined below. NOTE: Skilled experimentalists can accurately count all nuclei in a 3D image without double-counting or missing any nuclei. Alternately, one may count every nucleus in every z-plane, and the Marks-to-Cells R script3 may be used to remove multiply-counted nuclei.
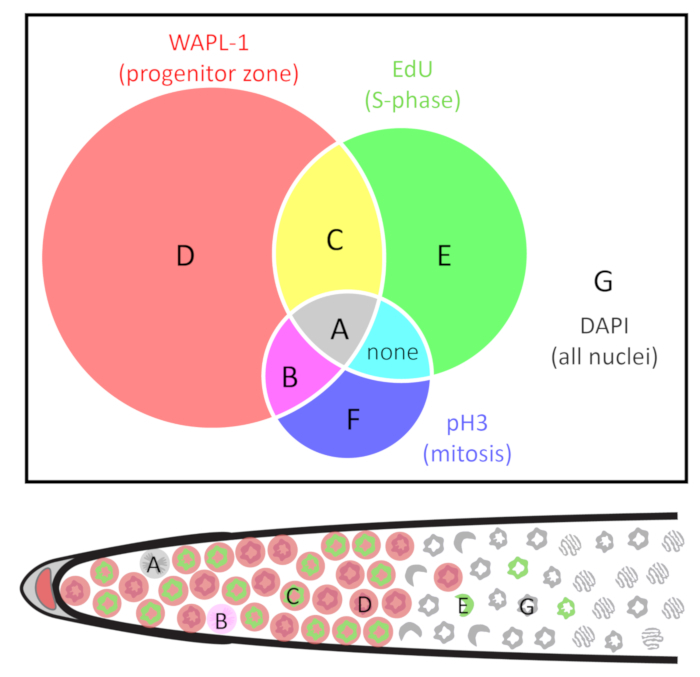
- Calculate cell numbers and frequencies from the above counts depending on the type of cell cycle measurement needed. The types of nuclei are defined in Table 1 and Figure 2. The calculations are summarized in Table 2.
- (Variation I) To identify the nuclei in S-phase, feed the animals EdU for 30 min. Any nuclei displaying EdU label are S-phase nuclei. To calculate, take the sum A and C nuclei, see Table 1 and Figure 2. NOTE: In a 30 min EdU pulse in wild-type adult hermaphrodites, all EdU labeled nuclei are co-labeled with progenitor zone markers1.
- To measure the progenitor zone, stain with a progenitor zone marker such as REC-8 or WAPL-1 antibody. The progenitor zone is defined here as all nucleoplasmic REC-818 or WAPL-1 immunoreactive germline nuclei. To calculate, sum all WAPL-1 immunoreactive nuclei (A+B+C+D, see Table 1 and Figure 2). NOTE: WAPL-1 also labels the DTC and somatic gonad nuclei which should not be counted. Somatic nuclei are easy to identify by extremely intense WAPL-1 signal, position slightly outside the germline, and a “fried-egg” morphology of the nuclei.
- To measure the S-phase index, perform a 30 min EdU experiment and co-label with REC-8 or WAPL-1 antibody. The S-phase index is defined as the proportion of the progenitor zone that is in S-phase. To calculate, count all S-phase nuclei, and then divide by the total number of progenitor zone nuclei (A+C / A+B+C+D, see Table 1 and Figure 2).
- (Variation II) To identify the nuclei in M-phase, stain with pH3 antibody. Any pH3 immunoreactive nuclei are M-phase nuclei. This works regardless of the duration of the EdU feed. To calculate, take the sum of A and B nuclei, see Table 1 and Figure 2. NOTE: WAPL-1 also labels the DTC and somatic gonad nuclei which should not be counted. Somatic nuclei are easy to identify by extremely intense WAPL-1 signal, position slightly outside the germline, and a “fried-egg” morphology of the nuclei.
- To measure the M-phase index, co-label with pH3 and REC-8 or WAPL-1 antibodies. The M-phase index is defined as the proportion of the progenitor zone that is in M-phase. To calculate, count all M-phase nuclei, and then divide by the total number of progenitor zone nuclei (A+B / A+B+C+D, see Table 1 and Figure 2).
- (Variation III) Nuclei in mitotic and meiotic S-phase both label with EdU. To tell the two populations apart, determine whether the S-phase was followed by mitosis or by meiosis. To determine whether nuclei are in mitotic or meiotic S-phase, feed EdU for 4 h and co-label for pH3 (an M-phase marker) and HIM-3 (a meiotic chromosome axis protein) by antibody staining. Record the nuclei that display both EdU and pH3 (type A, see Table 1 and Figure 2) as mitotic S-phase while nuclei that display both EdU and HIM-3 (type E, see Table 1 and Figure 2) as meiotic S-phase.
- (Variation IV) Calculate the duration of G2 phase. NOTE: G2-phase separates S-phase from M-phase. Although no marker has been reported to label G2 in the C. elegans germline, one can calculate the duration of G2 phase by combining data from several experiments that label M-phase (at the time of dissection) and S-phase (starting at several h before dissection). A cell that displays both M-phase and S-phase markers completed G2-phase during the course of the experiment. A cell that displays only the M-phase marker and not the S-phase marker was not in S-phase during the experiment.
- To calculate the duration of G2-phase, feed EdU for 2 h and co-label with pH3 antibody. Examine only nuclei that label with pH3 (these are in M-phase at the time of dissection) for the presence of EdU (these were in S-phase during the 2 h EdU label prior to dissection). Calculate the fraction of M-phase nuclei that completed G2-phase (A / A+B, see Table 1 and Figure 2).
- Repeat this experiment with a 3 h EdU label, and again with a 4 h EdU label (and optionally a 5 h EdU label). Plot the percent of pH3 positive nuclei that are EdU positive on the y-axis and the duration of EdU label on the x-axis, as shown in Figure 3A.
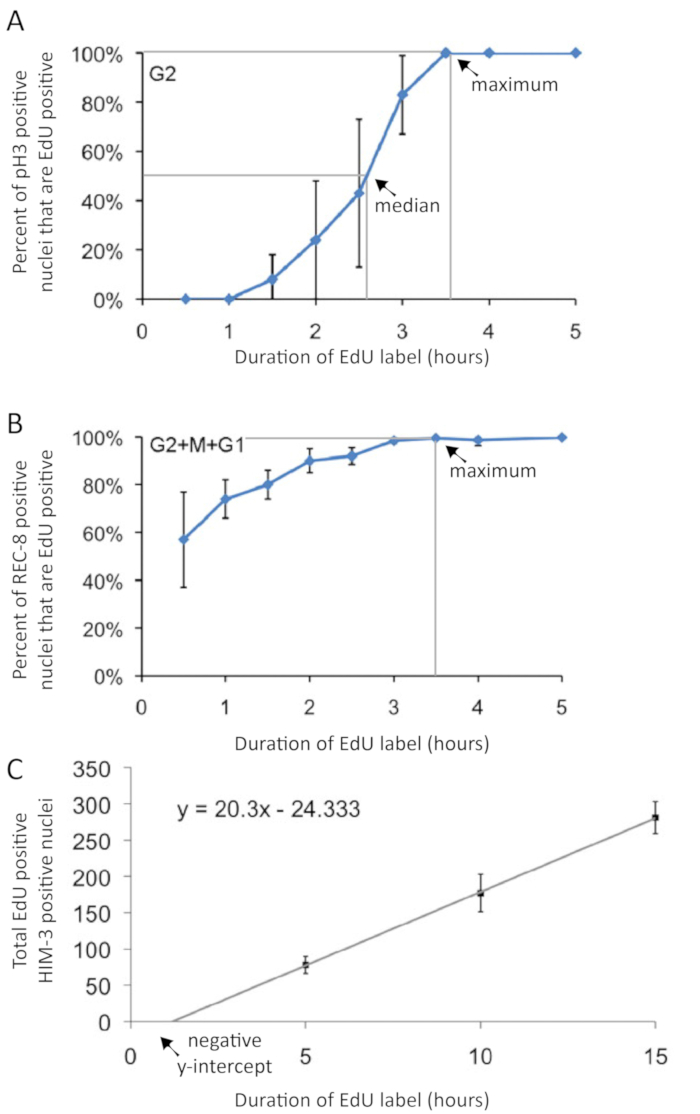
- Calculate the median duration of G2-phase by connecting the points on the graph and determining where the line crosses 50%, as shown in Figure 3A.
- Calculate the maximum duration of G2-phase by connecting the points on the graph and determining where the line crosses 99%, as shown in Figure 3A.
- (Variation V) Calculate the duraion of G2+M+G1. NOTE: In the C. elegans germline, G1 phase is unusually short. Although no marker has been reported to label G1 in the C. elegans germline, one can estimate the sum duration of G2, M, and G1 phase, and then compare this time with the G2-phase time described above. The maximum duration of G2+M+G1 is estimated from the percentage of all progenitor zone nuclei (WAPL-1 immunoreactive) that remain EdU negative (did not undergo S-phase) after EdU labeling for several hours.
- To calculate the duration of G2+M+G1, feed EdU for 2 h and co-label with REC-8 or WAPL-1 antibody. Determine the fraction of the progenitor zone that underwent S-phase during this time (A+C / A+B+C+D, see Table 1 and Figure 2).
- Repeat this experiment with a 3 h EdU label, and again with a 4 h EdU label (and optionally a 5 h EdU label). Plot the percent of REC-8 or WAPL-1 positive nuclei that are EdU positive on the y-axis and the duration of EdU label on the x-axis, as shown in Figure 3B.
- Calculate the maximum duration of G2+M+G1 by connecting the points on the graph and finding where the line crosses 99%, as shown in Figure 3B. NOTE: It is possible to perform the experiments to determine the duration of G2 and G2+M+G1 as a single set of 2, 3, 4, and 5 h EdU experiments by co-labeling with both rabbit-anti-WAPL-1 and mouse-anti-pH3 antibodies.
- (Variation VI) To identify the nuclei that replicated in the progenitor zone but have since entered meiosis, feed the animals EdU for 10 h and co-label with REC-8 or WAPL-1 antibodies. Any nuclei displaying EdU label were in S-phase during those 10 h. Any nuclei that do not display nucleoplasmic REC-8 or WAPL-1 staining were in meiosis. Simply count the nuclei with EdU labeling that do not display labeling with the progenitor zone marker (E, see Table 1). NOTE: Conversely, if gonads are stained for the meiotic prophase marker HIM-3 with anti-HIM-3 antibodies, count the number of nuclei with EdU labeling that are also positive for HIM-3.
- To calculate the rate of meiotic entry, perform the above experiment with a 5 h, 10 h, and 15 h label of EdU. Plot the number of nuclei that entered meiosis on the y-axis and the duration of the EdU label on the x-axis, as shown in Figure 3C. Then use a simple linear regression to calculate the slope (nuclei entered meiosis per h) from y=mx+b. NOTE: It is critical to use a linear regression to calculate the rate of meiotic entry. It would be incorrect to simply divide the number of nuclei that entered meiosis by the duration of the EdU label, because the y-intercept is not zero.
- (Variation VII) Measure the rate of meiotic progression. NOTE: Since EdU is covalently incorporated into DNA, it can be used to track a population of cells through differentiation. The cells that underwent S-phase in the progenitor zone retain the EdU label as they enter into meiosis, progress through meiosis, and undergo oogenesis. A pulse-chase experiment with EdU can be used to measure the rate of meiotic progression.
- Feed EdU-labeled bacteria to the animals for 4 h (the “pulse”). Transfer the animals to unlabeled OP50 bacteria for 48 h (the “chase”), then dissect and co-label with a progenitor zone marker such as REC-8 or WAPL-1 (or a meiotic prophase marker such as HIM-3) if desired.
- When imaging, look for the position of the most proximal EdU-labeled nucleus. The rate of meiotic progression is the distance (in cell diameters from the end of the progenitor zone) traveled by the most proximal EdU labeled nucleus during the 48 h chase.
Representative Results
Since DNA synthesis is required to incorporate EdU, one can conclude that EdU-labeled nuclei underwent S-phase during the EdU-labeling time window. One may interpret the nuclei that label in a 30 min feeding with EdU labeled bacteria as nuclei in S-phase at the time of dissection. Nuclei that label in a longer continuous EdU feeding experiment may have labeled early in the time window and since left S-phase, or may have labeled in the late part of the EdU time window. EdU signal co-localizes with DAPI signal. In some nuclei, EdU signal covers all chromosomes, while in other nuclei EdU signal localizes to 1–2 bright puncta (Figure 4). These puncta are likely the X-chromosome, which replicates late in S-phase13.
Here, the animals were fed with EdU continuously for 30 min and dissected, as described above and in Figure 5. One example of successful EdU staining in a young adult animal and one example of unsuccessful EdU staining in an older adult animal (see below) are shown in Figure 4. EdU signal from a 30 min labeling localizes to approximately half of the nuclei in the progenitor zone (defined by WAPL-1 antibody labeling but approximated by DAPI morphology26,27,28). S-phase index, the proportion of the progenitor zone that is EdU positive, was previously reported at 57 ±5% and as high as 70% in young adults1,2,3. M-phase index is approximately 2–3%1,29. In continuous feeding for 4 h or longer, all nuclei in the progenitor zone label with EdU, and some nuclei that labeled in the progenitor zone have since entered meiotic prophase1.
While the technique works consistently in wild-type young adult animals, a significant fraction of mated 5 day old hermaphrodites (even those containing sperm) failed to label in a 30 min EdU pulse (Figure 4E). However, with a 4 h EdU feeding, nearly all these animals label. Sporadic failure to label in genetic female animals with short pulses of EdU has also been reported30. There may be other situations that result in sporadic failure to label.
One can calculate the duration of the cell cycle by performing several EdU-labeling experiments with pH3 labeling in each. The duration of G2 was estimated by analyzing the percent of nuclei in M-phase (pH3 immunoreactive) that were EdU positive during the time course (Figure 6). This approach gives median and maximum duration of G2 (Figure 3A). The median time was interpolated, showing an approximate G2 duration of 2.5 h in young adult hermaphrodites. The duration of G2+M+G1 was estimated from the percentage of all progenitor zone nuclei (WAPL-1 immunoreactive) that were EdU positive (Figure 6). The G2+M+G1 method provides a maximum duration measure for the combined phases (Figure 3B). The 99th percentile time was interpolated, showing an approximate G2+M+G1 duration of 3.4 h in young adult hermaphrodites. Data from the same experiments were used to calculate the rate of meiotic entry (nuclei per h). The rate is the slope of the linear regression of the number of nuclei that entered meiosis (EdU positive, WAPL-1 negative or HIM-3 positive) over the duration of the EdU label (Figure 3C). The values for wild-type 1 day old adult hermaphrodites are shown in Table 2.
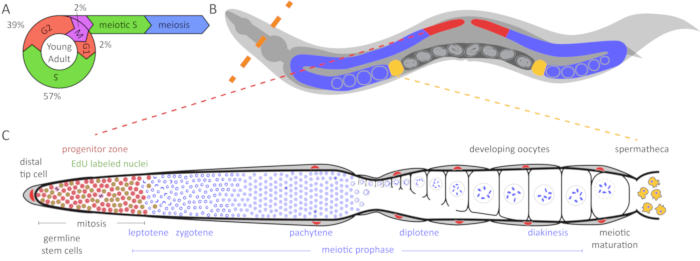
Figure 1: Diagram of C. elegans germline and cell cycle. (A) The cell cycle of germ cells in the young adult hermaphrodite germline. Numbers indicate the approximate percentage of time spent in each cell cycle stage. (B) C. elegans hermaphrodites have two U-shaped germlines (red and blue). The spermatheca is shown in yellow and the uterus with developing embryos is shown in dark gray. The dashed orange line indicates where animals are dissected to extrude the germlines. (C) Diagram of an unfolded C. elegans germline. DAPI (blue) is a DNA dye that highlights nuclear morphology. The distal progenitor zone (highlighted in red based on WAPL-1 antibody staining) contains mitotically cycling stem cells, progenitor cells, and cells in meiotic S-phase (WAPL-1 also labels somatic gonad nuclei). Cells in mitotic and meiotic S-phase label with a 30 min EdU pulse and are indicated in green. Two cells in M-phase label with pH3 antibody and are shown in black. The distal tip cell (DTC) provides the GLP-1/Notch ligand to maintain the stem cell fate of these cells. As the cells migrate away from the DTC, they exit the progenitor zone and enter meiotic prophase. Yellow cells are sperm in the spermatheca. Please click here to view a larger version of this figure.
Figure 2: Venn diagram of the classes of nuclei. Nuclei are grouped by the presence and absence of three markers: WAPL-1 indicates progenitor zone cells (red), EdU indicates S-phase cells (green), and pH3 indicates M-phase cells (blue). Cell types are identified as A-G. Note that in wild-type young adult hermaphrodites cells of type F are not found, and cells do not co-label with EdU and pH3 outside of the (WAPL-1 positive) progenitor zone. The distal gonad diagram below indicates one example of A-E and G nuclei. See Table 1 for more detail. Please click here to view a larger version of this figure.
Figure 3: Graphical presentation of cell cycle duration and rate of meiotic entry experimental data. (A) The duration of G2 phase is interpolated from pH3 and EdU co-labeling following varied-duration EdU pulses Gray lines indicate 50th and 99th percentiles used in interpolating median and maximum G2 durations, indicated by arrows. (B) A cell in G2, M, or G1 phase does not incorporate EdU. Thus, the maximum duration of G2+M+G1 phase can be estimated by measuring the maximum duration of EdU label that yields EdU-negative cells. The duration of G2+M+G1 phase is interpolated from EdU and REC-8 co-labeling following varied-duration EdU pulses. Gray line indicates 99th percentile used in interpolating maximum G2+M+G1 duration, indicated by an arrow. It is not possible to interpolate the median G2+M+G1 duration. (C) The rate of meiotic entry (in nuclei per h – see Table 2) is calculated from the slope of the regression line. Note that since the y-intercept intercept is not zero, a regression is necessary for an accurate calculation of the rate of meiotic entry (C). . Error bars indicate standard deviation. Figures modified and reprinted with permission from Fox et al. 20111. Please click here to view a larger version of this figure.
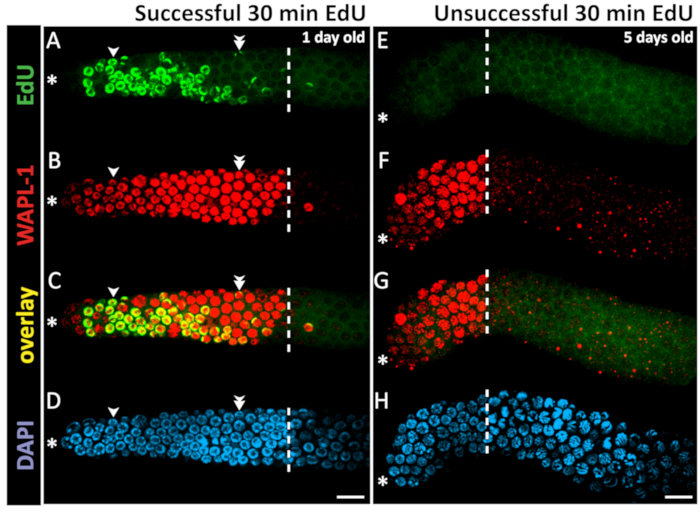
Figure 4: Example of successful and unsuccessful 30 min EdU staining. Confocal microscope images of a 1 day old (A-D) and a 5 day old (E-H) hermaphrodite gonad (not sperm depleted) after a 30 min EdU labeling experiment. The dashed white line marks the end of the progenitor zone. The asterisk marks the position of the distal tip. Green marks EdU staining visualized by click chemistry (A). Unsuccessful EdU labeling results in low-level background staining but no bright EdU+ nuclei (E). Red marks WAPL-1 immunofluorescence (B,F). Yellow indicates overlap (C, G). Blue marks DAPI staining for DNA (D, H). Single arrowheads indicate a nucleus with EdU staining throughout the chromatin. Double arrowheads indicate a nucleus with EdU puncta on only one pair of chromosomes. Images were obtained with a 63X objective. Scale bar = 10 µm (D, H). Please click here to view a larger version of this figure.
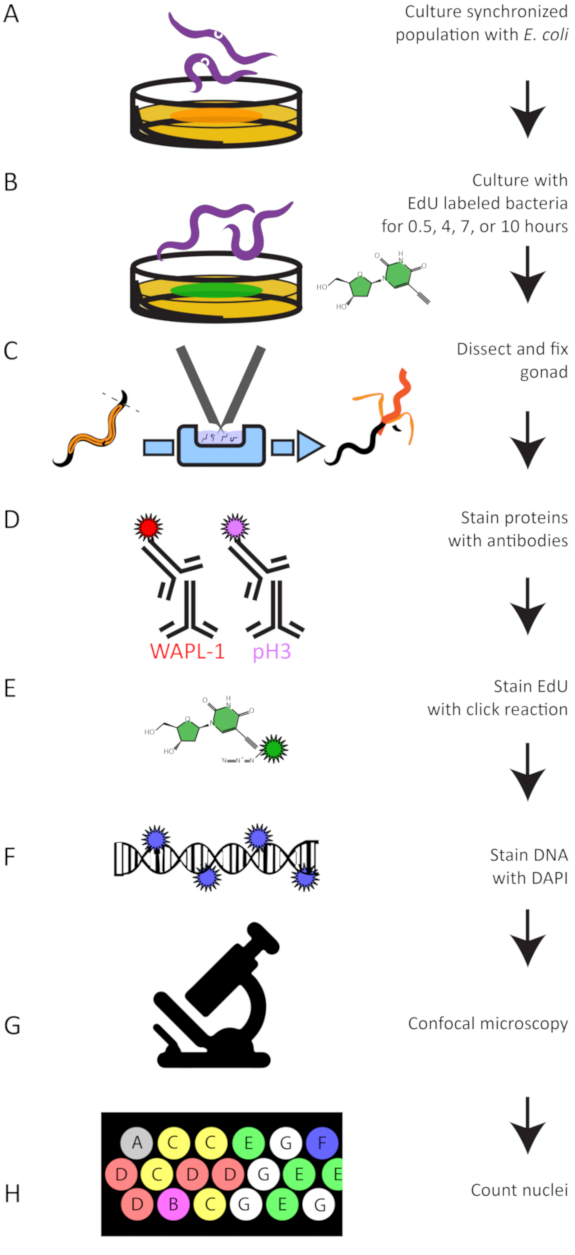
Figure 5: Experimental Workflow. A summary of the experimental protocol to grow (A), EdU label (B), dissect (C), antibody stain (D), perform the click reaction to attach a dye to EdU (E), stain DNA (F), image germlines (G), and quantify EdU labeled and antibody stained nuclei (H). Please click here to view a larger version of this figure.
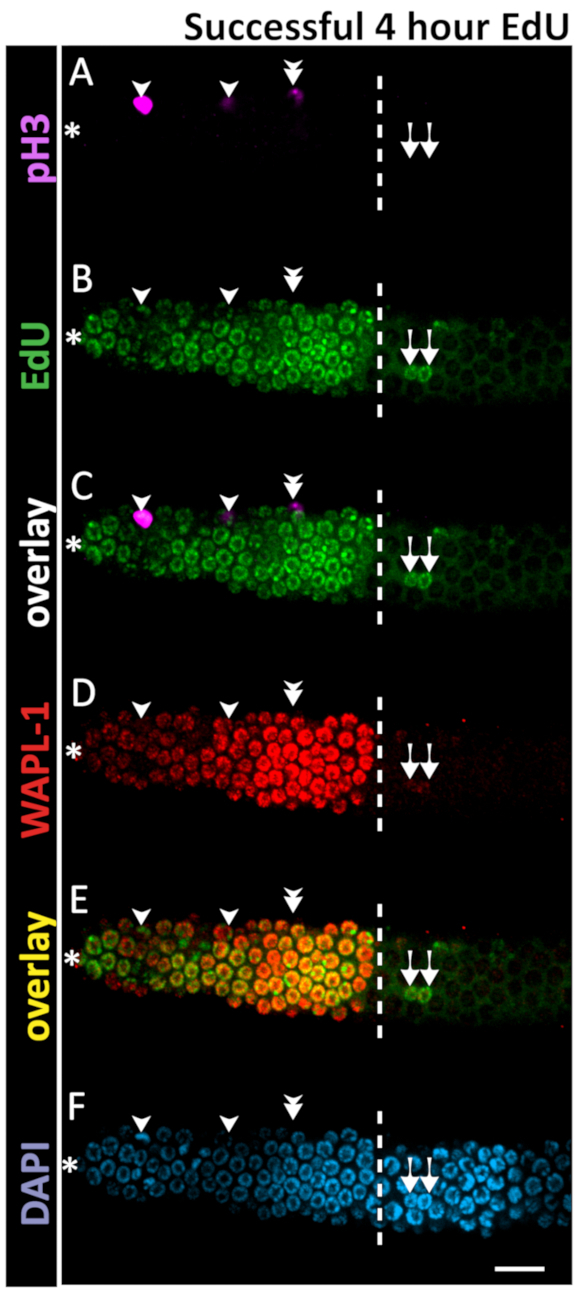
Figure 6: Example of successful 4 h EdU staining. Confocal microscope images of a 1 day old adult hermaphrodite gonad after a 4 h EdU labeling experiment. The dashed white line marks the end of the progenitor zone. The asterisk marks the position of the distal tip. Magenta marks pH3 immunofluorescence (A, C). Green marks EdU staining visualized by click chemistry (B,C). Red marks WAPL-1 immunofluorescence (D). Yellow indicates the overlap of EdU and WAPL-1 (E). Blue marks DAPI staining for DNA (F). Single arrowheads indicate nuclei co-labeled with EdU and pH3. Double arrowhead marks a pH3+ EdU- nucleus - a rare occurrence in a 4 h EdU labeling. Arrows mark EdU+ WAPL-1 - nuclei which have entered meiosis. Images were obtained with a 63X objective. A 10 µm scale bar is shown (F). Please click here to view a larger version of this figure.
| Marker: | pH3 | EdU* | WAPL-1 or REC-8 | HIM-3 | |
| Interpretation: | Mitosis | S-phase* | Progenitor Zone | Meiosis | |
| Class: | Combined Interpretation: | ||||
| A |
|
|
|
|
in M-phase, in progenitor zone, were in mitotic S-phase during EdU label (completed G2) |
| B |
|
|
|
|
in M-phase, in progenitor zone, were not in S-phase during EdU label |
| C |
|
|
|
|
in Interphase, in progenitor zone, were in S-phase during EdU label |
| D |
|
|
|
|
in Interphase, in progenitor zone, were not in S-phase during EdU label |
| E |
|
|
|
|
in meiosis, were in meiotic S-phase during EdU label (meiotic entry nuclei) |
| F |
|
|
|
|
return to mitosis (found in some mutants) or meiotic divisions (in spermatogenesis) |
| G |
|
|
|
|
in meiosis, were not in S-phase during EdU label |
| sum total pH3 positive; all cells in M-phase | sum total EdU positive; all cells in S-phase | sum total WAPL-1 positive; all cells in progenitor zone | sum total HIM-3 positive; all cells in meiotic prophase |
Table 1: Classes of nuclei. *Note that 30 min and 4 h EdU experiments differ in interpretation. In longer duration EdU experiments, cells have likely progressed beyond S-phase. See Introduction and Step 8 for duration of EdU labeling for relevant experiment.
| Cell cycle part | Operational Definition | Calculation* | Value** |
| Progenitor Zone nuclei | all WAPL-1 (or REC-8) positive, HIM-3 negative nuclei | A+B+C+D | 231 ± 23 nuclei |
| S-phase nuclei | nuclei EdU positive after 30 min EdU label and WAPL-1 positive | A+C | 133 ± 20 nuclei |
| M-phase nuclei | pH3 and WAPL-1 co-positive nuclei | A+B | 5.2 ± 2.3 nuclei |
| S-phase index | S-phase nuclei / Progenitor Zone nuclei | A+C/ A+B+C+D | 57% of cell cycle |
| M-phase index | M-phase nuclei / Progenitor Zone nuclei | A+B/ A+B+C+D | 2% of cell cycle |
| Meiotic Entry cells | EdU labeled nuclei in meiosis | E | varies by duration of EdU label |
| Meiotic Entry rate | Meiotic entry nuclei per h of EdU label | Slope from Figure 4C*** | 20.3 nuclei per h |
| G2 duration (median) | 50% intercept from Figure4A | 2.5 h | |
| G2 duration (maximum) | 99% intercept from Figure 4A | 3.5 h | |
| G2+M+G1 duration (maximum) | 99% intercept from Figure 4B | 3.5 h | |
| Cell cycle duration (median) | median G2 duration / G2-index**** | 6.5 h | |
| Cell cycle duration (maximum) | maximum G2 duration / G2-index**** | 8.1 h |
Table 2: Cell cycle calculations. *Letters represent the classes of nuclei defined in Table 1 and Figure 3. Calculations are modified from Fox et al. 20111. **Values (± standard deviation) for wild-type hermaphrodites raised at 20 °C aged to 24 h post mid-L4 stage. ***Note that since the y-intercept intercept is not zero, a regression is necessary for an accurate calculation of the rate of meiotic entry. ****The G2-index is determined by subtracting the S-phase index, M-phase index, and approximate G1-index (2%) from 100%, as described by Fox et al. 20111.
Discussion
Preparation of EdU-labeled bacteria (step 1) is critical for this protocol, and the first point for troubleshooting. Wild-type young adult hermaphrodites label very reliably in a 4 h EdU-pulse, making this a useful control for every new batch of EdU-labeled bacteria. Additionally, intact EdU-labeled bacteria that enter the intestine (in older animals or certain pharynx/grinder defective mutants) will label with click chemistry and appear as bright oblong puncta in the gut. An alternative technique for labeling hermaphrodites uses a “soak” in a high concentration (1 mM) of EdU3. This technique starves the animals for the duration of labeling, but provides a useful way to bypass making EdU-labeled bacteria when troubleshooting fixation and click chemistry. If an EdU “soak” experiment is successful while an EdU feed is not, then prepare fresh EdU-labeled bacteria. To reach a sufficient bacterial density while also achieving a high EdU content, one may need to adjust the concentrations of EdU and thymidine.
The main limitation of this technique for labeling of S-phase is in the need to feed EdU-labeled bacteria to animals. The animals that cannot feed (due to genotype or stage) may not be labeled with this technique. Nevertheless, nucleoside analogs are currently the only method to identify S-phase nuclei in the C. elegans germline, and their use does not require that any transgenes be present in the animals. Additionally, once incorporated, EdU remains in nuclei even as they exit S-phase, progress through the cell cycle, divide, or differentiate. The signal weakens by half with every cell division. This makes EdU perfect for tracking a cell’s history even through a few cell divisions.
The stability of EdU makes pulse-chase experiments straightforward; simply rinse excess EdU bacteria from the animals after the desired duration pulse is finished and transfer the animals to unlabeled bacteria. EdU remains in DNA and remains visible even after multiple cell divisions. However, the experiments are limited to a single type of S-phase label (a single pulse of EdU). Co-labeling with EdU and BrdU is possible in mammalian cells31 but has not been reported in C. elegans. Co-labeling of IdU and CldU is used in mammals32 but also has not been reported in C. elegans.
The main advantages of EdU labeling are that the method requires no transgenes, EdU can be fed to C. elegans during regular culture, the chemistry is compatible with immunofluorescent techniques, and EdU persists in DNA for a long time after feeding has stopped. These features make EdU a great tool to study many aspects of the cell cycle and germ cell dynamics.
Cell cycle and germ cell dynamics analysis with EdU can be applied to a variety of research questions. Just a few examples of further applications of this method: How do the dynamics of the cell cycle change in animals with cell cycle gene mutations? How do physiological conditions affect the cell cycle in stem cells, the rate of germ cell entry into meiotic prophase, and the rate of germ cell progression through meiotic prophase? How does the cell cycle change during larval development? How do major signaling pathway disruptions affect the cell cycle, in addition to changes in cell fate (such as ectopic proliferation)? This system can be modified to study what the cells are doing in many different conditions.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We are grateful to the E. coli stock center for MG1693; Wormbase; the Caenorhabditis Genetics Center which is funded by the National Institutes of Health Office of Research Infrastructure Programs (P40OD010440) for strains; Zach Pincus for statistical advice; Aiping Feng for reagents; Luke Schneider, Andrea Scharf, Sandeep Kumar, and John Brenner for training, advice, support, and helpful discussion; and the Kornfeld and Schedl labs for feedback on this manuscript. This work was supported in part by National Institutes of Health [R01 AG02656106A1 to KK, R01 GM100756 to TS] and a National Science Foundation predoctoral fellowship [DGE-1143954 and DGE-1745038 to ZK]. Neither the National Institutes of Health nor the National Science Foundation had any role in the design of the study, collection, analysis, and interpretation of data, nor in writing the manuscript.
References
- Fox PM, Vought VE, Hanazawa M, Lee M-H, Maine EM, Schedl T. Development. 11. Vol. 138. Cambridge, England: 2011. Cyclin E and CDK-2 regulate proliferative cell fate and cell cycle progression in the C. elegans germline; pp. 2223–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden SL, Leonhard KA, Byrd DT, Kimble J. Cellular analyses of the mitotic region in the Caenorhabditis elegans adult germ line. Molecular biology of the cell. 2006;17(7):3051–3061. doi: 10.1091/mbc.E06-03-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel HS, Kimble J. Cell-cycle quiescence maintains Caenorhabditis elegans germline stem cells independent of GLP-1/Notch. eLife. 2015;4 doi: 10.7554/eLife.10832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox PM, Schedl T. Analysis of Germline Stem Cell Differentiation Following Loss of GLP-1 Notch Activity in Caenorhabditis elegans. Genetics. 2015;201(9):167–184. doi: 10.1534/genetics.115.178061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocsisova Z, Kornfeld K, Schedl T. Cell cycle accumulation of the proliferating cell nuclear antigen PCN-1 transitions from continuous in the adult germline to intermittent in the early embryo of C. elegans. BMC Developmental Biology. 2018;18(1) doi: 10.1186/s12861-018-0171-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proceedings of the National Academy of Sciences. 2008;105(7):2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vanden Heuvel S, Kipreos ETC. elegans Cell Cycle Analysis. Methods in Cell Biology. 2012. pp. 265–294. [DOI] [PubMed]
- ThermoFisher. Click-iT EdU Imaging Kits. 2011. https://assets.thermofisher.com/TFS-Assets/LSG/manuals/mp10338.pdf.
- Pazdernik N, Schedl T. Germ Cell Development in C. elegans. Springer; 2013. pp. 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsh D, Oppenheim D, Klass M. Development of the reproductive system of Caenorhabditis elegans. Developmental Biology. 1976;49(1):200–219. doi: 10.1016/0012-1606(76)90267-0. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77(1):71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen D, Schedl T. Stem cell proliferation versus meiotic fate decision in Caenorhabditis elegans. Advances in Experimental Medicine and Biology. 2013;757:71–99. doi: 10.1007/978-1-4614-4015-4_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo-Lambert A, Ellefson M, Villeneuve AM, Engebrecht J. Differential timing of S phases, X chromosome replication, and meiotic prophase in the C. elegans germ line. Developmental Biology. 2007;308(1):206–221. doi: 10.1016/j.ydbio.2007.05.019. [DOI] [PubMed] [Google Scholar]
- Agarwal I, Farnow C, et al. HOP-1 presenilin deficiency causes a late-onset notch signaling phenotype that affects adult germline function in Caenorhabditis elegans. Genetics. 2018;208(2):745–762. doi: 10.1534/genetics.117.300605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumienny TL, Lambie E, Hartwieg E, Horvitz HR, Hengartner MO. Development. 5. Vol. 126. Cambridge, England: 1999. Genetic control of programmed cell death in the Caenorhabditis elegans hermaphrodite germline; pp. 1011–1022. [DOI] [PubMed] [Google Scholar]
- Hendzel MJ, Wei Y, et al. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106(6):348–360. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- Zetka MC, Kawasaki I, Strome S, Mü Ller F. Synapsis and chiasma formation in Caenorhabditis elegans require HIM-3, a meiotic chromosome core component that functions in chromosome segregation. Genes & development. 1999;13(17):2258–2270. doi: 10.1101/gad.13.17.2258. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC317003/pdf/x8.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen D, Hubbard EJA, Schedl T. Multi-pathway control of the proliferation versus meiotic development decision in the Caenorhabditis elegans germline. Developmental Biology. 2004;268(2):342–357. doi: 10.1016/j.ydbio.2003.12.023. [DOI] [PubMed] [Google Scholar]
- Crawley O, Barroso C, et al. Cohesin-interacting protein WAPL-1 regulates meiotic chromosome structure and cohesion by antagonizing specific cohesin complexes. eLife. 2016;5(2):1–26. doi: 10.7554/eLife.10851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Halicka HD, et al. DNA damage signaling, impairment of cell cycle progression, and apoptosis triggered by 5-ethynyl-2′-deoxyuridine incorporated into DNA. Cytometry Part A. 2013;83(11):979–988. doi: 10.1002/cyto.a.22396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiernagle T. Maintenance of C. elegans. WormBook. 2006. pp. 1–11. [DOI] [PMC free article] [PubMed]
- Gervaise AL, Arur S. Spatial and Temporal Analysis of Active ERK. in the C. elegans Germline. Journal of Visualized Experiments. 2016;117(117):e54901–e54901. doi: 10.3791/54901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos K. De Cell Counter Plugin. 2015. https://imagej.nih.gov/ij/plugins/cell-counter.html.
- Schindelin J, Arganda-Carreras I, et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband W. ImageJ. 2016. http://imagej.nih.gov/ij/>.
- Michaelson D, Korta DZ, Capua Y, Hubbard EJA. Development. 4. Vol. 137. Cambridge, England: 2010. Insulin signaling promotes germline proliferation in C. elegans; pp. 671–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z, Jane E, Hubbard A, Hubbard EJA. Non-autonomous DAF-16/FOXO activity antagonizes age-related loss of C. elegans germline stem/progenitor cells. Nature communications. 2015;6(5):7107. doi: 10.1038/ncomms8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, Kleemann GA, Ashraf JM, Shaw WM, Murphy CT. TGFB and Insulin Signaling Regulate Reproductive Aging via Oocyte and Germline Quality Maintenance. Cell. 2010;143(2):299–312. doi: 10.1016/j.cell.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narbonne P, Maddox PS, Labbe J-C. daf-18/PTEN locally antagonizes insulin signalling to couple germline stem cell proliferation to oocyte needs in C. elegans. Development. 2015. pp. 4230–4241. [DOI] [PMC free article] [PubMed]
- Cinquin A, Chiang M, et al. Intermittent Stem Cell Cycling Balances Self-Renewal and. Senescence of the C. elegans Germ Line. PLoS Genetics. 2016;12(4):1005985. doi: 10.1371/journal.pgen.1005985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Invitrogen EdU (5-ethynyl-2’-deoxyuridine) 2010. pp. 1–7. https://assets.thermofisher.com/TFS-Assets/LSG/manuals/mp10044.pdf.
- Tuttle AH, Rankin MM, et al. Immunofluorescent Detection of Two Thymidine Analogues (CldU and IdU) in Primary Tissue. Journal of Visualized Experiments. 2010;46(46):e2166–e2166. doi: 10.3791/2166. [DOI] [PMC free article] [PubMed] [Google Scholar]